
Biological and pharmacological activities of amaryllidaceae alkaloids
Maomao He†
,
Chunrong Qu†
,
Oude Gao,
Xianming Hu and
Xuechuan Hong
*
State Key Laboratory of Virology, Key Laboratory of Combinatorial Biosynthesis and Drug Discovery (Wuhan University), Ministry of Education, Wuhan University School of Pharmaceutical Sciences, Wuhan 430071, P. R. China. E-mail: xhy78@whu.edu.cn
First published on 29th January 2015
Abstract
The amaryllidaceae family consists of about 75 genera and 1100 species that are wide-spread in the tropics and warm temperate regions of the world. Since the first isolation of lycorine, more than 500 amaryllidaceae alkaloids have been isolated over the past three decades. The enormous numbers of diverse amaryllidaceae alkaloids are classified into different groups mainly according to their structural features. The representative alkaloids are norbelladine, lycorine, hippeastrine, narwedine, haemanthamine, pancratistatin, pretazettine, montanine, galanthindole, cherylline and ismine. Recently, more extensive studies have revealed that amaryllidaceae alkaloids exhibit a wide range of bioactivities, such as antitumor, antiviral, antibacterial, antifungal, antimalarial and analgesic. Acetylcholinesterase (AChE) inhibitory and cytotoxic activities have also been reported. The aim of the present review is to discuss the recent developments on the biological and pharmacological activities of amaryllidaceae alkaloids with IC50 or EC50 values since 2005, supporting the potential therapeutic possibilities for the use of these compounds.
 Maomao He | Maomao He obtained his B.Sc. degree in Pharmaceutical Engineering from Wannan Medical College in 2010. He joined Wuhan University School of Pharmaceutical Sciences for obtaining his M.Sc. in Medicinal Chemistry. He is currently pursuing his doctoral studies in Organic Biochemistry and Molecular Biology at the same institution under the guidance of Professor Xuechuan Hong. His doctoral work focuses on the total synthesis and anticancer evaluation of amaryllidaceae alkaloids and erythrina alkaloids. |
 Chunrong Qu | Chunrong Qu received her B.Sc. in Pharmacy from Southwest University in 2011. She then earned her M.Sc. from Wuhan University in 2013 under the supervision of Professor Xuechuan Hong. She is currently engaging in her Ph.D. research at the same group, where she obtained a sponsorship by “The Fundamental Research Funds for the Central Universities”. Her research interests include the total synthesis of medermycin, the synthesis of PBRs and applications in molecular imaging. |
 Xuechuan Hong | Xuechuan Hong studied chemistry and obtained his Ph.D. from University of Missouri-Columbia (Missouri, USA) in 2005. After a postdoc position at Emory University under the supervision of Professor Albert Padwa, he joined Albany Molecular Research Inc. as a senior research scientist. Since October 2010, he was appointed a Full Professor at Wuhan University (Wuhan, China). His research interests included targeted therapy for cancer, molecular imaging, drug development. |
1. Introduction
The family of amaryllidaceae takes its name from the genus Amaryllis and consists of about 75 genera, whose 1100 species are widely distributed in the tropics and warm temperate regions of the world.1 Plants of the amaryllidaceae family have been used for thousands of years in traditional herbal medicine. The earliest evidence of their therapeutic application was discovered in the fourth century B.C.E., when Hippocrates of Cos used the oil from the daffodil, Narcissus poeticus L. for the treatment of uterine tumors.2 Since the isolation of the first alkaloid lycorine (3) from Amaryllidaceae lycoris in 1877, more than 500 amaryllidaceae alkaloids representing 11 skeletal types (Fig. 1) have been isolated over the past three decades.3 The representative alkaloids are norbelladine (1), rystilline (2), lycorine (3), hippeastrine (4), narwedine (5), galanthamine (6), haemanthamine (7), pancratistatin (8), pretazettine (9), montanine (10), galanthindole (11), cherylline (12) and ismine (13) (Table 1). Such compounds exhibit a diversity of biological activities including antitumor, antiviral, antibacterial, antifungal, antimalarial, analgesic, acetylcholinesterase (AChE) inhibitory and cytotoxic activities. Among them, galanthamine (6) is one of the most significant amaryllidaceae alkaloids, which has been approved as a longer acting anticholinesterase drug in the treatment of Alzheimer's disease.4 Moreover, lycorine and its derivatives have shown potential therapeutical efficacy in cancers, viral and Alzheimer's disease.5–7 Their importance in medicinal chemistry has brought enormous attention to their synthesis and biological activity studies in the recent past. | ||
| Fig. 1 Main skeletal types of the amaryllidaceae alkaloids. | ||
| Framework-type | Ring type | Representative alkaloid | |
|---|---|---|---|
| I | Belladine-type | N-(3,4-Dioxybenzyl)-4-oxyphenethylamine | Norbelladine (1), rystilline (2) |
| II | Lycorine-type | Pyrrolo[d,e]phenanthridine | Lycorine (3) |
| III | Lycorenine-type | 2-Benzopyrano-[3,4-g]indole | Hippeastrine (4) |
| IV | Galanthamine-type | 6H-Benzofuro[3a,3,2-e,f]-2-benzazepine | Narwedine (5), galanthamine (6) |
| V | Crinine-type | 5,10b-Ethanophenanthridine | Haemanthamine (7) |
| VI | Narcilasine-type | Lycoricidine | Pancratistatin (8) |
| VII | Tazettine-type | 2-Benzopyrano[3,4-c]indole | Pretazettine (9) |
| VIII | Montanine-type | 5,11-Methanomorphanthridine | Montanine (10) |
| IX | Galanthindole-type | 7-Phenyloctahydroindole | Galanthindole (11) |
| X | Cherylline-type | Tetrahydroisoquinoline | Cherylline (12) |
| XI | Ismine-type | 2-Phenylcyclohexanamine | Ismine (13) |
In past decades, a variety of articles concerning the structure elucidation, biosynthesis and total synthesis and biological activities of amaryllidaceae alkaloids have been reviewed.1 The intent of this review is to provide an overview of their in vitro and in vivo biological activities of the amaryllidaceae alkaloid family with their IC50 or EC50 values since 2005. However, it is inevitable for this review to have some overlap with the contents in previous review articles or chapters of books, especially with several excellent reviews recently published in this area.1–3
2. Biological and pharmacological activities
2.1 Anticancer activities
