Cytotoxicity and slow release of the anti-cancer drug doxorubicin from ZIF-8†
Iane B.
Vasconcelos
a,
Teresinha G. da
Silva
b,
Gardenia C. G.
Militão
c,
Thereza A.
Soares
d,
Nailton M.
Rodrigues
e,
Marcelo O.
Rodrigues
df,
Nivan B. da
Costa
Jr.
e,
Ricardo O.
Freire
e and
Severino A.
Junior
*d
aPrograma de Pós-Graduação em Ciência dos Materiais, UFPE, 50590-470, Recife-PE, Brazil
bDepartamento de Antibióticos, UFPE, 50590-470, Recife-PE, Brazil
cDepartamento de Fisiologia e Farmacologia, UFPE, 50590-470, Recife-PE, Brazil
dDepartamento de Química Fundamental, UFPE, Av. Jornalista Aníbal Fernandes, s/n - Cidade Universitária, 50740-560, Recife-PE, Brazil. Tel: +55 81 21267475; Fax: +55 81 21268442; E-mail: salvesjr@ufpe.br
eDepartamento de Química, UFS, 49100-000, São Cristóvão-SE, Brazil
fInstituto de Química, Universidade de Brasília, 70910-900, Brasília–DF, Brazil
First published on 6th August 2012
Abstract
Metal–organic frameworks are emerging as a powerful platform for the delivery and controlled release of several drug molecules. Herein, we report the incorporation of the anti-cancer drug doxorubicin into the zeolitic imidazolate framework (ZIF-8) with high-load and progressive release. Adsorption measurements show that doxorubicin is incorporated into ZIF-8 with a load of 0.049 g doxorubicin g−1 dehydrated ZIF-8. Doxorubicin is released in a highly controlled and progressive fashion with 66% of the drug released after 30 days. We also characterize the antitumoral potential and cytotoxicity of the doxorubicin-ZIF-8 (DOXO-ZIF-8) complex towards the mucoepidermoid carcinoma of human lung (NCI-H292), human colorectal adenocarcinoma (HT-29), and human promyelocytic leukemia (HL-60) cell lines. It is shown that the complex doxorubicin-ZIF-8 exhibits lower cytotoxicity than pure doxorubicin for the tested cells, possibly due to the slower release of the incorporated drug. Furthermore, host–guest interactions have been addressed from a microscopic perspective through molecular docking simulations. In conjunction with our experimental characterization, the calculations suggest that doxorubicin binds preferentially to the surface rather than into the pores of ZIF-8, whose entry diameter is at least half the size of the shortest axis of the drug. These findings are also consistent with high-resolution X-ray crystallography and NMR spectroscopy studies of ZIF-8 which shows that this framework is very rigid under constant pressure in contrast to previous experimental and theoretical studies of ZIF-8 under gas pressure.
