
Electrospun gelatin nanofibers loaded with vitamins A and E as antibacterial wound dressing materials†
Heyu Lia,
Maochun Wanga,
Gareth R. Williamsb,
Junzi Wua,
Xiaozhu Suna,
Yao Lva and
Li-Min Zhu*a
aCollege of Chemistry, Chemical Engineering and Biotechnology, Donghua University, Shanghai, 201620, China. E-mail: lzhu@dhu.edu.cn; Tel: +86 2167792655
bUCL School of Pharmacy, University College London, 29-39 Brunswick Square, London, WC1N 1AX, UK
First published on 17th May 2016
Abstract
Vitamin A palmitate and vitamin E TPGS, common derivatives of the unstable vitamins A and E, were successfully incorporated into biodegradable gelatin nanofibers via electrospinning. Electron microscopy showed that smooth cylindrical fibers were produced, albeit with a small amount of beading visible for the vitamin-loaded systems. The diameters of the fibers decrease with the addition of vitamins. The presence of the vitamins in the fibers was confirmed by IR spectroscopy, and X-ray diffraction showed them to exist in the amorphous physical form post-electrospinning. The addition of vitamins did not affect the hydrophilic properties of the gelatin nanofibers. Fibers containing vitamin A or E alone showed a sustained release profile over more than 60 hours, and those incorporating both vitamins showed similar release characteristics, except that the extent of release for vitamin A was increased. Antibacterial tests demonstrated that materials loaded with vitamin E were effective in inhibiting the growth of E. coli and S. aureus. The fibers could promote the proliferation of fibroblasts during the early stages of culture, and enhance the expression of collagen-specific genes. In vivo tests determined that the fibers loaded with vitamins have better wound healing performance than a commercially used antiseptic gauze and casting films.
Introduction
Contemporary wound dressings are systematically designed to accelerate healing and ensure a pleasing aesthetic result. They maintain a suitable environment for healing, absorb excess exudates, provide thermal insulation, allow gaseous and fluid exchanges, and are nontoxic, nonallergenic, nonsensitizing, sterile, and non-scarring.1,2 Although various types of dressing have been fabricated, including films, foams, hydrogels, hydrocolloids and hydrofibers,3–6 there remains a major unmet need for new formulations with improved properties.Nanofibrous materials have attracted particular attention for wound healing because of their high surface area to volume ratio and porosity, and the great tunability of the systems. The electrospinning technique offers an effective method to fabricate nanofibers. Its products often resemble the morphological structure of the extra-cellular matrix (ECM) and have good biocompatibility.7–9 It has been found that wound dressings produced via this route have good hemostasis, absorbability, and semi-permeability.10,11 Multiple therapeutic compounds can easily be incorporated, leading to facile production of multifunctional dressings. For example, polycaprolactone fibers loaded with ZnO nanoparticles and poly(vinyl alcohol) nanofibers loaded with papain–urea have been prepared by electrospinning and found to have excellent antibacterial activity against common bacteria found on wounds.12–14 Epidermal growth factor and basic fibroblast growth factor have also been embedded into electrospun nanofibers, leading to gradual release of the growth factors and offering the potential for effective wound healing.15,16
It is well-known that vitamins play significant roles in wound healing. Usually, they are applied to the skin in the form of topical creams, lotions, or ointments.17 Vitamin A and E can increase collagen deposition in the repair zone of a wound.18,19 However, they suffer from stability issues. Thus, derivatives have been developed: vitamin A palmitate (VA) is a lipid-soluble substance used for the treatment of skin diseases,20 and offers greater stability than vitamin A.21 In physiological conditions, it can be converted into vitamin A (retinol).22 Vitamin E TPGS (VE) is a water-soluble derivative of vitamin E23 which is hydrolyzed in the intracellular environment releasing α-tocopherol; it has found applications in personal care and cosmetic products.24
Although vitamins have many benefits for skin function, few people have to date used them in the fabrication of wound dressings. In this work, we incorporated VA and VE into gelatin via electrospinning, with the aim of achieving their sustained release. The inclusion of VE should protect the VA from oxidation. In order to determine the functional performance of the vitamin-loaded nanofibers, their antibacterial activity, biocompatibility and promotion of collagen expression were probed in this study. Finally, a series of in vivo experiments was performed to verify the fibers' wound healing potential.
Experiments
Materials
Gelatin powder and glacial acetic acid were purchased from the Sinopharm Chemical Reagent Co., Ltd (China). Vitamin A palmitate and vitamin E TPGS (reagent grade) were obtained from the Aladdin Industrial Corporation (China). L929 cells were provided by the Institute of Biochemistry and Cell Biology (Chinese Academy of Sciences, China). Phosphate-buffered saline (PBS), sodium azide, penicillin, trypsin and thiazolyl blue (MTT) were obtained from Sigma-Aldrich Ltd. (USA). Dimethyl sulfoxide (DMSO) and DMEM culture medium were sourced from Jinuo Biological Medicine Technology Ltd. (China). RT-PCR kits and premix were purchased from Maibio Co. Ltd. (China), and primers were synthesized by Sangon Biotech Ltd. (China). All other chemicals used were analytical grade, and water was doubly distilled before use.Preparation of electrospinning solutions
A 14% (w/v) gelatin solution was first prepared in glacial acetic acid. Subsequently, 10% (w/w) vitamin E TPGS (VE) and 1% (w/w) vitamin A palmitate (VA), with respect to the mass of gelatin, were added (this ratio was selected based on the literature25). A solution of gelatin alone and gelatin solutions containing only 1% VA or 10% VE were prepared as controls. All solutions were stirred magnetically in closed bottles at room temperature for 12 h and degassed with a SK5200H ultrasonicator (350W, Shanghai Jinghong Instrument Co. Ltd., China) for 5 min immediately before electrospinning. The viscosity of the electrospinning solutions was measured using a viscometer (CAP 2000+, Brookfield Co. Ltd., USA). Full details of the solutions prepared are listed in Table 1.| Sample | Amount of gelatin (w/v) | Amount of vitamin A (w/w) | Amount of vitamin E (w/w) | Viscosity (cP) |