Many amaryllidaceae alkaloids have been reported to exhibit antitumor properties.5 The first well-known compound with cytostatic effect is lycorine (3) isolated in 1877 from the bulb of the Amaryllidaceae lycoris.6 Lycorine (3) has been intensively investigated in various preclinical models of human cancers both in vitro and in vivo.7 In general, lycorine (3) is recognized as a low micromolar antiproliferative agent against multidrug resistant and apoptosis-resistant cancers cells8–10 with selective cell type-dependent cytotoxicity in tumor cells by mitochondrial pathways and inducing apoptosis. Lycorine (3) can down-regulate Mcl-1 in human leukemia cells.11 The apoptosis process includes the regulation of the cell cycle in HL60 and KM3 cell lines, cytochrome-c release and caspase activation.11 The study has further revealed that lycorine can decrease HDAC enzymatic activities in K562 cells and up-regulate the expression of p53 and its target gene product p21, then inhibit the proliferation of K562 cells.12 Additionally, lycorine hydrochloride (20) can effectively suppress metastatic melanoma C8161 cell-dominant formation of capillary-like tubes in vitro and generation of tumor blood vessels in vivo with low toxicity. Mechanistic studies have revealed that lycorine hydrochloride (20) inhibits melanoma C8161 cell-dominant vasculogenic mimicry by reducing VE-cadherin gene expression and diminishing cell surface exposure of the protein.8 Recently, lycorine derivative 4-ethyl-5-methyl-5,6-dihydro-[1,3]dioxolo [4,5-j] phenanthridine (HLY78) is identified as an activator of the Wnt/β-catenin signaling pathway in a Wnt (Wingless and INT-1) ligand-dependent manner. HLY78 can promote LRP6 phosphorylation and Wnt signaling transduction, which has been recognized for its function in breast prostate cancer, and glioblastoma.13The structure–activity relationship (SAR) of lycorine (3) has been systematically evaluated by methodically changing different parts of the structure (Fig. 2). It can be seen that conformational freedom of the C-ring, stereochemistry of the C/D-ring junction and free diol functionality in the C-ring in its original configuration in lycorine (3) are very crucial for anticancer activities (Table 2).10,14,15 Lycorine (3) has exhibited cytostatic effects rather than cytotoxic effects through impairing the actin cytoskeleton organization in a large panel of apoptosis-resistant cancer cell lines (Table 2). Similarly, the structure feature of an open dioxole ring in pseudolycorine (14) is not essential for the antitumor activity. Haemanthamine (7) and haemanthidine (16) can decrease cell viability, mitochondrial membrane potential and induce apoptosis by declining the percentage of cells in the S phase of the cell cycle.16 In addition, the biological results have shown that the activity against apoptosis-resistant cancers is also shared by lycorine natural congeners10,17–19 as well as a number of synthetic analogues.20
 | ||
| Fig. 2 Structures of lycorine-type and haemanthamine-type amaryllidaceae alkaloids. | ||
| Entry | Comp. | Cell lines, IC50a (μM) | Ref. | |||||
|---|---|---|---|---|---|---|---|---|
| A549 | OE21 | Hs683 | U373 | SKMEL-28 | B16F10 | |||
| a All cell lines were cultured in RPMI medium supplemented with 10% heat-inactivated fetal bovine serum. MEM and RPMI cell culture media were supplemented with mM glutamine, 100 μg mL−1 gentamicin, and penicillin–streptomycin (200 U mL−1 and 200 μg mL−1). | ||||||||
| 1 | Lycorine (3) | 4.3 ± 0.3 | 5.1 ± 0.4 | 6.7 ± 0.3 | 7.6 ± 0.2 | 8.5 ± 0.3 | 6.3 ± 0.4 | 10 |
| 2 | Haemanthamine (7) | 4.5 ± 0.6 | 6.8 ± 0.7 | 7.0 ± 0.3 | 7.7 ± 0.5 | 8.5 ± 0.2 | 6.8 ± 0.2 | 17 |
| 3 | Pseudolycorine (14) | 7.5 ± 0.4 | 7.7 ± 0.3 | 7.9 ± 0.2 | 7.8 ± 0.3 | >10 | 7.5 ± 0.3 | 10 |
| 4 | Amarbellisine (15) | 7.2 ± 0.3 | 6.7 ± 0.2 | 8.3 ± 0.3 | 7.3 ± 0.2 | 8.3 ± 0.2 | 6.7 ± 0.3 | 17 |
| 5 | Haemanthidine (16) | 4.0 ± 0.4 | 3.7 ± 0.2 | 4.3 ± 0.2 | 3.8 ± 0.2 | 4.2 ± 0.2 | 3.1 ± 0.2 | 10 |
| 6 | Lycorine chlorohydrine (17) | 3.8 ± 0.2 | 9.6 ± 0.7 | 3.1 ± 0.3 | 2.3 ± 0.1 | >10 | 6.9 ± 0.5 | 10 |
| 7 | Lycorin-2-one (18) | 9.9 ± 0.5 | >10 | >10 | >10 | >10 | >10 | 10 |
| 8 | 1,2-α-Epoxy lycorine (19) | 3.4 ± 0.1 | 8.5 ± 0.5 | 3.3 ± 0.2 | 2.4 ± 0.1 | 9.5 ± 0.4 | 4.6 ± 0.2 | 10 |
| 9 | Lycorine hydrochloride (20) | 4.3 ± 0.2 | 4.6 ± 0.1 | 6.5 ± 0.2 | 8.6 ± 0.3 | 8.3 ± 0.3 | 5.5 ± 0.2 | 10 |
| 10 | Anhydro lycorine (21) | 4.5 ± 0.1 | 8.8 ± 0.2 | 7.1 ± 0.3 | 5.1 ± 0.1 | >10 | >10 | 10 |
Furthermore, lycorine provides significant therapeutic benefit in mice bearing brain grafts of the B16F10 melanoma model at non-toxic doses.10 Its potential (in vitro) therapeutic ratio has been shown (>15 times more active against cancer than normal cells) in the literature21 and its therapeutic potential has been demonstrated in a number of mouse models of human cancers, such as Hey1B ovarian cancer,22 LLC lung carcinoma,23 and HL-60 leukemia.24
The strong relationship between the compound's lipophilicity and anticancer activities is evidenced in the design of lycorine-based anticancer agents (Table 3). A series of C1, C2-esters are synthesized and many of them have retained the anticancer activity possibly due to intracellular hydrolysis to release the parent lycorine inside the cells.20 The increase in lipophilicity of C1, C2-esters leads to restoration of activity (3a–3d). Non-hydrolyzable C1, C2-ethers lycorine analogues are evaluated as well against a panel of cancer cell lines.25 Although the SAR analysis does not reveal the activity dependence on any specific structural features present in C1- or C2-ethers or esters, diallyllycorine (3e) and silyl ether analogue 3g were equipotent with lycorine throughout the tested cell lines. Diallyllycorine (3e) even is 100 times more potent against the apoptosis-resistant U373 glioblastoma (Table 3).20 However, C1- or C2-hydroxyls are derivatizated as methyl ethers 3f leading to a complete loss of the activity apparently due to reduced cell permeability through non-facilitated diffusion.
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Comp. | R1 | R2 | log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Pb Pb |
GI50 in vitro valuesa (μM) | Mean ± SEM | Ref. | ||||||
| Glioma | Carcinoma | Melanoma | |||||||||||
| Hs683 | T98G | U373 | A549 | MCF7 | SKMEL-28 | B16 | |||||||
| a The cells were cultured in RPMI media supplemented with 10% heat-inactivated fetal calf, 4 mM glutamine, 100 mg mL−1 gentamicin and penicillin, streptomycin (200 U mL−1 and 200 mg mL−1). The overall growth level of each cell line was determined using the colorimetric MTT (3-[4,5-dimethyl thiazol-2-yl]-diphenyl tetrazolium bromide) assay. Each experimental condition was performed in six replicates.b The logP values were calculated using the Calculate Molecular Properties protocol, launched from within Discovery Studio 3.5. 25.c TIPS = triisopropylsilyl. |
|||||||||||||
| 1 | 3 | H | H | — | 0.9 | 3 | 3 | 0.9 | 4 | 4 | 2 | 3 ± 1 | 20 |
| 2 | 3a | H | Bz | — | 4 | 32 | 0.6 | 32 | 5 | 4 | 8 | 11 ± 5 | 20 |