Introduction
Cancer therapies are curbed by the their unspecificity towards tumor cells, leading to high doses, rapid clearance, poor pharmacokinetics and serious side effects.1–3 Nanoparticle-based therapeutics can alleviate many of the pitfalls associated with free drug therapeutics while improving the efficacy of conventional drugs. The clinical success of nanoparticle therapies is illustrated by the large number of nanoparticle–drug conjugates under different stages of clinical development.2–5 Metal–organic frameworks (MOF) or coordination polymers are crystalline solids assembled by the connection of metal ions or clusters through tunable organic linkers whose structures are held together either by strong metal–ligand bonding or by weaker bonding forces (e.g. hydrogen bonding and π–π interactions).6–8 The modular nature of MOFs coupled with the many different types of bridging ligands allows for a multitude of frameworks with desirable topologies, architectures, and properties inherent to the building blocks, such as geometric rigidity, chemical functionality, or chirality.9 The versatility of MOFs has led to their broad application in gas separation and/or storage, sensors, non-linear optics, catalysis, and forensic chemistry.10–19Over the past five years, a promising application for MOFs has emerged in the controlled delivery of several drug molecules.5,20–22 These applications rely on the several desirable properties of MOFs as potential drug carriers: their remarkably high surface areas and large pore sizes for drug encapsulation, their intrinsic biodegradability, their versatile functionality for post-synthesis grafting of drug molecules, and their scalability to the nanoregime.5,20 In particular, non-toxic porous iron(III)-based MOFs have been reported as superior nanocarriers for the controlled delivery of several antitumoral and retroviral drugs.21,22 Another class of MOFs, the zeolitic imidazolate framework (ZIF), also exhibits interesting properties as a carrier.23–26 ZIFs are comprised of tetrahedral transition metal ionsconnected by imidazolate units arranged in topologies with large cages and small apertures. ZIFs exhibit both high thermal and chemical stability, thus overcoming two of the main issues when considering the use of MOFs in biomedical applications.23,24,26 Recently, it has been reported that ZIF-8 can function as a pH-triggered carrier for the anticancer drug 5-fluorouracil.27
In this report, we describe the incorporation of the antitumoral drug doxorubicin (DOXO) into ZIF-8 with high-load and controlled release via Fourier transform infrared (FTIR) spectroscopy, thermogravimetric analysis (TGA), X-ray diffraction (XRD), and confocal microscopy. It is further shown that the DOXO–ZIF-8 complex has a higher antitumoral potential and lower cytotoxicity towards the HL-60 and MCF-7 cell lines compared with doxorubicin in the absence of ZIF-8. Host–guest interactions were characterized from a macroscopic perspective via computational simulations. These simulations offer insights into the ZIF-8 binding conformation and preferences of doxorubicin, and are consistent with the powder synchrotron X-ray diffraction measurements. These findings suggest that ZIF-8 can be a potential carrier system for doxorubicin, whose short half-life and low bioavailability in biological media, coupled with low membrane permeability, presents a much improved therapeutic profile when entrapped in nanocarriers.21,28
Results and discussion
A high-resolution X-ray powder pattern was acquired at room temperature for DOXO–ZIF-8 (Fig. 1). The collected data could be readily indexed with the presence of one impurity line via DICVOL 04.11 Due to a lack of crystallographic data for doxorubicin, the Rietveld refinement for the DOXO–ZIF-8 complex was performed considering only the atomic coordinates of the ZIF-8. The refinement of the XRD data sets shows that the structural integrity of ZIF-8 remains unaltered after adsorption of the drug. Doxorubicin in solution has a pallid orange color, which changes to purple upon its incorporation into ZIF-8, thus providing further evidence for the formation of the DOXO–ZIF-8 complex.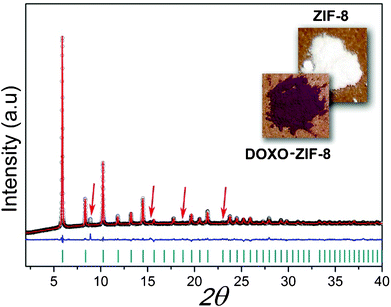 | ||
| Fig. 1 Final Rietveld refinement of DOXO–ZIF-8. Observed data points are indicated as black circles, the best-fit profile (upper trace) and the difference pattern (lower trace) are drawn as solid red and blue lines, respectively. Dark cyan vertical bars and red arrows indicate the angular positions of the allowed Bragg reflections and impurity lines from lactose. Reliability factors for refinement: Rp: 1.4; Rwp: 2.07; χ2: 5.28; RF2: 18.52. | ||
FTIR spectra were obtained for ZIF-8, doxorubicin, and the complex DOXO–ZIF-8 (ESI†, Fig. S1). In the spectrum corresponding to ZIF-8, two bands at 3135 and 2928 cm−1 can be observed for the aromatic C–H stretch and the aliphatic C–H stretch of the imidazole, respectively. The 1606 cm−1 band is for the C–C stretch, and the peak at 1580 cm−1 is for the C–N stretch. The C–N absorption bands are found in the 1100–1400 cm−1 region. The absorption band at 421 cm−1 is associated with the Zn–N stretching mode. These assignments are in agreement with the FTIR measurements from Park et al.29 Several bands are observed for doxorubicin: the band at 3441 cm−1 is due to an axial strain of the N–H bond, at 2936 cm−1 to C–H axial deformation, at 1635 cm−1 to the axial deformation of the C–O bond, and at 100–1260 cm−1 to the absorption associated with the stretching of the alcohol group. In the the region between 675–900 cm−1 there is an out-of-plane bending of the –OH group that has also been reported by Chouhan et al.30 The FTIR spectrum analysis for the system DOXO–ZIF-8 does not undoubtedly show the adsorption of doxorubicin into ZIF-8, but the detection of characteristic bands for both ZIF-8 and DOXO indicates the presence of both compounds. Moreover, the incorporation of doxorubicin into ZIF-8 is supported by the color change of the ZIF-8 crystals, which were initially colorless, and after the incorporation process, exhibited a purple color.
