
Curcumin loaded nano graphene oxide reinforced fish scale collagen – a 3D scaffold biomaterial for wound healing applications†
Tapas Mitraa,
Piyali Jana Mannaa,
S. T. K. Rajab,
A. Gnanamanib and
P. P. Kundu*a
aDepartment of Polymer Science & Technology, University of Calcutta, University College of Science & Technology, 92, A.P.C. road, Kolkata 700009, West Bengal, India. E-mail: ppk923@yahoo.com
bMicrobiology Division, CSIR-Central Leather Research Institute, Adyar, Chennai 600020, Tamil Nadu, India
First published on 29th October 2015
Abstract
In recent years, graphene oxide (GO) has been functionalized to make GO a potential useful material in the biomedical field. GO is a one-atom thick planar sheet of sp2-bonded carbon atoms with functional groups containing oxygen attached to both the sides and surface area of the flake. Functionalized GO has attracted significant research interest based on its potential application in different fields including biomedicine. In the present work we prepare highly stabilized nano graphene oxide (NGO) in aqueous media. NGO is functionalized with type I collagen (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, NGO:collagen) to make a 3D scaffold as a novel platform for better tissue engineering research. The functionalization of NGO is achieved by a grafting process, an innovative method to modify the properties of NGO. The size of the prepared NGO is measured by dynamic light scattering, and the collagen functionalized NGO (CFNGO) is characterized by X-ray diffraction, attenuated total reflectance (ATR)-FTIR, ultraviolet visible (UV-vis), atomic force microscopy (AFM) and Raman spectroscopy. The surface property of the CFNGO is characterized by employing transmission electron microscopy (TEM) and scanning electron microscopy (SEM). The mechanical stability of the CFNGO is three times greater than that of native collagen. An in vitro cell line study reveals no toxicity of CFNGO against NIH 3T3 fibroblast cell line. The antimicrobial study of curcumin loaded CFNGO shows Gram +ve and Gram −ve organism growth is reduced by a considerable amount, and in vivo wound healing studies showed faster wound healing efficiency of curcumin loaded CFNGO scaffold than that of collagen alone. These findings suggest that curcumin loaded CFNGO could serve as a better platform for wound healing applications.
:1, NGO:collagen) to make a 3D scaffold as a novel platform for better tissue engineering research. The functionalization of NGO is achieved by a grafting process, an innovative method to modify the properties of NGO. The size of the prepared NGO is measured by dynamic light scattering, and the collagen functionalized NGO (CFNGO) is characterized by X-ray diffraction, attenuated total reflectance (ATR)-FTIR, ultraviolet visible (UV-vis), atomic force microscopy (AFM) and Raman spectroscopy. The surface property of the CFNGO is characterized by employing transmission electron microscopy (TEM) and scanning electron microscopy (SEM). The mechanical stability of the CFNGO is three times greater than that of native collagen. An in vitro cell line study reveals no toxicity of CFNGO against NIH 3T3 fibroblast cell line. The antimicrobial study of curcumin loaded CFNGO shows Gram +ve and Gram −ve organism growth is reduced by a considerable amount, and in vivo wound healing studies showed faster wound healing efficiency of curcumin loaded CFNGO scaffold than that of collagen alone. These findings suggest that curcumin loaded CFNGO could serve as a better platform for wound healing applications.
1. Introduction
The major aim of tissue engineering (TE) is to restore or regenerate tissues and organs using biomimetic fabrication of scaffolds with a similar structure and functionality to the extracellular matrix.1 There has been enormous progress in TE in recent years; many techniques have been developed to design suitable scaffolds for tissue repair. The role of the scaffold is to modulate differentiation and growth behavior, and to control the attachment and sitting patterns, of cells.Collagen, an extracellular protein abundant in animal tissues, is of great interest in TE because of its biological origin, excellent biodegradability and biocompatibility. These characteristics have also contributed to the safe use of collagen in biomedical applications.2,3 Although it has been one of the most preferred biomaterials for in vivo medical applications, difficult processing and poor mechanical properties have been the barriers to its wide use in biomedical and engineering applications. To improve the poor mechanical properties reinforcement with fillers or cross-linking has been used, although the presence of residual cross-linking agents could lead to toxic side effects.4 Among the various types, type I collagen has been extensively used as a TE scaffold and wound dressing system due to its low antigenic and high direct cell adhesion properties. At present, the main sources of type I collagen are bovine or porcine dermis.5 However, due to Bovine Spongiform Encephalopathy (BSE), Transmissible Spongiform Encephalopathy (TSE), and Foot and Mouth Disease (FMD) in pigs and cattle, the use of collagen and collagen derived products from these sources have been limited.6 Type I collagen has also been extracted from skin, bone, fins, and scales of fresh water and marine fishes, chicken skin and other marine animals like squid, octopus, jellyfish, starfish, etc.7–11 Collagen from these sources has been evaluated for their potential application as an alternative to mammalian collagen.
Graphene oxide (GO) is a graphene sheet with carboxylic groups at its edges and phenol hydroxyl and epoxide groups on its basal plane.12 It has been reported that GO is a good TE substrate due to its unique physicochemical properties, including large surface area, high dispersibility and hydrophilicity.13 GO can promote biological interactions due to its many surface functional groups.14,15 Several researchers have reported that GO can serve as a carrier for drugs and other biomolecules.16,17 In addition, GO regulates the proliferation and differentiation of cultured mesenchymal stem cells.18–20 GO will likely be used in combination with other materials21–23 or growth factors24,25 in future medical applications, playing a facilitative role with other TE materials. Furthermore, nano graphene oxide (NGO) has been widely investigated for biomedical applications due to its unique physical, mechanical, and optical properties.26,27 These benefits suggest that if GO has been properly exploited it would solve most of the challenges of clinicians. If collagen has been functionalized with GO, the resulting GO–collagen scaffold can efficiently deliver both hydrophobic and hydrophilic drugs simultaneously. Hydrophobic drugs can be tethered to GO with π–π interactions, whereas, the hydrophilic drug can interact with the collagen through its free primary amine and carboxylic functional groups. The diverse functional groups in GO–collagen scaffolds further facilitate the conjugation of growth factors – DNA, SiRNA, etc. – implying the suitability for a multiple targeted drug delivery system, which is in demand currently.
Curcumin (diferuloylmethane) is an orange-yellow component of the perennial herb Curcuma longa. Curcumin is a naturally occurring multifunctional polyphenolic phytoconstituent which presents anti-inflammatory,28 antimicrobial,29 antiviral,30 anticancer,31 antioxidant,32 and wound healing activities.33 Despite all these promising features, a common problem with curcumin is the very low solubility in aqueous solutions, which limits its bioavailability and clinical efficacy. To overcome the problems of solubility and bioavailability of curcumin, the development of novel delivery systems has attracted significant attention.34 One method is the topical formulation of curcumin to support dermal wound healing.35 Topical applications of curcumin provide antibacterial, anti-inflammatory, and antioxidant (free radical scavenging) activity and patients have shown to have significantly improved wound healing.35 Therefore, development of novel curcumin delivery systems is required.
In the present study we have selected curcumin as a model hydrophobic drug to show the ionic as well as π–π stacking interactions with a GO–collagen scaffold. After the incorporation of the drug, the material’s use as a wound healing material has also been explored.