| 3 | 3b | Bz | H | — | 6 | 70 | 1 | 70 | 23 | 60 | 21 | 36 ± 10 | 20 |
| 4 | 3c | H | Ac | — | 11 | 2 | 4 | 3 | 4 | 3 | 43 | 9 ± 5 | 20 |
| 5 | 3d | TIPS | Ac | — | 36 | 6 | 15 | 18 | 26 | 27 | 34 | 30 | 20 |
| 6 | 3e | Allyl | Allyl | 3.1 | 2 | 4 | 0.03 | 4 | 0.2 | 6 | 4 | 3 ± 1 | 20 |
| 7 | 3f | Me | H | −0.4 | 36 | 84 | — | 31 | >100 | 92 | 40 | >50 | 25 |
| 8 | 3g | TIPS | Allyl | 5.9 | 7.6 | 6.6 | — | 4.5 | 5 | 2.4 | 0.8 | 4.5 | 25 |
| 9 | 3h | H | TIPSc | 4.8 | 24 | 20 | — | 15 | 23 | 24 | 9.5 | 19.5 | 25 |

The β-crinane distichamine (22) as well as the phenanthridone narciprimine (23), two rare components of amaryllidaceae alkaloids, as shown in Fig. 3, are evaluated for cytotoxic activities against acute lymphoblastic leukemia (CEM) and other cancerous cell lines.26 As seen in Table 4, distichamine (22) is active against all cancer cell lines with IC50 values ranging from 2.2 to 14.7 μM. Narciprimine (23) is active against CEM cells (IC50 value = 13.3 μM), while homolycorine (25) is active on CEM, K562 and G-361 cells. As expected from prior observations, lycorine inhibits all six cell lines in a dose-dependent manner (IC50 values ranging from 1.6 to 13.0 μM) after 72 h. Similar observations have previously been made for haemanthamine (7), which is active across the cell lines with IC50 values ranging from 2.1 to 8.1 μM. According to the previous demonstrations, distichamine (22) and narciprimine (23) can increase the proportion of G2/M phase cells in a dose-dependent manner. Besides, narciprimine (23) and arolycoricidine (24) are effective in both type I and type II DNA topoisomerase (cellular targets of a number of chemotherapeutical drugs) reactions in a dose-dependent manner.27
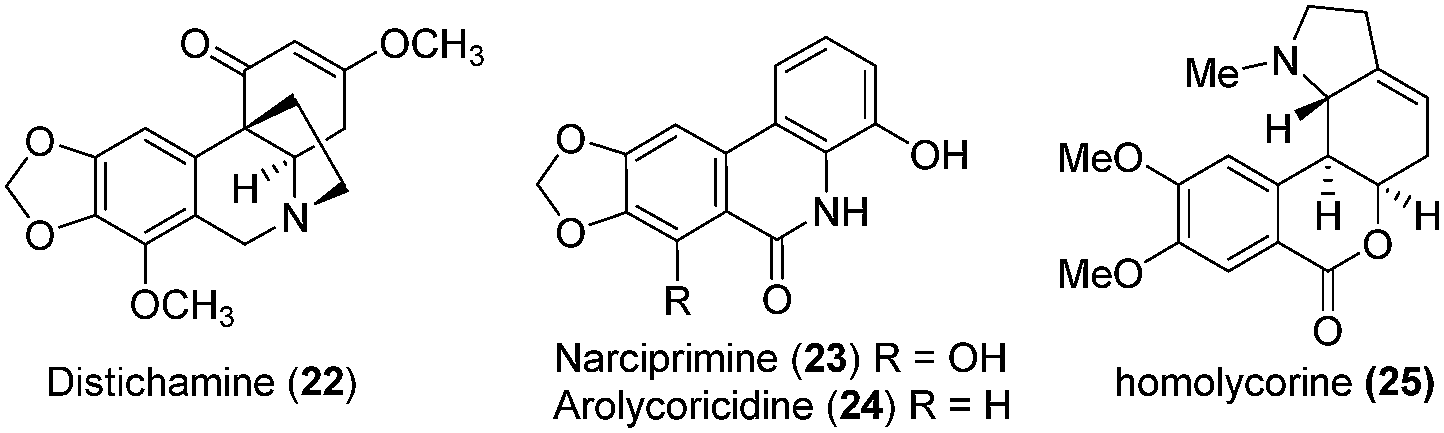 | ||
| Fig. 3 Structure of distichamine (22), narciprimine (23), arolycoricidine (24) and homolycorine (25). | ||
| Entry | Comp. | Cell lines, IC50a,b (μM) | Ref. | |||||
|---|---|---|---|---|---|---|---|---|
| CEM | K562 | MCF7 | HeLa | G-361 | BJ | |||
| a All cells were treated for 72 h with serial concentrations of samples.b Values are means of at least three independent experiments performed in triplicate, with standard deviation as indicated (nt = not tested).c Staurosporine and galanthamine used as positive and negative controls, respectively. | ||||||||
| 1 | Lycorine (3) | 1.6 ± 0.0 | 3.6 ± 1.2 | 13.0 ± 2.9 | 10.6 ± 0.9 | 5.0 ± 0.3 | 1.9 ± 0.1 | 26 |
| 2 | Galanthamine (6)c | >50 | >50 | >50 | >50 | >50 | >50 | 26 |
| 3 | Haemanthamine (7) | 2.1 ± 0.4 | 3.4 ± 1.6 | 8.1 ± 3.3 | 7.0 ± 2.2 | 3.7 ± 0.4 | 2.7 ± 0.2 | 26 |
| 4 | Distichamine (22) | 4.5 ± 1.6 | 4.1 ± 0.9 | 2.3 ± 0.8 | 2.2 ± 0.1 | 14.7 ± 0.1 | 10.5 ± 1.9 | 26 |
| 5 | Narciprimine (23) | 13.3 ± 2.5 | >50 | >50 | >50 | >50 | 7.9 ± 0.2 | 26 |
| 6 | Homolycorine (25) | 15.0 ± 5.3 | 19.4 ± 0.8 | >50 | >50 | 32.9 ± 6.0 | 20.8 ± 2.3 | 26 |
| 7 | Staurosporine (26)c | 0.023 ± 0.002 | nt | 0.064 ± 0.002 | 0.175 ± 0.007 | nt | 2.2 ± 0.0 | 26 |
Isocarbostyril amaryllidaceae alkaloids (Fig. 4) are represented by hydroxylated benzophenathridones or isoquinolinone types of structure without basic nitrogen atoms. Pancratistatin (8), narciclasine (27) and lycoricidine (28) are the most widely known compounds against cancer cell lines among this category.28 The cytotoxicity of narciclasine (27) has been evaluated in 60 cancer cell lines by the NCI, and the mean IC50 value was 0.046 μM.29 Lycoricidine (28) is 10 times weaker (mean IC50 value = 0.33 μM) and pancraistatin (8) is 5 times weaker (mean IC50 value = 0.26 μM).
 | ||
| Fig. 4 Structure of pancretistatin (8), narciclasine (27) and lycoricidine (28). | ||
Apart from foregoing alkaloids in the family of amaryllidaceae, narciclasine (27) has been demonstrated to possess antitumor efficacy, which is originally isolated from Narcissus pseudonarcissus, with its antimitotic and displaying colchicine-like effects in 1967.30 Narciclasine (27) is a potentially promising GTPase agent against brain tumors including gliomas and brain metastases. It has displayed the cytostatic activity instead of cytotoxic activities in vivo (IC50 values = 30–90 nM) in experimental models of brain cancers. The possible mechanism for the cytostatic activity involves the impairment of actin cytoskeleton organization by targeting both Rho pathway and the elongation factor eEF1A.31 Robert Kiss et al. have reported that narciclasine can impair cancer cell proliferation and migration at concentrations >1 μM and is approximately 250-fold less sensitive to normal human fibroblast cell. Additionally, narciclasine (27) can induce apoptosis-mediated cytotoxic effects by triggering the activation of initiator caspases (caspase-8 and caspase-10) of the death receptor pathway in human PC-3 prostate and MCF-7 breast cancer cells.32 The molecular docking data have shown that narciclasine (27) directly binds to human recombinant and yeast-purified eEF1A.33,34
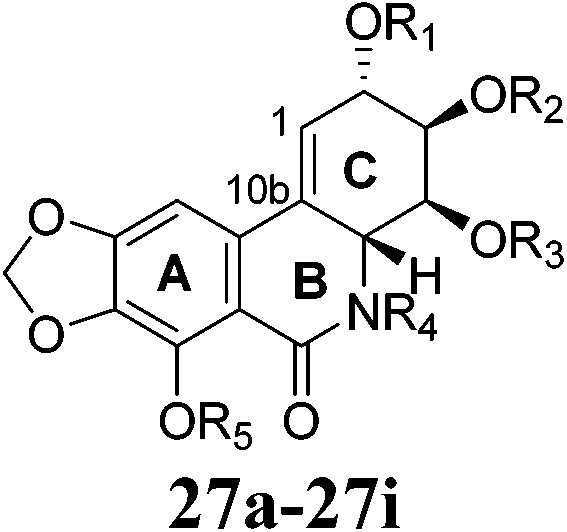
To explore the structure–activity relationship of narciclasine and related products, reactive positions of narciclasine (R1, R2, R3, R4, R5) have been modified and partial in vitro antitumor activity data are summarized in Table 5. The data suggest that the double bond between C-10b and C1, a free lactam carbonyl function at position R4 and a free phenolic hydroxyl group at position R5 seem therefore to be necessary for antitumor activities. Any modifications made to positions R4 and R5 lead to compounds devoid of antitumor activity in vitro (27a, 27b). Though ester 27c has a ∼10 fold weaker activity, mono-esterification of the hydroxyl at position R1 (27d, 27e) possibly improves or maintains in vitro antitumor activity of narciclasine (Table 5). This may be attributed to the hydrolysis of esters to narciclasine. The elimination of the ester group in the allylic position, subsequent aromatization of ring C possibly results in the formation of narciprimine in vitro.31 Accordingly, Kiss and co-workers32 have evaluated potential prodrugs of narciclasine with respect to their high oral bioavailability and anti-tumor activity in vivo. Indeed narciclasine 4-O-β-D-glucopyranoside (27i) has comparable anticancer activity to narciclasine35 and has <20% degradation in 1 h at pH 2 and 92% stability at physiological pH 7.4. After oral administration, prodrug (27i) (81132 ng min mL−1) is found to increase the absolute bioavailability of narciclasine (54812 ng min mL−1) to ∼52%. Compound (27i) significantly increases survival times in two human GBM models (Hs683 and GL-19) at a dose level of 1 mg per kg per day while narciclasine (27) failed in either model at this dose.31 Thus, it is considered to be a suitable candidate for further evaluation by both the intravenous and oral routes.
