
Recent advances in crosslinking chemistry of biomimetic poly(ethylene glycol) hydrogels
Chien-Chi Lin*
Department of Biomedical Engineering, Purdue School of Engineering and Technology, Indiana University-Purdue University Indianapolis, Indianapolis, IN 46202, USA. E-mail: lincc@iupui.edu; Fax: +1-317-278-2455; Tel: +1-317-274-0760
First published on 17th April 2015
Abstract
The designs and applications of biomimetic hydrogels have become an important and integral part of modern tissue engineering and regenerative medicine. Many of these hydrogels are prepared from synthetic macromers (e.g., poly(ethylene glycol) or PEG) as they provide high degrees of tunability for matrix crosslinking, degradation, and modification. For a hydrogel to be considered biomimetic, it has to recapitulate key features that are found in the native extracellular matrix, such as the appropriate matrix mechanics and permeability, the ability to sequester and deliver drugs, proteins, or nucleic acids, as well as the ability to provide receptor-mediated cell–matrix interactions and protease-mediated matrix cleavage. A variety of chemistries have been employed to impart these biomimetic features into hydrogel crosslinking. These chemistries, such as radical-mediated polymerizations, enzyme-mediated crosslinking, bio-orthogonal click reactions, and supramolecular assembly, may be different in their crosslinking mechanisms but are required to be efficient for gel crosslinking and ligand bioconjugation under aqueous reaction conditions. The prepared biomimetic hydrogels should display a diverse array of functionalities and should also be cytocompatible for in vitro cell culture and/or in situ cell encapsulation. The focus of this article is to review recent progress in the crosslinking chemistries of biomimetic hydrogels with a special emphasis on hydrogels crosslinked from poly(ethylene glycol)-based macromers.
Introduction
Biomimetic hydrogels are a class of water-imbibing but insoluble polymer networks that present aspects of native extracellular matrix (ECM) to the surrounding or encapsulated cells. These aspects include the ability to emulate native matrix mechanics, sequester and deliver growth factors, as well as provide cell–matrix interactions such as ligand–receptor binding and protease-medicated matrix cleavage.1–4 To mimic matrix mechanics, one can simply adjust the degree of network crosslinking of a hydrogel.5–11 Growth factor sequestration is often achieved by the covalent immobilization of ‘affinity ligands’ (e.g., heparin, affinity peptides, small molecular weight ligands, and aptamers) in the network.2,12–17 The presentation of receptor-binding ligands (e.g., Arg–Gly–Asp peptide, often as network-immobilized pendent motifs) induces receptor-mediated intracellular signaling that is important for maintaining or guiding cell viability and function.3,18,19 Furthermore, the presence of protease-sensitive substrates (e.g. matrix metalloproteinases (MMPs) cleavable peptides, often serve as a gel crosslinker) permits cell-mediated local matrix cleavage and subsequent cell fate processes, which include migration, extension of cellular processes, and proliferation.4,20,21 It is known that matrix mechanics profoundly affect cell fate processes through regulating intracellular tensions.22–24 Emerging work has also demonstrated that along with matrix degradation, the mechanics of the matrix influence cell spreading and cell fate determination.25–27 Hydrogels are ideal matrices for this type of study as the mechanical properties of these water-swollen matrices can be easily and sometimes independently tuned to mimic native tissue elasticity and bio-functionality.28 The overarching goal of creating biomimetic hydrogels is to recapitulate local cell–matrix interactions for improving the outcome of global tissue regeneration and/or to understand fundamental mechanisms by which specific extracellular signals influence cell fate determination.In the past few decades, significant efforts have been dedicated to the design and synthesis of biomimetic hydrogels for tissue engineering and regeneration medicine applications.1,3,4,29 Many of these hydrogels are fabricated to present soluble or immobilized proteins/peptides as well as controllable matrix elasticity to cells that are encapsulated within or adhered onto the biomimetic hydrogels. Biomimetic hydrogels can be tailored to allow both two-dimensional (2D) and three-dimensional (3D) cell cultures.30 Compared with flat, rigid, and 2D tissue culture polystyrene (TCPS) or animal-based 3D matrices (e.g., Matrigel or collagen gel), biomimetic hydrogels composed of synthetic polymers, such as poly(ethylene glycol) or PEG, are more flexible and have tunable material properties. Notable applications of biomimetic hydrogels include preservation and differentiation of stem/progenitor cells, exploration of tumor cell migration, invasion, drug responsiveness, and epithelial–mesenchymal transition (EMT), as well as tissue/organ regeneration. The goal of this article is to review some important hydrogel crosslinking chemistries and to provide an update on recent advances in the fabrication of PEG-based biomimetic hydrogels.
Functionalization of PEG-based biomimetic hydrogels using photopolymerizations
 | ||
| Fig. 1 (A) Schematic of chain-growth photopolymerization for forming peptide-immobilized hydrogels (PI: photopolymerization, hv: light source). (B) Acrylated RGDS peptide for co-polymerization into PEGDA hydrogels. (C) Schematic of an acrylate-PEG–GPQGIWGQK–PEG-acrylate crosslinker. | ||
The co-polymerization of pendent peptides within PEG hydrogels provides binding sites in the gels for cell surface receptor activation. To render PEG-based hydrogels truly cell responsive, protease sensitivity must also be integrated in the design of a biomimetic hydrogel. PEG hydrogels crosslinked from PEGDA or PEGDMA are hydrolytically and proteolytically stable on therapeutically relevant time scales. To render the covalently crosslinked PEG hydrogels degradable, segments of degradable motifs can be incorporated in the macromer backbone.32,37 For example, the polymerization of acrylated PLA–PEG–PLA macromers yields hydrolytically degradable hydrogels with predictable gel degradation rates.32,38,39 Protease responsiveness can be integrated into PEG-based hydrogels through crosslinking with known peptide substrates for selective proteases. For example, hetero-bifunctional PEG macromers such as acrylate-PEG–NHS were used to react with MMP-sensitive peptides (e.g., NH2-GPQG↓IWGQK) through nucleophilic addition to primary amines, thus producing a homopolymerizable PEG macromer with protease sensitivity (Fig. 1C).40 Cells encapsulated within this type of hydrogel network were able to remodel their local matrix through protease secretion. While the protease sensitivity of this type of chain-growth PEG hydrogel can be tuned by adjusting the amount of protease sensitive macromer (i.e., acryl-peptide–PEG–peptide-acryl) added during network crosslinking, the accessibility of protease sensitive sections of the macromer to the co-encapsulated cells might be limited because of the presence of poly(acrylate) kinetic chains following crosslinking. Another limitation of this system is that the NHS on acrylate-PEG–NHS might react with bioactive lysine residues that are present within the protease sensitive sequence.