Thus, the present work has been designed to: (i) improve the mechanical properties of collagen by reinforcing NGO; (ii) exploit the functional groups (oxygen-containing groups including hydroxyls, epoxides, diols, ketones and carboxyls on the surface) of NGO for the conjugation of hydrophobic drugs; and finally (iii) to have a suitable TE material for wound healing applications.
2. Experimental section
2.1. Materials
All the major reagents used in this research were as follows: graphite nanopowder was purchased from SIsco research laboratories and Type I collagen extracted from fish scale; N-(3-dimethylaminopropyl-N′-ethylcarbodiimide)hydrochloride (EDC·HCl), N-hydroxysuccinimide (NHS), picrylsulfonic acid [2,4,6-trinitrobenzene sulfonic acid (TNBS)], 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT), dialysis bags (MWCO = 14000), and curcumin were purchased from Sigma-Aldrich Co.; 2-(N-morpholino)ethanesulfonic acid (MES buffer) was obtained from HiMedia (India); DMEM cell culture medium, fetal bovine serum (FBS), calcein AM, and a cell-permeant dye were purchased from Invitrogen, India. All of the other reagents were of analytical reagent grade and used as received.
2.2. Preparation of graphene oxide sheets
Graphite nano powder was oxidized to nano graphite oxide according to the method followed by Hummers36 with slight modification. In brief, to a 1000 ml beaker, graphite (0.5 g), NaNO3 (0.25 g), and concentrated H2SO4 (50 ml) were added and stirred vigorously in an ice bath. After dissolution of the sodium nitrate, solid KMnO4 (1.5 g) was added slowly under continuous stirring over 30 min to prevent dangerous overheating (i.e., >30 °C). Upon addition of all the KMnO4, the ice bath was replaced with an oil bath (35–38 °C) and the solution was stirred vigorously for the next 3.5 h. During this time, the solution became highly viscous and turned dark brown. The reaction flask was then cooled in an ice bath, and double distilled water (75 ml) was slowly added to the solution, ensuring that the temperature remained below 40 °C, it was then stirred for another 2 h and 40 ml of 5% H2O2 was added, yielding a light yellow suspension of nano graphite oxide. The resulting graphite oxide was purified by three cycles of centrifugation (5500 rpm 15 min in centrifuge model REMI R-8C, INDIA), at first decantation, and then 5% H2SO4 was added to the resuspension with further centrifugation in the second step, followed by freshly prepared 5% H2O2 was added to the precipitate and washed thoroughly and centrifuged. One M HCl was also added to the precipitate collected from the second step and centrifuged. In the following step, the precipitate was washed five times with distilled water. The as-synthesized purified graphite oxide was suspended in double distilled water (100 ml) and exfoliated into individual GO nano sheets using a titanium-alloy solid probe ultrasonicator (125 W Qsonica Sonicators Q125 Sonicator, Qsonica, LLC, Newtown, CT, USA) set at 40% intensity for 6 h ON time. Any residual unexfoliated graphite oxide was removed by centrifugation at 5500 rpm for 20 min with the precipitate discarded. The resulting GO solution was then dialyzed in double distilled water to remove the remaining salts (water changed every 4 h).2.3. Double oxidation of graphene oxide
An aqueous dispersion of the as-prepared GO was lyophilized and (OPERON FDU-8606, 6 liter, KOREA) the water free dried GO sample (0.5 g) was subjected again to the oxidation process as described above with proper scaling of all other reagents. All the other operations were repeated as described above to have double oxidized GO.2.4. Extraction of collagen from fish scales
Extraction of collagen from fish scales involves two sequential steps, viz.Step I: fish scales were demineralized using EDTA (see Section 2.5).
Step II: collagen was isolated by dilute acetic acid treatment (see Section 2.6).
Both processes were carried out at 4 °C to avoid the denaturation of the helical pattern of collagen.
2.5. Demineralization
Fish scales of Labeo rohita (Rohu) and Catla catla were collected from the local market and washed thoroughly with distilled water. Next, the scales were washed with a solvent system described elsewhere,37 containing 1 M NaCl, 0.05 M Tris HCl and 20 mM EDTA at pH 7.5 for a period of 48 h to remove unwanted proteins from the surface. Demineralization of the scales was achieved by treating with 0.5 M EDTA solution for 48 h (pH 7.4). The demineralized fish scale was washed with distilled water three times.2.6. Isolation of collagen
Demineralized fish scales were treated with 0.5 M acetic acid solution at pH 2.5 over a period of 48 h and the insoluble part of the scales was filtered out. Sodium chloride was added to the filtrate to a final concentration of 5% (w/v) to induce salting out of collagen and was left undisturbed for 24 h at 4 °C. The suspension was centrifuged at 10000 rpm for 30 min and the precipitate was re-solubilized in 0.05 M acetic acid. The salting out and centrifugation steps were repeated three times for better purification of collagen. The final solution was then dialyzed using a dialysis membrane against 0.005 M acetic acid and distilled water for 24 h each and freeze-dried.
2.7. Preparation of the GO–collagen 3D scaffold
GO solution (2 mg ml−1) was prepared with the addition of 100 mg of GO in 50 ml of 0.1 M MES buffer to maintain the pH of the solution at 6.5, and then it was treated with a probe sonicator (125 W Qsonica Sonicators Q125 Sonicator, Qsonica, LLC, Newtown, CT, USA) set at 30% intensity for 30 min in an ice bath. To activate the carboxyl groups of the GO flakes, EDC·HCl, and NHS were added into the GO solution (50 ml) at a GO:EDC:NHS molar ratio of 1:2:2 (ref. 38) and stirred with a magnetic bar for 24 h. About 0.5 g of fish scale type I collagen was dissolved in 50 ml of 1% acetic acid solution and added to the EDC–NHS activated GO solution, reaction was allowed to proceed further for another 24 h at room temperature to obtain the final product of collagen functionalized NGO (CFNGO) (Scheme 1). The resultant solution was purified by exhaustive dialysis against double distilled water for 48 h, and lyophilized in the form of sponge.