The thermal analysis TG/DTG curve for DOXO–ZIF-8 shows four weight loss events (%) at temperature ranges of 80–100, 190–390, and 390–620 °C (ESI†, Fig. S2). The first event is related to the loss of hydration water molecules trapped in the pores of ZIF-8. The second event is typical of the loss of the DOXO molecules since the decomposition profile of ZIF-8 has not displayed any weight loss in this temperature range. The third decomposition event corresponds to the simultaneous thermal decomposition of DOXO and ZIF-8 followed by carbonization of the material. The decomposition of ZIF-8 and DOXO–ZIF-8 samples yields residual zinc oxide (ZnO) (ca. 37 and 21% respectively). A similar ratio between MIL-53(Cr, Fe) and MIL-53(Cr, Fe)–ibuprofen has been reported in the literature by Horcajada and co-authors.22 The SEM image for ZIF-8 shows nanometer-sized crystals with an average diameter around 200 nm (ESI†, Fig. S3B). Comparison of this value against our XRD results indicates the formation of nanoparticle microclusters. The SEM image for DOXO–ZIF-8 shows crystalline nanoparticles with sizes of about 300 nm, which are similar to the crystallite sizes estimated by XRD (ESI†, Fig. S3C). The photomicrograph of doxorubicin is also shown in Fig. S3A to assist with data interpreting. Adsorption measurements for the system DOXO–ZIF-8 show that drug incorporation was carried out successfully with a load of 0.049 g of doxorubicin g−1 of dehydrated ZIF-8. This load is less than the value obtained for the 5-fluorouracil–ZIF-8 system, i.e. 0.660 g of 5-fluorouracil g−1 of ZIF-8.27 However, given the much smaller size of 5-fluorouracil (5.42 × 4.50 × 0.00 Å) compared to doxorubicin (14.64 × 10.02 × 6.90 Å) it is likely that the former is incorporated inside the framework pores, which we argue is not the case for the latter (see below).