|---|---|---|---|---|
| G | 14% | — | — | 214.4 ± 12.3 |
| G/A | 14% | 1% | — | 281.2 ± 10.9 |
| G/E | 14% | — | 10% | 254.4 ± 15.1 |
| G/A + E | 14% | 1% | 10% | 321.4 ± 11.1 |
Preparation of nanofibers
The solution to be used for electrospinning was loaded into a 5 mL syringe fitted with a metal needle (I.D. 0.5 mm) and mounted on a syringe pump (KDS100, Cole-Parmer, USA). The solution was expelled from the syringe at a rate of 0.6 mL h−1 and a high voltage power supply (ZGF-2000, Shanghai Sute Electrical Co. Ltd., China) used to apply a voltage of 16 kV between the needle and a grounded collector (a flat piece of aluminum foil of 10 × 10 cm). The distance between the needle and collector was 15 cm. The relative humidity was ca. 40%, and the temperature 25 °C. After electrospinning for 8 hours, the products were stored in a vacuum oven (DZF-6050, Shanghai Laboratory Instrument Work Co. Ltd., China) at room temperature for 24 h to remove residual solvent. Subsequently, the fibers were crosslinked. 10 mL of a 25% v/v aqueous glutaraldehyde solution was added to a Petri dish and placed at the bottom of a brown desiccator. The fiber mats were placed on a porous ceramic shelf above the Petri dish before the desiccator was sealed and stored at room temperature. This led to the fibers being crosslinked in the glutaraldehyde vapor.26 After 3 days, the samples were removed from the desiccator and stored in a vacuum drying oven at room temperature.27Electron microscopy
The morphological structure of the nanofibers was determined using a scanning electron microscope (SEM; JSM-5600, JOEL, Japan) at a voltage of 10 kV. The average fiber diameter for each sample was calculated by the analysis of around 100 fibers in SEM images, using the Image J software (National Institutes of Health, USA). The fibers were also assessed using a transmission electron microscope (TEM, H-800 instrument, Hitachi, Japan). The TEM was fitted with a field emission electron gun and operated at 200 kV accelerating voltage.Additional fiber characterization
Fourier transform infrared spectroscopy (FTIR) was conducted using a Nicolet-Nexus 670 FTIR spectrometer (Nicolet Instrument Corporation, WI, USA) over the scanning range 500–4000 cm−1 with a resolution of 2 cm−1. X-ray diffraction (XRD) patterns were obtained on a D/Max-BR diffractometer (Rigaku, Japan) with Cu Kα radiation (40 kV/30 mA) over the 2θ range 5–60°.A contact angle system (322W, Thermo Cahn Co., USA) was used for the measurement of water contact angles. Static contact angles were measured at room temperature using the sessile drop method. A 5 μL drop of water was placed onto the dry surface of each sample with a microsyringe and measurements recorded at least 5 times.
Thermogravimetric analysis (TGA) was performed on a TG209F1 thermogravimetric analyzer (TA Instruments Corp., Delaware, USA) from 30 to 900 °C, at a heating rate of 20 °C min−1 and under a nitrogen atmosphere.
In vitro vitamin release
Before the release experiment, the loadings of vitamins in the gelatin fibers were measured. Because the cross-linked fibers cannot be dissolved in glacial acetic acid, three samples of the vitamin-loaded fiber mats (before cross-linking) were cut into circular discs of 5.0 cm in diameter and dissolved in 10 mL of glacial acetic acid to measure the vitamin loadings in the fibers. The amount of dissolved VA and VE was then determined using a UV spectrophotometer (UV-1800, SHJH Company, China) at 330 and 285 nm, respectively. The UV spectra of the pure vitamins are shown in Fig. S1 (ESI†). The amount of vitamin present in the nanofibers was back-calculated against a predetermined calibration curve for each vitamin. The results are reported as mean ± S.D. (n = 3).Vitamin release experiments were conducted at 37 °C and 100 rpm in a thermostatic shaking incubator (Jintan Instrument Co. Ltd., China). Three cross-linked fiber samples (cut into circular discs of 5.0 cm in diameter) were separately immersed in 30 mL of the release medium (pH 7.4 phosphate buffered saline (PBS) with 0.5% (v/v) Tween 80). At specific times, 1 mL of the test medium was withdrawn and an equal amount of fresh preheated PBS was added. The amount of vitamin released was determined using UV spectroscopy. Experiments were carried out in triplicate and the results are reported as mean ± S.D. The cumulative release can be calculated as follows:
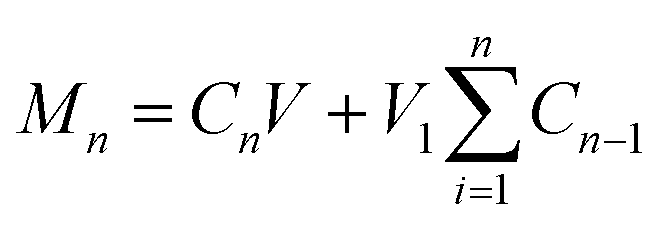
| W = Mn/M × 100% |
Antibacterial activity
The shake flask method was used to examine the antibacterial activity of the cross-linked fibers. E. coli and S. aureus were selected as representative microorganisms, and Luria–Bertani medium used as the culture medium.28 Circular samples (diameter 5 cm) were cut from each fiber mat, placed into a 150 mL Erlenmeyer flask, and cultured at 37 °C with 1.0 mL of a nutrient broth culture containing 1–2 × 105 CFU of bacteria. A negative control was established by undertaking the same experiment with no fibers in the flask. The number of bacteria after incubation for 24 h and 48 h were indirectly measured by optical density at 625 nm in a UV-vis spectrometer (UV-1800, SHJH Company, China), and the antibacterial activity was evaluated quantitatively as follows:| Antibacterial activity = (A − B)/A × 100% |
Cell growth
Vitamin loaded fibers were electrospun directly onto cover slips. 30 cover slips were placed onto the collector, and 5 mL of solution was dispensed at 0.6 mL h−1. The fibers were then cross-linked as detailed above. These slips were placed in the wells of 24-well plates, with an untreated cover slip used as a negative control. Prior to cell culture experiments, the culture plates were sterilized by alcohol steam for 24 h. 200 μL of dissociated L929 fibroblast cells (2.0 × 104 cells per mL, in DMEM supplemented with 10% v/v FBS and 1% v/v penicillin–streptomycin) was added to each well before the plate was placed in an incubator (37 °C, 5% CO2). After 1, 3, or 5 days, the culture medium in each well was replaced by 360 μL of fresh DMEM and 40 μL of MTT solution (5 mg mL−1 thiazolyl blue in PBS). After incubation for 6 h, 400 μL DMSO was added to each well and the plates shaken for 30 min at room temperature. The purple solution in each well was transferred into a 96-well plate and the number of L929 cells assessed via the OD values at 570 nm, measured using a microplate reader (Multiskan, ThermoFisher, USA). Experiments were carried out in triplicate.PT-PCR analysis