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) GPQG↓IWGQ). The additional cysteines permit a radical-mediated thiol–norbornene ‘photo-click’ reaction, while the presence of the MMP-sensitive sequence allows cell-mediated network cleavage to accommodate cellular processes such as migration, proliferation, and differentiation.46–48 Other bioactive motifs (e.g., cell adhesive ligands RGDS) can be incorporated within these hydrogels through the conjugation of mono-cysteine peptides during network crosslinking. The incorporation of pendent bioactive motifs can be easily achieved using peptides bearing a cysteine residue (Fig. 2B). Compared to chain-growth acrylate homopolymerizations, step-growth thiol–norbornene gelation has been shown to exhibit better cytocompatibility for radical sensitive cells such as pancreatic β-cells41 and chondrocytes.49 In addition to UV light mediated cross-linking, our own group has developed a visible light (400–700 nm) mediated thiol–norbornene photo-crosslinking method aided by a non-cleavage type photosensitizer eosin-Y.50–52 Upon visible light exposure, excited eosin-Y molecules deprotonate thiols to give thiyl radicals that subsequently initiate thiol–norbornene reactions to produce step-growth hydrogels with protease sensitivity and cell adhesive properties. The use of visible light eliminates the concerns that UV light, even at long wavelengths and low intensity, could induce cellular damage with biological complications. Readers are directed to a recent review that summarizes the progress of thiol–norbornene hydrogels to-date.53
GPQG↓IWGQ). The additional cysteines permit a radical-mediated thiol–norbornene ‘photo-click’ reaction, while the presence of the MMP-sensitive sequence allows cell-mediated network cleavage to accommodate cellular processes such as migration, proliferation, and differentiation.46–48 Other bioactive motifs (e.g., cell adhesive ligands RGDS) can be incorporated within these hydrogels through the conjugation of mono-cysteine peptides during network crosslinking. The incorporation of pendent bioactive motifs can be easily achieved using peptides bearing a cysteine residue (Fig. 2B). Compared to chain-growth acrylate homopolymerizations, step-growth thiol–norbornene gelation has been shown to exhibit better cytocompatibility for radical sensitive cells such as pancreatic β-cells41 and chondrocytes.49 In addition to UV light mediated cross-linking, our own group has developed a visible light (400–700 nm) mediated thiol–norbornene photo-crosslinking method aided by a non-cleavage type photosensitizer eosin-Y.50–52 Upon visible light exposure, excited eosin-Y molecules deprotonate thiols to give thiyl radicals that subsequently initiate thiol–norbornene reactions to produce step-growth hydrogels with protease sensitivity and cell adhesive properties. The use of visible light eliminates the concerns that UV light, even at long wavelengths and low intensity, could induce cellular damage with biological complications. Readers are directed to a recent review that summarizes the progress of thiol–norbornene hydrogels to-date.53
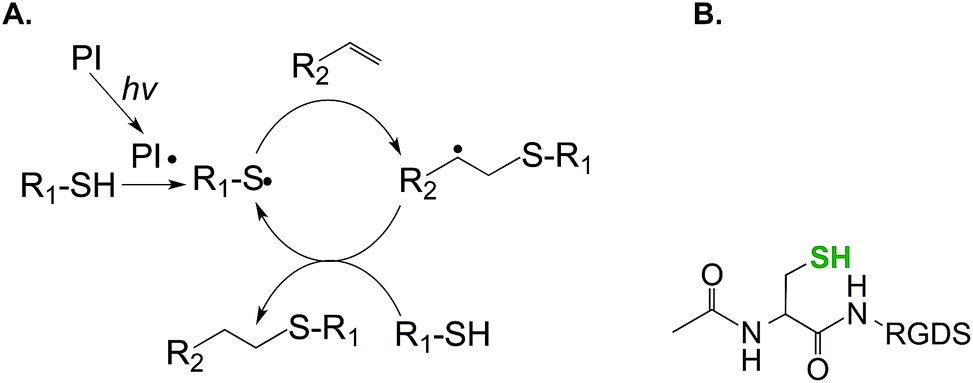 | ||
| Fig. 2 Schematic of step-growth photopolymerization for forming hydrogels (PI: photopolymerization, hv: light source). (B) Cysteine-containing RGDS peptide for co-polymerization into the step-growth hydrogel (N-terminal acetylated peptide). | ||
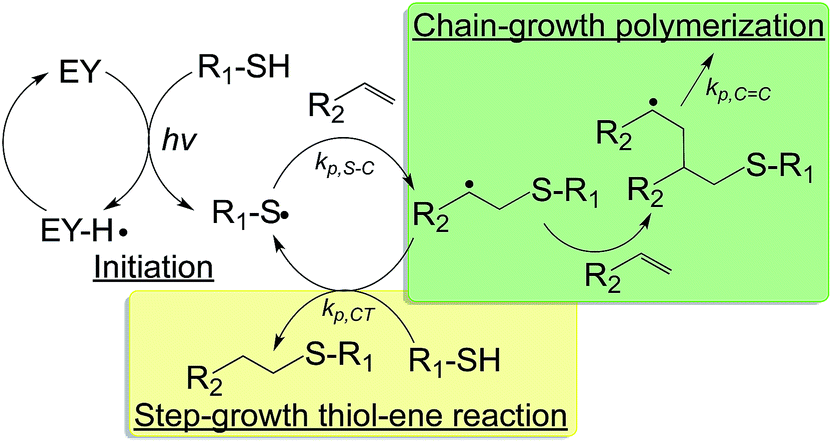 | ||
Fig. 3 Schematic of a visible light initiated mixed-mode photopolymerization for forming hydrogels (EY: eosin-Y; hv: visible light source; kp,S–C, kp,C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, kp,CT: kinetic constants for thiol–carbon radical propagation, carbon–carbon radical propagation, and chain-transfer, respectively).36 C, kp,CT: kinetic constants for thiol–carbon radical propagation, carbon–carbon radical propagation, and chain-transfer, respectively).36 | ||
Enzyme-mediated crosslinking of biomimetic hydrogels
Several enzymes have been employed for fabricating hydrogels in biomedical applications. For example, horseradish peroxidase (HRP) catalyzes the formation of carbon–carbon bond or carbon–oxygen bond in substrates, such as phenols or anilines, in the presence of hydrogen peroxide (H2O2). Similarly, glucose oxidase (GOX), when mixed with glucose and dissolved oxygen, generates gluconolactone and H2O2 that is further reduced into hydroxyl ions (OH−) and hydroxyl radicals (˙OH) in the presence of ferrous ions (Fe2+). When sufficient vinyl monomers are present in the solution, hydroxyl radicals initiate chain-growth polymerization to form a covalently crosslinked hydrogel.59–61 Except for a few examples,62,63 the cyto- and bio-compatibility of hydrogels crosslinked by HRP or GOX is adversely affected due to the requirement for (in the case of HRP) or generation of (in the case of GOX) H2O2. While past efforts have demonstrated the unique biomedical applications of PEG-based hydrogels crosslinked by enzymatic activity of HRP or GOX, the use of these enzymes to prepare biomimetic hydrogels that have both cell adhesiveness and protease sensitivity has attracted less attention.Thrombin is a critical enzyme in the coagulation cascade. In the presence of factor XIIIa, an activated transglutaminase, thrombin converts soluble fibrinogen into an insoluble fibrin clot. Factor XIIIa catalyzes an acyl-transfer reaction between the γ-carboxamide group of protein bound glutaminyl residues and the amino group of lysine residues to form covalent isopeptide bridges. Lutolf et al. harnessed this efficient reaction and developed biomimetic hydrogels capable of being degraded by enzymatic reactions (Fig. 4).64 In particular, factor XIIIa was utilized to simultaneously cross-link peptide-functionalized PEG and incorporate bioactive peptides. The fusion peptides used contained substrates for factor XIIIa and MMP (e.g., Ac-FKGG-GPQGIWGQ-ERCG-NH2 and H-NQEQVSPL-ERCG-NH2). Some sequences also contained the cell adhesive ligand such as H-NQEQVSPL-RGDSPG-NH2. Among these peptides, the sequence NQEQVSPL was derived from the N-terminus of α2-plasmin inhibitor (α2PI1–8), whereas the sequence Ac-FKGG was optimized for rapid transglutaminase reaction.65 Upon the addition of factor XIIIa and Ca2+, the two segments containing Lys and Gln residues (i.e., Ac-FKGG and NQEQVSPL, respectively) were catalytically incorporated into a covalent linkage that either has MMP sensitivity (from sequence GPQGIWGQ) or cell adhesiveness (from sequence RGDS). Depending on the gel formulations and enzyme concentrations, the gelation could occur within several minutes and the resulting gels supported spreading, proliferation, and migration of human dermal fibroblasts. Hydrogels crosslinked by factor XIIIa-mediated enzymatic reactions have been used in a variety of functional tissue engineering applications. For instance, diverse 3D peptide (e.g., RGD) or protein (e.g., fibrin, VEGF, or PDGF) patterns could be created within PEG-based hydrogels through selective light-activated enzymatic reactions.66 hMSCs encapsulated within these dynamically patterned hydrogels showed pattern shape-guided invasion66 and pattern gradient-induced morphogenesis.67
 | ||
| Fig. 4 Schematic of factor XIIIa-catalyzed formation of a PEG–peptide biomimetic hydrogel. Factor XIIIa was used to cross-link two PEG–peptide conjugates (n-PEG–MMP–Lys and n-PEG–Gln) in combination with a cell adhesion peptide (TG–Gln–RGD) to form multifunctional biomimetic hydrogels. Reprinted with permission from.64 Copyright 2007, American Chemical Society. | ||
Tyrosinase, an enzyme that oxidizes phenols, is another useful enzyme for the crosslinking of hydrogels and underwater bioadhesives.68 For example, Messersmith and colleagues synthesized 3,4-dihydroxyphenylalanine (DOPA)-modified PEGs,69 which were cross-linked into hydrogels in the presence of tyrosinase (Fig. 5). In another example, Park et al. prepared tyramine-functionalized Pluronic F-127 tri-block copolymers, which were utilized to form self-assembled micelles.70 The tyramine-conjugated micelles were converted to highly reactive catechol conjugated micelles by tyrosinase. Stable hydrogels were formed due to the cross-linking of Pluronic copolymer micelles. Although these hydrogels did not contain peptide linkers sensitive to cell-secreted proteases or cell adhesion ligands, it will be possible to create such biomimetic matrices using macromers pre-conjugated with cell adhesive and/or protease sensitive peptides.