UV-vis spectroscopy was used to measure the doxorubicin release profile based on the amount of drug in phosphate buffer at pH 7.4 (Fig. 2). ZIF-8 has been shown to be very stable in these conditions.27 After 30 days, only 66% of the drug was released suggesting strong chemical interactions with the ZIF-8 framework. The release profile from ZIF-8 showed a pattern of zero-order, similar to the release profile previously reported for the system ibuprofen–MIL-100.31 It has been previously shown that ibuprofen exhibits different release profiles depending on the nature of the metal present in the MOF used for adsorption. For ZIF-8 containing Zn2+ ions, the release profile still followed zero-order kinetics after 30 days. This is an excellent rate compared with doxorubicin release from MIL-100, which is 100% in 14 days.21
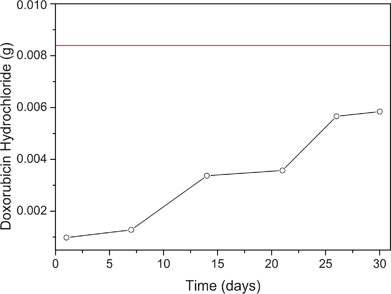 | ||
| Fig. 2 Graphical representation of the amount of drug released from ZIF-8 in 30 days. The red line in the graph represents the total amount of drug incorporated into the ZIF-8. | ||
The cytotoxicities of ZIF-8 and the complex DOXO–ZIF-8 were assessed by the colorimetric MTT assay (Table 1). The method is based on the conversion of 3-(4,5-dimethyl-2-thiazole)-2,5-diphenyl-2H-tetrazolium bromide (MTT) into blue formazan from mitochondrial enzymes present only in metabolically active cells.32 It has been used in the screening program of the National Cancer Institute of the United States (NCI), which tests more than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 samples each year.33 The MTT assay offers a fast and sensitive approach for the analysis of the viability and metabolic state of the cell. The concentrations causing 50% cell growth inhibition (IC50) at three lineages ranged from 0.01 to 7.9 μg mL−1 for DOXO, and 0.79 to14.96 μg mL−1 for DOXO–ZIF-8. CNI-H292 proved to be the most sensitive lineage with an IC50 value of 0.01 μg mL−1 for DOXO and 0.79 μg mL−1 for DOXO–ZIF-8. ZIF-8 was not cytotoxic at the tested concentration (25 μg mL−1). The reduced cytotoxicity of DOXO–ZIF-8 compared to DOXO may be explained by the slow release of the drug.
000 samples each year.33 The MTT assay offers a fast and sensitive approach for the analysis of the viability and metabolic state of the cell. The concentrations causing 50% cell growth inhibition (IC50) at three lineages ranged from 0.01 to 7.9 μg mL−1 for DOXO, and 0.79 to14.96 μg mL−1 for DOXO–ZIF-8. CNI-H292 proved to be the most sensitive lineage with an IC50 value of 0.01 μg mL−1 for DOXO and 0.79 μg mL−1 for DOXO–ZIF-8. ZIF-8 was not cytotoxic at the tested concentration (25 μg mL−1). The reduced cytotoxicity of DOXO–ZIF-8 compared to DOXO may be explained by the slow release of the drug.
Confocal microscopy analysis showed the fluorescence pattern and intracellular fate of the DOXO and DOXO–ZIF-8 systems. The DOXO fluorescence was primarily detected in intracellular vesicles and nuclei, consistent with its intercalation within genomic DNA. The complex DOXO–ZIF-8 was dispersed in the cell. Some of DOXO–ZIF-8-treated cells presented stained-tubular processes towards the nuclear matrix (Fig. 3E, 3F, 3I, 3J). No fluorescence signal could be found in the control cells (Fig. 3A, top left) and only a weak signal could be detected in ZIF-treated cells (Fig. 3B, top right). However, cells treated with this complex showed an evident shift in the red signal when compared with DOXO alone (Fig. 3C, 3D, 3G, 3H). Both DOXO and DOXO–ZIF-8 complexes induced considerable morphological changes in HL-60 cells in a time- and dose-dependent manner, with induction of bubbles from the cellular surface resembling apoptotic bodies. Luminescent properties were more pronounced in cells treated with DOXO–ZIF-8, which may indicate that complexation with ZIF-8 facilitated the entry of doxorubicin in the cells (Fig. 3). Macromolecular pro-drugs such as doxorubicin enter cells via endocytosis, and are compartmentalized in the endosome and lysosome.34 Hence, it is plausible that free and ZIF-8-complexed doxorubicin exhibit different mechanisms of entry into cells.