 | ||
| Scheme 1 Covalent amide interaction between NGO and collagen – a representative image. | ||
2.8. Preparation of curcumin loaded CFNGO scaffold
Curcumin loaded CFNGO was prepared by a simple, non-covalent interaction method. The loading of curcumin onto CFNGO scaffold was carried out by mixing 10 ml of curcumin solution (60 mg of curcumin) in acetone with 60 ml of a freshly prepared solution of CFNGO (60 mg of CFNGO scaffold) with constant stirring for 24 h, at room temperature. The suspension was then squeezed through a muslin cloth to remove any precipitate formed during the process and finally the solution was lyophilized (Scheme 2). | ||
| Scheme 2 A representative image of the noncovalent interaction (ionic/H-bond and π–π stacking interactions) between curcumin and CFNGO scaffold. | ||
2.9. Dispersion study
Graphite and GO (single and doubly oxidized) (1 mg ml−1) were dispersed in water and sonicated for only 10 min in order to preserve their characteristic layers. All dispersions were left to settle for 24 h at room temperature.2.10. Morphology of the scaffold
The surface morphology of the collagen and CFNGO scaffolds was assessed using scanning electron microscopy (SEM) (ZEISS EVO-MA 10 Scanning Electron Microscope) at a high voltage of 15 kV.2.11. Transmission electron microscopy (TEM)
The morphology and size of NGO, collagen and CFNGO were examined by using a JEOL JEM 2100 HR with EELS TEM (JEOL, Japan) operated at 200 kV.2.12. Atomic force microscopy analysis
Tapping-mode atomic force microscopy (AFM) experiments were performed (Innova, Bruker AXS Pvt Ltd) to detect the morphology and size of NGO, collagen and CFNGO. To perform the experiment the dilute aqueous dispersion of the samples (10 μl, 0.1 mg ml−1) were cast on a freshly cleaved mica surface and dried at room temperature, the plates were washed again with double distilled water and dried completely at room temperature.2.13. Dynamic light scattering
Dynamic light scattering (DLS) measurements were performed on a Zetasizer Nano Series (Malvern), they were used to analyze GO hydrodynamic diameters and size distribution.2.14. Spectroscopic analysis
UV-vis absorption spectra were obtained on a Perkin Elmer Lambda 25 UV-vis spectrophotometer.2.15. Analysis of functional groups by FTIR spectroscopy
The analysis of the functional groups present in NGO, collagen and CFNGO were made by a Fourier transform infrared spectrophotometer (Alpha, Bruker, Germany). All spectra were recorded at the resolution of 4 cm−1 in the range of 400–4000 cm−1.2.16. Raman spectroscopy
Raman spectra of NGO were obtained by use of a back scattering geometry using a micro-Raman setup consisting of a spectrometer (model LabRAM HR, JobinYvon) and a Peltier-cooled charge-coupled device (CCD) detector. An air cooled argon ion laser with a wavelength of 633 nm was used as the excitation light source. Raman spectra of all the samples were recorded at room temperature in the frequency range 0–4000 cm−1.2.17. X-ray diffraction analysis
The X-ray diffraction of graphite nanopowder, NGO, collagen and CFNGO was performed using a wide angle X-ray scattering diffractometer (Panalytical X-ray diffractometer, model-X’pert Pro) with Cu-Kα radiation (λ = 1.54060 Å) in the range of 5–69° (2θ) at 40 kV and 30 mA.2.18. Analysis of mechanical properties
Mechanical properties, viz., Young’s modulus, tensile strength and the maximum stretching length of the scaffolds were measured using a Universal Testing Machine (LLOYD model no LR10k Plus) at a crosshead speed of 5 mm min−1 at 25 °C and at 65% relative humidity. Length and width of the dumbbell shaped test samples were maintained at 20 and 5 mm, respectively. All of the mechanical tests were performed with dried samples and were examined in triplicate.2.19. Thermo gravimetric analysis
Thermo gravimetric analysis (TGA) of collagen and CFNGO were carried out under nitrogen flow (40 and 60 ml min−1) with ramp of 20 °C min−1 using a TGA Q 50 (V20.6 build 31) instrument.2.20. Estimation of amine group interaction with collagen in CFNGO by TNBS assay
The analysis of the interacting amine groups was quantified using a TNBS assay according to the procedure summarized by Manna et al.38 In brief, native and interacting (collagen and CFNGO) scaffolds were cut into small pieces of size 4.5 mm. Six milligram cut pieces were immersed in a 2 ml solution consisting of 1 ml of 4% (w/v) di-sodium hydrogen orthophosphate and 1 ml of 0.5% (v/v) TNBS, and incubated at 40 °C for 2 h. Reaction termination was achieved with the addition of 3 ml of 6 M (v/v) HCl solution and incubation was continued at 60 °C for 90 min. The absorbance of the resulting solution was measured at 345 nm and the interaction of the amine groups was calculated from the difference in the absorbance divided by the absorbance of the native material and then multiplied with 100. The absorbance of the resulting mixture was measured at A345 nm using UV-vis.2.21. Swelling ratio test
The swelling ratio test was carried out as follows. A total of 2–5 mg of dry sample was placed in 1.5 ml of physiological saline buffer at 37 °C for a period of 72 h. Upon equilibrium with water, the swelling ratio was calculated according to the following equation:| SR (%) = (WS − Wd/Wd) × 100, |
2.22. Hemolytic assay
Blood compatibility was examined by hemolytic assay. Fresh human blood was used in this study. Following approval by the CSIR-CLRI ethical committee and prior informed consent, blood was collected from the patients. The sample size was fixed at 12, comprising of six male and six female healthy patients. Red blood cell (RBC) lysis investigates the protein denaturing effect of curcumin loaded CFNGO using a biological material (RBC) as a substrate. Freshly isolated RBCs were incubated in phosphate buffer saline. An irritating substance will cause lysis of the RBCs, leading to the release of hemoglobin in the sample. The cell debris and intact cells were separated by centrifugation and the amount of hemoglobin, which corresponded to the number of cells lysed by the curcumin loaded CFNGO, was assayed by spectrophotometry. At first, different volumes of samples in tubes (10, 20, 30, 50, 75, 100 μl) were taken and made up to 950 μl with PBS, followed by the addition and mixing of 50 μl of RBC sample. The samples were incubated in the dark for 10 min and then centrifuged for 10 min at 6000 rpm. The OD value of the supernatant was measured at 540 nm using a spectrophotometer. The obtained results were compared with the positive control (50 μl RBC + 950 μl H2O) and the negative control (50 μl RBC + 950 μl PBS). Each concentration was evaluated in triplicate. The percentage of lysis calculated by the formula; %lysis = (OD of sample − OD of positive control) × 100/(OD of negative control − OD of positive control).382.23. Cell culturing and maintenance
NIH 3T3 embryonic mouse fibroblast cells, procured from NCCS, Pune, India, were used. The cultures were maintained in DMEM supplemented with 10% FBS, 200 mM glutamine, 2 mg ml−1 sodium bicarbonate, and 1× antibiotic and antimycotic solution. Periodically the medium was replaced. The cells were cultured in tissue culture flasks and incubated at 37 °C in a humidified atmosphere of 5% CO2. Trypsin at 0.05% was used to detach the cells.2.24. Cell adherence and proliferation
NIH 3T3 cell adherence and proliferation assessment of collagen and curcumin loaded CFNGO was carried out with collagen and curcumin loaded CFNGO pre-coated culture plates. In brief, 2 ml of collagen and the prepared curcumin loaded CFNGO solution were added to each well of twelve-well culture plates and subjected to air drying at 40 °C. The control wells were not coated with anything. The dried plates were then surface sterilized with 70% alcohol for 30 min followed by UV sterilization for 1 h. Next, the plates were washed with sterile PBS for 1 h. A cell density of 3 × 104 cells per well was seeded and incubated with the growth medium for the period of 12, 24 and 48 h, and the observations on cell viability, adherence and proliferation were made as described below. The cell adherence and proliferation of fibroblast cells were visualized and quantified by live cell tracker assay and MTT assay methods. With respect to the cell tracker assay, after scheduled time intervals (12, 24, 48 h), the medium was removed and washed with PBS. Calcein AM solution (4 mM; 500 ml) was added and the sample was incubated for 30 min. The plates were then washed with PBS and viewed at fluorescence excitation and emission wavelengths of 495/515 nm, respectively, using a fluorescence microscope with a blue filter (Euromex, Holland). With regard to the MTT assay, the culture medium of each well was replaced with MTT (5 mg ml−1 diluted in serum-free medium) and incubated at 37 °C for 4 h. After the removal of MTT, dimethyl sulfoxide was added and the medium was left at room temperature for 2 min and the absorbance was recorded at 570 nm using a plate reader (Epoch, BIOTEK).2.25. Cell migration assay
A cell migration assay was performed according to the method summarized by Liang et al.39 NIH 3T3 cells were harvested by trypsinization and loaded in a 48-well plate at a concentration of 5 × 104 cells per well, and allowed to form a uniform monolayer. Followed by attaining 80 to 85% cell confluence, a scratch was introduced using a sterile 200 ml tip. The plates were then washed with PBS to remove the dead cells and supplemented with DMEM medium containing collagen and CFNGO. Wells filled with the medium alone served as a control. Migration of cells and the reduction in empty space were measured at 0, 12 and 24 h of incubation using Image J software.2.26. Cell morphology of NIH 3T3 cells in CFNGO scaffold
Curcumin loaded CFNGO scaffold (2 × 2 × 1 cm) was placed individually in 6 well culture plates (Tarson, India) and ethylene oxide (ETO) sterilized. Culture media was added to the scaffold overnight. NIH 3T3 fibroblast cells seeded on to the scaffold at a density of 5 × 104 cells and incubated in an atmosphere of 5% CO2 at 37 °C. The medium was changed every 24 h. Morphology of the cells was examined after 12 days according to the following procedure. The cell-scaffold construct was fixed in 2.5% glutaraldehyde and dehydrated through graded ethanol series. The dried cell-scaffold was coated with gold and examined under SEM (ZEISS EVO-MA 10 Scanning Electron Microscope).2.27. In vitro curcumin release
A known amount of curcumin loaded CFNGO scaffold (70 mg) was immersed in 3 ml of phosphate buffer solution, pH 7.4 at 37 °C, followed by transfer into 7 centrifuge tubes and was kept in a shaker. Free curcumin is not soluble in water; therefore, at predetermined time intervals, the solution was centrifuged at 5000 rpm for 9 min to separate the released curcumin from the CFNGO scaffold. The released curcumin was redissolved in 3 ml of ethanol and its concentration was determined by UV-vis at 425 nm. The concentration of released curcumin was then calculated using a standard curve of curcumin in ethanol. The percentage of curcumin released was determined from the following equation: curcumin release (%) = 100 × curcumin released in time (t)/total curcumin loaded in CFNGO scaffold.2.28. Evaluation of antimicrobial activity
The antimicrobial activity of the prepared curcumin loaded/not loaded CFNGO scaffold was tested by the disc diffusion method against Gram-negative bacteria, P. aeruginosa, and Gram-positive bacteria, S. aureus. The scaffolds were cut into a disc shape of 5 mm diameter followed by sterilization under UV light for 10 min and were then placed on agar plates with cultures. The plates were incubated for 24 h at 37 °C in an incubator and the zone of inhibition was then calculated. The zone of inhibition was not observed for native CFNGO scaffolds (not loaded with curcumin). This experiment was performed in triplicate with each organism and an average diameter of zone of inhibition was noted.2.29. Evaluation of wound healing efficacy of curcumin loaded CFNGO scaffold
In order to assess the efficacy of curcumin loaded CFNGO scaffold as a wound cover, an animal model study was taken up with prior approval from the CSIR-CLRI ethical committee; with vide approval no. 466/01a/CPCSEA for open wound model studies. Male albino rats (Wistar strain) of weight ranging from 125–150 g were used. They were housed individually in standardized environmental conditions, fed with a pellet rodent diet and had access to water ad libitum. Animals were randomly grouped into three groups with six animals per group. Group-1 (control-wound covered with cotton soaked in phosphate buffered saline); group-II (wound covered with scaffold prepared from collagen alone); group-III (wound covered with scaffold of curcumin loaded CFNGO scaffold). An incision of 4 cm (2 cm)2 was created on the dorsal part of all the rats with the help of scissors and surgical blade and the wound area was sterilized with surgical spirit. Respective dressing scaffolds were applied to animals and closed with gauze. All the rats received regular dressing changes on alternate days.2.30. Rate of wound contraction area
Wound contraction was measured as a percentage reduction in wound size at 4 day intervals and photographed. Progressive decrease in the wound size was measured by tracing the wound margin on a transparent paper having a millimeter scale, and the reduction in wound size was measured planimetrically.The biodegradability of the CFNGO and native collagen scaffold are summarized in detail in ESI S1.†
3. Results and discussion
It has been observed that graphite and NGO (single and double oxidized) look different because of their distinct structural and physicochemical properties. Black particles are visible in the graphite dispersion after sonication for 0.5 h. Most graphite particles precipitate after 0.5 h of graphite dispersion. The singly oxidized NGO dispersion is blackish-yellow in color. Some small NGO particles can also be identified in the NGO dispersion. A significant portion of NGO particles precipitated after the NGO dispersion was idle for 2 h. However, a clear and homogeneous yellow colored dispersion is observed for doubly oxidized NGO. The NGO dispersion was stable after standing still for several days, this could be due to the large amount of hydrophilic functional groups, such as carboxyl, hydroxyl, and epoxy groups, on the NGO nanosheets.403.1. Morphology of the scaffold
The surface morphology of collagen and CFNGO scaffolds is shown in Fig. 1a. Compared to the native collagen scaffold, the CFNGO scaffold is highly porous (the porosity of CFNGO was observed at around 10–20 μm) and the pore structures of the membranes are well-distributed and interconnected. The conjugation of NGO with collagen increases the scaffold pore size in CFNGO, as determined by SEM images. It is obvious that most of the space of the membranes were covered by the interconnecting pore space. The high porosity suggests the suitability of this scaffold for biomedical applications, including serving as absorption sponges and matrices for cell proliferation.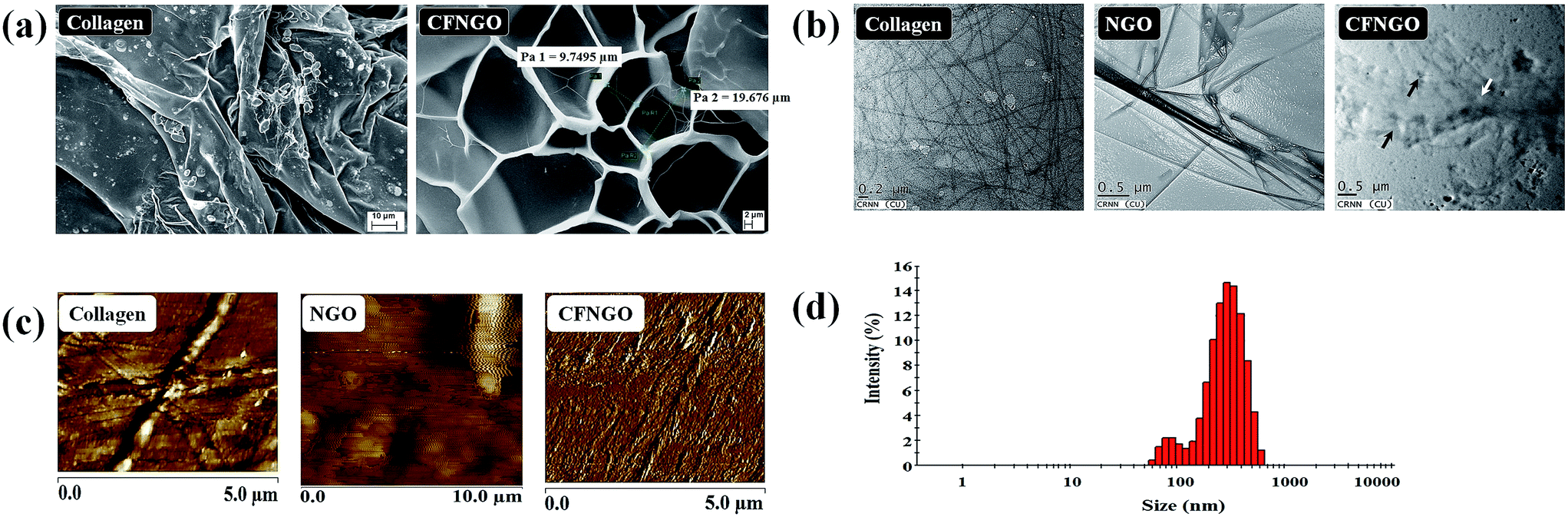 | ||
| Fig. 1 (a) SEM micrographs of native collagen and CFNGO. (b) TEM images of collagen, NGO and CFNGO (the black arrow indicates the existence of NGO and the white arrow indicates the collagen fiber). (c) AFM images of collagen, NGO and CFNGO. (d) DLS based size distribution of NGO. | ||
3.2. Transmission electron microscopy
TEM images of collagen show the existence of collagen fiber, whereas NGO shows that it is fully exfoliated into individual sheets (Fig. 1b). In the CFNGO image, the NGO nano sheet is observed randomly distributed inside the collagen fiber, and enhances the interaction between the amine group of collagen and the carboxylic group of NGO. As a result, all the randomly distributed collagen fibers (as shown in Fig. 1b) come close to each other which could be the reason for the high mechanical strength of the CFNGO scaffold. Scheme 3 shows how collagen fiber and NGO co-exist in the CFNGO scaffold, the white arrow indicates the presence of NGO and the black arrow indicates the collagen fiber existence inside the TEM image of CFNGO. | ||
| Scheme 3 The scheme represents the pattern of NGO and collagen conjugation. | ||
3.3. Atomic force microscopy analysis
The AFM images (Fig. 1c), show a 2D surface morphology of collagen fiber and ultrathin (1.5 nm thick) NGO sheets which is typical for a one-atom-thick GO nano layer.40,41 Fig. 1c also presents the representative AFM image of the CFNGO scaffold and it confirms the presence of uniform layers of NGO and collagen fibers, suggesting the successful interaction between NGO and collagen.3.4. Size distribution by dynamic light scattering
Fig. 1d depicts the hydrodynamic size of the prepared doubly oxidized NGO nano sheet in the range 60–600 nm with a major peak in the 220–350 nm range. All results reflect the average of three measurements and the difference between each measurement was <5%.3.5. Spectroscopic analysis
In the UV-vis spectrum (Fig. 2a), fish scale collagen showed one peak at 213 nm due to the presence of amide linkage, and one small hump at around 280 nm due to the presence of a very small amount of aromatic residues (phenylalanine and tyrosine).42,43 NGO shows a characteristic peak at 231 nm (corresponding to π–π* transitions of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds) and a shoulder at 300 nm (due to n–π* transitions of –COOH groups).44 CFNGO shows one peak at 216 nm which could be due to the presence of CO, –COOH and –CONH2 groups, and one peak at 300 nm, as observed in the NGO spectrum, has disappeared due to the interaction of the –COOH group of NGO and the free primary amine groups of collagen.
C bonds) and a shoulder at 300 nm (due to n–π* transitions of –COOH groups).44 CFNGO shows one peak at 216 nm which could be due to the presence of CO, –COOH and –CONH2 groups, and one peak at 300 nm, as observed in the NGO spectrum, has disappeared due to the interaction of the –COOH group of NGO and the free primary amine groups of collagen.
 | ||
| Fig. 2 (a) UV-vis absorption spectra of collagen, NGO and CFNGO. (b) FTIR spectra of collagen, NGO and CFNGO scaffolds. (c) Raman spectrum of NGO. (d) XRD patterns of (A) graphite powder, (B) NGO, (C) collagen, and (D) CFNGO. (e) TGA of native collagen and CFNGO scaffolds. | ||
3.6. FTIR assessment
FTIR experiments were carried out to investigate the covalent amide (–CONH2) interaction between NGO and collagen. Fig. 2b illustrates the FTIR spectrum of collagen, NGO and CFNGO scaffolds. The FTIR spectrum of NGO and collagen are similar to those previously presented in literature.45–47 In the collagen spectrum the peak at 3282 cm−1 is attributed to the –NH2 stretching vibration, and at 3078 cm−1 to the Fermi resonance overtone of the 1539 cm−1 band, 2927 cm−1 is assigned to C–H stretching. The bands at 1626, 1539 and 1390 cm−1 correspond to CO stretching (amide I), N–H bending (amide II) and C–N stretching (amide III) of amide (–CONH2) linkages in collagen protein. The peak at 1236 cm−1 is due to the C–N stretching of amine.47
In the NGO spectrum, the peaks at 3384 cm−1, 1700 cm−1, 1640 cm−1, 1035 cm−1 are assigned to –OH stretching vibration, CO stretching vibration of the carboxylic group, CC stretching mode of the sp2 network and C–O stretching vibration, respectively,45 demonstrating the presence of –COOH and –OH groups in the NGO.
By comparing the spectra of the CFNGO scaffold, it has been found that there are no additional peaks, excluding the specific bands of collagen and NGO, but they have a greater intensity and are accordingly ascribed to the EDC–NHS coupling interaction between NGO and collagen through the –CONH2 (amide) linkage between the –COOH group of NGO and the free primary –NH2 group of collagen.
3.7. Raman spectroscopy
Raman spectroscopy gives useful information related to the structural properties of NGO. A typical Raman spectra of the sample is shown in Fig. 2c. The spectra shows three distinct signals: a band close to 1590 cm−1 (G band), another one around 1330 cm−1 (D band) and a 2D band in the region of 2740 cm−1.48,49 The G band originates from in-plane vibrations of the sp2 carbon atoms of the graphite lattice.50,51 D band is due to the out-of-plane breathing mode of the sp2 atoms, it reveals the presence of certain defects, whereas 2D is the second order of this vibrational mode.44,52 These observations confirm the layer separation of the NGO structure, and the corresponding 2D band indicates considerable defects in the graphene sheet produced by oxidation.523.8. X-ray diffraction analysis
Fig. 2d depicts the X-ray diffractograms of graphite nanopowder, NGO, collagen and CFNGO scaffolds. It has been observed that the most intense peak for graphite is at 2θ = 26.5° and d spacing is 3.3 Å, whereas the NGO spectrum exhibits a characteristic peak at 2θ = 10.4° with the d spacing of 8.5 Å which is within the range of values that has been previously reported.53 The above results indicate that the interlayer distance for NGO is significantly larger than that of the graphite. These observations suggest that the successful oxidation of the graphite to NGO resulted in the oxygenated functional groups on the carbon sheets which ultimately increases the interlayer space of NGO. The oxidation of graphite to NGO has already been confirmed by attenuated total reflectance (ATR)-FTIR spectra as described earlier.The characteristic peak of collagen appeared at the positions 2θ = 22° with average intermolecular distance of 3.9 Å, this broad peak indicates a typical diffraction pattern for collagen.
The diffraction spectrum of the CFNGO scaffold displays a broad spectrum at 2θ = 23° indicating its amorphous nature and the intermolecular distance is observed at 3.8 Å which is less than that of the NGO (8.5 Å), this could be due to the aggregation of NGO with collagen.
3.9. Mechanical properties
The mechanical property of the CFNGO scaffold shows significant enhancement compared to the collagen scaffold alone. The tensile strength of the CFNGO scaffold is 3.19 MPa, whereas the tensile strength of native collagen is only 0.09 MPa. The strain at maximum load for CFNGO is observed much higher than that of the native collagen and may be due to the covalent interaction between NGO and collagen, which has already been confirmed by ATR-FTIR analysis. The ultimate tensile strength, strain at maximum load and Young’s modulus of native collagen and the CFNGO scaffold are summarized in Table 1. All results reflect the average of three measurements, and the difference between each measurement was <5%.| Sample name | Strain at maximum load (%) | Tensile strength (MPa) | Young’s modulus (MPa) |
|---|---|---|---|
| Collagen scaffold | 6.58 (±0.5) | 0.09 (±0.004) | 1.36 (±0.4) |
| CFNGO scaffold | 23.73 (±1.2) | 3.19 (±0.5) | 25.85 (±1.76) |
3.10. Thermo gravimetric analysis
The thermal stability of the scaffolds is evaluated with TGA. Major thermal decomposition of native collagen occurs between 300–400 °C (Fig. 2e), the corresponding thermal degradation values are listed in Table 2. The major decomposition of the CFNGO scaffold initiated after 350 °C (at 400 °C 41% weight loss is observed and 50% weight loss is at 800 °C) and only 9% of weight loss is observed at 800 °C suggesting that the CFNGO scaffold is much more thermally stable compared to native collagen due to the covalent interaction between NGO and collagen. The thermal stability also influences the durability of the scaffolds.| Temperature (°C) | % of weight loss (heating rate 20 °C min−1) | |
|---|---|---|
| Native collagen scaffold | CFNGO scaffold | |
| 100 | 13 | 11 |
| 200 | 15 | 15 |
| 300 | 26 | 23 |
| 400 | 62 | 41 |
| 500 | 70 | 45 |
| 600 | 74 | 47 |
| 700 | 77 | 49 |
| 800 | 80 | 51 |
The percentage of the interacted amine group is observed as 47% for the CFNGO scaffold.
3.11. Hemolysis
The hemolysis assay is a significant index of materials for application in the biomedical field because the material is usually exposed to blood and damages the erythrocytes to a certain extent. In the present study, the assay is carried out to evaluate the blood compatibility of the CFNGO scaffold. As shown in Fig. 3a, no damage to the erythrocytes was caused by the CFNGO scaffold inferring the hemocompatibility of the prepared scaffold. All the results reflect the average of three measurements, and the difference between each measurement was <5%. | ||
| Fig. 3 (a). Blood compatibility studies of curcumin loaded CFNGO where 1–6. 10, 20, 30, 50, 75 and 100 μl of CFNGO was made up to 950 μl with PBS and then 50 μl of RBC sample was added and mixed. Number 7 shows the positive control (50 μl RBC + 950 μl H2O), and 8 the negative control (50 μl RBC + 950 μl PBS). (b) In vitro fluorescence images of NIH 3T3 fibroblast cells showing adherence and proliferation of the curcumin loaded CFNGO scaffold, native collagen and control, to the cell walls at time intervals of 12, 24 and 48 h. A blue filter was used for fluorescence excitation and emission. The scale bar measures 10 mm. (c) Cell viability assessment based on MTT quantification of NIH 3T3 cell population upon being exposed to the curcumin loaded CFNGO in comparison with native collagen and control (all the values are the mean SD of triplicate measurements). (d) Influence of the curcumin loaded CFNGO scaffold on the migration of fibroblast cells (NIH 3T3) assessed from a scratch assay (in vitro conditions). Image J analysis was used to measure the space covered by the migrated cells at incubation periods of 0, 12 and 24 h. The scale bar measures 10 mm. (e) Attachment of fibroblast cells on the curcumin loaded CFNGO scaffold (white arrow indicates the adhered cells on the scaffold). | ||
3.12. Water holding capacity
The water holding capacity of the scaffold plays an important role in TE. The swelling ratio of the native collagen scaffold is 1925% (±12.1) and for the CFNGO scaffold it is 3579% (±10.3). It has been observed that the water holding capability of CFNGO scaffold is much higher than that of the native collagen scaffold due to the incorporation of NGO. The swelling ratio of the scaffold strongly depends on the hydrophilic nature and microstructure of the scaffold. Since collagen (–NH2 and –COOH groups) and NGO (numerous –COOH and –OH groups) are both hydrophilic materials, the CFNGO scaffold has a higher water retention capability with appreciable mechanical properties. The poor mechanical properties of collagen led to the collapse of the porous structure when it is taken out of PBS solution. On the contrary, NGO possesses higher mechanical stability which is helpful for the retention of the scaffolds original porous structure. All the obtained results are the average of three measurements, and the difference between each measurement was <5%.3.13. Th cell adhesion and proliferation profile of the CFNGO scaffold
It is essential to evaluate the cytotoxicity of the prepared curcumin loaded CFNGO scaffold, for use in biological and biomedical applications, such as drug delivery and gene transfection. In the present study, the cell adherence and cell proliferation potentials of the curcumin loaded CFNGO scaffold are assessed using NIH 3T3 cells. Fig. 3b shows the fluorescence images of the cells observed at different time intervals. When cells are stained with a calcein fluorescent probe, live cells release a green-fluorescence caused by acetoxymethyl ester hydrolysis by intracellular esterases. In the present study, no significant difference is seen in fluorescence intensity in the curcumin loaded CFNGO scaffold-treated cells compared to the control. Furthermore, no morphological changes are observed. Results on cell viability and quantification suggest that more than 90% of the cells are viable and incorporation of curcumin and NGO into collagen does not exert adverse effects on cell viability (Fig. 3c). It has already been reported that the cytotoxicity of NGO is dose-dependent and a small quantity of NGO does not negatively affect the viability of cells.54–56 The improved hydrophilicity due to the hydrophilic groups of NGO and collagen provides higher cell adhesion on collagen and curcumin loaded CFNGO scaffolds than the control since cells adhere, spread and grow more easily on moderately hydrophilic surfaces than on hydrophobic ones.57 It is considered that relatively higher hydrophilicity of collagen and curcumin loaded CFNGO scaffolds promote the adhesion of cells.3.14. In vitro cell migration assay
Results on cell migration studies of the curcumin loaded CFNGO scaffold using NIH 3T3 cells suggest that compared to the control (untreated) curcumin loaded CFNGO scaffold-treated cells migrate at the fastest rate and more than 90% of the scratch area is covered by the cells within 24 h (Fig. 3d) similar to the collagen scaffold-treated cells. These observations suggest that the curcumin loaded CFNGO scaffold influences cell migration, and this property adds value to the material for its application to wound healing studies.3.15. Cell morphology of NIH 3T3 cells in CFNGO scaffolds
SEM images of the cell seeded curcumin loaded CFNGO scaffold are displayed in Fig. 3e demonstrating that after being cultured for a prolonged time (12 days), fibroblasts were detected in the scaffolds (curcumin loaded CFNGO) with typical spindle shaped morphology suggesting that the cells infiltrated into the scaffolds and proliferated there.The release of curcumin from the CFNGO scaffold is conducted in phosphate buffer solution to evaluate its sustained delivery. The average concentration of curcumin release from the curcumin loaded CFNGO scaffold is approximately 12.1, 14.5, 18.9, 20.6, 27.7, 36.1, 52.7, 78.7 and 82.5% in the respective duration of 1, 3, 6, 9, 12, 24, 48, 72 and 96 h. Comparing all of these results, it is confirmed that the curcumin loaded CFNGO scaffold prepared in the present study has better sustained release properties. A material with the above said property will be very helpful as a wound dressing material because the curcumin will release on the wound site at a slower rate and this will restrict the bacterial growth for a prolonged time, subsequently leading to quicker wound healing.
The antibacterial activity of curcumin loaded CFNGO scaffolds are obtained as 15 (±3) and 16 (±2) mm against Gram-negative P. aeruginosa and Gram-positive bacteria S. aureus. Due to this antibacterial property, it can again be applied as an efficient wound healing material because the inherent antimicrobial property of the material will not allow microbial growth on the wound site. Nguyen et al. reported a similar kind of observation for the preparation of curcumin loaded chitosan/gelatin composite sponge for wound healing applications.58
The curcumin loaded CFNGO scaffold was also subjected to a test as a wound dressing material (open wound) using an animal model (Wistar strain). Fig. 4 illustrates the progressive wound closure of three different experimental groups. The control group (group-I) took more than 20 days for complete closure, whereas, complete wound closure was evidenced within 16 days for group-II (collagen alone) and 12 days for group-III (curcumin loaded CFNGO scaffold) animals. Similarly, wound contraction measurements (Table 3) also showed accelerated wound closure in both group-II and group-III animals compared to group-I. The results discussed over here were the representative results obtained for the curcumin loaded CFNGO scaffold.
 | ||
| Fig. 4 Open wound upon treatment with curcumin loaded CFNGO scaffold. | ||
| Days | Control (group-I) | Native collagen (group-II) | Curcumin loaded CFNGO (group-III) | |||
|---|---|---|---|---|---|---|
| Wound areaa (mm2) | % of wound contractiona | Wound areaa (mm2) | % of wound contractiona | Wound areaa (mm2) | % of wound contractiona | |
| a Mean ± SD values. | ||||||
| 0th | 333.3 ± 42.5 | 0 | 358.5 ± 62 | 0 | 368.67 ± 48.5 | 0 |
| 4th | 250 ± 37 | 10.8 ± 11.28 | 301.17 ± 68 | 18.3 ± 16.18 | 329.5 ± 68.5 | 21.71 ± 20.86 |
| 8th | 145.83 ± 44.5 | 55.61 ± 17.39 | 103.67 ± 31.5 | 70.28 ± 16.47 | 100.33 ± 90 | 74.19 ± 26.95 |
| 12th | 33.5 ± 11.5 | 89.99 ± 2.29 | 12.17 ± 5.9 | 96.57 ± 1.88 | 41.67 ± 45 | 89.3 ± 11.81 |
| 16th | 7.67 ± 7 | 97.74 ± 1.91 | 1.67 ± 1.37 | 99.52 ± 0.725 | Healed | 100 |
| 20th | 2.83 ± 3.19 | 99.18 ± 1.31 | Healed | Healed | ||
4. Conclusions
Current biomaterials research demands new materials with advanced functional properties. Although biocompatibility and biodegradability properties have been incorporated into the material via functionalization and modification procedures, materials for drug delivery studies lack the ability to simultaneously release both hydrophobic and hydrophilic drugs. Since incorporation of GO demonstrates an appreciable role in imparting strength to the materials, cross-linking of NGO with collagen may be suitable for the delivery of drugs, growth factors or cells as a unique delivery system irrespective of the nature of the delivery molecules. Hence, an attempt was made in the present study to reinforce NGO with fish scale collagen via EDC–NHS coupling, and curcumin was incorporated into CFNGO by non-covalent interactions as a model drug. The resulting curcumin loaded CFNGO has appreciable mechanical strength (when 2% of NGO was mixed with 1% of collagen), cell adherence and proliferation, biocompatibility, etc. The antimicrobial study reveals that the curcumin loaded CFNGO scaffold also has appreciable antibacterial activity. Animal model wound healing studies further explored curcumin loaded CFNGO scaffold as a wound dressing material. The ESI file †proves that the obtained material is enzymatically degradable under physiological conditions. The presence of various functional groups in NGO–collagen scaffolds suggests the possibility to use of the scaffolds for multidrug delivery. A study on the process of delivery of multidrugs using NGO–collagen scaffold has been initiated and the observations will soon be in the public domain.Acknowledgements
One of he authors Dr Tapas Mitra acknowledges UGC, New Delhi, for financial assistance provided in the form of Dr D. S. Kothari Post-Doctoral Fellowship. All authors thank Dr Achintya Singha, Department of Physics, Bose Institute, 93/1, Acharya Prafulla Chandra Road, Kolkata 700009, India, for his assistance with Raman spectroscopy. Authors also acknowledge Mr Sourav Chakraborty, Calcutta University – DBT Interdisciplinary Program in Life Science (DBT-IPLS), 35 Ballygunge Circular Road Kolkata-700019, for AFM analysis. Authors also acknowledge Miss Urmila Goswami, CRNN, University of Calcutta, JD-2, Salt Lake City, Kolkata 700098, West Bengal, India for her help with the TEM images of the samples.References
- C. P. Barnes, S. A. Sell, E. D. Boland, D. G. Simpson and G. L. Bowlin, Adv. Drug Delivery Rev., 2007, 59, 1413–1433 CrossRef CAS PubMed.
- M. F. Desimone, C. Helary, G. Mosser, M.-M. Giraud-Guille, J. Livage and T. Coradin, J. Mater. Chem., 2010, 20, 666–668 RSC.
- Z. Xia, X. Yu, X. Jiang, H. D. Brody, D. W. Rowe and M. Wei, Acta Biomater., 2013, 9, 7308–7319 CrossRef CAS PubMed.
- B. Balakrishnan, M. Mohanty, P. R. Umashankar and A. Jayakrishnan, Biomaterials, 2005, 26, 6335–6342 CrossRef CAS PubMed.
- M. Ogawa, M. W. Moody, R. J. Portier, J. Bell, M. A. Schexnayder and J. N. Losso, J. Agric. Food Chem., 2003, 51, 8088–8092 CrossRef CAS PubMed.
- A. Jongjareonrak, S. Benjakul, W. Visessanguan, T. Nagai and M. Tanaka, Food Chem., 2005, 93, 475–484 CrossRef CAS.
- G. Bilgen, G. Oktay, Z. TokgoZ, G. Guner and S. Yalcin, Turk. J. Vet. Anim. Sci., 1999, 23, 483–488 Search PubMed.
- T. Nagai, Y. Araki and N. Suzuki, Food Chem., 2002, 78, 173–177 CrossRef CAS.
- T. Nagai, M. Izumi and M. Ishii, Int. J. Food Sci. Technol., 2004, 39, 239–244 CrossRef CAS.
- T. Nagai and N. Suzuki, Food Chem., 2000, 68, 277–281 CrossRef CAS.
- D. Swatschek, W. Schatton, J. Kellermann, W. E. G. Muller and J. Kreuter, Eur. J. Pharm. Biopharm., 2002, 53, 107–113 CrossRef CAS PubMed.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS.
- M. Tang, Q. Song, N. Li, Z. Jiang, R. Huang and G. Cheng, Biomaterials, 2013, 34, 6402–6411 CrossRef CAS PubMed.
- J. Hong, N. J. Shah, A. C. Drake, P. C. DeMuth, J. B. Lee, J. Chen and P. T. Hammond, ACS Nano, 2011, 6, 81–88 CrossRef PubMed.
- L. Zhang, J. Xia, Q. Zhao, L. Liu and Z. Zhang, Small, 2010, 6, 537–544 CrossRef CAS PubMed.
- G. Y. Chen, D.-P. Pang, S. M. Hwang, H. Y. Tuan and Y. C. Hu, Biomaterials, 2012, 33, 418–427 CrossRef CAS PubMed.
- W. C. Lee, C. H. Y. X. Lim, H. Shi, L. A. L. Tang, Y. Wang, C. T. Lim and K. P. Loh, ACS Nano, 2011, 5, 7334–7341 CrossRef CAS PubMed.
- T. R. Nayak, H. Andersen, V. S. Makam, C. Khaw, S. Bae, X. Xu, P.-L. R. Ee, J.-H. Ahn, B. H. Hong and G. Pastorin, ACS Nano, 2011, 5, 4670–4678 CrossRef CAS PubMed.
- H. Bao, Y. Pan, Y. Ping, N. G. Sahoo, T. Wu, L. Li, J. Li and L. H. Gan, Small, 2011, 7, 1569–1578 CrossRef CAS PubMed.
- X. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano Res., 2008, 1, 203–212 CrossRef CAS PubMed.
- K. Yang, J. Wan, S. Zhang, Y. Zhang, S.-T. Lee and Z. Liu, ACS Nano, 2011, 5, 516–522 CrossRef CAS PubMed.
- W.-G. La, S. Park, H.-H. Yoon, G.-J. Jeong, T.-J. Lee, S. H. Bhang, J. Y. Han, K. Char and B.-S. Kim, Small, 2013, 9, 4051–4060 CrossRef CAS PubMed.
- X. Shi, H. Chang, S. Chen, C. Lai, A. Khademhosseini and H. Wu, Adv. Funct. Mater., 2012, 22, 751–759 CrossRef CAS.
- C. Cha, S. R. Shin, N. Annabi, M. R. Dokmeci and A. Khademhosseini, ACS Nano, 2013, 7, 2891–2897 CrossRef CAS PubMed.
- K. P. Loh, Q. Bao, G. Eda and M. Chhowalla, Nat. Chem., 2010, 2, 1015–1024 CrossRef CAS PubMed.
- E.-M. Strasser, B. Wessner, N. Manhart and E. Roth, Biochem. Pharmacol., 2005, 70, 552–559 CrossRef CAS PubMed.
- S. Han and Y. Yang, Dyes Pigm., 2005, 64, 157–161 CrossRef CAS.
- O. Vajragupta, P. Boonchoong, G. M. Morris and A. J. Olson, Bioorg. Med. Chem. Lett., 2005, 15, 3364–3368 CrossRef CAS.
- B. B. Aggarwal, A. Kumar and A. C. Bharti, Anticancer Res., 2003, 23, 363–398 CAS.
- S.-Y. Park, H.-S. Kim, E.-K. Cho, B.-Y. Kwon, S. Phark, K.-W. Hwang and D. Sul, Food Chem. Toxicol., 2008, 46, 2881–2887 CrossRef CAS PubMed.
- M. Panchatcharam, S. Miriyala, V. S. Gayathri and L. Suguna, Mol. Cell. Biochem., 2006, 290, 87–96 CrossRef CAS.
- S. Bisht and A. Maitra, Curr. Drug Discovery Technol., 2009, 6, 192–199 CrossRef CAS.
- D. Gopinath, M. R. Ahmed, K. Gomathi, K. Chitra, P. K. Sehgal and R. Jayakumar, Biomaterials, 2004, 25, 1911–1917 CrossRef CAS.
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- F. Pati, B. Adhikari and S. Dhara, Bioresour. Technol., 2010, 101, 3737–3742 CrossRef CAS PubMed.
- P. J. Manna, T. Mitra, N. Pramanik, V. Kavitha, A. Gnanamani and P. P. Kundu, Int. J. Biol. Macromol., 2015, 75, 437–446 CrossRef CAS PubMed.
- C.-C. Liang, A. Y. Park and J.-L. Guan, Nat. Protoc., 2007, 2, 329–333 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- H. C. Schniepp, J.-L. Li, M. J. McAllister, H. Sai, M. Herrera-Alonso, D. H. Adamson, R. K. Prud’homme, R. Car, D. A. Saville and I. A. Aksay, J. Phys. Chem. B, 2006, 110, 8535–8539 CrossRef CAS.
- S. Yunoki, T. Suzuki and M. Takai, J. Biosci. Bioeng., 2003, 96, 575–577 CrossRef CAS PubMed.
- P. Bama, M. Vijayalakshimi, R. Jayasimman, P. T. Kalaichelvan, M. Deccaraman and S. Sankaranarayanan, Int. J. Pharm. Pharm. Sci., 2010, 2, 133–137 CAS.
- J. I. Paredes, S. Villar-Rodil, A. Martinez-Alonso and J. M. D. Tascon, Langmuir, 2008, 24, 10560–10564 CrossRef CAS.
- L. Liu, C. Li, C. Bao, Q. Jia, P. Xiao, X. Liu and Q. Zhang, Talanta, 2012, 93, 350–357 CrossRef CAS PubMed.
- V. K. Malesu, D. Sahoo and P. L. Nayak, Int. J. Appl. Biol. Pharm. Technol., 2011, 2, 402–411 Search PubMed.
- T. Mitra, G. Sailakshmi and A. Gnanamani, J. Chem. Sci., 2014, 126, 127–140 CrossRef CAS.
- S. Wakeland, R. Martinez, J. K. Grey and C. C. Luhrs, Carbon, 2010, 48, 3463–3470 CrossRef CAS.
- S. Park, J. An, R. D. Piner, I. Jung, D. Yang, A. Velamakanni, S. T. Nguyen and R. S. Ruoff, Chem. Mater., 2008, 20, 6592–6594 CrossRef CAS.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud’Homme, I. A. Aksay and R. Car, Nano Lett., 2008, 8, 36–41 CrossRef CAS PubMed.
- D. Yang, A. Velamakanni, G. L. Bozoklu, S. Park, M. Stoller, R. D. Piner, S. Stankovich, I. Jung, D. A. Field and C. A. Ventrice, Carbon, 2009, 47, 145–152 CrossRef CAS.
- T. V. Cuong, V. H. Pham, Q. T. Tran, S. H. Hahn, J. S. Chung, E. W. Shin and E. J. Kim, Mater. Lett., 2010, 64, 399–401 CrossRef CAS.
- J. Wang, X. Wang, C. Xu, M. Zhang and X. Shang, Polym. Int., 2011, 60, 816–822 CrossRef CAS.
- K. Wang, J. Ruan, H. Song, J. Zhang, Y. Wo, S. Guo and D. Cui, Nanoscale Res. Lett., 2011, 6, 1–8 Search PubMed.
- K.-H. Liao, Y.-S. Lin, C. W. Macosko and C. L. Haynes, ACS Appl. Mater. Interfaces, 2011, 3, 2607–2615 CAS.
- E. L. K. Chng and M. Pumera, Chem.–Eur. J., 2013, 19, 8227–8235 CrossRef CAS PubMed.
- Y. Arima and H. Iwata, Biomaterials, 2007, 28, 3074–3082 CrossRef CAS PubMed.
- V. C. Nguyen, V. B. Nguyen and M.-F. Hsieh, Int. J. Polym. Sci., 2013, 2013 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra15726a |
| This journal is © The Royal Society of Chemistry 2015 |