|
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Comp. | Reaction sitea | Cell lines, IC50b (μM) | Stabilityc (%) | Ref. | ||||||||||
| R1 | R2 | R3 | R4 | R5 | PC-3 | U373 | BxPC3 | LoVo | A549 | MCF-7 | Median | ||||
| a Chemical modification to each of 1's reaction sites.b The in vitro antiproliferative activities of the compounds are reported as IC50 values (nM) determined using the MTT colorimetric assay.c The stability of products was measured by HPLC analysis following incubation in a physiological solution at 37 °C over 7 days. Results are expressed as the % of the incubated compound recovered.d nd: not determined. | |||||||||||||||
| 1 | 27 | H | H | H | H | H | 0.03 | 0.03 | 0.03 | 0.09 | 0.03 | 0.05 | 0.03 | 100 | 31 |
| 2 | 27a | H | H | H | H | Et | >10 | >10 | >10 | >10 | >10 | >10 | >10 | 96 | 31 |
| 3 | 27b | H | H | H | Et | H | >10 | >10 | >10 | >10 | >10 | >10 | >10 | 100 | 31 |
| 4 | 27c | Ac | H | H | H | H | 0.1 | 0.1 | 0.07 | 0.3 | 0.1 | 0.4 | 0.1 | 20 | 31 |
| 5 | 27d | Bz | H | H | H | H | 0.03 | 0.03 | 0.004 | 0.05 | 0.04 | 0.2 | 0.04 | 82 | 31 |
| 6 | 27e | iPrCO | H | H | H | H | 0.005 | 0.03 | 0.003 | 0.05 | 0.03 | 0.03 | 0.03 | 29 | 31 |
| 7 | 27f | Ac | H | H | H | Ac | 0.3 | 0.06 | 0.4 | 0.3 | 0.3 | 0.4 | 0.3 | 56 | 31 |
| 8 | 27g | H | H | SO3Na | H | H | >10 | >10 | >10 | >10 | >10 | >10 | >10 | 100 | 31 |
| 9 | 27h | OTBS | C(Me)2 | H | H | >10 | 2 | 8 | 3 | >10 | 4 | >10 | ndd | 31 | |
| 10 | 27i | H | H | H | H |  |
0.8 | 0.7 | 1 | 2 | 2 | 2 | 1.5 | 92 | 31 |
Another important isocarbostyril amaryllidaceae alkaloid, pancratistatin (8), discovered by Pettit in 1984 (ref. 36) exhibits potential anticancer activities. So far, the mechanism of pancratistatin's anticancer potential is not exactly elucidated. Pancratistatin (8) can induce the apoptosis in cancer cells, produce less cytotoxic effect on the normal cells, associate with the up-regulation of Fas, increase in caspase-3, flip phosphatidyl serine and destabilize mitochondrial membrane potential.37 Kekre's group has reported that caspase-3 activation and exposure of phosphatidyl serine on the outer leaflet of the plasma membrane are earlier than ROS and DNA fragmentation, which means pancratistatin wouldn't cause DNA double-strand breaks or DNA damage prior to the execution phase of apoptosis in cancer cells.38 Instead, pancratistatin can selectively induce the cell death in human colon tumor xenografts' study independent of Bax and caspase activation by targeting HT-29 cancer cell in vivo.39
The intensive research work has been done in the searching of higher anti-cancer pancratistatin derivatives (Fig. 5). The pancratistatin's SAR data indicate that the presence of large hydrophobic C-1 substituents can increase anticancer activities, and the 7-hydroxy group is an important part of the cytotoxic pharmacophore as well as the full amino inositol motif (Table 6).40 Removal of another oxygen in ring A of pancratistatin further lowers the potency 10 times relative to 7-deoxypancratistatin and 100 times relative to pancratistatin. Ingrassia and co-workers have confirmed that a trans-B/C ring junction and C2-, C3-, C4-hydroxyl groups are necessary to maintain pancratistatin's potent cytotoxicity. Otherwise, the cis B/C fusion stereochemistry leads to the reduction or complete loss of potency in isocarbostyril compounds. However, methoxy-substuituted crinane skeleton instead of polygydroxylated lycorane part of pancratistatin, has shown higher binding affinity to target proteins by caspase-3 activity in jurkat cells.41
 | ||
| Fig. 5 Structure of pancratistatin analogues. | ||
| Entry | Comp. | Cell lines, IC50a (μM) | Ref. | |||
|---|---|---|---|---|---|---|
| BXPC-3 | DU-145 | NCI-H460 | MCF-7 | |||
| a Concentration required to reduce the viability of cells by 50%, after 48 h of treatment with indicated compounds, relative to DMSO control; ±SD from two independent experiments, each performed in four replicates, determined by MTT assay. | ||||||
| 1 | 8 | 0.061 | 0.046 | 0.098 | 0.071 | 28 |
| 2 | 27 | 0.05 ± 0.01 | 0.05 ± 0.04 | 0.04 ± 0.01 | 0.04 ± 0.01 | 40 |
| 3 | 28 | 0.77 ± 0.01 | 1.10 ± 0.20 | 0.40 ± 0.01 | 0.86 ± 0.06 | 40 |
| 4 | 29a | 0.34 ± 0.05 | 0.72 ± 0.27 | 0.53 ± 0.01 | 1.81 ± 1.20 | 40 |
| 5 | 29b | 0.22 ± 0.01 | 0.09 ± 0.01 | 0.09 ± 0.01 | 0.24 ± 0.01 | 40 |
| 6 | 29c | 0.07 ± 0.01 | 0.06 ± 0.01 | 0.07 ± 0.01 | 0.52 ± 0.47 | 40 |
| 7 | 30 | 0.039 | 0.021 | 0.03 | 0.017 | 28 |
| 8 | 31 | 0.0044 | 0.00049 | 0.00023 | 0.00072 | 28 |
| 9 | 32 | 14 | >10.2 | 10.8 | 10.8 | 28 |
2.2 Acetylcholinesterase (AChE) inhibitory activities
Acetylcholinesterase enzyme serves as a major position in transmissions of nerve pulse by hydrolysis of acetylcholine (ACh). It has been demonstrated that ACh plays a great role in functioning human's brain with releasing basal forebrain choline. ACh deficit will lead to several neurodegenerative disorders such as Alzheimer's disease (AD). Although huge efforts have been made to investigate its pathophysiology, AD remains incurable and affects more than 36 million people around the world.42 The typical pathological hallmarks of AD are intracellular neurofibrillary tangles, amyloid β-peptide (Aβ) plaques and loss of central cholinergic function. The current medications used for the symptomatic AD treatment such as tacrine, galanthamine, rivastigmine and donepezil mainly belong to cholinesterase inhibitors. Because cholinesterase inhibitors can slow down neurodegenerative progression via inhibiting AChE-induced Aβ aggregation, inhibitors of AChE and BuChE also provide additional benefits for AD treatment.43,44Galanthamine (6), an amaryllidaceae alkaloid, is originally isolated from Galanthus nivalis L. in the 1940s. Galanthamine hydrobromide, under the generic name Reminyl, is the first amaryllidaceae alkaloid to be approved as a prescription drug by FDA due to its high inhibitory efficacy and both reversible and selective activity on AChE. Compared to most of the amaryllidaceae alkaloids, galanthamine displays IC50 value of 1.5 μM, exceeds over 20 folds to other alkaloids in inhibitory potency (Table 7). As an exception, sanguinine (6d), a 9-hydroxy analogue, shows 10 times more active than galanthamine (Table 7, entry 5). The others (entry 2–4, 6 and 7) show a relative lower inhibitory activities.45–47 It seems that a –OH group or protected –OH group in its allylic position (R1) is crucial for the activities. However, its clinical application is impeded by rare nature existence. To date, a considerable amount of sanguinine has been extracted from Pancratium Illyricum L. in Italy by Iannello group.45 Nevertheless, the unexpected significant inhibitory effect of sanguinine has brought some hypotheses toward the binding mechanism between galanthamine and AChE with potential modifications of its analogues.
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Compounds | R1 | R2 | R3 | R4 | R5 | R6 | R7 | IC50 (μM) | Ref. |
| 1 | Galanthamine (6) | OH | H | H | H | H | OMe | Me | 1.5 ± 0.2 | 45 |
| 2 | Leucotamine (6a) | OCOCH2CHOHCH3 | H | H | H | H | H | Me | 5.3 ± 0.9 | 45 |
| 3 | 6b | OCOCH2CHOHCH3 | H | H | H | H | OMe | Me | 6.0 ± 0.7 | 45 |
| 4 | 6c | OCOCH2CHOHCH3 | H | H | OH | H | OMe | Me | 3.5 ± 1.1 | 46 |
| 5 | Sanguinine (6d) | OH | H | H | H | H | OH | Me | 0.1 ± 0.01 | 46 |
| 6 | 6e | OH | H | H | H | OH | OMe | Me | 1.61 ± 0.21 | 46 |
| 7 | 6f | H | OH | H | H | H | OMe | H | 9.60 ± 0.65 | 46 |

Among the subclasses of amaryllidaceae alkaloids, lycorine (3) and its derivatives are investigated intensively for their anti-AChE activities (Fig. 6). Ungiminorine (3i) is found to be inhibitory against AChE with IC50 value of 0.35 μM, which is about 6–10 times stronger than galanthamine.47 Assoanine (3j) also shows very strong inhibitory activity against AChE with IC50 values of 3.87 μM, another analogue oxoassoanine (3k) shows moderate inhibitory activity against AChE (IC50 = 47.21 μM).46
 | ||
| Fig. 6 Structure of lycorine analogues. | ||
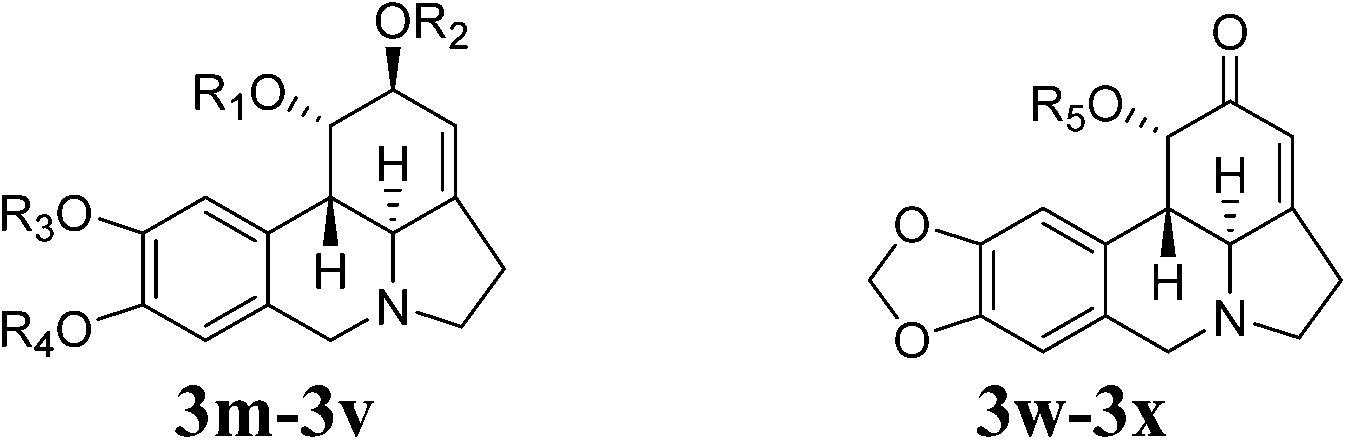
Subsequent modification of lycorine has been reported by several groups.48–51 Some of results are summarized in Table 8. The data have shown that compound 2-O-tert-butyldimethylsilyl-1-O-(methylthio)methyllycorine (3p) is a dual inhibitor of human acetylcholinesterase (hAChE) and butyrylcholinesterase (hBChE) with IC50 values of 11.40 ± 0.66 μM and 4.17 ± 0.29 μM respectively (entries 1–6, 16–18) and the acylated/etherified derivatives of lycorine and lycorin-2-one are more potent against hBChE than hAChE.48 New C1 and C2 functionalized analogs in the lycorine subset are synthesized and systematically studied.49,50 A pronounced spike in activity from the inactive parent lycorine 3 to the 2-TBS analog 3q (Ki = 0.86 μM) is observed. Acetylation or benzoylation of 3q afforded 3r (Ki = 0.34 μM) and 3s (Ki = 0.39 μM) respectively. The lipophilic C2 silyl group binding at the enzyme active site apparently plays an important role against AChE. Desilylation of 3t to the alcohol 3a (Ki = 0.54 M) and subsequent acetylation to compounds 3t (Ki = 0.97 μM), seems to be the least efficacious of the series. Moreover, 1-acetyllycorine (3c) has shown a potent inhibitor of AChE (Ki = 0.43 μM) while lycorine (3), 2-acetyllycorine (3c), and 1,2-diacetyllycorine (3u) exhibits no or very weak inhibitory activity.49–51
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Comp. | R1 | R2 | R3 | R4 | R5 | IC50/Ki (μM) | Ref. |
| a Values are means of three experiments, na = not active at >10 μM, determined by Ki.b nd = not detected. | ||||||||
| 1 | 3l | Benzoyl | Benzoyl | –CH2– | — | >50 | 48 | |
| 2 | 3b | H | Benzoyl | –CH2– | — | >50 | 48 | |
| 3 | 3m | Cinnamoyl | Cinnamoyl | –CH2– | — | 46.76 ± 0.95 | 48 | |
| 4 | 3n | H | Cinnamoyl | –CH2– | — | >50 | 48 | |
| 5 | 3o | Cinnamoyl | H | –CH2– | — | >50 | 48 | |
| 6 | 3p | MeSCH2– | TBS | –CH2– | — | 11.4 ± 0.66 | 48 | |
| 7 | 3 | H | H | –CH2– | — | na | 49a | |
| 8 | 3c | Ac | H | –CH2– | — | 0.43 ± 0.02 | 49 | |
| 9 | 3q | H | TBS | –CH2– | — | 0.86 ± 0.03 | 49 and 50 | |
| 10 | 3r | Ac | TBS | –CH2– | — | 0.34 ± 0.08 | 49 and 50 | |
| 11 | 3s | Bz | TBS | –CH2– | — | 0.39 ± 0.03 | 49 and 50 | |
| 12 | 3t | Bz | Ac | –CH2– | — | 0.97 ± 0.10 | 49 and 50 | |
| 13 | 3a | Bz | H | –CH2– | — | 0.54 ± 0.03 | 49 and 50 | |
| 14 | 3u | Ac | Ac | –CH2– | — | 211 ± 10 | 49 | |
| 15 | Pseudolycorine (14) | H | H | Me | H | — | 152.32 ± 32.06 | 46 |
| 16 | 3v | H | Ac | Me | H | — | ndb | 46 |
| 17 | Lycorin-2-one (18) | — | — | — | — | H | >50 | 48 |
| 18 | 3w | — | — | — | — | Ac | >50 | 48 |
| 19 | 3x | — | — | — | — | Cinnamoyl | >50 | 48 |
Amaryllidaceae alkaloids having several different ring types are evaluated for their AChE inhibitory activities (Table 9 and Fig. 7). As known before, lycorine-type alkaloids are the most active alkaloids with 1-O-acetyllycorine exhibiting inhibitory effects two-fold more potent than that of galanthamine. In addition, crinine (33), crinamidine (35), epivittatine (37), 6-hydroxycrinamine (38a), N-desmethyl-8α-ethoxypretazettine (39), N-desmethyl-8β-ethoxypretazettine (39a), lycorine (3), tazettine (9a) and cherylline (12) have weak activity.52
| Entry | Compounds | IC50 (μM) | Ref. |
|---|---|---|---|
| 1 | Crinine (33) | 461 ± 14 | 52 |
| 2 | Epibuphanisine (34) | 547 ± 5 | 52 |
| 3 | Crinamidine (35) | 300 ± 27 | 52 |
| 4 | Hamayne (36) | 553 ± 3 | 52 |
| 5 | 3-O-Acetylhamayne (36a) | 594 ± 8 | 52 |
| 6 | Epivittatine (37) | 239 ± 9 | 52 |
| 7 | Crinamine (38) | 697 ± 12 | 52 |
| 8 | 6-Hydroxycrinamine (38a) | 490 ± 7 | 52 |
| 9 | 8a-Ethoxycrinamine (38b) | 1145 ± 87 | 52 |
| 10 | N-Desmethyl-8α-ethoxyprecriwelline (39) | 234 ± 13 | 52 |
| 11 | N-Desmethyl-8β-ethoxyprecriwelline (39a) | 419 ± 8 | 52 |
| 12 | Tazettine (9a) | 705 ± 63 | 52 |
| 13 | Cherylline (12) | 407 ± 32 | 52 |
| 14 | 1-O-Acetyllycorine (3c) | 0.96 ± 0.04 | 52 |
| 15 | Lycorine (3) | 213 ± 1 | 52 |
| 16 | Galanthamine (6) | 1.9 ± 0.16 | 52 |
 | ||
| Fig. 7 Other types of amaryllidaceae alkaloids. | ||
2.3 Antiviral activities
The preliminary evaluation of lycorine, pancratistatin, narciclasines and their derivatives gives antiviral screening data that indicate the presence of in vitro/in vivo activities against Flaviviridae, Bunyaviridae and Japanese encephalitis.53 Several lycorine analogues are further screened for anti-West Nile virus activity and cytotoxicity (Table 10). When C7 of lycorine is oxidized to a carbonyl group (entry 2), compound 3y has lower antiviral potency. However, when one or two hydroxyl groups at the C1 and C2 positions of lycorine are substituted with other functional groups, all analogues show higher CC50 values than that of parental lycorine. Among them, compound 3z (entry 5) has higher potency (EC50 value = 0.19 μM) and lower cytotoxicity (CC50 > 300 μM, the highest tested concentration). It is reported that lycorine inhibited flaviviruses mainly through suppression of viral RNA synthesis. Furthermore, the data reveal that the 2K peptide plays a direct role in flavivirus RNA replication and lycorine strongly hinders RNA synthesis in flaviviruses and weakly inhibits viral protein translation.54
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Comp. | R1 | R2 | R3 | EC50a (μM) | CC50b (μM) | Ref. |
| a EC50 values were derived from viral titer reduction assays. Vero cells were infected with West Nile virus (0.1 MOI) in the presence of various concentrations of each compound. Viral titers at 42 h.b CC50 values were derived from Vero cells using an MTT assay. | |||||||
| 1 | Lycorine (3) | H | OH | H | 0.23 | 24 | 54 |
| 2 | 3y | Ac | OAc | O | >300 | >300 | 54 |
| 3 | 3u | Ac | OAc | H | 1.49 | 110 | 54 |
| 4 | 3c | Ac | OH | H | 0.86 | 66 | 54 |
| 5 | 3z | Ac | O | H | 0.19 | >300 | 54 |
| 6 | 3q | H | OTBS | H | 0.73 | 78 | 54 |

Amaryllidaceae alkaloids lycorine (3), hippeastrine (4), hemanthamine (7) and their analogous (Table 11) exhibit moderate to good anti-influenza activities. Particularly, lycorine and hemanthamine derivative 7a show strong activities against influenza A virus N5H1 in vitro. Compound 4 has lower antiviral activity than other five compounds. Mechanistic studies show that none of these amaryllidaceae alkaloids affected the activity of the ribonucleoprotein (RNP) complex in the viral generation and replication. Instead, lycorine (3) delays the export of nucleoprotein from nuclear and compound 7 can block the migration from nuclear to cytoplasm in single and multiple replications.55,56 It has been demonstrated that lycorine can inhibit the cytopathic effect (CPE) induced by severe acute respiratory syndrome-associated coronavirus (SARS-CoV). The EC50 value is 15.7 ± 1.2 nM.57 Lycorine (3) is also screened for antiviral activity against poliovirus (PV) using a cellular fluorescence resonance energy transfer (FRET) assay and the results have shown that it can reduce 1log10 unit of virus titer at 2.5 μM without cytotoxicity.58 Zhang and his co-workers have reported that lycorine can inhibit human enterovirus 71 (EV71) infection in rhabdomyosarcoma (RD) cells.59

2.4 Antibacterial activities
The CH2Cl2/MeOH extract, two isolated compounds 40 and 40a from leaves of Crinum purpurascens are screened for antibacterial activity. The extracts have shown remarkable potencies against all the bacteria strains used (Table 12). However, none of isolated compounds have better activity than that of the crude extract.60
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Comp. | Parameters | Bacteria straina | Ref. | |||||
| EC | PA | KP | SA | ST | SPB | ||||
| a EC = Escherichia coli; KP = Klebsiella pneumoniae; ST = Salmonella typhi; PA = Pseudomonas aeruginosa; SA = Staphylococcus aureus; SPB = Salmonella paratyphi B; NT = not tested; NA = not active; ND = not determined; MIC = Minimum Inhibitory Concentration; MBC = Minimum Bactericidal Concentration. | |||||||||
| 1 | R1 = H, R2 = Me (40) | MIC | NA | NA | NA | NA | NA | NA | 60 |
| MBC | NA | NA | NA | NA | NA | NA | |||
| 2 | R1 + R2 = –CH2– (40a) | MIC | 250 | 250 | 200 | 200 | 250 | 250 | 60 |
| MBC | 300 | 300 | 300 | 300 | 300 | 300 | |||
| 3 | DCM/MeOH extract | MIC | 4 | 3 | 4 | 4 | 6 | 6 | 60 |
| MBC | 8 | 7 | 10 | 7 | 12 | 10 | |||
| 4 | Ciprofloxacin (41) | MIC | 2 | 2 | 2 | 2 | NT | NT | 60 |
| MBC | 5 | 5 | 8 | 5 | NT | NT | |||

The Gram-negative bacterium Flavobacterium columnare which occurs in channel catfish is able to cause columnaris disease. Lycorine (3), ungeremine (42) and their analogues are evaluated using a rapid bioassay for antibacterial activity against two isolates (ALM-00-173 and BioMed) of Flavobacterium columnare (Table 13). It has been found that substitution at the C1-O- or C2-O-position is pivotal to antibacterial efficacy of these compounds. The disubstituted lycorine O-analogues have better antibacterial activities than that with only one substitution at either carbon. Notably, the carbamate analogue 3ac possesses the stronger antibacterial activity toward both F. columnare isolates with 24 h IC50 value of 3.0 mg L−1. On the basis of the 24 h IC50 results, ungeremine has higher antibacterial activities than that of lycorine and their analogues, but none of the ungeremine analogues are more active than itself against F. columnare. Possibly, the aromatization of the C ring as well as the oxidation to azomethine group of C-7 is the structural feature important to antibacterial activity against F. columnare. Besides, the presence of the 1,3-dioxole ring joined to the A ring together with the position of the oxygenation of the C ring also plays a significant role in providing antibacterial activity.61–64
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Comp. | R1 | R2 | R3 | R4 | 24 h IC50a,b | MICb | 24 h IC50 | MIC | Ref. | ||
| RDCFc | RDCOd | RDCFc | RDCOd | |||||||||
| a 24 h 50% inhibition concentration in mg L−1.b Minimum inhibitory concentration in mg L−1.c Relative to drug control florfenicol.d Relative to drug control oxytetracycline. | ||||||||||||
| 1 | 3u | Ac | Ac | — | — | 2 | 4 | 3 | 3 | 10 | 11 | 61 and 62 |
| 2 | 3 | H | H | — | — | 27 | 29 | 49 | 47 | 100 | 108 | 63 |
| 3 | 3c | Ac | H | — | — | 7.3 | 3.3 | 10.4 | 9.9 | 10 | 10.8 | 63 |
| 4 | 3v | H | Ac | — | — | 17.7 | 16.6 | 27.8 | 26.4 | 50.5 | 54.3 | 63 |
| 5 | 3aa | H | p-MePhCO | — | — | 14.6 | 4.7 | 16.4 | 15.6 | 10 | 10.8 | 63 |
| 6 | 3aa | H | o-MePhNCO | — | — | 11.2 | 23.2 | 13.7 | 13.1 | 55 | 59.2 | 63 |
| 7 | 3ac | o-MePhNCO | o-MePhNCO | — | — | 3.0 | 5.5 | 2.8 | 2.6 | 10.0 | 10.8 | 63 |
| 8 | 42 | Me | Me | H | OH | 0.5 | 1 | 0.28 | 0.27 | 1 | 1.1 | 64 |
| 9 | 43 | Me | Me | OH | H | 14.6 | 10.0 | 7.6 | 7.2 | 10.0 | 10.8 | 64 |
| 10 | 44 | H | Me | H | OH | 45.5 | 100 | 18.4 | 19.9 | 100 | 107.5 | 64 |

2.5 Other potentially useful activities
 | ||
| Fig. 8 Structures of lycorine derivatives 45, 45a–45c. | ||
3. Conclusion
In summary, amaryllidaceae has played a significant role for phytochemical based drug discovery. The commercialization of the Alzheimer's drug galanthamine has captured a significant share of the neurodegenerative disease market. Lycorine, pancratistatin and their series of analogues have been intensively investigated in various preclinical models of human cancers both in vitro and in vivo. In addition, the increasing lipophilic substitution at the C2 group of lycorine apparently enhances the AChE inhibitory activity sharply, some resulting adducts have exhibited inhibitory effects several-fold more potent than that of galanthamine. They may be open a new era on the treatments of various cancers and Alzheimer diseases. Though novel approaches such as phylogenetic method and CADD to screen and obtain targeted alkaloids from amaryllidaceae plants have been reported, further studies of amaryllidaceae alkaloids should focus on SAR and QSAR analysis as well as searching for synthetic methods for the metabolites and their derivatives. To date, literatures have elucidated the mechanisms of amaryllidaceae alkaloids to some extent. However, the fully understanding the mechanism of the amaryllidaceae alkaloids are still a long way to go in the future.Conflicts of interest
The authors hereby declare no conflicts of interest pertaining to this work.Acknowledgements
This work was supported, in part, by Technology Major Project of the Ministry of Science and Technology of China (no. 2012ZX10004801-003-011), the National Natural Science Foundation of China (no. 81373254 and 21390402), the Natural Science Foundation of Hubei Province (no. 2014CFB704), the Fundamental Research Funds for the Central Universities (no. 2014306020206) and Innovation Seed Fund of Wuhan University School of Medicine.Notes and references
- Z. Jin, Nat. Prod. Rep., 2013, 30, 849–868 RSC.
- A. Kornienko and A. Evidente, Chem. Rev., 2008, 108, 1982–2014 CrossRef CAS PubMed.
- Z. Jin, Nat. Prod. Rep., 2005, 22, 112–126 Search PubMed.
- N. Unver, Phytochem. Rev., 2007, 6, 125–135 CrossRef CAS.
- A. Evidente and A. Kornienko, Phytochem. Rev., 2009, 8, 449–459 CrossRef CAS.
- Y. Nakagawa, S. Uyeo and H. Yajima, The Double Bond in Lycorine, Soc Chemical Industry, 14 Belgrave Square, London, SW1X 8PS, England, 1956, pp. 1238–1239 Search PubMed.
- Z. Cao, P. Yang and Q. Zhou, Sci. China: Chem., 2013, 56, 1382–1391 CrossRef CAS.
- R. Liu, Z. Cao, J. Tu, Y. Pan, B. Shang, G. Zhang, M. Bao, S. Zhang, P. Yang and Q. Zhou, Pigm. Cell Melanoma Res., 2012, 25, 630–638 CrossRef CAS PubMed.
- R. E. Hayden, G. Pratt, M. T. Drayson and C. M. Bunce, Haematologica, 2010, 95, 1889–1896 CrossRef CAS PubMed.
- D. Lamoral-Theys, A. Andolfi, G. Van Goietsenoven, A. Cimmino, B. Le Calvé, N. Wauthoz, V. Mégalizzi, T. Gras, C. Bruyère and J. Dubois, J. Med. Chem., 2009, 52, 6244–6256 CrossRef CAS PubMed.
- X.-S. Liu, J. Jiang, X.-Y. Jiao, Y.-E. Wu, J.-H. Lin and Y.-M. Cai, Cancer Lett., 2009, 274, 16–24 CrossRef CAS PubMed.
- L. Li, H.-J. Dai, M. Ye, S.-L. Wang, X.-J. Xiao, J. Zheng, H.-Y. Chen, Y. Luo and J. Liu, Cancer Cell Int., 2012, 12, 1–6 CrossRef PubMed.
- S. Wang, J. Yin, D. Chen, F. Nie, X. Song, C. Fei, H. Miao, C. Jing, W. Ma and L. Wang, Nat. Chem. Biol., 2013, 9, 579–585 CrossRef CAS PubMed.
- D. Lamoral-Theys, E. Fattorusso, A. Mangoni, C. Perinu, R. Kiss and V. Costantino, J. Nat. Prod., 2011, 74, 2299–2303 CrossRef CAS PubMed.
- J. McNulty, J. J. Nair, J. Bastida, S. Pandey and C. Griffin, Phytochemistry, 2009, 70, 913–919 CrossRef CAS PubMed.
- R. Havelek, M. Seifrtova, K. Kralovec, L. Bruckova, L. Cahlikova, M. Dalecka, J. Vavrova, M. Rezacova, L. Opletal and Z. Bilkova, Phytomedicine, 2014, 21, 479–490 CrossRef CAS PubMed.
- G. Van Goietsenoven, A. Andolfi, B. Lallemand, A. Cimmino, D. Lamoral-Theys, T. Gras, A. Abou-Donia, J. Dubois, F. Lefranc and V. Mathieu, J. Nat. Prod., 2010, 73, 1223–1227 CrossRef CAS PubMed.
- D. Lamoral-Theys, C. Decaestecker, V. Mathieu, J. Dubois, A. Kornienko, R. Kiss, A. Evidente and L. Pottier, Mini-Rev. Med. Chem., 2010, 10, 41–50 CrossRef CAS.
- P. Cao, D.-S. Pan, S. Han, C.-Y. Yu, Q.-J. Zhao, Y. Song and Y. Liang, Arch. Pharmacal Res., 2013, 36, 927–932 CrossRef CAS PubMed.
- N. M. Evdokimov, D. Lamoral-Theys, V. Mathieu, A. Andolfi, L. V. Frolova, S. C. Pelly, W. A. van Otterlo, I. V. Magedov, R. Kiss and A. Evidente, Bioorg. Med. Chem., 2011, 19, 7252–7261 CrossRef CAS PubMed.
- J. McNulty, J. J. Nair, M. Singh, D. J. Crankshaw, A. C. Holloway and J. Bastida, Bioorg. Med. Chem. Lett., 2009, 19, 3233–3237 CrossRef CAS PubMed.
- Z. Cao, D. Yu, S. Fu, G. Zhang, Y. Pan, M. Bao, J. Tu, B. Shang, P. Guo and P. Yang, Toxicol. Lett., 2013, 218, 174–185 CrossRef CAS PubMed.
- B. S. Min, J. J. Gao, N. Nakamura, Y. H. Kim and M. Hattori, Chem. Pharm. Bull., 2001, 49, 1217–1219 CrossRef CAS.
- J. Liu, Y. Li, L. Tang, G. Zhang and W. Hu, Biomed. Pharmacother., 2007, 61, 229–234 CrossRef CAS PubMed.
- R. Dasari, L. M. Y. Banuls, M. Masi, S. C. Pelly, V. Mathieu, I. R. Green, W. A. van Otterlo, A. Evidente, R. Kiss and A. Kornienko, Bioorg. Med. Chem. Lett., 2014, 24, 923–927 CrossRef CAS PubMed.
- J. J. Nair, L. Rárová, M. Strnad, J. Bastida and J. van Staden, Bioorg. Med. Chem. Lett., 2012, 22, 6195–6199 CrossRef CAS PubMed.
- B. B. Sarikaya, S. Zencir, N. U. Somer, G. I. Kaya, M. A. Onur, J. Bastida, A. Berenyi, I. Zupko and Z. Topcu, Rec. Nat. Prod., 2012, 6, 381–385 Search PubMed.
- L. Ingrassia, F. Lefranc, V. Mathieu, F. Darro and R. Kiss, Clin. Transl. Oncol., 2008, 1, 1–13 CrossRef.
- G. R. Pettit, N. Melody and D. L. Herald, J. Nat. Prod., 2004, 67, 322–327 CrossRef CAS PubMed.
- G. Ceriotti, Nature, 1967, 213, 595–596 CrossRef CAS.
- L. Ingrassia, F. Lefranc, J. Dewelle, L. Pottier, V. Mathieu, S. Spiegl-Kreinecker, S. Sauvage, M. El Yazidi, M. Dehoux and W. Berger, J. Med. Chem., 2009, 52, 1100–1114 CrossRef CAS PubMed.
- P. Dumont, L. Ingrassia, S. Rouzeau, F. Ribaucour, S. Thomas, I. Roland, F. Darro, F. Lefranc and R. Kiss, Neoplasia, 2007, 9, 766–776 CrossRef CAS.
- G. Van Goietsenoven, J. Hutton, J.-P. Becker, B. Lallemand, F. Robert, F. Lefranc, C. Pirker, G. Vandenbussche, P. Van Antwerpen and A. Evidente, FASEB J., 2010, 24, 4575–4584 CrossRef CAS PubMed.
- G. Van Goietsenoven, V. Mathieu, F. Lefranc, A. Kornienko, A. Evidente and R. Kiss, Med. Res. Rev., 2013, 33, 439–455 CrossRef CAS PubMed.
- A. H. Abou-Donia, A. De Giulio, A. Evidente, M. Gaber, A.-A. Habib, R. Lanzetta and A. A. Seif El Din, Phytochemistry, 1991, 30, 3445–3448 CrossRef CAS.
- G. R. Pettit, V. Gaddamidi, G. M. Cragg, D. L. Herald and Y. Sagawa, J. Chem. Soc., Chem. Commun., 1984, 24, 1693–1694 RSC.
- R. M. Patel and R. N. Prajapati, Biotechnol. Mol. Biol. Rev., 2011, 6, 59–68 CAS.
- N. Kekre, C. Griffin, J. McNulty and S. Pandey, Cancer Chemother. Pharmacol., 2005, 56, 29–38 CrossRef CAS PubMed.
- C. Griffin, A. Karnik, J. McNulty and S. Pandey, Mol. Cancer Ther., 2011, 10, 57–68 CrossRef CAS PubMed.
- S. Vshyvenko, J. Scattolon, T. Hudlicky, A. E. Romero and A. Kornienko, Bioorg. Med. Chem. Lett., 2011, 21, 4750–4752 CrossRef CAS PubMed.
- C. Griffin, N. Sharda, D. Sood, J. Nair, J. McNulty and S. Pandey, Cancer Cell Int., 2007, 7, 1–7 CrossRef PubMed.