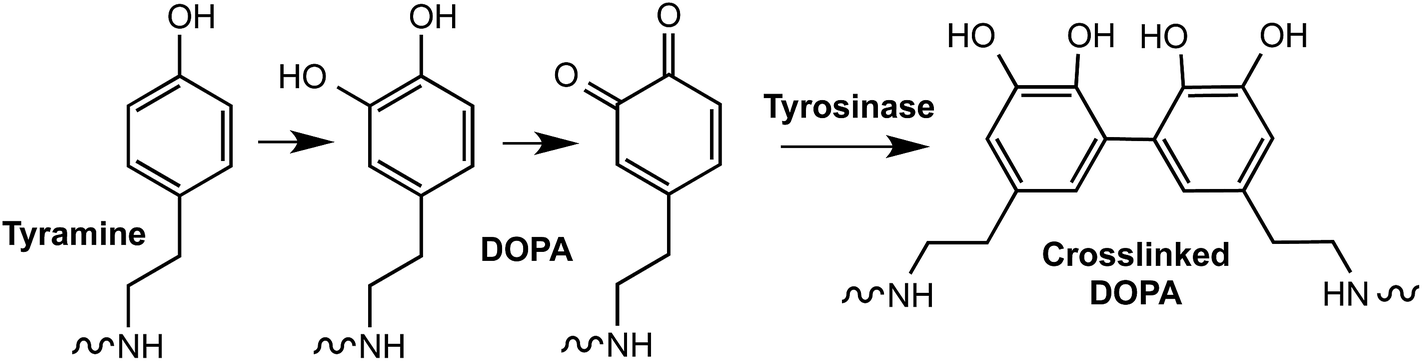 | ||
| Fig. 5 Oxidative conversion of tyramine to a catechol and subsequent crosslinking by tyrosinase. | ||
Click hydrogels as biomimetic matrices
‘Click’ chemistry is used to describe highly efficient, quantitative, and orthogonal reactions between mutually reactive functional groups and it has been used to create functional polymers and network hydrogels for biomedical applications.71,72 For example, Hubbell and colleagues pioneered the development of PEG-based click hydrogels. They incorporated elegant MMP-sensitive peptide sequences in the hydrogels using nucleophilic Michael-type addition reactions between multi-arm PEG-vinylsulfone and MMP-sensitive peptide crosslinkers with terminal cysteines.20 Cell adhesive ligands could be easily conjugated using the same Michael-type addition chemistry. Methacrylate, acrylate, and maleimide can also be used to react with bis-cysteine peptides (or multifunctional thiol macromers) for forming cell responsive hydrogels. The major benefit of biomimetic hydrogels formed by nucleophilic conjugation addition reactions is that it does not involve the generation of radicals, which poses major cytocompatibility concerns for radical-sensitive cells. Some of the other notable click chemistries useful in creating biomimetic hydrogels include native chemical ligation,69,73 oxime-ligand,74 azide–alkyne addition,75–78 Diels–Alder reaction,79 and tetrazine chemistry.80,81 Similar to the Michael-type conjugation reaction, these chemoselective chemistries are not light dependent and do not require initiators to initiate gel crosslinking.82 In general, this gelation chemistry lacks spatial-temporal control in gelation kinetics. Furthermore, the reaction rates may be slow at neutral pH values.The cross-linking chemistries discussed above have been highly useful for creating biomimetic hydrogels for 3D cell studies. However, most of these chemistries do not permit the dynamic modification of biophysical or biochemical gel properties. The ability to dynamically control gel properties is especially important if one considers that stem and progenitor cells receive complex and dynamic extracellular signals during morphogenesis. Pathological processes in many diseases are also induced by the deregulation of biological signals. It has become increasingly evident that biomaterials capable of mimicking dynamic changes of biological cues are powerful tools for studying tissue regeneration. Significant efforts have been dedicated to formulating such dynamic matrices. For example, Shoichet et al. incorporated a nitrobenzyl-protected cysteine in agarose hydrogels to guide 3D cell growth and migration.83 Upon UV exposure, the nitrobenzyl group is removed, revealing the free sulfhydryl group for additional thiol–maleimide conjugation. Biomolecules can be patterned in 3D to guide cell migration in a spatial-temporally regulated manner.
It will be beneficial if the modification of hydrogel properties can be performed in the presence of cells. For example, Fairbanks et al. developed step-growth thiol–norbornene hydrogels that were crosslinked with an excess amount of norbornene functionalities during network crosslinking.45 Due to the lack of homopolymerization between norbornene groups and the step-growth nature of the thiol–norbornene reaction, additional thiol-bearing molecules can be patterned within the gel network in the presence of cells. In addition to immobilizing pendent ligands in the presence of cells, one may wish to ‘exchange’ the ligands to truly recapitulate a dynamic developmental process during tissue morphogenesis. In this regard, an addition–fragmentation-chain transfer reaction was developed to allow controlled and reversible exchange of biochemical ligands within an allyl sulfide functionalized PEG hydrogel (Fig. 6).84
 | ||
| Fig. 6 Mechanism of addition fragmentation chain transfer of an allyl sulfide functional group upon attack by a thiyl radical. Redrawn with permission from.84 Copyright 2014, John Wiley and Sons. | ||
Significant achievements have been made on using orthogonal ‘click’ chemistry to synthesize biomimetic and dynamically tunable hydrogels. Anseth and colleagues created photolabile hydrogels by incorporating nitrobenzyl groups to the PEG or peptide crosslinkers.85–87 Hydrogels were firstly formed via redox crosslinking and the gel crosslinking density was decreased in a spatial-temporally controlled manner by adjusting the dosage and location of UV light exposure. When the photolabile group was incorporated on pendent peptides, UV light exposure caused the liberation of these peptides. The system is cytocompatible and the modification of hydrogel properties could be performed in the presence of cells.
Dynamically tunable hydrogels can be prepared by copper-catalyzed azide–alkyne Huisgen cycloaddition reactions (CuAAC). Anseth et al. used this chemistry to fabricate PEG-based hydrogels with patternability.88 Unfortunately, this reaction is not suitable for in situ cell encapsulation due to the cytotoxicity of copper ions. Furthermore, they developed an alternative approach wherein cyclooctyne, a macromer synthesized originally by the Bertozzi group,89 was used to react with azides free of cytotoxic metal ions.76 The metal-free and orthogonal reactivity between a strained cyclooctyne and an azide has allowed researchers to design multifunctional macromers that can be cleaved by cell-secreted proteases and for spatial-temporally controlled conjugation of bioactive motifs. For example, DeForest and Anseth designed a ‘sequential-click’ approach in a step-growth network to allow the formation of a hydrogel network and modification of its properties through orthogonal conjugation and cleavage of biomimetic peptides.76–78,90 To expand the utility of dynamic patterning of bioactive motifs in 3D, DeForest and Tirrell recently reported the conjugation and removal of the whole protein within the orthogonally crosslinked network.91 Two bioorthogonal photochemistries, oxime ligation (Fig. 7A) and ortho-nitrobenzyl ester photoscission (Fig. 7B), were employed to permit user-defined spatial-temporal photo-patterning and removal of whole proteins (Fig. 7C). The Anseth group has expanded the toolkits of bio-orthogonal click hydrogels to include: (1) tetrazine–norbornene click reaction (Fig. 8A)80 and (2) ligation between aliphatic hydrazine with a benzaldehyde- or an aliphatic-aldehyde (Fig. 8B).92 The hydrazine–aldehyde click reaction is particularly intriguing as it results in a covalently adaptable network that can respond to cell-induced stress through breaking/reforming elastically active crosslinks while maintaining a macroscopically stable material.
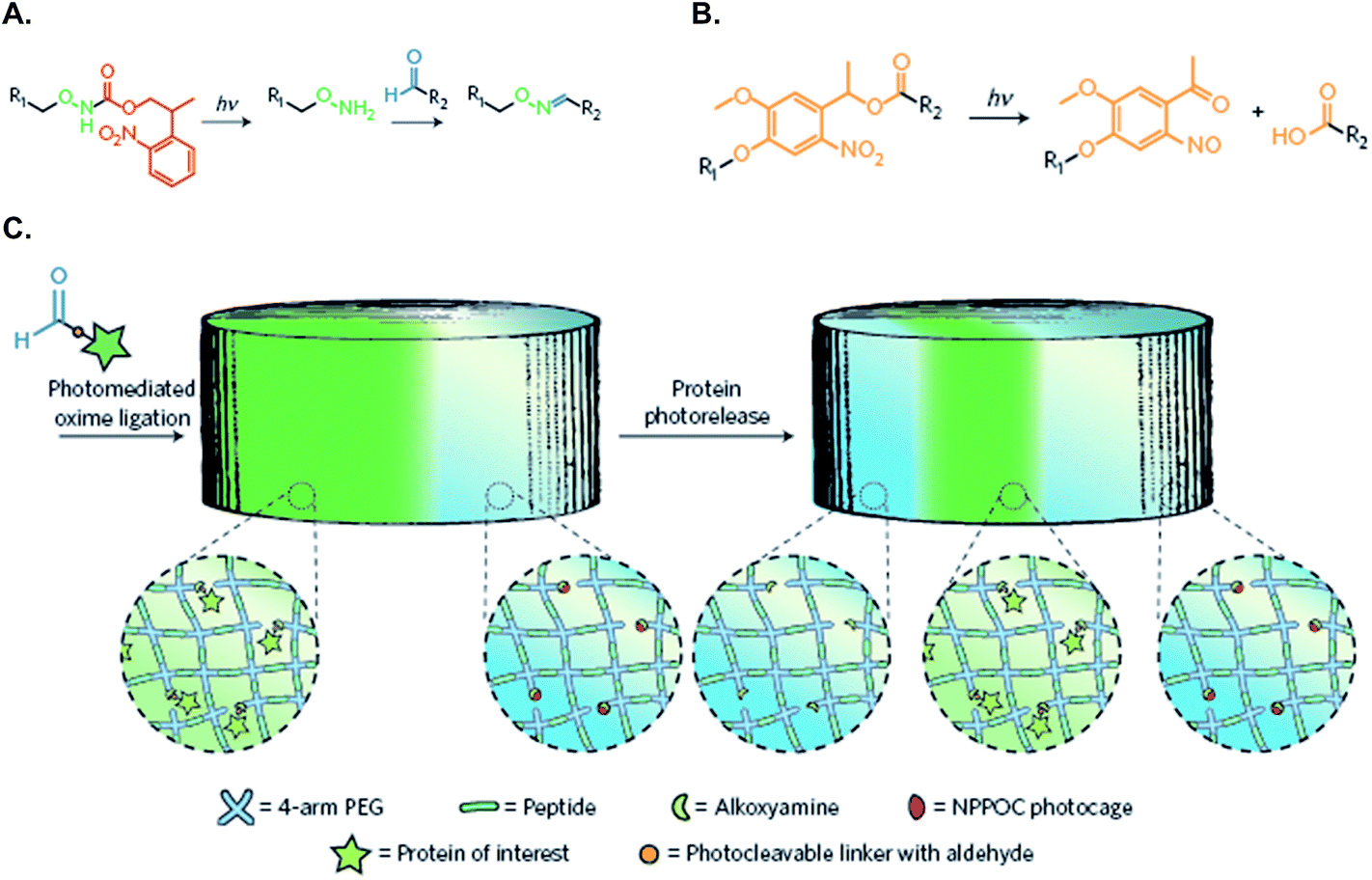 | ||
| Fig. 7 (A) Caged alkoxyamines undergo irreversible β-elimination upon exposure to 365 or 740 nm light. The liberated alkoxyamines react with aldehyde-functionalized proteins (R2) to form oxime linkages. (B) o-Nitrobenzyl ester (oNB) moieties linking the protein of interest (R1) and the hydrogel (R2) undergo photocleavage upon exposure to 365 nm or 740 nm light. (C) Schematic of the photo-reversible patterning strategy. NHS–oNB–CHO-functionalized proteins are first tethered to the gel through photo-mediated oxime ligation and subsequently removed on secondary light exposure. Reprinted with permission from.91 Copyright 2015, Nature Publishing Group. | ||
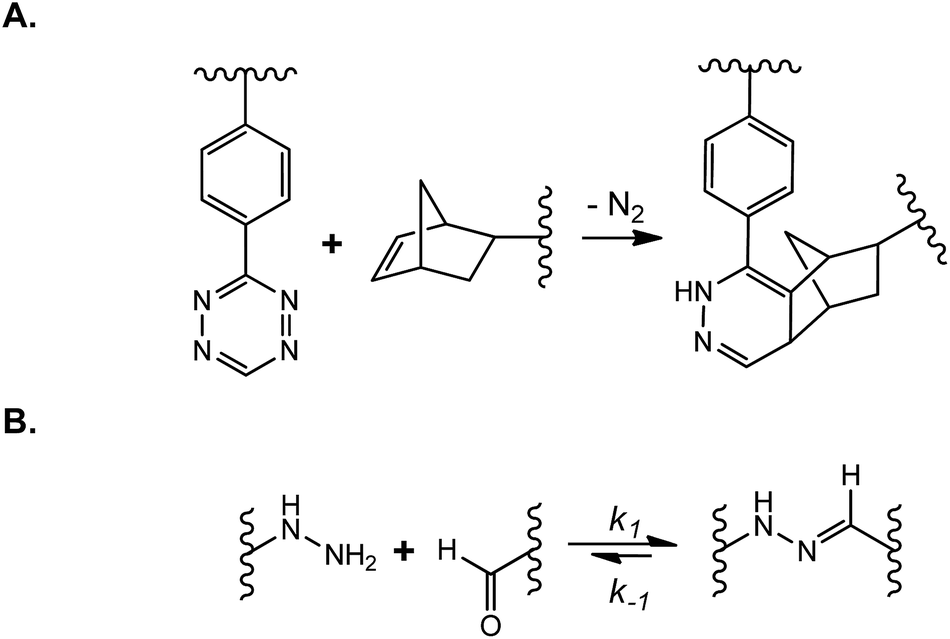 | ||
| Fig. 8 (A) Tetrazine–norbornene click reaction; (B) covalently adaptable network formed by N-methyl hydrazine–butyraldehyde ligation. | ||
Hydrogels formed from supramolecular assembly
In addition to the aforementioned covalent crosslinking strategies, PEG-based biomimetic hydrogels can be prepared through macromolecular or supramolecular self-assembly. Macromolecular/supramolecular assembled hydrogels are favorable in many applications because gelation is induced by a purely physical process, which does not rely on radical species.93 However, compared with covalently crosslinked hydrogels, these gels can have weaker mechanical properties due to the instability of the physical interactions between macromolecules. The most commonly used functionality in macromolecules suitable for forming self-assembled hydrogels are amphiphilic cyclodextrins (CD), including α-CD, β-CD, and γ-CD, which are composed of 6, 7, and 8 cyclic saccharides, respectively.94,95 The inner hydrophobic cavity of CDs affords physical interactions with hydrophobic molecules, while the hydrophilic outer surface facilitates the dissolution of the molecules in an aqueous environment. The hydroxyl groups on CDs provide handles for facile chemical modifications, which expand the utility of CDs in biomaterials, drug delivery, and tissue engineering applications.Supramolecular polymers are increasingly being used as ‘building blocks’ to fabricate diverse 3D crosslinked polymer networks. For example, macrocyclic CDs and cucurbit[n]urils are routinely used with specific ‘guest’ molecules to form poly-rotaxanes or catenanes. In particular, CD forms inclusion complexes with hydrophobic guest molecules such as adamantane, azobenzene, ferrocene, and stilbene.93 In one example, Burdick and colleagues harnessed inclusion complex formation between CD and adamantane (Ad), which were separately conjugated to hyaluronic acid (HA), to prepare shear-thinning hydrogels for tissue engineering applications.96–98 The inclusion complexes formed between CD and Ad led to HA hydrogel formation but the complexes disassembled under shear force, leading to a gel–sol transition. Upon the removal of shear force, the host–guest complexes, and hence the crosslinked hydrogel, re-formed. This new class of supramolecular polymer offers high injectability for implanted biomaterials in a minimally invasive manner. Similar to CDs that bind to hydrophobic molecules, cucurbit[n]uril (CB) binds strongly to guest molecules such as naphthalene and viologen. The complexation between CB and naphthyl-/viologen-functionalized polymers has been used to prepare protein-loaded hydrogels for controlled release applications.99–101 The aforementioned examples have demonstrated the great utilities of supramolecular assembly in hydrogel formation for biomedical applications.
Supramolecular assembly strategies have also been integrated in the design of PEG-based biomimetic hydrogels. For example, Elisseeff and colleagues have developed α-CD-threaded PEGDA hydrogels for tuning the biophysical and biochemical properties of chain-growth PEG-based hydrogels.102 Fig. 9 illustrates how supramolecular polymers can serve as ‘carriers’ to impart multiple and orthogonal functionalities to a hydrogel network. The PEG ‘necklaces’ are decorated/threaded with functionalized α-CD to produce hydrogels with tunable mechanics (by changing PEGDA concentration), adhesion (by adjusting the concentration of functionalized α-CD but not PEGDA), or chemistry (by introducing α-CD with different functional groups, e.g., –CH3 or –PO4−). By manipulating the compositions of these supramolecules, multifunctional hydrogels were created for promoting adhesion, proliferation, and differentiation of hMSCs102 or for studying the roles of matrix mechanics and functions on cancer cell invasion.103 While not demonstrated in this publication, protease-sensitivity can be integrated into PEG–CD hydrogels by replacing PEGDA with diacrylated PEG-peptide macromers (Fig. 1C).
 | ||
| Fig. 9 Schematic of a molecular-necklace system to create multifunctional hydrogels with independent control of gel mechanics, cell adhesiveness, and chemical functionality. (a) Inclusion complex between PEGDA and α-CD (R = hydroxyl or other functional groups). (b) Tuning the mechanical properties of an α-CD–PEG hydrogel independent of α-CD concentration. (c) Tuning the concentration of a cell adhesive peptide through threading different amount of functionalized α-CD independent of gel cross-linking density. (d) Tuning the chemical functionality of a hydrogel through threading α-CD with different functional groups (i.e., hydrophobic, hydrophilic or charged groups). Reprinted with permission from.102 Copyright 2013, John Wiley and Sons. | ||
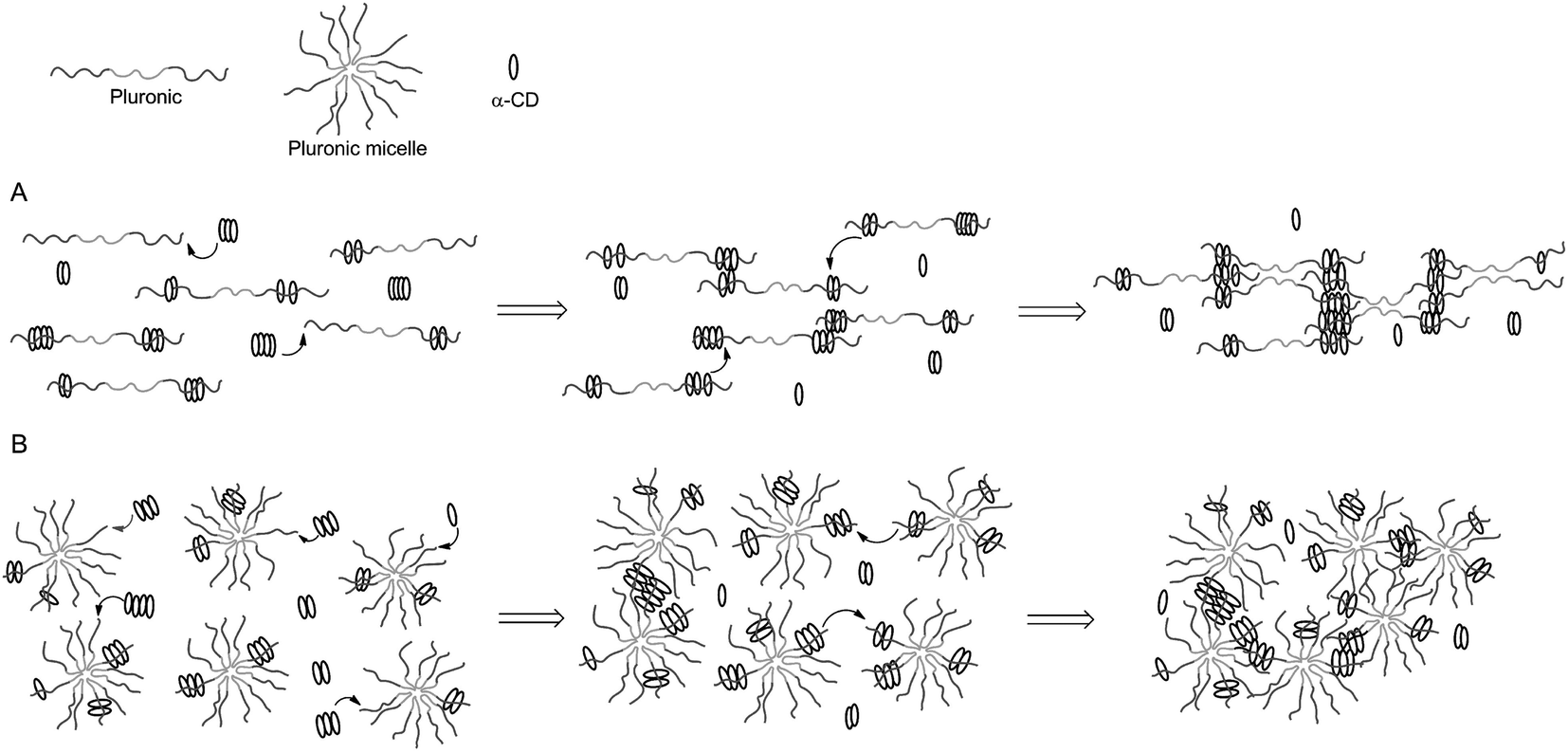
The example shown in Fig. 9 exploited modified CD as carriers of functional or bioactive motifs. The self-assembly between CD and PEG has also been utilized to prepare ‘pseudopolyrotaxane’ hydrogels. For instance, Cooper-White et al. explored the gelation kinetics and viscoelastic properties of such hydrogels formed by assembly of α-CD and Pluronic polymers (F68 and F127) without or with micelle formation.104 The formation of ‘poly-CDs’ at a concentration of over 40 mM of α-CD (25 °C) is critical in this physical gelation/assembly system. The threading of Pluronic polymers into pre-assembled poly-CDs leads to gelation (Fig. 10A). In the presence of Pluronic micelles, however, the threading of poly-CDs onto Pluronic polymer becomes more difficult due to the presence of steric hindrance (Fig. 10B). The gelation was slower and the gels were weaker in the presence of Pluronic micelles. Although these hydrogels could be formed with strong mechanical properties (G′ ∼ 106 Pa), they were not stable and dissociated rapidly when immersed in a liquid containing no pseudopolyrotaxane complexes. Cooper-White and colleagues recently reported an improved design of pseudopolyrotaxane hydrogels in which Pluronic/CD assembly was combined with enzyme-mediated crosslinking of tyramine-modified PEG.105 The gelation was facilitated through a horseradish peroxidase (HRP) and hydrogen peroxide (H2O2) mediated tyramine oxidation (similar to Fig. 5). However, careful optimization of enzymatic crosslinking parameters is critical for maintaining acceptable cell viability since H2O2 is toxic to cells.
 | ||
| Fig. 10 Temporal gelation mechanism of linear Pluronic (e.g.: F6810 and F6820) or Pluronic micelles (e.g.: F12710) and α-CD. (A) Gelation without micelle formation. (B) Gelation in the presence of micelles. Reprinted with permission from.104 Copyright 2013, American Chemical Society. | ||
Conclusions
The diversity of biomimetic PEG-based hydrogels has been expanded greatly in recent years owing to the discovery/adaptation of bioconjugation chemistries suitable for biomedical applications. Past efforts have also demonstrated that a hydrogel matrix with multiple functionalities outperforms a single-purpose one for most tissue regeneration applications. While no single chemistry is perfect for all applications, one can certainly adopt and integrate the available chemistries to create suitable biomimetic matrices for a particular tissue regeneration need. It is expected that due to its simplicity, diversity, and proven cytocompatibility in many cell types, radical-mediated hydrogel crosslinking (whether initiated by UV or visible light) will continue to serve as an indispensable chemistry for the design and synthesis of biomimetic hydrogels. The rise of bio-orthogonal chemistry, enzyme-mediated crosslinking, and supramolecular chemistries offer great opportunities for scientists to create complex multifunctional hydrogel matrices for addressing biological questions that are otherwise difficult to answer. Lastly, the combination of two or more of these diverse chemistries is anticipated to significantly increase the applicability of biomimetic hydrogels in tissue engineering and regenerative medicine applications.Acknowledgements
This study was supported in parts by the National Institutes of Health (R21CA188911) and a Faculty Startup Fund from Indiana University-Purdue University Indianapolis.References
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS PubMed.
- C. C. Lin and K. S. Anseth, J. Pharm. Res., 2009, 26, 631–643 CrossRef CAS PubMed.
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS PubMed.
- M. P. Lutolf and J. A. Hubbell, Nat. Biotechnol., 2005, 23, 47–55 CrossRef CAS PubMed.
- D. A. Wang, C. G. Williams, Q. A. Li, B. Sharma and J. H. Elisseeff, Biomaterials, 2003, 24, 3969–3980 CrossRef CAS.
- S. J. Bryant, K. S. Anseth, D. A. Lee and D. L. Bader, J. Orthop. Res., 2004, 22, 1143–1149 CrossRef CAS PubMed.
- S. J. Bryant, R. J. Bender, K. L. Durand and K. S. Anseth, Biotechnol. Bioeng., 2004, 86, 747–755 CrossRef CAS PubMed.
- S. J. Bryant, T. T. Chowdhury, D. A. Lee, D. L. Bader and K. S. Anseth, Ann. Biomed. Eng., 2004, 32, 407–417 CrossRef.
- X. Z. Shu, Y. C. Liu, F. S. Palumbo, Y. Lu and G. D. Prestwich, Biomaterials, 2004, 25, 1339–1348 CrossRef CAS PubMed.
- S. R. Peyton, C. B. Raub, V. P. Keschrumrus and A. J. Putnam, Biomaterials, 2006, 27, 4881–4893 CrossRef CAS PubMed.
- L. M. Weber, C. G. Lopez and K. S. Anseth, J. Biomed. Mater. Res., Part A, 2009, 90, 720–729 CrossRef PubMed.
- C.-C. Lin and K. S. Anseth, Adv. Funct. Mater., 2009, 19, 2325–2331 CrossRef CAS PubMed.
- C.-C. Lin and A. T. Metters, J. Biomed. Mater. Res., Part A, 2007, 83, 954–964 CrossRef PubMed.
- C.-C. Lin and A. T. Metters, Biomacromolecules, 2008, 9, 789–795 CrossRef CAS PubMed.
- C.-C. Lin, A. T. Metters and K. S. Anseth, Biomaterials, 2009, 30, 4907–4914 CrossRef CAS PubMed.
- C.-C. Lin, P. D. Boyer, A. A. Aimetti and K. S. Anseth, J. Controlled Release, 2010, 142, 384–391 CrossRef CAS PubMed.
- S. M. Willerth, P. J. Johnson, D. J. Maxwell, S. R. Parsons, M. E. Doukas and S. E. Sakiyama-Elbert, J. Biomed. Mater. Res., Part A, 2007, 80, 13–23 CrossRef PubMed.
- D. L. Hern and J. A. Hubbell, J. Biomed. Mater. Res., 1998, 39, 266–276 CrossRef CAS.
- D. L. Elbert and J. A. Hubbell, Biomacromolecules, 2001, 2, 430–441 CrossRef CAS PubMed.
- M. P. Lutolf, J. L. Lauer-Fields, H. G. Schmoekel, A. T. Metters, F. E. Weber, G. B. Fields and J. A. Hubbell, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5413–5418 CrossRef CAS PubMed.
- A. B. Pratt, F. E. Weber, H. G. Schmoekel, R. Muller and J. A. Hubbell, Biotechnol. Bioeng., 2004, 86, 27–36 CrossRef CAS PubMed.
- S. R. Peyton, C. M. Ghajar, C. B. Khatiwala and A. J. Putnam, Cell Biochem. Biophys., 2007, 47, 300–320 CrossRef CAS.
- A. M. Kloxin, C. J. Kloxin, C. N. Bowman and K. S. Anseth, Adv. Mater., 2010, 22, 3484–3494 CrossRef CAS PubMed.
- K. A. Mosiewicz, L. Kolb, A. J. van der Vlies and M. P. Lutolf, Biomater. Sci., 2014, 2, 1640–1651 RSC.
- N. Huebsch, P. R. Arany, A. S. Mao, D. Shvartsman, O. A. Ali, S. A. Bencherif, J. Rivera-Feliciano and D. J. Mooney, Nat. Mater., 2010, 9, 518–526 CrossRef CAS PubMed.
- S. Khetan, M. Guvendiren, W. R. Legant, D. M. Cohen, C. S. Chen and J. A. Burdick, Nat. Mater., 2013, 12, 458–465 CrossRef CAS PubMed.
- C. Yang, M. W. Tibbitt, L. Basta and K. S. Anseth, Nat. Mater., 2014, 13, 645–652 CrossRef CAS PubMed.
- X. Tong and F. Yang, Biomaterials, 2014, 35, 1807–1815 CrossRef CAS PubMed.
- M. C. Cushing and K. S. Anseth, Science, 2007, 316, 1133–1134 CrossRef CAS PubMed.
- C. A. DeForest and K. S. Anseth, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 421–444 CrossRef CAS PubMed.
- B. V. Slaughter, S. S. Khurshid, O. Z. Fisher, A. Khademhosseini and N. A. Peppas, Adv. Mater., 2009, 21, 3307–3329 CrossRef CAS PubMed.
- K. T. Nguyen and J. L. West, Biomaterials, 2002, 23, 4307–4314 CrossRef CAS.
- C. R. Nuttelman, M. A. Rice, A. E. Rydholm, C. N. Salinas, D. N. Shah and K. S. Anseth, Prog. Polym. Sci., 2008, 33, 167–179 CrossRef CAS PubMed.
- J. L. West and J. A. Hubbell, React. Polym., 1995, 25, 139–147 CrossRef CAS.
- J. L. West and J. A. Hubbell, Macromolecules, 1999, 32, 241–244 CrossRef CAS.
- Y. T. Hao, H. Shih, Z. Munoz, A. Kemp and C. C. Lin, Acta Biomater., 2014, 10, 104–114 CrossRef CAS PubMed.
- S.-H. Lee, J. J. Moon, J. S. Miller and J. L. West, Biomaterials, 2007, 28, 3163–3170 CrossRef CAS PubMed.
- M. N. Mason, A. T. Metters, C. N. Bowman and K. S. Anseth, Macromolecules, 2001, 34, 4630–4635 CrossRef CAS.
- A. T. Metters, K. S. Anseth and C. N. Bowman, Polymer, 2000, 41, 3993–4004 CrossRef CAS.
- B. K. Mann, A. S. Gobin, A. T. Tsai, R. H. Schmedlen and J. L. West, Biomaterials, 2001, 22, 3045–3051 CrossRef CAS.
- C. C. Lin, A. Raza and H. Shih, Biomaterials, 2011, 32, 9685–9695 CrossRef CAS PubMed.
- B. D. Fairbanks, M. P. Schwartz, A. E. Halevi, C. R. Nuttelman, C. N. Bowman and K. S. Anseth, Adv. Mater., 2009, 21, 5005–5010 CrossRef CAS PubMed.
- H. Shih and C. C. Lin, Biomacromolecules, 2012, 13, 2003–2012 CrossRef CAS PubMed.
- A. Metters and J. Hubbell, Biomacromolecules, 2005, 6, 290–301 CrossRef CAS PubMed.
- B. D. Fairbanks, M. P. Schwartz, A. E. Halevi, C. R. Nuttelman, C. N. Bowman and K. S. Anseth, Adv. Mater., 2009, 21, 5005–5010 CrossRef CAS PubMed.
- C. S. Ki, T.-Y. Lin, M. Korc and C.-C. Lin, Biomaterials, 2014, 35, 9668–9677 CrossRef CAS PubMed.
- T.-Y. Lin, C. S. Ki and C.-C. Lin, Biomaterials, 2014, 35, 6898–6906 CrossRef CAS PubMed.
- C. Wang, X. Tong and F. Yang, Mol. Pharm., 2014, 11, 2115–2125 CrossRef CAS PubMed.
- J. J. Roberts and S. J. Bryant, Biomaterials, 2013, 34, 9969–9979 CrossRef CAS PubMed.
- H. Shih and C. C. Lin, Macromol. Rapid Commun., 2013, 34, 269–273 CrossRef CAS PubMed.
- H. Shih, A. K. Fraser and C. C. Lin, ACS Appl. Mater. Interfaces, 2013, 5, 1673–1680 CAS.
- H. Shih, R. G. Mirmira and C.-C. Lin, J. Mater. Chem. B, 2015, 3, 170–175 RSC.
- C.-C. Lin, C. S. Ki and H. Shih, J. Appl. Polym. Sci., 2015, 132, 41563 Search PubMed.
- C. N. Salinas and K. S. Anseth, Macromolecules, 2008, 41, 6019–6026 CrossRef CAS.
- A. E. Rydholm, C. N. Bowman and K. S. Anseth, Biomaterials, 2005, 26, 4495–4506 CrossRef CAS PubMed.
- S. J. Bryant, C. R. Nuttelman and K. S. Anseth, J. Biomater. Sci., Polym. Ed., 2000, 11, 439–457 CrossRef CAS PubMed.
- G. M. Cruise, O. D. Hegre, D. S. Scharp and J. A. Hubbell, Biotechnol. Bioeng., 1998, 57, 655–665 CrossRef CAS.
- Y. Hao and C.-C. Lin, J. Biomed. Mater. Res., Part A, 2014, 102, 3813–3827 CrossRef PubMed.
- L. M. Johnson, C. A. DeForest, A. Pendurti, K. S. Anseth and C. N. Bowman, ACS Appl. Mater. Interfaces, 2010, 2, 1963–1972 CAS.
- P. S. Hume, C. N. Bowman and K. S. Anseth, Biomaterials, 2011, 32, 6204–6212 CAS.
- R. Shenoy, M. W. Tibbitt, K. S. Anseth and C. N. Bowman, Chem. Mater., 2013, 25, 761–767 CrossRef CAS PubMed.
- L.-S. Wang, J. E. Chung, P. P.-Y. Chan and M. Kurisawa, Biomaterials, 2010, 31, 1148–1157 CrossRef CAS PubMed.
- D. J. Menzies, A. Cameron, T. Munro, E. Wolvetang, L. Grondahl and J. J. Cooper-White, Biomacromolecules, 2013, 14, 413–423 CrossRef CAS PubMed.
- M. Ehrbar, S. C. Rizzi, R. G. Schoenmakers, B. San Miguel, J. A. Hubbell, F. E. Weber and M. P. Lutolf, Biomacromolecules, 2007, 8, 3000–3007 CrossRef CAS PubMed.
- B. H. Hu and P. B. Messersmith, J. Am. Chem. Soc., 2003, 125, 14298–14299 CrossRef CAS PubMed.
- K. A. Mosiewicz, L. Kolb, A. J. van der Vlies, M. M. Martino, P. S. Lienemann, J. A. Hubbell, M. Ehrbar and M. P. Lutolf, Nat. Mater., 2013, 12, 1072–1078 CrossRef CAS PubMed.
- P. S. Lienemann, Y. R. Devaud, R. Reuten, B. R. Simona, M. Karlsson, W. Weber, M. Koch, M. P. Lutolf, V. Milleret and M. Ehrbar, Integr. Biol., 2015, 7, 101–111 RSC.
- M. Cencer, Y. Liu, A. Winter, M. Murley, H. Meng and B. P. Lee, Biomacromolecules, 2014, 15, 2861–2869 CrossRef CAS PubMed.
- B. P. Lee, J. L. Dalsin and P. B. Messersmith, Biomacromolecules, 2002, 3, 1038–1047 CrossRef CAS PubMed.
- S. H. Lee, Y. Lee, S.-W. Lee, H.-Y. Ji, J.-H. Lee, D. S. Lee and T. G. Park, Acta Biomater., 2011, 7, 1468–1476 CrossRef CAS PubMed.
- Y. Jiang, J. Chen, C. Deng, E. J. Suuronen and Z. Zhong, Biomaterials, 2014, 35, 4969–4985 CrossRef CAS PubMed.
- S. Liu, K. T. Dicker and X. Jia, Chem. Commun., 2015, 51, 5218–5237 RSC.
- B.-H. Hu, J. Su and P. B. Messersmith, Biomacromolecules, 2009, 10, 2194–2200 CrossRef CAS PubMed.
- G. N. Grover, J. Lam, T. H. Nguyen, T. Segura and H. D. Maynard, Biomacromolecules, 2012, 13, 3013–3017 CrossRef CAS PubMed.
- D. D. McKinnon, T. E. Brown, K. A. Kyburz, E. Kiyotake and K. S. Anseth, Biomacromolecules, 2014, 15, 2808–2816 CAS.
- C. A. DeForest, B. D. Polizzotti and K. S. Anseth, Nat. Mater., 2009, 8, 659–664 CrossRef CAS PubMed.
- C. A. DeForest, E. A. Sims and K. S. Anseth, Chem. Mater., 2010, 22, 4783–4790 CrossRef CAS PubMed.
- C. A. DeForest and K. S. Anseth, Nat. Chem., 2011, 3, 925–931 CrossRef CAS PubMed.
- K. C. Koehler, D. L. Alge, K. S. Anseth and C. N. Bowman, Biomaterials, 2013, 34, 4150–4158 CrossRef CAS PubMed.
- D. L. Alge, M. A. Azagarsamy, D. F. Donohue and K. S. Anseth, Biomacromolecules, 2013, 14, 949–953 CrossRef CAS PubMed.
- H. Zhang, K. T. Dicker, X. Xu, X. Jia and J. M. Fox, ACS Macro Lett., 2014, 3, 727–731 CrossRef CAS PubMed.
- A. J. García, Ann. Biomed. Eng., 2014, 42, 312–322 CrossRef PubMed.
- Y. Luo and M. S. Shoichet, Nat. Mater., 2004, 3, 249–253 CrossRef CAS PubMed.
- N. R. Gandavarapu, M. A. Azagarsamy and K. S. Anseth, Adv. Mater., 2014, 26, 2521–2526 CrossRef CAS PubMed.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59–63 CrossRef CAS PubMed.
- A. M. Kloxin, J. A. Benton and K. S. Anseth, Biomaterials, 2010, 31, 1–8 CrossRef CAS PubMed.
- A. M. Kloxin, M. W. Tibbitt, A. M. Kasko, J. A. Fairbairn and K. S. Anseth, Adv. Mater., 2010, 22, 61–66 CrossRef CAS PubMed.
- B. D. Polizzotti, B. D. Fairbanks and K. S. Anseth, Biomacromolecules, 2008, 9, 1084–1087 CrossRef CAS PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- C. A. DeForest and K. S. Anseth, Angew. Chem., Int. Ed., 2012, 51, 1816–1819 CrossRef CAS PubMed.
- C. A. DeForest and D. A. Tirrell, Nat. Mater., 2015, 14, 523–531 CrossRef PubMed.
- D. D. McKinnon, D. W. Domaille, J. N. Cha and K. S. Anseth, Adv. Mater., 2014, 26, 865–872 CrossRef CAS PubMed.
- R. Dong, Y. Zhou, X. Huang, X. Zhu, Y. Lu and J. Shen, Adv. Mater., 2015, 27, 498–526 CrossRef CAS PubMed.
- J. Zhang and P. X. Ma, Adv. Drug Delivery Rev., 2013, 65, 1215–1233 CrossRef CAS PubMed.
- B. V. K. J. Schmidt, M. Hetzer, H. Ritter and C. Barner-Kowollik, Prog. Polym. Sci., 2014, 39, 235–249 CrossRef CAS PubMed.
- M. Guvendiren, H. D. Lu and J. A. Burdick, Soft Matter, 2012, 8, 260–272 RSC.
- C. B. Rodell, A. L. Kaminski and J. A. Burdick, Biomacromolecules, 2013, 14, 4125–4134 CrossRef CAS PubMed.
- C. B. Rodell, J. W. MacArthur Jr, S. M. Dorsey, R. J. Wade, L. L. Wang, Y. J. Woo and J. A. Burdick, Adv. Funct. Mater., 2015, 25, 636–644 CrossRef CAS PubMed.
- E. A. Appel, F. Biedermann, U. Rauwald, S. T. Jones, J. M. Zayed and O. A. Scherman, J. Am. Chem. Soc., 2010, 132, 14251–14260 CrossRef CAS PubMed.
- E. A. Appel, X. J. Loh, S. T. Jones, F. Biedermann, C. A. Dreiss and O. A. Scherman, J. Am. Chem. Soc., 2012, 134, 11767–11773 CrossRef CAS PubMed.
- E. A. Appel, X. J. Loh, S. T. Jones, C. A. Dreiss and O. A. Scherman, Biomaterials, 2012, 33, 4646–4652 CrossRef CAS PubMed.
- A. Singh, J. Zhan, Z. Ye and J. H. Elisseeff, Adv. Funct. Mater., 2013, 23, 575–582 CrossRef CAS PubMed.
- J. N. Beck, A. Singh, A. R. Rothenberg, J. H. Elisseeff and A. J. Ewald, Biomaterials, 2013, 34, 9486–9495 CrossRef CAS PubMed.
- C. Pradal, K. S. Jack, L. Grondahl and J. J. Cooper-White, Biomacromolecules, 2013, 14, 3780–3792 CrossRef CAS PubMed.
- C. Pradal, L. Grondahl and J. J. Cooper-White, Biomacromolecules, 2015, 16, 389–403 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |