DOI:
10.1039/C6RA08022G
(Paper)
RSC Adv., 2016,
6, 44506-44513
PLA-based thermogel for the sustained delivery of chemotherapeutics in a mouse model of hepatocellular carcinoma†
Received
28th March 2016
, Accepted 25th April 2016
First published on 26th April 2016
Abstract
A thermogelling poly(ester urethane) comprising poly(ethylene glycol) (PEG), poly(propylene glycol) (PPG) and poly(lactic acid) (PLA) blocks was synthesized. Drug release studies of the thermogel were carried out using paclitaxel (PTX). The release rate of the drug can be achieved by changing the concentration of the gel, greatly prolonging the release and elimination times to afford long-term effects. The thermogels showed very low toxicity on HEK293 cells. A nude mice model of hepatocellular carcinoma was developed and intratumoural injection of drug-loaded thermogel showed that PTX-loaded thermogel effectively inhibited the growth of tumours.
Introduction
There has been an enormous increase in human cancers in the last three decades. In the United States, more than 140 million new patients were diagnosed with cancer annually, among which nearly 40% of those diagnosed died from various cancers.1 Liver cancer is the sixth-most common cancer worldwide.2 Hepatoma occurrences has increased dramatically in Europe and the United States recently as a result of the widespread occurrence of hepatitis C.3,4 It has very poor prognosis and has recorded the third-most incidences of death from cancer. Typical survival rates range from 3–5%. (1) Even with surgical therapies, the prognosis for hepatocellular carcinoma (HCC) is fairly bleak for many patients.5 A complicating factor is that the number of suitable patients ranges from between 20% and 35% amongst all the cases diagnosed. Furthermore, the recurrence of cancer still poses a risk and requires local radiotherapy or systemic chemotherapy for complete removal. The therapeutic activity of antineoplastic drugs can be improved through using direct intratumoural injection in systemic chemotherapy.6,7 An ideal drug delivery system need to be able to deliver drugs at sufficiently high concentrations to the tumour to achieve better therapeutic effect. Paclitaxel (PTX) is one of the most effective anticancer drugs. It originates from the bark of the Pacific yew, Taxus brevifolia. PTX is widely used to treat breast, lung and gastro-intestinal cancers.8 The drug polymerizes microtubule in the G2 phase of the mitosis and functions with β-subunit of tubulin. Through the formation of stable microtubules that inhibit mitosis, cell death of the cancer cells can be achieved.9 PTX is particularly effective against breast, ovarian, head-and-neck, acute leukemia and lung cancer.10 PTX has also been used as a drug in the treatment of HCC.11 However, a problem could be the acquisition of drug resistance in HCC patents, rendering the PTX therapy less effective.12 In the delivery of PTX, one of the main challenge is that PTX is poorly water-soluble. As a result, PTX is used mainly as Taxol, which is made up of PTX in a mixture of polyethoxylated castor oil, Cremophor EL, and ethanol. However, the formulation contains Cremophor EL which cause severe side effects, such as hypersensitivity reactions, nephrotoxicity and neurotoxicity, which could lead to death of the patient.13 In order to overcome the challenges associated with the administration of PTX, many researchers have been focused on the development of new delivery systems for PTX. Recent micelle systems encapsulating PTX appear to have completely released all the drug within 40 h to 80 h.14,15 However, administration of micellar formulations would be challenging as the drug formulation could be circulated throughout the body and need to be shielded from the macrophages in the body to avoid being destroyed before achieving therapeutic efficacy.16,17 Thermogels have been used for various biomedical applications.18–22 Jeong et al. showed that they can be used for encapsulation and differentiation of stem cells.23–28 Recently, these thermogels have achieved excellent results as polymeric drug carriers.29–34 Recently, another paper described a formulation with irinotecan (IRN) and poly(D,L-lactide-co-glycolide)-b-poly(ethylene glycol)-b-poly(D,L-lactide-co-glycolide) (PLGA–PEG–PLGA).35 Subcutaneous administration of the drug loaded thermogels into nude mice with xenografted SW620 human colon tumours resulted in excellent in vivo antitumour efficacy as compared with controls. In another report, PEG–PLGA–PEG triblock copolymer was used as an embolic agent and sustained drug delivery system for radio-labeled Lipiodol to form a new radio-thermogelling emulsion.36 The therapeutic potential of this system was evaluated in a rodent hepatoma model. The studies demonstrated protracted reductions in tumour volumes and 75% of the hepatoma-bearing rats survived. Chitosan thermogels have been developed for the local expansion and delivery of tumour-specific T lymphocytes for enhanced cancer immunotherapies.37 Hydrogels, developed as stimuli-responsive materials, exhibit phase change with a change of environmental conditions such as temperature or pH are widely used in the medical field.38 Poly[(R)-3-hydroxybutyrate] (PHB), a member of the polyhydroxyalkanoate family, have been drawing attention as a useful biomaterial because of its biodegradability and biocompatibility.39 We have previously reported the synthesis of poly(PEG/PPG/PHB urethane)s.40–42 Associated micelles are formed by the amphiphilic multiblock copolymers at a highly diluted solution (99.9% water, 0.1% polymer). These self-assembled micelles have PEG hydrophilic tails that interact with water and hydrophobic cores that consist of PPG and PHB. A minimum polymer concentration (2–5 wt%) and optimum micelle concentration, is necessary for gel formation. At low temperature, the aqueous solution is clear because the PPG segments behave more hydrophilic causing the polymers to be well-solvated in water. Increasing the temperature resulted in the dehydration of PEG segments, and the entire polymer becomes more hydrophobic above its LCST. Micellar aggregation due to self-association of PEG corona and increased hydrophobicity of PPG drives the gel formation. PHB, added as a third component in this system, enhanced the hydrophobicity, leading to more mechanically robust gel as compared to Pluronics F127, a commercially available thermogelling polymer. In this paper, we report the incorporation of PLA into the polymer backbone to enhance the hydrolytic degradability of the polymer. PLA is a hydrolytically more liable than PHB and we expect the degradation rate to be enhanced significantly. PLA is FDA approved and to date, there has been no studies on the in vivo efficacy of drug delivery using the PLA-based thermogels. Most of the work has been limited to vesicle/micelle drug delivery, polymer blends and insoluble polymers for packaging.43–48 We use these gels to demonstrate the effective anticancer drug delivery in vivo and show that this material has the potential to be used to inhibit the growth of hepatocellular carcinoma.
Experimental
1. Materials
L-Lactide, stannous octoate, hexa(ethylene glycol) was supplied by Aldrich. Poly(ethylene glycol) (PEG) and poly(propylene glycol) (PPG) was purchased from Aldrich. The Mn and Mw of PEG were found to be 1890 and 2060, respectively. The Mn and Mw of PPG were found to be 2180 and 2290, respectively. Dibutyltin dilaurate (95%) 1,6-hexamethylene diisocyanate (HMDI) (98%), methanol, diethyl ether, 1,2-dichloroethane (99.8%) and 1,6-diphenyl-1,3,5-hexatriene (DPH) were purchased from Aldrich. 1,2-Dichloroethane was distilled over CaH2 before use. All starting chemicals were used as received.
2. Molecular characterization
NMR spectra were measured at room temperature using Bruker AV-400 and JEOL 500 MHz NMR spectrometers in CDCl3 solvent. Gel permeation chromatography (GPC) analysis was carried out with a Tosoh EcoSEC GPC System equipped with two Styragel® 5μ HR 1 and HR 5E columns (size: 300 × 7.80 mm) in series and a dual flow RI detector. Chloroform was used as eluent at a flow rate of 1.0 mL min−1 at column temperature of 40 °C. Gel permeation chromatography (GPC) analysis was also carried out with a Viscotek GPC max module equipped with two phenogel columns (103 and 105 Å) (size: 300 × 7.80 mm) in series and a Viscotek TDA 305 triple detector. Tetrahydrofuran was used as eluent at a flow rate of 1.0 mL min−1 at column temperature of 40 °C. Mono-dispersed poly(methyl methacrylate) standards were used to obtain calibration curves in both machines.
3. Synthesis of PLA
PLA was prepared by a ring-opening polymerization of L-lactide using stannous octoate as a catalyst. Hexa(ethylene glycol) was used as an initiator. As an example for the synthesis of PLA (Mn = 1000), 1 g of hexa(ethylene glycol) was dissolved in 50 mL of anhydrous toluene in a 250 mL two-neck flask. The solvent was distilled off to a final volume of 20 mL. 5 g of L-lactide and 10 mL of stannous octoate were added to the reaction mixture and stirred at 120C for 6 h. The molecular weight of the polymer was continuously monitored by GPC. When the desired molecular weight was attained, the product was isolated by precipitation into n-hexane. The crude polymer was redissolved in methylene chloride and purified by precipitation in diethyl ether. The yield was 75% after purification. 1H NMR (CDCl3) of PLA: δ (ppm) 1.55–1.64 (–OCH(C![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) 3)CO–), 3.62 (–OC2C2O–), 4.24–4.28 (–O(CH3)CCO–; end group; quartet), 5.12–5.20 (–O(CH3)CCO–).
3)CO–), 3.62 (–OC2C2O–), 4.24–4.28 (–O(CH3)CCO–; end group; quartet), 5.12–5.20 (–O(CH3)CCO–).
4. Synthesis of poly(PEG/PPG/PLA urethane)s
Poly(PEG/PPG/PLA urethane)s were synthesized from PLA, PEG and PPG with PLA content at 10 wt% using HMDI as a coupling reagent. The amount of HMDI added was equivalent to the reactive hydroxyl groups in the solution. 0.3 g of PLA–diol, 1.8 g of PEG and 0.9 g of PPG were dried in a 250 mL two-neck flask at 50 °C under high vacuum overnight. Then, 20 mL of anhydrous 1,2-dichloroethane was added to the flask and any trace of water in the system was removed through azeotropic distillation with only 1 mL of 1,2-dichloroethane being left in the flask. When the flask was cooled down to 75 °C, 0.27 g of HMDI and two drops of dibutyltin dilaurate (∼8 × 10−3 g) were added sequentially. The reaction mixture was stirred at 75 °C under a nitrogen atmosphere for 48 h. The resultant copolymer was precipitated from diethyl ether, and further purified by redissolving into 1,2-dichloroethane followed by precipitation in a mixture of methanol and diethyl ether to remove remaining dibutyltin dilaurate. The yield was 80% and above after isolation and purification. 1H NMR (CDCl3) of poly(PEG/PPG/PLA urethane): δ (ppm) 1.12 (–O(C3)CHCH2O–), 1.31 (–OOCNHCH2CH2C2C2CH2CH2NHCOO–), 1.47 (–OOCNHCH2C2CH2CH2C2CH2NHCOO–), 1.56 (–O(C3)CHCO–), 3.13 (–OOCNHC2CH2CH2CH2CH2C2NHCOO–), 3.38 (–O(CH3)CCH2O–), 3.45 (–O(CH3)CHC2O–), 3.63 (–OC2C2O–), 4.19 (–OOCNCH2CH2CH2CH2CH2CH2NCOO–), 5.05–5.15 (–O(CH3)CCO–). 13C NMR (CDCl3) of poly(PEG/PPG/PLA urethane)s: δ (ppm) 17.42 (–O(![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) H3)CHCO–), 17.71 (–O(H3)CHCH2O–), 26.69 (–OOCNHCH2CH2H2H2CH2CH2NHCOO–), 30.26 (–OOCNHCH2H2CH2CH2H2CH2NHCOO–), 64.25 (–OOCNHH2CH2CH2CH2CH2H2NHCOO–), 69.32 (–O(CH3)HCO–), 70.99 (–OH2H2O–), 72.28 (–O(CH3)CHH2O–), 75.77 (–O(CH3)HCH2O–), 156.84 (–OONHCH2CH2CH2CH2CH2CH2NHOO–), 170.15 (–O(CH3)CHO–).
H3)CHCO–), 17.71 (–O(H3)CHCH2O–), 26.69 (–OOCNHCH2CH2H2H2CH2CH2NHCOO–), 30.26 (–OOCNHCH2H2CH2CH2H2CH2NHCOO–), 64.25 (–OOCNHH2CH2CH2CH2CH2H2NHCOO–), 69.32 (–O(CH3)HCO–), 70.99 (–OH2H2O–), 72.28 (–O(CH3)CHH2O–), 75.77 (–O(CH3)HCH2O–), 156.84 (–OONHCH2CH2CH2CH2CH2CH2NHOO–), 170.15 (–O(CH3)CHO–).
5. Sol–gel transition
The sol–gel transition was determined by a test tube inverting method with temperature increments of 2 °C per step. Each sample of a given concentration was prepared by dissolving the polymer in distilled water in a 2 mL vial. After equilibration at 4 °C for 24 h, the vials containing samples were immersed in a water bath at a constant designated temperature for 15 min. The gelation temperature was characterized by the formation of a firm gel that remained intact for 2 min when the tube was inverted by 180°. The critical gelation concentration (CGC) is defined as the minimum copolymer concentration in aqueous solution at which the gelation behavior could be observed.
6. Preparation of PTX loaded thermogel
The copolymer was added in normal saline (n.s.) with at a concentration of 20 wt% and 25 wt% at 4 °C for 24 h to form blank thermogels. PTX was mixed directly into the thermogelling solution. 0.2 mg of drug was added into the thermogel (20 wt% or 25 wt%, 2 mL) to form an injectable solution at 4 °C. The prepared drug delivery system was kept at 4 °C before use.
7. In vitro drug release studies of thermogel
The drug release behavior of the thermogels were studied with PBS (pH = 7.4) as a release medium. 0.1 mg of PTX were added in 1 mL of 20 wt% or 25 wt% thermogel at 4 °C. In a typical example, 1 mL of polymer solution was injected into a porous cellulose cassette (pore size: ∼100 μm) and left to equilibrate at 37 °C. The polymer hydrogel obtained had dimensions of 10 mm × 25 mm × 4 mm and was placed in 25 mL of phosphate buffer release solutions in a test tube, which was incubated and shaken at 50 rpm in a water bath at 37 °C. The buffer solutions were replaced with fresh ones at predetermined time intervals, and the experiments were done in triplicate. The collected buffer solutions were lyophilized and kept at −80 °C for further analysis. PTX was assayed by HPLC with a mobile phase of H2O/acetonitrile (ACN) (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 v/v) at 273 nm.
:50 v/v) at 273 nm.
8. Cell and animals
Cells were grown in Dulbecco's Modified Eagle's Medium (DMEM) containing 10% fetal bovine serum (FBS), penicillin (100 U mL−1) and streptomycin (100 μg mL−1) in a 5% CO2 incubator at 37 °C. Female BALB/c mice (6–8 weeks old) were purchased from the Xiamen University Laboratory Animals Centre. Animal experiments were performed in Xiamen University Laboratory Animal Centre. All animal treatments and surgical procedures followed approved protocols and were performed in accordance with the Animal Care Guidelines of Xiamen University.
9. Cytotoxicity studies
For the cytotoxicity study of the thermogel, 1 × 104 HEK293 cells (ATCC) were plated in a 96 well plate and incubated 12 h at 37 °C incubator containing 5% CO2. Cells were treated with the copolymer solution of ranging from 1 μg mL−1 to 100 μg mL−1. After 24 h, MTT was added in each well with a volume of 10 μL. The plate was incubated at 37 °C for 4 h, and samples were tested with microplate reader at absorbance of 495 nm.
10. In vivo tumour growth
Nude mice were fed in sterilized cages with sterile food and water and filtered air, which were handled under a laminar flow hood using aseptic techniques. 2 × 106 cells in a 0.2 mL suspension were injected subcutaneously into the back of each animal to induce tumour formation. When the tumours reached 140 mm3, the experimental day 0 was defined. Eighteen hairless mice (6 weeks old) were divided randomly into six groups. Each group contains three mice. The six solutions were prepared in advance, which were (1) normal saline, (2) PTX only, (3) 20 wt% thermogel only, (4) 25 wt% thermogel only, (5) PTX-loaded 20 wt% thermogel (0.2 mg PTX), (6) PTX-loaded 25 wt% thermogel. On the day 0, 100 μL of these solutions was subcutaneously injected into the tumour with a 1 mL syringe. The tumour diameters were measured in two dimensions every day with vernier calipers. The tumour volume (V) was calculated based on the formula: V = [length × (width)2]/2. On day 7, the mice were sacrificed and the weights of tumours were measured.
11. Histological analysis
On day 7, the mice were sacrificed and the tumours were removed from the subcutaneous dorsum. The tumours were dehydrated with sucrose solution for 24 h. Later, the tumours were sliced into frozen section with a thickness of 2 μm and with H & E.
12. Statistical analysis
The data of the cytotoxicity, tumour size and tumour weight studies were evaluated as means ± standard deviations (SD). The results were analyzed by Origin 8 and GraphPad Prism 5. Analysis of variance (ANOVA), followed by Student's t-test, was used to determine the significant differences among the groups, and p-values less than 0.05 were considered significant.
Results and discussion
1. Synthesis of poly(PEG/PPG/PLA urethane) copolymer and its gel properties
The structure of poly(PEG/PPG/PLA urethane) copolymer used in this study is shown in Scheme 1. This polymer was synthesized by the coupling of di-isocyanates with polyols and has been previously reported by us.49 Telechelic PLA–diol was prepared by the ring-opening polymerization of L-lactide using hexa(ethylene glycol) (Scheme S1†). Poly(PEG/PPG/PLA urethane)s were prepared by the randomly coupling PLA, PEG and PPG segment blocks using 1,6-hexamethlyene diisocyanate (HMDI) in the presence of dibutyltin dilaurate (Scheme S1†).
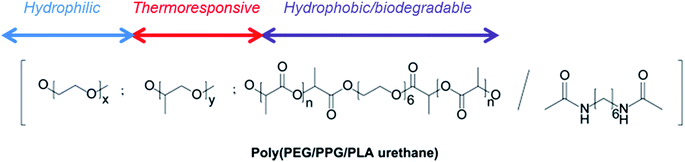 |
| | Scheme 1 Structure of poly(PEG/PPG/PLA urethane) copolymer. | |
The thermogel used in this system is made from PEG and PPG of ∼2000 kDa and PLA of ∼1000 kDa. The NMR of the PLA is shown in Fig. S1.† The GPC profile of PLA is shown in Fig. S2.† FTIR of PLA is presented in Fig. S3,† showing the presence of the ester bonds in PLA. The PLA content is 10 wt%, PEG content is 60 wt% and PPG content is 30 wt%. The molecular weight (Mn) of the polymer was 34.5 kDa with a PDI of 1.3. The FTIR of the poly(PEG/PPG/PLA urethane) is shown in Fig. S3.† Thermogravimetric analysis (TGA) was used to evaluate the thermal stability of poly(PEG/PPG/PLA urethane). The degradation of pure PLA–diol starts at 220 °C and completes at 320 °C, PPG starts to degrade at 350 °C while pure PEG starts at 400 °C, at which pure PLA–diol has completed the degradation.49 The copolymer undergoes a three-step thermal degradation with the first step occurring between 290 and 310 °C and the second and third steps between 320 and 460 °C. These polymers were soluble in water. At a concentration of 1 wt%, the polymers formed micelles with an average size of ∼50 nm, according to dynamic light scattering experiments (Fig. S4†). The thermogelling behavior of the copolymer was next evaluated. The phase diagram of the polymer in aqueous solution is presented in Fig. 1. The phase diagram of the poly(PEG/PPG/PLA urethane) in aqueous solutions was determined by the test tube inverting method. The lower soluble region, gel region and the upper soluble region can be observed in the diagram. As the temperature increased monotonically from 4 to 80 °C, the aqueous polymer solution underwent a sol–gel–sol transition. The reversible change also took place upon cooling from 80 to 4 °C. The critical gelation concentration (CGC) is defined as the minimum copolymer concentration in aqueous solution at which the gelation behavior could be observed. The CGCs of the copolymer in this work was 10 wt%.
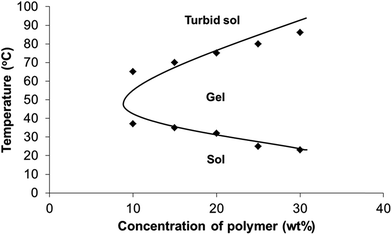 |
| | Fig. 1 Phase diagram of the poly(PEG/PPG/PLA urethane) thermogel used in this study. | |
2. Thermogels for the delivery of drugs and cytotoxicity effects
In our study, we loaded PTX into the thermogel system and assessed the therapeutic effects of the drug delivery in a BALB/c nude mice model. Current cancer therapy includes a combination therapy involving surgery, radiation and chemotherapy to reduce any existing cancer. It is aimed at eradicating the residual tumour cells with chemotherapy and radiation. In this study, we use thermogelling poly(PEG/PPG/PLA urethane)s, with good biocompatibility, as a delivery system of chemotherapeutics for tumour therapy. Before loading drugs into the thermogel, we examined the biocompatibility of the block copolymer. The in vitro cytotoxicity was first evaluated using a MTT assay. According to the results in Fig. 2, the thermogelling polymer exhibited slight cytotoxicity against HEK293 cells with a cell viability of over 90% in the culture medium with polymer concentrations as high as 1000 μg mL−1. In contrast, cells usually do not survive in that concentration in common surfactants. Here, the thermogel has very low toxicity in vitro, suggesting that it will not kill normal cells and can possibly be safely applied in vivo. PTX was used as model drugs for the chemotherapeutic treatment in vivo. Control of the release rate of the drug can be achieved by changing the thermogel concentration. The release profile is presented in Fig. 3. The PTX concentration was within the reported concentration of administration.50,51
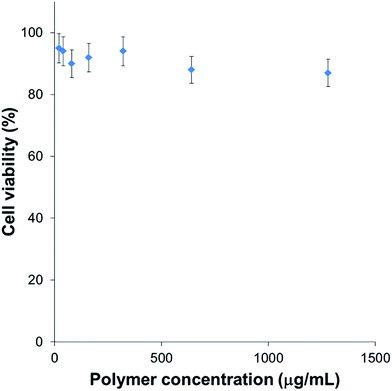 |
| | Fig. 2 Toxicity of polymer against HEK293 cells. | |
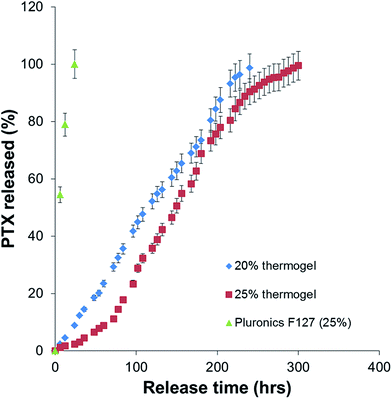 |
| | Fig. 3 PTX release profiles for poly(PEG/PPG/PLA urethane) hydrogels of different copolymer compositions (20 and 25 wt%) in comparison with PEG–PPG–PEG triblock copolymer (25 wt%). | |
Sustained release profiles of the gels with the different polymer concentrations were compared. The period of sustained release can be controlled by adjustment of the concentration of the polymer in the thermogelling solution from 20 wt% (240 h) to 25 wt% (300 h). When an extended sustained release is desired, the thermogelling solution can be made more concentrated, leading to greater packing in the gel structure. This demonstration is consistent with other reported drug release studies. Muley et al. reported an almost complete release of paclitaxel after just 40 h whereas Ma et al. showed that the release of PTX from a Pluronic F127 based micelle could only be prolonged for up to 80 hours.14,15 In our case, we have extended the duration of the release by at least 300% by simply creating a gel semi-solid form which acts as a physical barrier for the slow release of the drug in solution. Comparatively, the control thermogel Pluronics F127 dissolves completely within 24 h and releases its entire payload of drug during that interval. This short burst release is not suitable for our next section of the study which was the intratumoural delivery of the drug in vivo. This study also shows that the entire drug content loaded in the gel can be released during the course of the drug release study. This is a distinct advantage from other gel systems which are unable to achieve complete release of the loaded drug. The drug release profiles has two distinct phases, (1) erosion sub-stage 1 (period of no mass loss of the thermogel) and (2) linear polymer erosion phase (sub-stage 2). At the initial stages of the release experiment (erosion sub-stage 1), the gel exists as a tightly packed structure. There is an induction period whereby there is little release of PTX. After the induction period (24 h for 20 wt% thermogel and 72 h for 25 wt% thermogel), PTX starts to be released via the diffusion through water-rich regions of the gel structure. Diffusivity of a solute through physically crosslinked hydrogels decreases with an increase in the crosslinking density and with a decrease of the volume fraction of solvent within the hydrogel. Increasing the concentration of the polymer in our thermogelling formulation reduced the proportion of the solvent-rich regions, leading to a decrease in the release of PTX as observed. After some time, erosion of the polymer gel structure follows. During the linear polymer erosion phase (sub-stage 2), a linear release of PTX with respect to time was observed. The release of PTX is dominated by the erosion of the polymer gel leading to a linear profile in the release of the protein. The rate of release of PTX is affected by the concentration of the polymer in the formulation. This example shows that the packing of the gel structure is an important factor in the determination of the release rate of PTX. Our experimental results showed that better packing tends to result in greater resistance against erosion and manifests itself as a longer lasting sustained release profile.
3. Delivery of drugs in vivo to hepatocellular carcinoma
We found that the thermogel drug delivery system containing 20 to 25 wt% polymer could persist for at least 7 days in vivo by subcutaneous administration. The initial tumour volumes were all pre-designed to be approximately 140 mm3 before drug administration; the actual experimental values were 147 mm3 on average. The final tumour volume of the NS group was 590 mm3 ± 138 mm3. For the group in which only PTX was administered, the growth rate was slower than that of the normal saline (NS) group (p < 0.05). The tumour volume increased continuously during the entire therapy, reaching a final volume of about 315 mm3 ± 77 mm3. For the PTX/thermogel groups, the tumour volume decreased significantly with the average of the final values of the relative tumour volume being about 170 mm3, indicating significant inhibition of the growth of the tumour after treatment with the PTX/thermogel (p < 0.05). The tumour growth curve is shown in Fig. 4a. The weights of the tumours treated with PTX/thermogel were also significantly lower than the NS group after the 7 day therapy, with the weight at about 30% of that in the control group (p < 0.05) (Fig. 4b). The gels were injected nicely into the tumour in the mice and remained in place 1 day after administration (Fig. 4c). The mice were sacrificed after 7 days and tumours were extracted. It is clear that the drug loaded thermogels inhibited the growth of the tumour significantly (p < 0.05) (Fig. 4d).
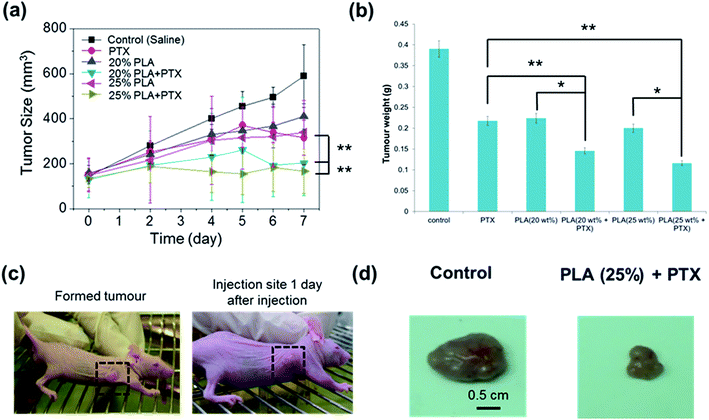 |
| | Fig. 4 (a) Inhibition of tumour volume by intratumoural injection of saline, PTX or PTX-loaded thermogels. Dorsal subcutaneous implantation of HepG2 cancer cells into mice was followed by administration of each solution after tumours had reached a volume of ∼140 mm3. (b) Inhibition of tumour weight by intratumoural injection of 100 μL of saline, PTX or PTX-loaded PLA gels. Dorsal subcutaneous implantation of HepG2 cancer cells into mice was followed by administration of each solution after tumours had reached a volume of ∼140 mm3. The excised tumour removed after 7 days was weighted for evaluation. (c) Induction of tumour growth in mice and the injection site 1 day after injection. (d) The excised tumour removed after 7 days treated with or without PTX treatment. Scale bars represent 0.5 cm *p < 0.01 vs. non-drug loaded thermogel; **p < 0.001 vs. direct injection of PTX without thermogel. | |
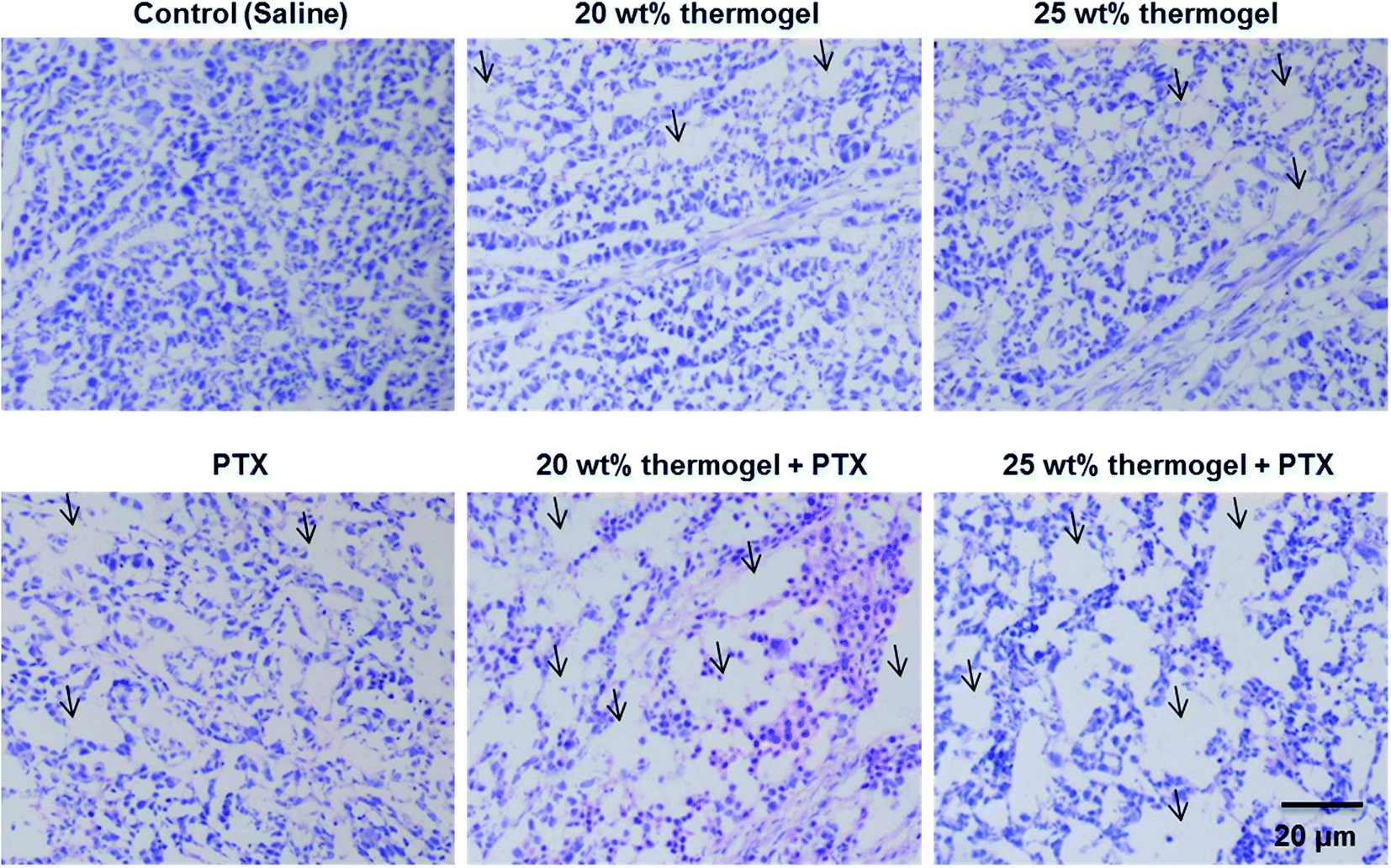
It is also interesting to note that while the administration of PTX alone did not work very well in inhibiting the growth of the tumour, the use of the thermogel as a carrier for PTX enhanced the tumour growth inhibition significantly. The biocompatibility and biodegradability of these PLA-based thermogels makes them a useful biomaterial delivery platform for the development of localized chemotherapy. In this study we focused the application of thermogels in the treatment of hepatocellular-carcinoma, and we expect that the use of these thermogels could be easily translated to a number of other cancers where local minimally invasive treatment is desirable. The thermogelling solutions were injectable at room temperature and formed gel depots containing the chemotherapeutic drugs at the injection site. These thermogels exhibit minimal batch to batch variation, easy storage, handling and preparation in the procedure and they fit the odd shapes of the tumour sites perfectly. Gel systems such as these present the potential for long term delivery of drugs in comparison to micelle systems which typically are unable to sustain the release of drug for more than 5 days.52 The local inflammatory response was evaluated by histological observation 7 days after a subcutaneous injection of the thermogel. H & E staining is a standard method used for the diagnosis of various diseases. It is widely used in the observation and identification of cell apoptosis and tissue necrosis. In normal tissues, the cell nuclei will be stained by alkaline hematoxylin which renders it blue-violet, while the extracellular matrix and cytoplasm show up in pink due to the reaction between the protein and acidic eosin. Necrotic cells tend to lose their normal cellular morphologies. Signs of nuclear pyknosis and fragmentation can also be detected. Interestingly, few apoptotic or necrotic tumour cells in the center of tumors from the saline group could be observed. Distinctively, both the periphery and the center of tumors from the PTX, 20 wt% thermogel and 25 wt% thermogel group displayed more apoptosis or necrosis area (as indicated in the Fig. 5), whereas the largest area of cell damage could be observed in 20 wt% thermogel + PTX or 25 wt% thermogel + PTX group, confirming the excellent in vivo anti-tumour efficacy by taking the advantage of sustained release thermogel depot. Similar reports could be found in other reports.53,54 This experimental evidence indicates that the application of thermogel only might act as a blockage of nutrient supply of solid tumour and causes some necrosis or apoptosis, which might account for the influence of the PLA thermogel in tumour growth. Moreover, one thing that needs to be mentioned was that the appearance of this interesting phenomenon might to due to the injection of large amount of thermogel into solid tumors, considering the injection of 100 μL (or 100 mm3) thermogel into tumour with initial volume of around 140 mm3. We also observed the clear expansion of tumour volume after hydrogel or saline injection. As the absorption and metabolism of liquid saline is much faster than that of solid hydrogel, we could observe fast shrinkage of tumour volume after saline injection but not in the case of hydrogel, which provided the possible physical barrier of nutrient supply to tumor. Hence, we considered that the tumour growth inhibition mechanism of thermogel only was very different from PTX, which caused the cell cycle arrest and apoptosis.
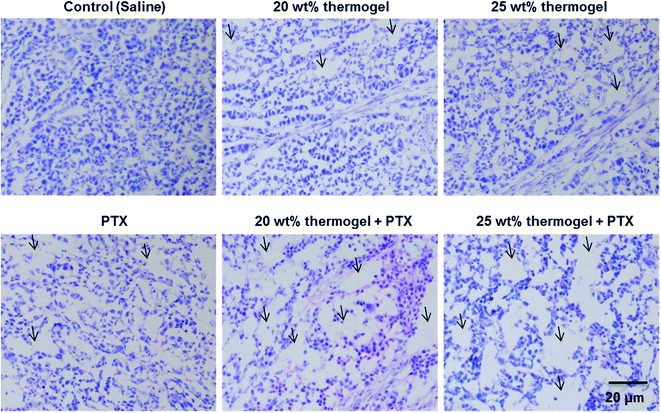 |
| | Fig. 5 H & E-stained histologic section of tumour treated with saline, PTX, 20 wt% thermogel, 25 wt% thermogel or PTX-loaded thermogels 7 days after administration. Arrows indicate the inflammatory cells. | |
Conclusion
Drug-loaded thermogels were studied for the potential application of using them for chemotherapeutic treatment of hepatocellular-carcinoma. We showed that the use of a sustained release thermogel depot enhanced the localisation of the drug at the tumour site leading to a greater inhibition of the growth of the tumour. This work represents the first time that the poly(PEG/PPG/PLA urethane)s have been used for the delivery of drugs to tumours in vivo and the encouraging results points to the potential for further development of this thermogel platform for anti-cancer applications.
Acknowledgements
The authors would like to express gratitude to the A*STAR Personal Care Grant (Project no. 1325400026) for support of this project. The authors would also acknowledge the financial support from the Natural Science Foundation (Grant No. 21303145), the Science and Technology Project of Fujian Province, China (Grant No. 2014J01063).
References
- A. Jemal, R. Siegel, E. Ward, Y. Hao, J. Xu, T. Murray and M. J. Thun, Ca-Cancer J. Clin., 2008, 58, 71–96 CrossRef PubMed.
- D. M. Parkin, F. Bray, J. Ferlay and P. Pisani, Ca-Cancer J. Clin., 2005, 55, 74–108 CrossRef PubMed.
- R. Capocaccia, M. Sant, F. Berrino, A. Simonetti, V. Santi and F. Trevisani, Am. J. Gastroenterol., 2007, 102, 1661–1670 CrossRef PubMed.
- P. Jepsen, H. Vilstrup, R. E. Tarone, S. Friis and H. T. Sørensen, Int. J. Cancer, 2007, 121, 1624–1626 CrossRef CAS PubMed.
- T. Y. Lin, C. S. Lee, K. M. Chen and C. C. Chen, Br. J. Surg., 1987, 74, 839–842 CrossRef CAS PubMed.
- E. P. Goldberg, A. R. Hadba, B. A. Almond and J. S. Marotta, J. Pharm. Pharmacol., 2002, 54, 159–180 CrossRef CAS PubMed.
- T. Parvez, Technol. Cancer Res. Treat., 2008, 7, 241–248 CrossRef PubMed.
- B. Y. Ong, S. H. Ranganath, L. Y. Lee, F. Lu, H. S. Lee, N. V. Sahinidis and C. H. Wang, Biomaterials, 2009, 30, 3189–3196 CrossRef CAS PubMed.
- P. Singh, K. Rathinasamy, R. Mohan and D. Panda, IUBMB Life, 2008, 60, 368–375 CrossRef CAS PubMed.
- A. K. Singla, A. Garg and D. Aggarwal, Int. J. Pharm., 2002, 235, 179–192 CrossRef CAS PubMed.
- Y. Chao, W. K. Chan, M. J. Birkhofer, O. Y. Hu, S. S. Wang, Y. S. Huang, M. Liu, J. Whang-Peng, K. H. Chi, W. Y. Lui and S. D. Lee, Br. J. Cancer, 1998, 78, 34–39 CrossRef CAS PubMed.
- A. S. Meena, A. Sharma, R. Kumari, N. Mohammad, S. V. Singh and M. K. Bhat, PLoS One, 2013, 8, e61524 Search PubMed.
- C. Chun, S. M. Lee, S. Y. Kim, H. K. Yang and S. C. Song, Biomaterials, 2009, 30, 2349–2360 CrossRef CAS PubMed.
- P. Muley, S. Kumar, F. El Kourati, S. S. Kesharwani and H. Tummala, Int. J. Pharm., 2016, 500, 32–41 CrossRef CAS PubMed.
- Y. Ma, X. Fan and L. Li, Carbohydr. Polym., 2016, 137, 19–29 CrossRef CAS PubMed.
- V. P. Torchilin, Pharm. Res., 2007, 24, 1–16 CrossRef CAS PubMed.
- H. Otsuka, Y. Nagasaki and K. Kataoka, Adv. Drug Delivery Rev., 2003, 55, 403–419 CrossRef CAS PubMed.
- Y.-L. Wu, X. Chen, W. Wang and X. J. Loh, Macromol. Chem. Phys., 2016, 217, 175–188 CrossRef CAS.
- S. S. Liow, Q. Dou, D. Kai, A. A. Karim, K. Zhang, F. Xu and X. J. Loh, ACS Biomater. Sci. Eng., 2016, 2, 295–316 CrossRef CAS.
- Z. B. Li and X. J. Loh, Chem. Soc. Rev., 2015, 44, 2865–2879 RSC.
- E. Y. Ye, P. L. Chee, A. Prasad, X. T. Fang, C. Owh, V. J. J. Yeo and X. J. Loh, Mater. Today, 2014, 17, 194–202 CrossRef CAS.
- Q. Q. Dou, S. S. Liow, E. Y. Ye, R. Lakshminarayanan and X. J. Loh, Adv. Healthcare Mater., 2014, 3, 977–988 CrossRef CAS PubMed.
- M. Patel, H. J. Moon, B. K. Jung and B. Jeong, Adv. Healthcare Mater., 2015, 4, 1565–1574 CrossRef CAS PubMed.
- M. H. Park, H. J. Moon, J. H. Park, U. P. Shinde, D. Y. Ko and B. Jeong, Macromol. Biosci., 2015, 15, 464–472 CrossRef CAS PubMed.
- J. Park, I. Y. Kim, M. Patel, H. J. Moon, S. J. Hwang and B. Jeong, Adv. Funct. Mater., 2015, 25, 2573–2582 CrossRef CAS.
- M. H. Park, Y. Yu, H. J. Moon, D. Y. Ko, H. S. Kim, H. Lee, K. H. Ryu and B. Jeong, Adv. Healthcare Mater., 2014, 3, 1782–1791 CrossRef CAS PubMed.
- S. J. Kim, M. H. Park, H. J. Moon, J. H. Park, D. Y. Ko and B. Jeong, ACS Appl. Mater. Interfaces, 2014, 6, 17034–17043 CAS.
- B. Yeon, M. H. Park, H. J. Moon, S. J. Kim, Y. W. Cheon and B. Jeong, Biomacromolecules, 2013, 14, 3256–3266 CrossRef CAS PubMed.
- A. Vintiloiu and J.-C. Leroux, J. Controlled Release, 2008, 125, 179–192 CrossRef CAS PubMed.
- L. Yu, K. Li, X. J. Liu, C. Chen, Y. C. Bao, T. Y. Ci, Q. H. Chen and J. D. Ding, J. Pharm. Sci., 2013, 102, 4140–4149 CrossRef CAS PubMed.
- L. Yu, T. Y. Ci, S. C. Zhou, W. J. Zeng and J. D. Ding, Biomater. Sci., 2013, 1, 411–420 RSC.
- L. Yu, T. Y. Ci, S. C. Zhou and J. D. Ding, J. Controlled Release, 2013, 172, E53 CrossRef.
- T. Li, L. Chen, T. Y. Ci, L. Yu and J. D. Ding, J. Controlled Release, 2013, 172, E68–E69 CrossRef CAS.
- K. Li, L. Yu, X. J. Liu, C. Chen, Q. H. Chen and J. D. Ding, Biomaterials, 2013, 34, 2834–2842 CrossRef CAS PubMed.
- T. Y. Ci, L. Chen, L. Yu and J. D. Ding, Sci. Rep., 2014, 4, 5473 CAS.
- Y. H. Shih, X. Z. Lin, C. H. Yeh, C. L. Peng, M. J. Shieh, W. J. Lin and T. Y. Luo, Int. J. Nanomed., 2014, 9, 4191–4201 Search PubMed.
- A. Monette, C. Ceccaldi, E. Assaad, S. Lerouge and R. Lapointe, Biomaterials, 2016, 75, 237–249 CrossRef CAS PubMed.
- A. K. Bajpai, S. K. Shukla, S. Bhanu and S. Kankane, Prog. Polym. Sci., 2008, 33, 1088–1118 CrossRef CAS.
- I. S. Aldor and J. D. Keasling, Curr. Opin. Biotechnol., 2003, 14, 475–483 CrossRef CAS PubMed.
- X. J. Loh, S. H. Goh and J. Li, J. Phys. Chem. B, 2009, 113, 11822–11830 CrossRef CAS PubMed.
- X. J. Loh, S. H. Goh and J. Li, Biomacromolecules, 2007, 8, 585–593 CrossRef CAS PubMed.
- X. J. Loh, S. H. Goh and J. Li, Biomaterials, 2007, 28, 4113–4123 CrossRef CAS PubMed.
- J. Bonilla, E. Fortunati, M. Vargas, A. Chiralt and J. M. Kenny, J. Food Eng., 2013, 119, 236–243 CrossRef CAS.
- T. Jin and H. Zhang, J. Food Sci., 2008, 73, M127–M134 CrossRef CAS PubMed.
- J.-W. Rhim, H.-M. Park and C.-S. Ha, Prog. Polym. Sci., 2013, 38, 1629–1652 CrossRef CAS.
- F. Ahmed and D. E. Discher, J. Controlled Release, 2004, 96, 37–53 CrossRef CAS PubMed.
- S. A. Hagan, A. G. A. Coombes, M. C. Garnett, S. E. Dunn, M. C. Davis, L. Illum, S. S. Davis, S. E. Harding, S. Purkiss and P. R. Gellert, Langmuir, 1996, 12, 2153–2161 CrossRef CAS.
- E. S. Lee, K. T. Oh, D. Kim, Y. S. Youn and Y. H. Bae, J. Controlled Release, 2007, 123, 19–26 CrossRef CAS PubMed.
- X. J. Loh, Y. X. Tan, Z. Y. Li, L. S. Teo, S. H. Goh and J. Li, Biomaterials, 2008, 29, 2164–2172 CrossRef CAS PubMed.
- E. K. Rowinsky, E. A. Eisenhauer, V. Chaudhry, S. G. Arbuck and R. C. Donehower, Semin Oncol., 1993, 20, 1–15 CAS.
- N. Siddiqui, A. Boddy, H. Thomas, N. Bailey, L. Robson, M. Lind and A. Calvert, Br. J. Cancer, 1997, 75, 287 CrossRef CAS PubMed.
- O. Mezghrani, Y. Tang, X. Ke, Y. Chen, D. Hu, J. Tu, L. Zhao and N. Bourkaib, Int. J. Pharm., 2015, 478, 553–568 CrossRef CAS PubMed.
- H. Chen, B. Li, M. Zhang, K. Sun, Y. Wang, K. Peng, M. Ao, Y. Guo and Y. Gu, Nanoscale, 2014, 6, 12580–12590 RSC.
- L. Shan, S. Cui, C. Du, S. Wan, Z. Qian, S. Achilefu and Y. Gu, Biomaterials, 2012, 33, 146–162 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra08022g |
|
| This journal is © The Royal Society of Chemistry 2016 |