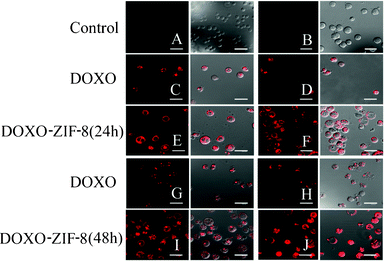 | ||
| Fig. 3 Confocal microscopy of HL-60 cells incubated in the absence (A) or presence of ZIF8 (B); IC50 (C) and 2 × IC50 (D) of DOXO; IC50 (E) and 2 × IC50 (F) of DOXO–ZIF-8 for 24 h; IC50 (G) and 2 × IC50 (H) of DOXO; IC50 (I) and 2 × IC50 (J) of DOXO–ZIF-8 for 48 h. The left column represents the fluorescence of the red channel and the right column represents the merging of the red channel and the differential interference contrast image. | ||
The experimental measurements presented in this report show that doxorubicin is loaded onto ZIF-8 and released at slow rate. Such release behavior may be interpreted as indicative of doxorubicin being incorporated into the pores. However, such a conclusion is not reconcilable with the fact that ZIF-8 has been shown by X-ray crystallography and NMR spectroscopy to be very rigid. Additionally, the largest pore size in the ZIF-8 structure has a diameter of ca. 11.6 Å interconnected by 6-ring windows of a diameter of 3.40 Å whereas doxorubicin has dimensions of 14.64 × 10.02 × 6.90 Å, and XDR analysis of the complex shows unchanged lattice parameters for ZIF-8 previous to and after doxorubicin loading. Hence, doxorubicin appears overly large to pass through even the largest pore entrance. To ascertain such an assessment, flexible molecular docking calculations were performed for doxorubicin on the structure of ZIF-8. Could doxorubicin adsorb onto the ZIF-8 surface so effectively to explain the measured release rates?
We have addressed this question by the means of molecular docking simulations of doxorubicin onto/into the ZIF-8 structure. The treatment of doxorubicin dihedral torsions as flexible allows for different conformations of the molecule to be taken into account during the conformational sampling and binding energy calculations. A total of 1.35 × 108 conformations of doxorubicin were sampled and ranked according to their interaction energies to the ZIF-8 structure. Among the sampled conformations, the 100 lowest conformations bind exclusively only to the framework surface (Fig. 4). Doxorubicin interacts with the Zn2+ cations via chelating sites comprised of the quinone and the phenolic oxygens on both sides of the anthracycline aromatic moiety (Fig. 4). The Zn2+ cations in the ZIF-8 structure exhibit tetrahedral geometry coordinated by four neighboring imidazolate groups. It is expected that the Zn2+ cations on the surface of the ZIF-8 structure will have two imidazolate ligands replaced by water molecules. Our molecular docking results show that doxorubicin binds to the Zn2+ cation, thus maintaining its tetrahedral coordination geometry, possibly by replacing two water molecules acting as ligands to the cation. A similar binding pattern has been previously reported for doxorubicin complexed with Fe3+ cations via absorption and circular dichroism measurements in aqueous solution and in semi-aqueous MeOH.34 Limiting the conformational search exclusively to the largest pore cavity did not yield conformations with favorable interaction energies. It is interesting to note that both X-ray diffraction and NMR spectroscopy structural characterization of ZIF-8 show that the framework is very rigid.35 This is in contrast with previous theoretical and experimental studies under gas pressure, which is of great relevance for applications in gas storage.36,37 Because such conditions are not expected in the cell, the pressure-induced flexibility of ZIF-8 should not be relevant for the present work.
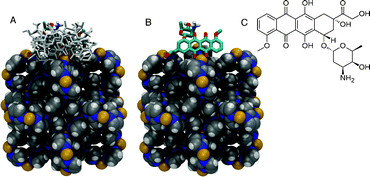 | ||
| Fig. 4 Representation of the ZIF-8 (in van der Waals spheres) crystallographic unit cell and the docked conformations of doxorubicin in stick. (A) Ten lowest energy conformations of doxorubicin bound to the X-ray structure of ZIF-8. (B) The lowest energy conformation with an occurrence of ca. 70% among the 100 lowest energy conformers selected from a total of 1.35 × 108 sampled conformations. (C) Chemical structure of doxorubicin. Carbon atoms in grey, nitrogen in blue, hydrogen in white, and Zn2+ cations in yellow. | ||
Conclusions
Doxorubicin is an anthracycline antitumoral drug widely used in the treatment of various cancers. Despite its efficacy in the treatment of carcinomas, sarcomas and hematological cancers, doxorubicin exhibits serious cumulative dose-dependent cardiotoxicity. Its therapeutical efficiency is further compromised due to the drug poor stability in biological media and low membrane permeability. However, it has been shown that Zn2+ can competitively inhibit the binding interaction of the anthracyclines with the major contractile protein cardiac myosin, thus exerting a cardioprotective effect against the drug.38 It has also been shown that the therapeutic profile of doxorubicin is much improved when the drug is entrapped in nanocarriers.21,28 The use of ZIF-8 as a carrier for doxorubicin may alleviate cardiotoxic side effects while increasing its therapeutical efficiency. We have demonstrated that doxorubicin can be incorporated in ZIF-8 with a load of 0.049 g of doxorubicin g−1 of dehydrated ZIF-8 and a very slow release rate with a pattern of zero-order (which lasts for 30 days). We have also shown that the DOXO–ZIF-8 system is internalized by HL-60 cells, and exhibits reduced cytotoxicity compared to pure doxorubicin, likely due to its the slow release from ZIF-8. Refinement of XRD showed that the lattice parameters for ZIF-8 are unaltered after adsorption of doxorubicin, which suggests that doxorubicin interacts predominantly with the surface of ZIF-8. This assumption is supported by molecular docking calculations, which show that doxorubicin binds to the Zn2+ cations on the framework surface via quinone and phenolic oxygens on the anthracycline aromatic moiety.Experimental and computational procedures
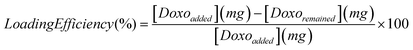 | (1) |
A sample of DOXO–ZIF-8 was added to a dissolution medium containing 900 ml of deionized water at 37.5 ± 0.5 °C with a stirring speed of 96 rpm until full release or within 48 h of the onset of testing.11 A volume aliquot was collected for every 2 mL of dissolution medium. The sample was collected at different intervals during a period of 48 h.
000 generations, and mutation and crossover rates of 0.02 and 0.08, respectively. An optional elitism parameter equal to 1 was applied. A maximum of 300 iterations per local search was allowed. The lowest energy docked conformations were sorted in order of increasing energy and the root-mean-square deviation (RMSD) of each conformation was calculated and compared in order to cluster together conformations with a RMSD smaller than 1.5 Å. A detailed description of the LGA parameters and procedures employed here can be found elsewhere.55,56
Acknowledgements
We appreciate the financial support from the following Brazilian agencies, institutes, and networks: CNPq, CAPES, FACEPE, FAPITEC-SE, INAMI, RENAMI and nBioNet. Computational resources were provided by CENAPAD (Centro Nacional de Processamento de Alto Desempenho) in Campinas, Brazil and the Environmental Molecular Sciences Laboratory at Pacific Northwest National Laboratory. Dr Regina Figueiredo is acknowledged for the confocal microscopy imaging.References
- J. R. Heath and M. E. Davis, Annu. Rev. Med., 2008, 59, 251–265 CrossRef CAS.
- M. Ferrari, Nat. Rev. Cancer, 2005, 5, 161–171 CrossRef CAS.
- M. E. Davis, Z. G. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS.
- R. C. Huxford, J. D. Rocca and W. B. Lin, Curr. Opin. Chem. Biol., 2010, 14, 262–268 CrossRef CAS.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319–330 CrossRef CAS.
- M. Eddaoudi, H. Li and O. M. Yaghi, J. Am. Chem. Soc., 2000, 122, 1391–1397 CrossRef CAS.
- S. L. James, Chem. Soc. Rev., 2003, 32, 276–288 RSC.
- A. Corma, H. Garciĺa and F. X. Llabreés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS.
- A. Boultif and D. Louer, J. Appl. Crystallogr., 2004, 37, 724–731 CrossRef CAS.
- K. L. Mulfort, O. K. Farha, C. D. Malliakas, M. G. Kanatzidis and J. T. Hupp, Chem.–Eur. J., 2010, 16, 276–281 CrossRef CAS.
- O. K. Farha, C. D. Malliakas, M. G. Kanatzidis and J. T. Hupp, J. Am. Chem. Soc., 2009, 132, 950–952 CrossRef.
- D. J. Tranchemontagne, J. L. Mendoza-Cortes, M. O'Keeffe and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1257–1283 RSC.
- D. Farrusseng, S. Aguado and C. Pinel, Angew. Chem., Int. Ed., 2009, 48, 7502–7513 CrossRef CAS.
- A. M. Shultz, O. K. Farha, J. T. Hupp and S. T. Nguyen, J. Am. Chem. Soc., 2009, 131, 4204–4205 CrossRef CAS.
- A. P. Nelson, O. K. Farha, K. L. Mulfort and J. T. Hupp, J. Am. Chem. Soc., 2008, 131, 458–460 CrossRef.
- S. Natarajan and P. Mahata, Chem. Soc. Rev., 2009, 38, 2304–2318 RSC.
- I. T. Weber, A. J. G. de Melo, M. A. D. Lucena, M. O. Rodrigues and S. Alves, Anal. Chem., 2011, 83, 4720–4723 CrossRef CAS.
- A. C. McKinlay, R. E. Morris, P. Horcajada, G. Ferey, R. Gref, P. Couvreur and C. Serre, Angew. Chem., Int. Ed., 2010, 49, 6260–6266 CrossRef CAS.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J. S. Chang, Y. K. Hwang, V. Marsaud, P. N. Bories, L. Cynober, S. Gil, G. Ferey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172–178 CrossRef CAS.
- P. Horcajada, C. Serre, G. Maurin, N. A. Ramsahye, F. Balas, M. Vallet-Regi, M. Sebban, F. Taulelle and G. Ferey, J. Am. Chem. Soc., 2008, 130, 6774–6780 CrossRef CAS.
- X.-C. Huang, Y.-Y. Lin, J.-P. Zhang and X.-M. Chen, Angew. Chem., Int. Ed., 2006, 45, 1557–1559 CrossRef CAS.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS.
- S. Keskin and S. Kızılel, Ind. Eng. Chem. Res., 2011, 50, 1799–1812 CrossRef CAS.
- C. Y. Sun, C. Qin, X. L. Wang, G. S. Yang, K. Z. Shao, Y. Q. Lan, Z. M. Su, P. Huang, C. G. Wang and E. B. Wang, Dalton Trans., 2012 Search PubMed.
- R. R. Patil, S. A. Guhagarkar and P. V. Devarajan, Crit. Rev. Ther. Drug Carrier Syst., 2008, 25, 1–61 CrossRef CAS.
- K. S. Park, Z. Ni, A. P. Cote, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS.
- R. Chouhan and A. Bajpai, J. Nanobiotechnol., 2009, 7, 5 CrossRef.
- P. Horcajada, C. Serre, M. Vallet-Regi, M. Sebban, F. Taulelle and G. Ferey, Angew. Chem., Int. Ed., 2006, 45, 5974–5978 CrossRef CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. Mcmahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef CAS.
- X. W. Dai, Z. L. Yue, M. E. Eccleston, J. Swartling, N. K. H. Slater and C. F. Kaminski, Nanomed.: Nanotechnol., Biol. Med., 2008, 4, 49–56 CrossRef CAS.
- W. Morris, C. J. Stevens, R. E. Taylor, C. Dybowski, O. M. Yaghi and M. A. Garcia-Garibay, J. Phys. Chem. C, 2012, 116, 13307–13312 CAS.
- C. O. Ania, E. Garcia-Perez, M. Haro, J. J. Gutierrez-Sevillano, T. Valdes-Solis, J. B. Parra and S. Calero, J. Phys. Chem. Lett., 2012, 3, 1159–1164 CrossRef CAS.
- D. Fairen-Jimenez, S. A. Moggach, M. T. Wharmby, P. A. Wright, S. Parsons and T. Duren, J. Am. Chem. Soc., 2011, 133, 8900–8902 CrossRef CAS.
- B. Liu, H. Shioyama, T. Akita and Q. Xu, J. Am. Chem. Soc., 2008, 130, 5390–5391 CrossRef CAS.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- A. C. Larson and R. B. Von Dreele, General Structure Analysis System (GSAS), Los Alamos National Laboratory Report LAUR 86-748 (1994).
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- D. W. Lewis, A. R. Ruiz-Salvador, A. Gómez, L. M. Rodriguez-Albelo, F.-X. Coudert, B. Slater, A. K. Cheetham and C. Mellot-Draznieks, CrystEngComm, 2009, 11, 2272–2276 RSC.
- M. Jarvinen, J. Appl. Crystallogr., 1993, 26, 525–531 CrossRef CAS.
- P. Thompson, D. E. Cox and J. B. Hastings, J. Appl. Crystallogr., 1987, 20, 79–83 CrossRef CAS.
- R. A. Young and P. Desai, Archiwun Nauki o Materialach, 1989, 10, 71–90 Search PubMed.
- V. Sidey, J. Appl. Crystallogr., 2004, 37, 1013–1014 CrossRef CAS.
- M. Tim, J. Immunol. Methods, 1983, 65, 55–63 CrossRef.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS.
- D. W. Lewis, A. R. Ruiz-Salvador, A. Gomez, L. M. Rodriguez-Albelo, F.-X. Coudert, B. Slater, A. K. Cheetham and C. Mellot-Draznieks, CrystEngComm, 2009, 11 Search PubMed.
- D. Fairen-Jimenez, S. A. Moggach, M. T. Wharmby, P. A. Wright, S. Parsons and T. Duüren, J. Am. Chem. Soc., 2011, 133, 8900–8902 CrossRef CAS.
- R. Huey, G. M. Morris, A. J. Olson and D. S. Goodsell, J. Comput. Chem., 2007, 28, 1145–1152 CrossRef CAS.
- P. J. Goodford, J. Med. Chem., 1985, 28, 849–857 CrossRef CAS.
- M. Valiev, E. J. Bylaska, N. Govind, K. Kowalski, T. P. Straatsma, H. J. J. van Dam, D. Wang, J. Nieplocha, E. Apra, T. L. Windus and W. A. de Jong, Comput. Phys. Commun., 2010, 181, 1477–1489 CrossRef CAS.
- M. K. Rana, F. G. Pazzona, G. B. Suffritti and F. Demontis, J. Chem. Theory Comput., 2010, 7, 1575–1582 CrossRef.
- T. A. Soares, D. S. Goodsell, R. Ferreira, A. J. Olson and J. M. Briggs, J. Mol. Recognit., 2000, 13, 146–156 CrossRef CAS.
- T. A. Soares, D. S. Goodsell, J. M. Briggs, R. Ferreira and A. J. Olson, Biopolymers, 1999, 50, 319–328 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: infrared spectra, TGA/DTGA curves, and SEM analysis. See DOI: 10.1039/c2ra21087h |
| This journal is © The Royal Society of Chemistry 2012 |