After L929 cells seeded on nanofibers were incubated for 5 days (see above), a reverse transcriptase-polymerase chain reaction (RT-PCR) was performed to confirm the level of collagen type I expressed. This was undertaken using a RevertAid First Strand cDNA synthesis kit (Maibio Co. Ltd. China). 1 μg of the RNA templates and the primers (3 μL) were mixed in an RT-PCR premix tube and up to 20 μL of RNase-free distilled water was added. cDNA synthesis was carried out with one cycle at 25 °C for 5 min, 42 °C for 60 min and 70 °C for 5 min. Amplification was performed using 50 cycles of 95 °C for 30 s, 56 °C for 60 s, and 72 °C for 60 s. The primer sequences of collagen type I were 5′-CACCCGACCCACCCTCCCTT-3′ (forward primer) and 5′-AGTGTACCTGAAACCTTTTG-3′ (reverse primer). The PCR primer sequences for a comparator gene, GAPDH, were 5′-GCACCGTCAAGGCTGAGAAC-3′ (forward primer) and 5′-ATGGTGGTGAAGACGCCAGT-3′ (reverse primer). The PCR products were analyzed by 2% agarose gel electrophoresis.In vivo wound healing test
All animal experiments were performed under certificate SYZK 2014-0022 issued by the Shanghai Science and Technology Committee authority, in full accordance with their rules and regulations. Male SD (Sprague Dawley) rats from SLAC Laboratory Animals Inc. (China) weighing from 220–250 g were selected for animal experiments. The animals were anesthetized with an intraperitoneal injection of pentobarbitone sodium (50 mg kg−1) and then a 3 cm × 3 cm (900 mm2) open excision wound was created to the depth of loose subcutaneous tissue on their upper backs.Nanofiber mats and casting films (3 cm × 3 cm) which had been pre-sterilized in alcohol steam for 24 h were used to cover the skin wounds. An antiseptic gauze loaded with ampicillin (Shanghai Yinjing Medical Supplies Company Ltd., China) was placed on each mat/film, and the gauze edge sutured to the skin around the wound area.16 Two control groups of rats were respectively treated with a conventional drug-free gauze and the same gauze soaked with a vitamin solution (1.4% w/v VE and 0.14% w/v VA). As an additional control, a cross-linked gelatin casting film loaded with VA and VE was prepared by pouring the G/A + E electrospinning solution into a mould and allowing the solvent to evaporate in air. The film was then cross-linked using the same glutaraldehyde method as for the fibers, and the film sterilised and sutured using the same procedures as detailed above.
Each group contained seven rats. After recovery from anesthesia, the animals were housed individually in disinfected cages at room temperature. The mats, films and gauzes in all the groups were replaced every three days. On the 0th, 5th, 14th and 21st days post-operation, the appearance of the wound was photographed. The area of unhealed wound was measured from these images using the Photoshop software (Adobe, USA). The relative wound area was determined by the following equation:
| Relative wound area = At/A0 × 100% |
Histological examinations
21 days post-operation, three rats from each group were sacrificed using a large dose of anesthetic. The wound areas were excised, and collected, and cut into small sections. These sections were fixed with 10% formaldehyde, rinsed in water and placed in a transparent block of melted paraffin, making sure that they was completely immersed. Sections were then block fixed on a slicer for hematoxylin–eosin (HE) staining. For HE staining, the sections were washed with distilled water, and stained with hematoxylin. They were then rinsed in running tap water, differentiated with 0.3% acid alcohol, rinsed in running tap water, stained with eosin for 2 min, then dehydrated, cleared and mounted. HE-stained sections were observed with a light microscope (Nikon Eclipse E400, Japan).Results and discussion
Fiber morphology
SEM images of the electrospun materials before crosslinking are shown in Fig. 1(a)–(d), together with their diameter distributions. It is clear that the gelatin solutions could be electrospun into nanofibers under the conditions used in this work. Gelatin is a protein biopolymer derived from partial hydrolysis of native collagens.29 However, it is water soluble and hence the fibers will be destroyed by water.27 To render the fiber mats suitable for use as wound dressings, a crosslinking treatment is required to impart them with water-resistant properties. This was undertaken using glutaraldehyde, which is widely used for gelatin crosslinking due to its high efficiency in stabilizing collagenous materials. Crosslinking of gelatin with glutaraldehyde involves the reaction of free amino groups on amino acid residues of the polypeptide chains with the aldehyde groups of GTA30,31 (see ESI, Fig. S2†). After crosslinking, the fibers become thicker and slightly shrunken (Fig. 1(e)–(h)). Visually, they can be seen to change from colorless to a yellowish color upon crosslinking (data not shown), as a result of aldimine linkages (CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) forming.
N) forming.
 | ||
| Fig. 1 SEM images and diameter distributions of electrospun nanofibers prepared with (a) pure gelatin (G); (b) gelatin and VA (G/A); (c) gelatin and VE (G/E); (d) gelatin, VA and VE (G/A + E); and the same fibers after cross-linking: (e) G; (f) G/A; (g) G/E; (h) post-crosslinking G/A + E. | ||
The addition of vitamins increases the viscosity of the electrospinning solutions (Table 1), increasing the electrospinnability and aiding the formation of smooth fibers.32 However, higher viscosities can also result in the formation of beaded fibers,33 and there is evidence for a small amount of beading in the vitamin loaded fibers depicted in Fig. 1. The diameters of the vitamin loaded fibers are rather narrower than that of the pure gelatin material, both before and after cross-linking. The post-crosslinking diameters of the G/A (gelatin/vitamin A), G/E (gelatin/vitamin E), and G/A + E (gelatin/vitamins A and E) fibers are 538 ± 176 nm, 582 ± 131 nm and 566 ± 153 nm, respectively, as compared to 757 ± 161 nm for the pure gelatin fibers. This may result from an increase in conductivity following the addition of vitamins; this is known to make elongation of the polymer jet easier and to generate narrower nanofibers.34
Transmission electron microscopy images of the four samples before cross-linking are depicted in Fig. 2. Smooth fibers are observed in all cases, with the gelatin fibers (Fig. 2(a)) having larger diameters than the other samples (Fig. 2(b)–(d)). This is in accordance with the results of SEM. There are some black spots on the surfaces of the vitamin-loaded fibers which cannot be seen on the surface of the pure gelatin fibers. These could possibly be aggregates of vitamins which formed on the surface during electrospinning.
 | ||
| Fig. 2 TEM images of (a) G; (b) G/A; (c) G/E; and (d) G/A + E before cross-linking. | ||
Further characterization
Further characterization was undertaken on the cross-linked fiber samples. X-ray diffraction (XRD) patterns are presented in Fig. 3(a). Vitamin A palmitate (VA) is a liquid at room temperature, and thus is not shown here. It is clear that vitamin E TPGS (VE) exists as a crystalline material, displaying a number of characteristic reflections between 10° and 40° 2θ in its diffraction pattern. The gelatin fibers show only the characteristic and weak humps of amorphous systems. For the fibers loaded with vitamins, again no Bragg reflections can be observed in the XRD patterns, demonstrating that VE was rendered amorphous by the electrospinning process. These results agree with the literature.34–37 It has been reported that the very rapid solvent evaporation which occurs during electrospinning prevents crystallization from occurring, because there is insufficient time for the molecular rearrangement required for development of a crystal lattice. The random arrangement of molecules in the solution phase is thus propagated into the solid state, leading to amorphous materials.38–40 | ||
| Fig. 3 (a) XRD patterns of vitamin E TPGS and the cross-linked nanofibers prepared in this work; (b) FTIR spectra of the cross-linked fibers; (c) the in vitro vitamin release profiles from the G/A, G/E and G/A + E fibers. | ||
Fourier transform IR (FTIR) spectra are shown in Fig. 3(b). The neat gelatin nanofibers exhibit distinctive protein bands at approximately 1650 cm−1 (amide I) and 1540 cm−1 (amide II),41 corresponding to the stretching of CO, and the combined bending of N–H and stretching of C–N, respectively.42 The existence of the amide I band is attributable to there being both random coil and α-helix conformations present.43 The characteristic peaks of gelatin can also be seen in the G/A, G/E and G/A + E fibers. However, there are some differences. Weak peaks can be seen at 1710 cm−1 in the vitamin-loaded materials (marked with arrows in Fig. 3(b)). These are characteristic peaks of ester bonds (O–CO), present in both in VA and VE. In addition, for G/E and G/A + E, the characteristic ether linkage (C–O–C) peak of VE appears at 1120 cm−1. The observation of these peaks demonstrates that the vitamins were incorporated into the gelatin fibers during the electrospinning process.
Contact-angle data for all the samples are listed in Table 2. As expected, the gelatin nanofibers have good wettability, as a result of its hydrophilic nature. The contact angles of the vitamin-loaded systems G/A, G/E, and G/A + E are all very close to those for gelatin alone (30.4 ± 3.6°). VE is water-soluble, and thus the addition of vitamin E should not reduce the hydrophilicity of the fibers. Although VA is lipid-soluble, the amount added is small and thus is not expected to have any major influence on hydrophilicity. Wound dressing materials with hydrophilic properties have been found to be extremely useful for medical hemostatic applications,44 indicating that the systems prepared in this work may have potential in this regard.
| Sample | G | G/A | G/E | G/A + E |
| Contact-angle | 30.4 ± 3.6° | 32.1 ± 5.5° | 28.3 ± 1.6° | 29.6 ± 2.0° |
In vitro release behavior
The amount of intact vitamin incorporated in the fibers before cross-linking was determined prior to the investigation of their release behavior. The actual amounts of VA and VE in the fibers G/A and G/E are 65.5 ± 3.8% and 78.6 ± 4.6% of the theoretical loading, respectively. This can be attributed to the vulnerability of the vitamins to oxygen, heat, light and humidity. In G/A + E, the contents of VA and VE were determined to be 71.4 ± 5.5% and 73.1 ± 3.2% of the theoretical loading. This indicates that there has been less VA degradation than in the G/A fibers; vitamin E is an antioxidant, and thus some VA was presumably protected from oxidization because of its presence. As the cross-linking method is performed under vacuum and the fibers are protected from light during it, the amounts of vitamins loaded in the fibers are expected to be unchanged by cross-linking. It is not possible to dissolve the cross-linked fibers to verify this, and thus we employed TGA to check for the loss of any vitamin content. The TGA data for G/A + E before and after cross-linking (see ESI, Fig. S3(a)†) show that the thermal mass loss profile is unchanged. The vitamin content of the cross-linked systems was estimated from the masses remaining in TGA at 900 °C (at which point the pure vitamins show 0% residual mass). The results (VA in G/A 0.68%; VE in G/E 7.50%; VA in G/A + E 0.70%; VE in G/A + E 6.95%) agree well with the results measured by UV for the fresh, non-cross-linked fibers.The vitamin release profiles from the cross-linked fibers are depicted in Fig. 3(c). The cumulative release is expressed as a percentage of the measured loading. VA is freed from the G/A fibers with an initial burst release of 20% during the first 8 h, followed by further gradual release over more than 50 h. After immersion for 60 h, the release of VA reached a plateau at ca. 64%. The G/E fibers exhibit a burst release of about 30% in the first 10 h, after which gradual release ensues for about 60 hours. After immersion for 68 h, the cumulative release reached approximately 72%.
The initial bursts of release observed with VA and VE arise from the presence of some vitamin at the surface of the fibers. Drug release behavior from these types of systems is controlled by diffusion, polymer erosion, or a combination of the two.45 After crosslinking for 3 days, gelatin nanofibers have been shown to preserve their fibrous structure even after immersion in 37 °C water for 6 days.27 Therefore, it is believed that diffusion will be the dominant factor in drug release from G/A and G/E. It is clear that more than 20% of the incorporated vitamins were not released into the medium. It is postulated that this is a result of some vitamin being encapsulated deep inside the fibers (and thus not diffusing to the surface during the timescale of the experiment), and/or the degradation of some of the vitamin content during the release experiment.
The release profiles of the vitamins from G/A + E are also shown in Fig. 3(c) and can be seen to be similar to those of G/A and G/E, with an initial burst release of both VA and VE followed by sustained release for more than 60 h. However, the VA release form G/A + E took place over a longer time period (75 h) and reached a higher cumulative release percentage (69%) than from G/A. As indicated above, this can be explained by VE protecting VA from oxidation, resulting in less degradation during the release process.
Antibacterial activity
Fig. 4 illustrates the antibacterial effects of the vitamin-loaded cross-linked fibers, as measured by the UV absorbance of bacterial suspensions at 625 nm. The G and G/A fibers led to an increase in the number of E. coli and a small decrease in population of S. aureus after incubation for 2 days. However, after this time G/E and G/A + E were found to inhibit the growth of both E. coli and S. aureus. The antibacterial activities of G/E and G/A + E against E. coli are calculated to be 82.2 ± 5.9% and 79.1 ± 8.8% respectively after incubation for 2 days. The activities against S. aureus are 75.7 ± 7.4% and 67.3 ± 8.2%. It is thus clear that the fibers loaded with vitamin E TPGS have potent antibacterial properties.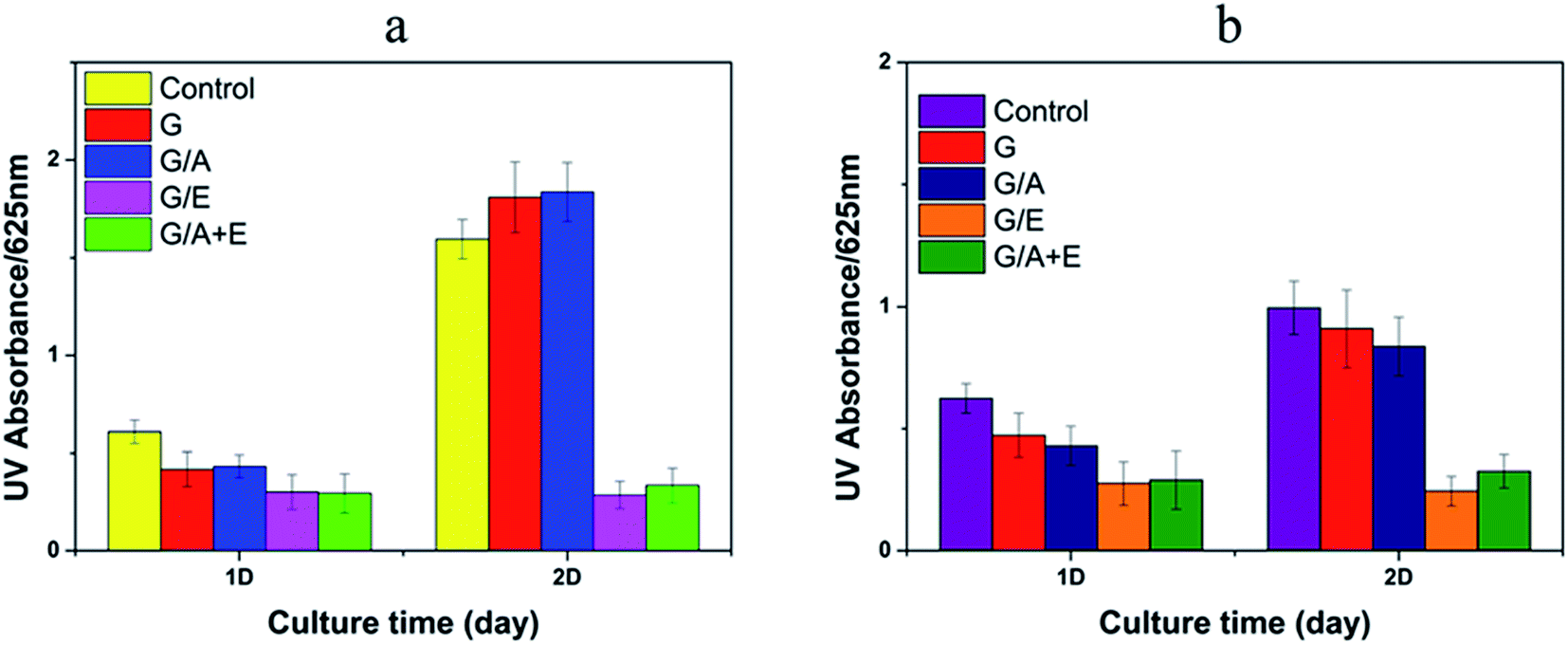 | ||
| Fig. 4 The antibacterial activity of the cross-linked fibers against (a) E. coli; (b) S. aureus. | ||
Cell growth
Fibroblast cells play a vital role in the process of wound repair.46 The cross-linked fibers were thus assessed for their abilities to stimulate L929 fibroblast adhesion and proliferation in vitro. The results of these experiments are given in Fig. 5(a). All the fibers show increased cell viability compared to the negative control after 1 and 3 days' culture. This is due to their moderately hydrophilic surfaces; many studies have demonstrated that cells adhere, spread and grow more easily on moderately hydrophilic substrates than on hydrophobic or very hydrophilic ones.47 On the 5th day of culture, only G/A + E shows greater cell numbers than the control. More cells could be observed on G/A + E and G/A than on G; the addition of vitamin A thus appears to improve the proliferation of fibroblasts.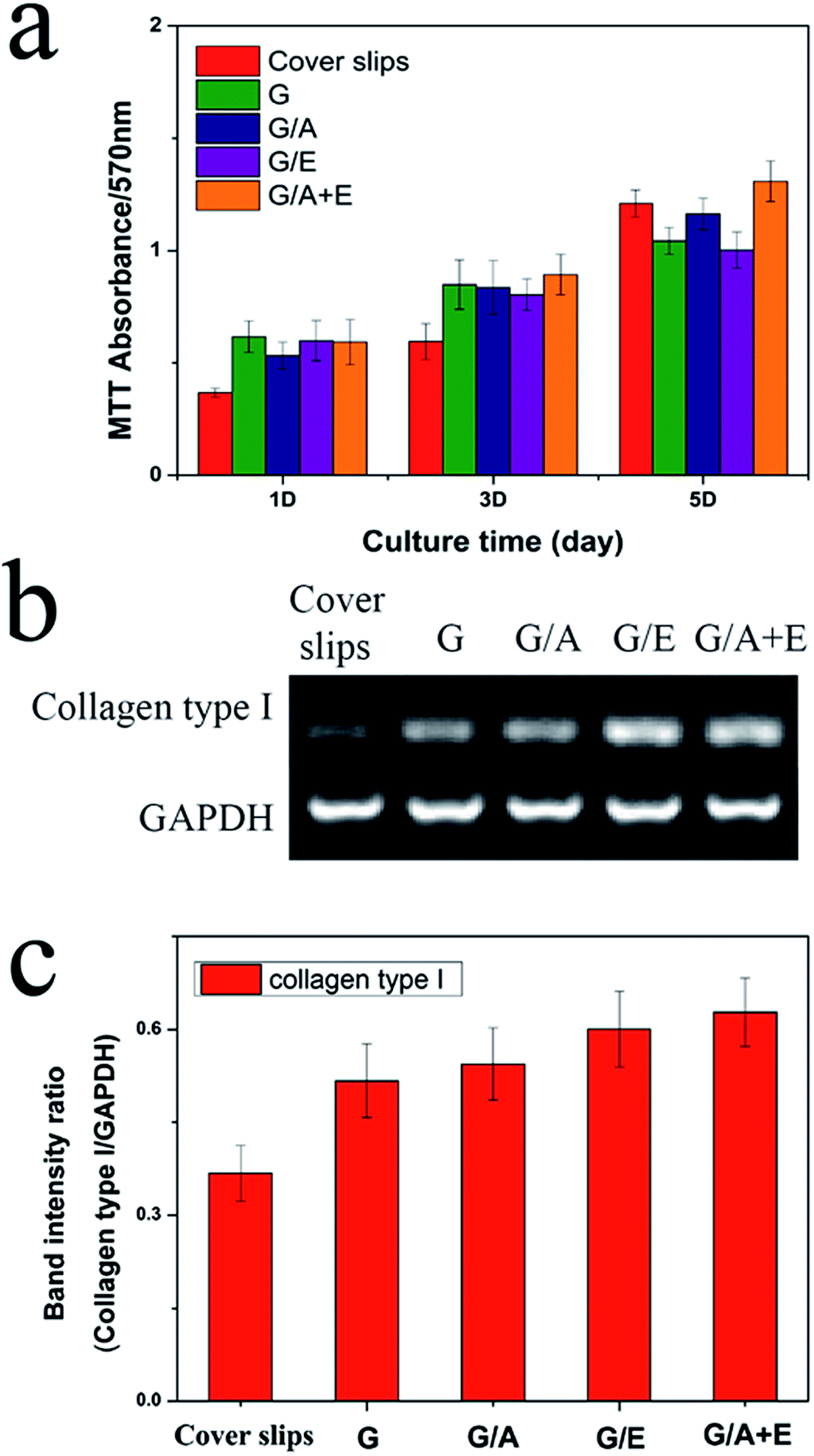 | ||
| Fig. 5 (a) Cell viability after exposure to the electrospun fibers for 1, 3, or 5 days; (b) mRNA expression levels and (c) normalized expression levels of collagen type I with respect to GADPH in L929 cells. Measurements were recorded after three independent experiments and the results reported as mean ± S.D. | ||
RT-PCR analysis
Gene expression levels of collagen type I were examined for the fibroblasts cultivated on each cross-linked fiber sample and on fiber-free cover slips as a negative control. Collagen is the main component of the extra-cellular matrix secreted by fibroblasts, so its production is important for wound repair. After incubation for 5 days, RNA was extracted from the cells and 1 μg used for RT-PCR. Fig. 5(b) shows the RT-PCR results for collagen type I in comparison with a housekeeping gene, glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The band intensities were also quantified, as depicted in Fig. 5(c). The image analysis results clearly demonstrate that the expression of collagen type I is higher for the fibroblasts seeded on the electrospun nanofibers than those grown on fiber-free cover slips. This is in agreement with the literature, where both vitamins are reported to promote the secretion of collagen.18,19 Considering the promising properties in vitro of the G/A + E cross-linked gelatin fibers, they were selected to be tested in vivo.In vivo wound healing experiments
Fig. 6 depicts representative images of wounds made to the skin of male rats at various times after infliction. After operation, the rats were allowed to recuperate and then photographed after ca. 3 hours. As shown in Fig. 6, slight inflammation can be seen in the rats treated with the commercial gauze only. Throughout the healing time, the areas of wounds treated by the commercial gauze or the gauze immersed in vitamin solution were larger than those treated with the fibers or the film. Compared to the gelatin (G) fibers and gelatin/VA/VE (G/A + E) film, the wound areas of the G/A + E fiber-treated rats were smaller, indicating a better healing performance of the latter. | ||
| Fig. 6 Representative images of the skin wound recovery process after 0, 5, 14 and 21 days. The wound surfaces were treated with (a) commercial gauze, (b) the same type of gauze soaked with a vitamin solution (1.4% w/v VE and 0.14% w/v VA), (c) cross-linked gelatin fibers (G), (d) cross-linked gelatin films loaded with VA and VE, (e) cross-linked gelatin fibers loaded with VA and VE (G/A + E). | ||
The relative wound areas calculated at different stages after the wounds were first inflicted are given in Fig. 7. The results agree with the conclusions drawn from the images in Fig. 6. After 5 days, the wound areas in each group are almost unchanged from those immediately after operation. After 14 days, the relative wound areas have declined to different extents in all groups, with the G/A + E fiber mats and films promoting faster healing. After 21 days, rats treated with the commercial gauze show the highest relative area at 38.8 ± 2.2%, much higher than those treated with the gauze soaked in vitamin solution (30.6 ± 2.3%), G fiber mats (22.5 ± 1.8%), G/A + E films (18.9 ± 1.7%) and G/A + E mats (10.3 ± 1.6%). The G nanofibers clearly have a better healing performance than the gauze groups. Moreover, the G/A + E mats perform better than the G/A + E films. These results are attributed to the electrospun nanofibers efficiently absorbing wound exudates and closely attaching to the wound surface,16,48 vital for the wound healing process and which the films cannot effectively do. The fact that the healing performance of G/A + E mats and films is better than that of the G fibers indicates that the sustainable release of vitamins is beneficial in this regard.
 | ||
| Fig. 7 Relative wound areas measured 0, 5, 14 and 21 days post-operation. | ||
Histological examinations
Histological staining results are given in Fig. 8. The epidermis layers are labeled with circles. In Fig. 8(a), the new epidermis in the group treated with gauze alone can be seen to be discontinuous and irregular. In the group treated by the gauze soaked in vitamin solution (Fig. 8(b)) and that with the cross-linked gelatin fibers (G; Fig. 8(c)), the wounds have been covered by an epidermis layer, which is much thicker than that of the drug-free gauze group and inflammatory reactions (black arrows) can also be observed. The regenerated epidermis in the G/A + E film treated group (Fig. 8(d)) is similar to that in G, but no obvious inflammatory reactions can be seen, which could result from the presence of the vitamins. Compared with the other groups, the G/A + E fibers have by far the best healing results (Fig. 8(e)). After 21 days, the regenerated epidermis is regular and thick, and very similar in appearance to the natural unwounded skin (Fig. 8(f)). The basal cells (white arrows) are also closely arranged. These results hence demonstrate the potent healing effects of cross-linked gelatin fibers loaded with vitamins A and E.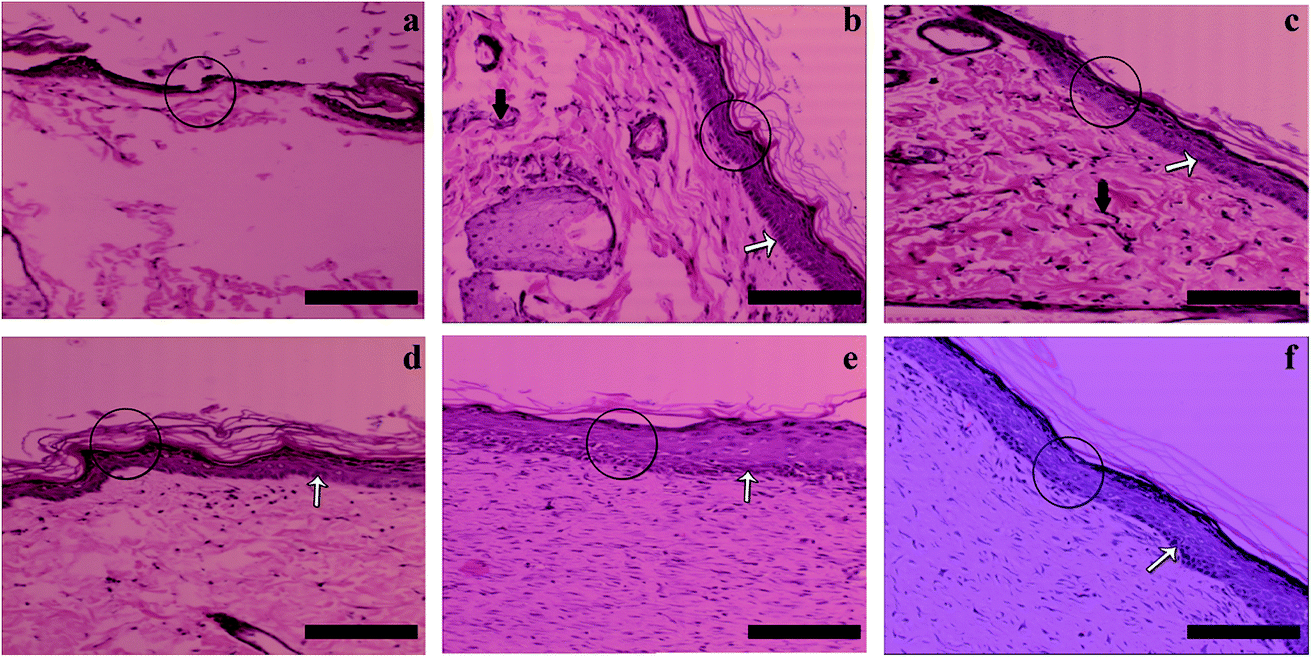 | ||
| Fig. 8 HE staining for epithelialization of skin wounds treated for 21 days with (a) commercial gauze, (b) the same type of gauze soaked with a vitamin solution (1.4% w/v VE and 0.14% w/v VA), (c) cross-linked gelatin fibers (G), (d) cross-linked gelatin films loaded with VA and VE, (e) cross-linked gelatin fibers loaded with VA and VE (G/A + E), (f) healthy skin where no wound has been inflicted nor any treatment applied. Scale bars represent 200 μm. | ||
Conclusions
In this study, gelatin nanofibers loaded with vitamin A palmitate and vitamin E TPGS were successfully prepared via electrospinning and cross-linked through a glutaraldehyde treatment. Electron microscopy showed that smooth fibers were produced, and their structure remained intact after cross-linking. The fiber diameters decreased with the addition of vitamins (cf. gelatin alone), and the incorporation of vitamins was confirmed by infrared spectroscopy. X-ray diffraction showed that the vitamins were present in the amorphous physical form after electrospinning. Contact angle tests showed the fibers to have moderately hydrophilic properties. Sustained release of both vitamins A (VA) and E (VE) was observed to proceed over more than 60 hours. Fibers loaded with VE or both VA and VE greatly inhibited the growth of E. coli and S. aureus. The fibers were also found to promote the adhesion and proliferation of L929 fibroblasts in the early stages of culture (1–2 days). The expression of collagen-specific genes by L929 fibroblasts was enhanced when these cells were exposed to the fibers. The electrospun fibers prepared in this work can thus inhibit bacterial growth, enhance the proliferation of fibroblasts, and increase their production of collagen. Preliminary in vivo investigations further demonstrate that the fibers have promising wound healing effects. Taken together, these results show that gelatin nanofibers loaded with vitamin A palmitate and vitamin E TPGS can act as effective wound dressing materials.Acknowledgements
This investigation was supported by the Biomedical Textile Materials “111 Project” of the Ministry of Education of China (No. B07024), and the UK–China Joint Laboratory for Therapeutic Textiles (based at Donghua University).Notes and references
- P. Zahedi, I. Rezaeian, S. O. Ranaei-Siadat, S. H. Jafari and P. Supaphol, Polym. Adv. Technol., 2010, 21, 77 CAS.
- J. S. Boateng, K. H. Matthews, H. N. E. Stevens and G. M. Eccleston, J. Pharm. Sci., 2008, 97, 2892 CrossRef CAS PubMed.
- Y. S. Cho, J. W. Lee, J. S. Lee, J. H. Lee, T. R. Yoon, Y. Kuroyanagi, M. H. Park and H. J. Kim, J. Mater. Sci.: Mater. Med., 2002, 13, 861 CrossRef CAS PubMed.
- L. Mi, H. Xue, Y. Li and S. Jiang, Adv. Funct. Mater., 2011, 21, 4028 CrossRef CAS.
- S. Meaume, P. Senet, R. Dumas, H. Carsin, M. Pannier and S. Bohbot, Br. J. Nurs., 2002, 11, 42 CrossRef PubMed.
- G. R. Newman, M. Walker, J. A. Hobot and P. G. Bowler, Biomaterials, 2006, 27, 1129 CrossRef CAS PubMed.
- X. Ma, J. Ge, Y. Li, B. Guo and P. X. Ma, RSC Adv., 2014, 4, 13652 RSC.
- P. Ke, X. N. Jiao, X. H. Ge, W. M. Xiao and B. Yu, RSC Adv., 2014, 4, 39704 RSC.
- R. Augustine, E. A. Dominic, I. Reju, B. Kaimal, N. Kalarikkal and S. Thomas, RSC Adv., 2014, 4, 51528 RSC.
- Y. Zhang, C. T. Lim and S. Ramakrishna, J. Mater. Sci.: Mater. Med., 2005, 16, 933 CrossRef CAS PubMed.
- H. F. Guo, Z. S. Li, S. W. Dong, W. J. Chen, L. Deng, Y. F. Wang and D. J. Ying, Colloids Surf., B, 2012, 96, 29 CrossRef CAS PubMed.
- R. Augustine, E. A. Dominic, I. Reju, B. Kaimal, N. Kalarikkal and S. Thomas, RSC Adv., 2014, 4, 24777 RSC.
- E. Shoba, R. Lakra, M. S. Kiran and P. S. Korrapati, RSC Adv., 2014, 4, 60209 RSC.
- R. Dastjerdi and M. Montazer, Colloids Surf., B, 2010, 79, 5 CrossRef CAS PubMed.
- J. S. Choi, K. W. Leong and H. S. Yoo, Biomaterials, 2008, 29, 587 CrossRef CAS PubMed.
- Y. Yang, T. Xia, W. Zhi, L. Wei, J. Weng, C. Zhang and X. Li, Biomaterials, 2011, 32, 4243 CrossRef CAS PubMed.
- P. Taepaiboon, U. Rungsardthong and P. Supaphol, Eur. J. Pharm. Biopharm., 2007, 67, 387 CrossRef CAS PubMed.
- S. Cho, L. Lowe, T. A. Hamilton, G. J. Fisher, J. J. Voorhees and S. Kang, J. Am. Acad. Dermatol., 2005, 53, 769 CrossRef PubMed.
- R. G. Santander, M. A. P. Arriba, G. M. Cuadrado, M. G. S. Martinez and M. M. D. L. Rosa, Int. J. Dev. Biol., 1996, 40, 181 Search PubMed.
- T. Tsunoda and K. Takabayashi, J. Soc. Cosmet. Chem., 1995, 46, 191 CAS.
- M. E. Carlotti, V. Rossatto and M. Gallarate, Int. J. Pharm., 2002, 240, 85 CrossRef CAS PubMed.
- S. Q. Wang, S. W. Dusza and H. W. Lim, J. Am. Acad. Dermatol., 2010, 63, 903 CrossRef PubMed.
- S. Somavarapu, S. Pandit, G. Gradassi, M. Bandera, E. Ravichandran and O. H. Alpar, Int. J. Pharm., 2005, 298, 344 CrossRef CAS PubMed.
- M. G. Traber, C. A. Thellman, M. J. Rindler and H. J. Kayden, Am. J. Clin. Nutr., 1988, 48, 605 CAS.
- J. Y. Lin, M. A. Selim, C. R. Shea, J. M. Grichnik, M. M. Omar, N. A. Monteiro-Riviere and S. R. Pinnell, J. Am. Acad. Dermatol., 2003, 48, 866 CrossRef PubMed.
- Y. P. Kato, D. L. Christiansen, R. A. Hahn, S. J. Shieh, J. D. Goldstein and F. H. Silver, Biomaterials, 1989, 10, 38 CrossRef CAS PubMed.
- Y. Z. Zhang, J. Venugopal, Z. M. Huang, C. T. Lim and S. Ramakrishna, Polymer, 2006, 47, 2911 CrossRef CAS.
- B. Son, B. Y. Yeom, S. H. Song, C. S. Lee and T. S. Hwang, J. Appl. Polym. Sci., 2009, 111, 2892 CrossRef CAS.
- S. Young, M. Wong, Y. Tabata and A. G. Mikos, J. Controlled Release, 2005, 109, 256 CrossRef CAS PubMed.
- E. Khor, Biomaterials, 1997, 18, 95 CrossRef CAS PubMed.
- L. O. Damink, P. J. Dijkstra, M. J. A. Van Luyn, P. B. Van Wachem, P. Nieuwenhuis and J. Feijen, J. Mater. Sci.: Mater. Med., 1995, 6, 460 CrossRef.
- X. Sheng, L. Fan, C. He, K. Zhang, X. Mo and H. Wang, Int. J. Biol. Macromol., 2013, 56, 49 CrossRef CAS PubMed.
- J. M. Deitzel, J. Kleinmeyer, D. Harris and N. C. Beck Tan, Polymer, 2001, 42, 261 CrossRef CAS.
- Y. Su, X. Li, S. Liu, X. Mo and S. Ramakrishna, Colloids Surf., B, 2009, 73, 376 CrossRef CAS PubMed.
- X. Xu, X. Chen, X. Xu, T. Lu, X. Wang, L. Yang and X. Jing, J. Controlled Release, 2006, 114, 307 CrossRef CAS PubMed.
- J. Hu, H. Y. Li, G. R. Williams, H. H. Yang, L. Tao and L. M. Zhu, J. Pharm. Sci., 2016, 105, 1104 CrossRef PubMed.
- S. Um-i-Zahra, X. X. Shen, H. Li and L. Zhu, J. Polym. Res., 2014, 21, 1 CAS.
- X. Zong, K. Kim, D. Fang, S. Ran, B. S. Hsiao and B. Chu, Polymer, 2002, 43, 4403 CrossRef CAS.
- J. M. Deitzel, J. Kleinmeyer, D. E. A. Harris and N. B. Tan, Polymer, 2001, 42, 261 CrossRef CAS.
- Y. T. Jia, J. Gong, X. H. Gu, H. Y. Kim, J. Dong and X. Y. Shen, Carbohydr. Polym., 2007, 67, 403 CrossRef CAS.
- D. M. Hashim, Y. B. Che Man, R. Norakasha, M. Shuhaimi, Y. Salmah and Z. A. Syahariza, Food Chem., 2010, 118, 856 CrossRef CAS.
- M. C. Chang, C. C. Ko and W. H. Douglas, Biomaterials, 2003, 24, 2853 CrossRef CAS PubMed.
- D. A. Prystupa and A. M. Donald, Polym. Gels Networks, 1996, 4, 87 CrossRef CAS.
- Y. Zhang, H. Ouyang, C. T. Lim, S. Ramakrishna and Z. M. Huang, J. Biomed. Mater. Res., Part B, 2005, 72, 156 CrossRef PubMed.
- Y. Dong, Z. Zhang and S. S. Feng, Int. J. Pharm., 2008, 350, 166 CrossRef CAS PubMed.
- H. Cook, P. Stephens, K. J. Davies, K. G. Harding and D. W. Thomas, J. Invest. Dermatol., 2000, 115, 225 CrossRef CAS PubMed.
- Y. Arima and H. Iwata, Biomaterials, 2007, 28, 3074 CrossRef CAS PubMed.
- B. Zou, X. Li, H. Zhuang, W. Cui, J. Zou and J. Chen, Polym. Degrad. Stab., 2011, 96, 114 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra05092a |
| This journal is © The Royal Society of Chemistry 2016 |