- D. S. Auld, T. J. Kornecook and S. Bastianetto, Prog. Neurobiol., 2002, 68, 209–245 CrossRef CAS.
- R. Schliebs and T. Arendt, Behav. Brain Res., 2011, 221, 555–563 CrossRef CAS PubMed.
- J. Zhu, C. Wu, X. Li, G. Wu, S. Xie, Q. Hu, Z. Deng, X. M. Zhu, H. Luo and X. Hong, Bioorg. Med. Chem., 2013, 21, 4218–4224 CrossRef CAS PubMed.
- C. Iannello, N. B. Pigni, F. Antognoni, F. Poli, A. Maxia, J. P. de Andrade and J. Bastida, Fitoterapia, 2014, 92, 163–167 CrossRef CAS PubMed.
- S. López, J. Bastida, F. Viladomat and C. Codina, Life Sci., 2002, 71, 2521–2529 CrossRef.
- K. Rhee, N. Appels, B. Hofte, B. Karabatak, C. Erkelens, L. M. Stark, L. A. Flippin and R. Verpoorte, Biol. Pharm. Bull., 2004, 27, 1804–1809 Search PubMed.
- Y.-H. Wang, Q.-L. Wan, C.-D. Gu, H.-R. Luo and C.-L. Long, Chem. Cent. J., 2012, 6, 1–6 CrossRef PubMed.
- J. J. Nair and J. van Staden, Nat. Prod. Commun., 2012, 7, 959–962 CAS.
- J. McNulty, J. J. Nair, J. R. Little, J. D. Brennan and J. Bastida, Bioorg. Med. Chem. Lett., 2010, 20, 5290–5294 CrossRef CAS PubMed.
- I. L Elisha, E. E. Elgorashi, A. A. Hussein, G. Duncan and J. N. Eloff, S. Afr. J. Bot., 2013, 85, 44–47 CrossRef PubMed.
- E. E. Elgorashi, G. I. Stafford and J. V. Staden, Planta Med., 2004, 70, 260–262 CrossRef CAS PubMed.
- B. Gabrielsen, T. P. Monath, J. W. Huggins, D. F. Kefauver, G. R. Pettit, G. Groszek, M. Hollingshead, J. J. Kirsi, W. M. Shannon, E. M. Schubert, J. DaRe, B. Ugarkar, M. A. Ussery and M. J. Phelan, J. Nat. Prod., 1992, 55, 1569–1581 CrossRef CAS.
- G. Zou, F. Puig-Basagoiti, B. Zhang, M. Qing, L. Chen, K. W. Pankiewicz, K. Felczak, Z. Yuan and P.-Y. Shi, Virology, 2009, 384, 242–252 CrossRef CAS PubMed.
- J. He, W. B. Qi, L. Wang, J. Tian, P. R. Jiao, G. Q. Liu, W. C. Ye and M. Liao, Influenza Other Respir. Viruses, 2013, 7, 922–931 CrossRef CAS PubMed.
- S.-D. Huang, Y. Zhang, H.-P. He, S.-F. Li, G.-H. Tang, D.-Z. Chen, M.-M. Cao, Y.-T. Di and X.-J. Hao, Chin. J. Nat. Med., 2013, 11, 406–410 CrossRef CAS.
- S.-Y. Li, C. Chen, H.-Q. Zhang, H.-Y. Guo, H. Wang, L. Wang, X. Zhang, S.-N. Hua, J. Yu and P.-G. Xiao, Antiviral Res., 2005, 67, 18–23 CrossRef CAS PubMed.
- Y.-C. Hwang, J. J.-H. Chu, P. L. Yang, W. Chen and M. V. Yates, Antiviral Res., 2008, 77, 232–236 CrossRef CAS PubMed.
- J. Liu, Y. Yang, Y. Xu, C. Ma, C. Qin and L. Zhang, Virol. J., 2011, 8, 483 CrossRef CAS PubMed.
- E. Nkanwen, D. Gatsing, D. Ngamga, S. Fodouop and P. Tane, Afr. Health Sci., 2009, 9, 264–269 CAS.
- O. Arrigoni, R. A. Liso and G. Calabrese, Nature, 1975, 256, 513–514 CrossRef CAS.
- K. K. Schrader, A. Andolfi, C. L. Cantrell, A. Cimmino, S. O. Duke, W. Osbrink, D. E. Wedge and A. Evidente, Chem. Biodiversity, 2010, 7, 2261–2280 CAS.
- C.-X. Tan, K. K. Schrader, C. S. Mizuno and A. M. Rimando, J. Agric. Food Chem., 2011, 59, 5977–5985 CrossRef CAS PubMed.
- K. K. Schrader, F. Avolio, A. Andolfi, A. Cimmino and A. Evidente, J. Agric. Food Chem., 2013, 61, 1179–1183 CrossRef CAS PubMed.
- W. H. Organization, Global prevalence and incidence of selected curable sexually transmitted infections: overview and estimates, 2001, pp. 1–120 Search PubMed.
- R. B. Giordani, P. C. D. B. Vieira, M. Weizenmann, D. B. Rosemberg, A. P. Souza, C. Bonorino, G. A. De Carli, M. R. Bogo, J. A. Zuanazzi and T. Tasca, J. Nat. Prod., 2010, 73, 2019–2023 CrossRef CAS PubMed.
- R. B. Giordani, P. D. B. Vieira, M. Weizenmann, D. B. Rosemberg, A. P. Souza, C. Bonorino, G. A. De Carli, M. R. Bogo, J. A. Zuanazzi and T. Tasca, Phytochemistry, 2011, 72, 645–650 CrossRef CAS PubMed.
- R. B. Giordani, M. Weizenmann, D. B. Rosemberg, G. A. De Carli, M. R. Bogo, J. A. S. Zuanazzi and T. Tasca, Parasitol. Int., 2010, 59, 226–231 CrossRef CAS PubMed.
- R. B. Giordani, C. O. Junior, J. P. de Andrade, J. Bastida, J. A. Zuanazzi, T. Tasca and M. V. de Almeida, Chem. Biol. Drug Des., 2012, 80, 129–133 CAS.
- R. W. Snow, C. A. Guerra, A. M. Noor, H. Y. Myint and S. I. Hay, Nature, 2005, 434, 214–217 CrossRef CAS PubMed.
- B. Şener, I. Orhan and J. Satayavivad, Phytother. Res., 2003, 17, 1220–1223 CrossRef PubMed.
- Y. Toriizuka, E. Kinoshita, N. Kogure, M. Kitajima, A. Ishiyama, K. Otoguro, H. Yamada, S. Ōmura and H. Takayama, Bioorg. Med. Chem., 2008, 16, 10182–10189 CrossRef CAS PubMed.
- J. C. Cedrón, D. Gutiérrez, N. Flores, Á. G. Ravelo and A. Estévez-Braun, Bioorg. Med. Chem., 2010, 18, 4694–4701 CrossRef PubMed.
- B. Hao, S.-F. Shen and Q.-J. Zhao, Molecules, 2013, 18, 2458–2468 CrossRef CAS PubMed.
- J. C. Cedrón, D. Gutiérrez, N. Flores, Á. G. Ravelo and A. Estévez-Braun, Bioorg. Med. Chem., 2012, 20, 5464–5472 CrossRef PubMed.
- J. C. Cedrón, D. Gutiérrez, N. Flores, Á. G. Ravelo and A. Estévez-Braun, Eur. J. Med. Chem., 2013, 63, 722–730 CrossRef PubMed.
- J. J. Nair and J. van Staden, Food Chem. Toxicol., 2013, 62, 262–275 CrossRef CAS PubMed.
- A. F. S. da Silva, J. P. de Andrade, L. R. Bevilaqua, M. M. de Souza, I. Izquierdo, A. T. Henriques and J. Â. S. Zuanazzi, Pharmacol., Biochem. Behav., 2006, 85, 148–154 CrossRef PubMed.
- G. I. Stafford, C. Birer, B. Brodin, S. B. Christensen, A. H. Eriksson, A. Jäger and N. Rønsted, S. Afr. J. Bot., 2013, 88, 101–106 CrossRef CAS PubMed.
- M. E. Pedersen, B. Szewczyk, K. Stachowicz, J. Wieronska, J. Andersen, G. I. Stafford, J. van Staden, A. Pilc and A. K. Jäger, J. Ethnopharmacol., 2008, 119, 542–548 CrossRef PubMed.
- J. S. Neergaard, J. Andersen, M. E. Pedersen, G. I. Stafford, J. V. Staden and A. K. Jäger, S. Afr. J. Bot., 2009, 75, 371–374 CrossRef CAS PubMed.
Footnote |
| † M. He and C. Qu contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |