DOI:
10.1039/C2RA21211K
(Paper)
RSC Adv., 2012,
2, 9114-9123
Nano thick poly(ε-caprolactone)-poly(ethylene glycol) coatings developed by catalyst-free plasma assisted copolymerization process for biomedical applications†
Received
19th June 2012
, Accepted 2nd August 2012
First published on 24th August 2012
1. Introduction
Over the past few decades, the use of poly(ε-caprolactone) (PCL), a versatile and resorbable polymer has significantly increased in the different domains of research, in perticularly in the field of biomaterials, biomedical and tissue engineering.1 The usage of PCL blends which present a higher mechanical preoperties as well as biocompatibility compared to the homopolymer itself, should mark a new era in the use of PCL in the drug delivery systems. PCL and its copolymers have been utilized for long-term biodegradable medical implants and can be easily modified biologically, physically and chemically to acquire tailorable degradation kinetics required for controlled release of medication, gene delivery etc. PCL having FDA approval and CE Mark registration makes it universally acceptable for in vitro and in vivo applications.2,3 PCL is a hydrophobic, semi-crystalline, mechanically compatible, biodegradable and bio-resorbable polymer which possesses exceptional blend-compatibility.4,5 The hydrophobic character and high degree of crystallinity of PCL limits its degradation rate and hence makes it less biocompatible with soft tissues. To change the properties of PCL, PEG can be a suitable copolymerization reagent because of its hydrophilicity, immunogenicity, nontoxicity and lack of antigenicity.6–8 In PCL-PEG copolymers, PEG can improve their hydrophilicity, biodegradability, mechanical properties and biocompatibility. PCL-PEG copolymers have been prepared by ring-opening polymerization (ROP) of ε-caprolactone (ε-CL) using PEG9–11 and amine-functionalized PEG12 as macro-initiators with stannous octanoate [Sn(Oct)2] as catalyst. Furthermore, a few new catalysts such as zinc,13 calcium,14 aluminum,15 yttrium tris(2,6-di-tert-butyl-4-methylphenolate) [Y(DBMP)3]16 and stannous chloride17 complexes were developed for the synthesis of amphiphilic PCL-PEG copolymers. However, the copolymers obtained by ROP using organometallic catalysts are not suitable because of incomplete catalyst removal, and because it is difficult and costly to remove the metal contaminant from the resultant polymers.18 Hence, the use of metal catalysts is not advisable in extending the use of PCL-PEG copolymers for practical biomedical purposes. In recent years, the application of metal-free strategies to perform ROP reactions has been developed for the synthesis of polymers which provide several advantages over those that require metals to initiate the process, the most obvious being the absence of the costly removal of metal impurities from the resultant polymers and the end products. Using wet processes such as hydrogel, spin casting etc, metal catalyst-free ROP of ε-CL was shown by tertiary amines in the synthesis of chitosan-graft-poly(ε-CL),19 nucleophilic phosphines at high-temperature anhydrous conditions20 and using mild organic acid (i.e. tartaric acid).21 Methoxy poly(ethylene glycol) (MePEG)-PCL diblock copolymers were synthesized by the catalyst-free ROP of ε-CL by MePEG in a vacuum oven at 185 °C followed by extraction of the copolymer by several filtration and washing runs, with the resultant product dried for three days at 40 °C (i.e. requirement of high temperatures and long processing time).22,23 Recently, ROP of ε-CL was observed by an activated monomer mechanism in the presence of HCl·Et2O (HCl solution in diethyl ether) which was performed to obtain copolymers with PEG.24–26 To summarize, there have been a limited number of studies which have been reported for the preparation of PCL-PEG copolymers without metal catalyst. The aim of the present study was to develop nanosized, amphiphilic, biodegradable and biocompatible PCL-co-PEG coatings by a single-step plasma co-polymerization process. To our knowledge, this is the first time that PCL-co-PEG coatings have been developed by catalyst-free ROP of ε-CL in the presence of DEGME in a low-pressure inductively excited radiofrequency (13.56 MHz) discharge in soft pulsed plasma mode to avoid excessive fragmentation of the two precursors used, and therefore retention of the EO and CL functionalities. The structural properties of plasma-polymerized PCL-co-PEG coatings have been characterized using two different physicochemical techniques (1H NMR and MALDI ToF).
2. Results and discusion
2.1 ROP of ε-CL in the presence of DEGME plasma
PCL-PEG copolymer coatings were synthesized by simultaneously introducing ε-CL and DEGME monomer vapours in an inductively excited radio frequency (13.56 MHz) discharge under soft plasma conditions (Fig. S1 and S2, ESI†). In the present study, PCL homopolymer coatings were developed by ROP of ε-CL via electron impact and/or Ar metastables (Ar*) in the pulsed plasma environment as shown in Scheme 1. During the time ON, electron impacts generate active radical species and Ar metastables responsible for the fragmentation of the DEGME monomer. Because of the nucleophilic character of DEGME radicals, copolymerization was accomplished with various ratios of ε-CL to DEGME monomer which can act as micro-initiators for the catalyst-free ROP. To summarize, the plasma polymerization can be initiated by electrons and active radicals created in the plasma which can lead to ring opening of ε-CL monomer but the latter can be caused also through DEGME radicals. During the time OFF, even if there are no electrons, the plasma-polymerization process can take place because of the presence of long-lived Ar metastables. Moreover graft polymerization can also take place either by adsorption of the fragments of the polymer formed during the ON time or by monomer adsorption on the reactive sites of the substrate surface. The co-polymer synthesis in the pulsed plasma discharge is shown in Scheme 2.
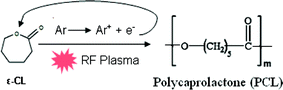 |
| | Scheme 1 ROP mechanism of ε-CL via electron impact. | |
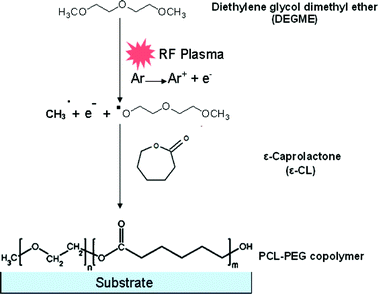 |
| | Scheme 2 PCL-co-PEG polymer synthesis in the pulsed plasma discharge. | |
2.2 FTIR-ATR analysis of plasma-polymerized PCL-co-PEG coatings
The series of PCL-co-PEG coatings were developed by catalyst-free ROP of ε-CL in the presence of DEGME as an initiator. The functional groups present in the coatings deposited onto Si wafer substrates were examined using FTIR-ATR spectroscopy. The reference spectra for both the cyclic ester (ε-CL) and ethylene oxide (DEGME) monomer was taken and reported (Fig. S3, ESI†). A precise volume of 40 μL was taken for each liquid monomer.
CW and PW plasma mode.
The spectral analyses shown in Fig. S4 (ESI†) allow comparative study of plasma deposited PCL-co-PEG coatings which were prepared under CW and PW plasma mode, using constant partial pressure ratios of monomer feed. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of the ester carbonyl group, the characteristic C–O–C stretching vibrations of the repeating –OCH2CH2 units of PEG and the C–O bonds stretching vibrations were observed at 1729, 1107 and 1246 cm−1, respectively. The absorption band at 3442 cm−1 is assigned to terminal –OH groups in the copolymer coatings. All the C–H stretching bonds are centered at 2946 and 2867 cm−1. The C–H bending was observed in the range from 1380 to 1440 cm−1. In the PCL-co-PEG copolymer coatings, the carbonyl group of ester linkages in PCL was observed at 1724 cm−1. A significant difference was observed in the intensity of the FTIR peaks of PCL-co-PEG (1
O stretching vibrations of the ester carbonyl group, the characteristic C–O–C stretching vibrations of the repeating –OCH2CH2 units of PEG and the C–O bonds stretching vibrations were observed at 1729, 1107 and 1246 cm−1, respectively. The absorption band at 3442 cm−1 is assigned to terminal –OH groups in the copolymer coatings. All the C–H stretching bonds are centered at 2946 and 2867 cm−1. The C–H bending was observed in the range from 1380 to 1440 cm−1. In the PCL-co-PEG copolymer coatings, the carbonyl group of ester linkages in PCL was observed at 1724 cm−1. A significant difference was observed in the intensity of the FTIR peaks of PCL-co-PEG (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) coatings prepared under CW mode as compared to that prepared in PW plasma mode, indicating the difference in the overall chemical composition. In the pulsed plasma polymerization, mainly two reactions can occur. First, during the plasma ON time, the fragmentation or the bond dissociation of the molecules and the formation of active species and ions. Secondly, chain growth of the polymers take place during the plasma OFF time and such reactions are more prominent when the monomers have a double or a triple bond because of their higher concentration of free radicals which could continue to contribute to the chain growth during the plasma OFF time.27 In the CW plasma mode, high-energy electrons continuously generate reactive species which cause more fragmentation and recombination of DEGME molecules and hence, leads to a reduced PEO character of PEG coatings obtained. ε-CL is a cyclic aliphatic polyester with a seven-membered ring of the lactone family. Its structure is comparatively stable as compared to other lactones which possess saturated bonds and which require high energy to obtain linear polymers. Such observations were reported for the plasma polymerization of aniline28 and octaflurotoluene.29 According to the plasma polymerization, ROP of ε-CL can be initiated by atomic polymerization27,30via high-energy electron impact or the nucleophilic character of DEGME radicals. FTIR-ATR spectral analysis of PCL-co-PEG (1:4) coatings deposited at 50 and 20 W, respectively, under the CW plasma mode clearly shows substantial changes in the chemical composition of the coatings with an increase of the applied power (Fig. S4a and b, ESI†). Indeed one can observe at 50 W the absence of 1107 and 1246 cm−1 bands corresponding to the C–O–C stretching vibrations of the repeated units of PEG and the C–O bond stretching vibrations, respectively. The intensity of C–O–C bonds is dramatically reduced as the effective plasma power employed during plasma polymerization is increased. It has been reported in the literature31,32 that the non-fouling behavior of PEG polymers can be attributed to the retention of ethylene oxide groups, as followed by the intensity of C–O–C stretching vibrations. In 50 W CW plasma deposited copolymer coatings, the CO stretching vibrations at 1729 cm−1 decreased as compared to 20 W CW plasma deposited coatings. By contrast, the intensity of aliphatic hydrocarbon C–H stretching bonds at 2946 and 2867 cm−1 was increased because of the decomposition and recombination processes occuring at high plasma powers. Conclusively, plasma-polymerized PCL-co-PEG (1:4) coatings obtained in CW plasma mode exhibited almost comparable chemical composition and surface characteristics with PCL homopolymers because the increase in the plasma power lead to a higher decomposition or fragmentation of DEGME molecules in the plasma discharge and therefore resulted in coatings with absence of PEO character. Hence, this is the reason why pulsed plasma polymerization under similar plasma deposition conditions was performed under PW mode. Plasma-polymerized PCL-co-PEG (1:4) coatings which were prepared under the PW plasma mode at 0.25 W (Ppk = 25 W, DC = 1%, ton = 1 ms, toff = 99 ms) and 1 W (Ppk = 25 W, DC = 4%, ton = 4 ms, toff = 96 ms) effective plasma power (Peff) (Fig. S4c and d, ESI†). The vibrational peak at 1107 cm−1 in the pulsed plasma deposited PCL-co-PEG (1:4) coatings indicated the incorporation of C–O–C groups while this peak was absent in the case of CW plasma deposited coatings.
:4) coatings prepared under CW mode as compared to that prepared in PW plasma mode, indicating the difference in the overall chemical composition. In the pulsed plasma polymerization, mainly two reactions can occur. First, during the plasma ON time, the fragmentation or the bond dissociation of the molecules and the formation of active species and ions. Secondly, chain growth of the polymers take place during the plasma OFF time and such reactions are more prominent when the monomers have a double or a triple bond because of their higher concentration of free radicals which could continue to contribute to the chain growth during the plasma OFF time.27 In the CW plasma mode, high-energy electrons continuously generate reactive species which cause more fragmentation and recombination of DEGME molecules and hence, leads to a reduced PEO character of PEG coatings obtained. ε-CL is a cyclic aliphatic polyester with a seven-membered ring of the lactone family. Its structure is comparatively stable as compared to other lactones which possess saturated bonds and which require high energy to obtain linear polymers. Such observations were reported for the plasma polymerization of aniline28 and octaflurotoluene.29 According to the plasma polymerization, ROP of ε-CL can be initiated by atomic polymerization27,30via high-energy electron impact or the nucleophilic character of DEGME radicals. FTIR-ATR spectral analysis of PCL-co-PEG (1:4) coatings deposited at 50 and 20 W, respectively, under the CW plasma mode clearly shows substantial changes in the chemical composition of the coatings with an increase of the applied power (Fig. S4a and b, ESI†). Indeed one can observe at 50 W the absence of 1107 and 1246 cm−1 bands corresponding to the C–O–C stretching vibrations of the repeated units of PEG and the C–O bond stretching vibrations, respectively. The intensity of C–O–C bonds is dramatically reduced as the effective plasma power employed during plasma polymerization is increased. It has been reported in the literature31,32 that the non-fouling behavior of PEG polymers can be attributed to the retention of ethylene oxide groups, as followed by the intensity of C–O–C stretching vibrations. In 50 W CW plasma deposited copolymer coatings, the CO stretching vibrations at 1729 cm−1 decreased as compared to 20 W CW plasma deposited coatings. By contrast, the intensity of aliphatic hydrocarbon C–H stretching bonds at 2946 and 2867 cm−1 was increased because of the decomposition and recombination processes occuring at high plasma powers. Conclusively, plasma-polymerized PCL-co-PEG (1:4) coatings obtained in CW plasma mode exhibited almost comparable chemical composition and surface characteristics with PCL homopolymers because the increase in the plasma power lead to a higher decomposition or fragmentation of DEGME molecules in the plasma discharge and therefore resulted in coatings with absence of PEO character. Hence, this is the reason why pulsed plasma polymerization under similar plasma deposition conditions was performed under PW mode. Plasma-polymerized PCL-co-PEG (1:4) coatings which were prepared under the PW plasma mode at 0.25 W (Ppk = 25 W, DC = 1%, ton = 1 ms, toff = 99 ms) and 1 W (Ppk = 25 W, DC = 4%, ton = 4 ms, toff = 96 ms) effective plasma power (Peff) (Fig. S4c and d, ESI†). The vibrational peak at 1107 cm−1 in the pulsed plasma deposited PCL-co-PEG (1:4) coatings indicated the incorporation of C–O–C groups while this peak was absent in the case of CW plasma deposited coatings.
The intensity of C–O–C groups detected at 1107 cm−1 was higher in the 1 W plasma deposited coatings as compared to 0.25 W coatings which indicated that 0.25 W effective plasma power might be sufficient for the DEGME homopolymerization but it is too low for the copolymerization. For the PCL-co-PEG (1:4) coatings, the evolution of C–O/CHx ratios (x = 1, 2, 3) obtained by FTIR-ATR spectral analysis, clearly showed that they decreased as Peff increased, suggesting a decrease of the retention of ethylene oxide groups in the copolymer composition with the increase of plasma power (Fig. 1). In this figure we have plotted the plasma-polymerized DEGME (pDEGME) deposited at very mild conditions (leading to a high retention of ethylene oxide groups) for comparison. The latter shows that in the case of copolymers deposited under the same conditions as PEG, one can retain the desired functionalities at mild conditions. Furthermore we have reported the detailed FTIR-ATR spectra of the homopolymers as well as the copolymers obtained under different partial pressure ratios of the precursors deposited at the same optimal power of 1 W (Fig. S5, ESI†).
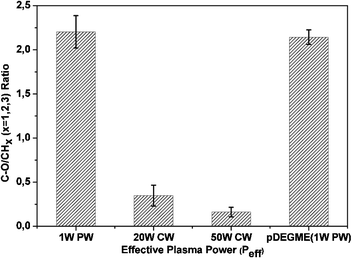 |
| | Fig. 1 C–O/CHx (x = 1, 2, 3) ratios determined from FTIR-ATR vs. effective plasma power (Peff) used to deposit PCL-co-PEG (1:4) coatings and pDEGME as a reference which was deposited under 1 W PW plasma (error bars indicate standard deviation on the mean of three measurements). | |
Fig. 2 shows the high-resolution XPS scan of the C1s peak of the pulsed plasma-polymerized PCL, PEG like coatings from DEGME (pDEGME), PCL-co-PEG (1:2) and PCL-co-PEG (1:4) coatings prepared under Peff = 1 W and designated by (a), (b), (c) and (d), respectively. The C1s XPS scan of plasma deposited PCL coatings was deconvoluted into four peaks attributed to C–C/C–H (C1: 285.0 eV), C–OH/C–O (C2: 286.6 eV), CO (C3: 287.8 eV) and OC–O (C4: 289.0 eV) (Fig. 2a). The C1, C2 and C4 peaks are observed in the case of a PCL obtained by a traditional wet process such as spin casting.33,34 The C3 peak observed at 287.8 eV in the case of plasma-polymerized PCL is probably due to ring opening and the rearrangement and/or oxidation of the resulting fragments. Fig. 2b shows the C1s XPS spectrum of the plasma-polymerized DEGME (pDEGME) coatings fitted with four components attributed to C–C/C–H (C1: 285.1 eV), C–O (C2: 286.6 eV), CO (C3: 288 eV) and OC–O (C4: 289.4 eV). One can observe in this spectrum a intense peak of the ether functions C2 at 286.6 eV35 which represents the fingerprint of pDEGME. Fig. 2c shows the C1s XPS spectrum of PCL-co-PEG (1:2) coatings, for which the intensity of the C–C/C–H (C1: 284.9 eV) peak is smaller as compared to the PCL homopolymer and the incorporation of OC–O (C4: 289.0 eV) groups indicated that the PCL-co-PEG (1:2) coatings have similar chemical functionalities as compared to plasma deposited PCL homopolymers. In the PCL-co-PEG (1:4) coatings, the intensity of C–C/C–H (C1: 285.0 eV) peak was further decreased and the incorporation of C–O (C2: 286.6 eV), CO (C3: 288.0 eV) and O–CO (C4: 289.2 eV) showed similar chemical composition as compared to the pDEGME coatings (Fig. 2d). In the PCL-co-PEG coatings (Fig. 2c and d), the presence of C–O (C2: 286.6 eV) groups, as explained before, are ascribed to PEG segments in the copolymer coatings. Since it is difficult to identify all the peaks of the copolymer, we have determined from the XPS C1s peak the C–O/C–C ratio which could be a good indicator for the estimation of the conjugation of PCL and PEG in the copolymer coatings. The evolution of the latter and the thickness of the coatings measured with the help of ellipsometry was plotted vs. ε-CL:DEGME partial pressure ratios for plasma-polymerized PCL, pDEGME and PCL-co-PEG coatings deposited under 1 W effective plasma power (Fig. 3a). According to the C–O/C–C ratios, two different characteristics of plasma deposited coatings were observed. Less hydrophilic surfaces were obtained for plasma-polymerized PCL and PCL-co-PEG (1:2) coatings for which the C–O/C–C ratios were less than one and around one respectively. In the PCL-co-PEG (1:4) coatings, C–H/C–C (C1s: 285 eV) groups decreased and C–O (C2: 286.6 eV) groups increased with the increase of the partial pressure of DEGME monomer in copolymer coatings, which confirmed what was expected, i.e. the introduction of PEG segments enhanced the surface hydrophilicity of PCL-co-PEG polymers. The C–O/C–C ratio was more than one for both the plasma-polymerized PCL-co-PEG (1:4) and pDEGME coatings, which proved the potential of soft plasma polymerization in the pulse mode to obtain a small fragmentation of monomer structure and more retention of ethylene oxide groups. Also, we have observed that the coating thicknesses decreased with the increase of the C–O/C–C ratio in the plasma-polymerized coatings, illustrating that the deposition rate decreased with the introduction of ethylene oxide groups.
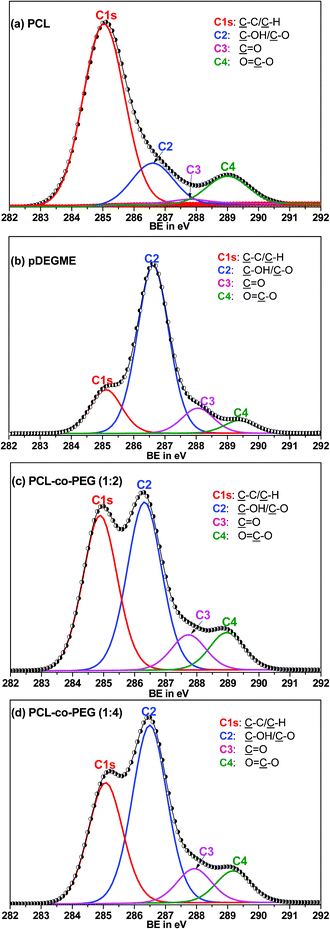 |
| | Fig. 2 High-resolution C1s XPS spectra of pulsed plasma-polymerized coatings prepared at Peff = 1 W: (a) PCL, (b) pDEGME, (c) PCL-co-PEG (1:2) and (d) PCL-co-PEG (1:4). | |
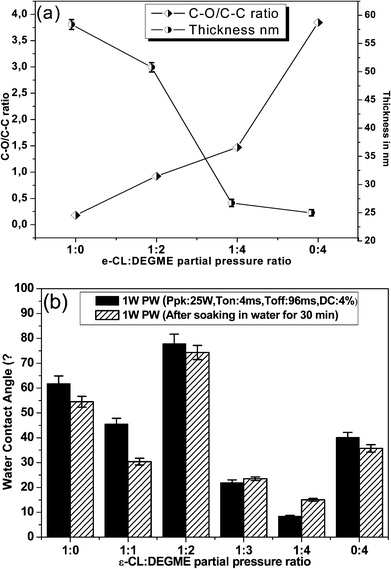 |
| | Fig. 3 (a) Evolution of the C–O/C–C ratio determined from C1s XPS spectra and coating thicknesses from ellipsometry vs. ε-CL:DEGME partial pressure ratio for plasma-polymerized PCL, pDEGME and PCL-co-PEG coatings deposited at 1 W effective plasma power (error bars designate standard deviation on the mean of three measurements) and (b) water contact angle measurement results for plasma-polymerized PCL, pDEGME and PCL-co-PEG coatings before and after soaking in water for 30 min (coating stability test). | |
2.4 Surface wetability measurement on PCL-co-PEG coatings
The surface hydrophilicity of the different plasma-polymerized coatings and their stability to water washing was determined by water contact angle (WCA) on coatings before and after being soaked in water for 30 min and quickly dried by air (Fig. 3b). PCL homopolymer, pDEGME and PCL-co-PEG coatings were deposited at 1 W effective plasma power on which the WCA was determined with DI water. The latter ranged from 8 to 78°. The WCA of PCL homopolymer coatings was around 60°. In the case of PCL-co-PEG (1:1) coatings, for the same ε-CL:DEGME partial pressure ratio, the WCA was lower than (45°) as measured on PCL homopolymer. It is noteworthy to point out that the WCA of PCL-co-PEG (1:2) coatings interestingly increased with the decrease of ε-CL:DEGME ratio obtained by increasing the partial pressure of DEGME monomer as compared to ε-CL. Indeed the DEGME monomer served as a catalyst for ROP of ε-CL initiated by the oxygen containing radicals resulting from the activation of DEGME monomer by collision with electrons or argon metastables. Because of their nucleophilic behaviour most of the cyclic ester monomer polymerized at this deposition condition and decreased the surface hydrophilicity of the copolymer. The WCA of PCL-co-PEG (1:2) was measured to be 78°. Further, the decrease of the WCA of plasma copolymerized coatings with the increased partial pressure of DEGME in copolymer coatings showed that the introduction of PEG segments enhanced the surface hydrophilicity of PCL-co-PEG coatings. The WCA of PCL-co-PEG (1:4) coating was shown to be 8° and the WCA of pDEGME coatings was measured to be 40°. The PCL-co-PEG coatings exhibited an excellent stability to soaking in water.
2.5
1H NMR spectral analysis of PCL-co-PEG coatings
In order to further confirm the formation of PCL-co-PEG polymers, 1H NMR spectra were recorded36 (Fig. 4a). In this figure, the characteristic absorption peaks at 2.3, 1.64, 1.4, 1.64 and 4.06 ppm corresponded to methylene protons in PCL repeat units which were designated by d, e, f, g and h. Additionally, the methylene proton of PEG which was directly linked to the PCL was observed at 4.2 ppm and was denoted by “c”. The terminal methoxy proton of PEG was observed at 3.38 ppm which was denoted by “a”. The broad peak at 3.64 ppm corresponded to the methylenes of the PEG and was designated by “b”. Two distinct peaks corresponding to non-polymerized ε-CL monomer were observed at 1.26 and 2.25 ppm.
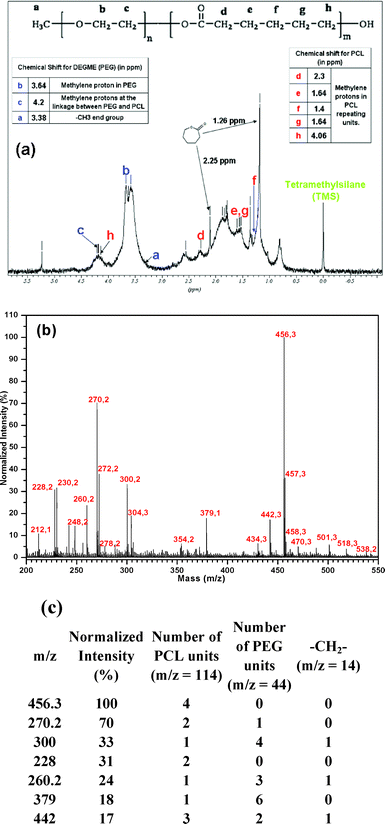 |
| | Fig. 4 (a) 1H NMR spectrum of PCL-co-PEG (1:2) coatings in CDCl3, (b) MALDI ToF mass spectrum of PCL-co-PEG (1:2) coatings prepared in alpha-matrix and (c) molecular weight of the most representative ion fragments measured by MALDI-ToF and estimated number average degrees of polymerization. | |
2.6 MALDI ToF mass spectral analysis of PCL-co-PEG coatings
The MALDI mass spectrum of the PCL-co-PEG (1:2) plasma-polymerized coatings dissolved in α-cyano-4-hydroxycinnamic acid (alpha-cyano or alpha-matrix)36,37 is shown in Fig. 4b. All the samples were deposited under the soft pulsed plasma discharge at Peff = 1 W and used for physicochemical analysis. The most intense ions detected have molecular weight ranging between 210 and 460 Da. The specific peaks of the copolymer chains were separated by m/z shifts of 114, 44 and 14 Da corresponding to one caprolactone unit (C6H10O2), one ethylene oxide unit (C2H4O) and one methylene unit, respectively. The number average molar masses of copolymer chains were calculated from the MALDI ToF data analysis. Polymer ion signals having normalized intensities higher than 15% are reported in Fig. 4c. Throughout the mass spectrum, 1 Da mass differences have been observed between two consecutive peaks and the intensive examination of the polymer ion signals suggests the presence of three different polymer series of 44, 88 and 114 Da coresponding to repeat units of –(CH2CH2O)–, –(CH2CH2O)2– and –(C6H10O2)–, respectively. From the mass spectrum, mass shifts of 32 Da were observed which is due to the incorporation of additional oxygen atoms during the plasma polymerization (Fig. S6 and S7, ESI†).
2.7 Cell adhesion and proliferation on plasma-polymerized coatings
The biological response to plasma deposited glass surfaces was investigated in vitro using human bone marrow endothelial cell line (HBMEC). 2 ml of cell suspension with a density of 1 × 105/well was injected into each well and was incubated at physiological conditions for 24, 48 and 72 h. As shown in Fig. 5, the fluorescent images of cytoskeleton stain demonstrated the cell adhesion and proliferation on the plasma deposited surfaces after 72 h of incubation (left side). For PCL and PCL-co-PEG (1:2) coatings, cells experienced some stretching across the cytoplasm resulting in flattening of the cells.
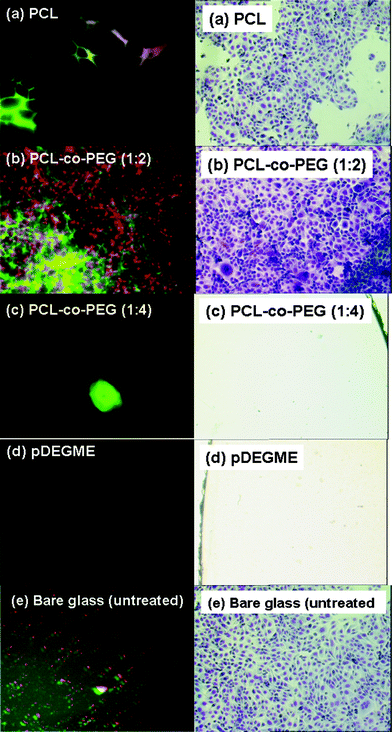 |
| | Fig. 5 Fluorescent images of cytoskeleton stain demonstrated human bone marrow endothelial cells (HBMEC) adhesion and proliferation on plasma-polymerized coatings after 72 h of incubation (image magnification: 60×) (left side) and optical microscopy images (900 μm × 600 μm) of stained human ovarian carcinoma cells (NIH:OVCAR-3) seeded for 120 h on plasma-polymerized coatings and on bare glass and polystyrene (PS) culture plates taken as positive controls (optical magnification: 10×) (right side). | |
Cells on the PCL-co-PEG (1:2) coated glass surfaces were well spread and developed a network required for better cell adhesion as compared to the PCL homopolymers.
In this case, the introduction of the ethylene oxide functionalities in the copolymer enhanced the overall rate of cell migration, cell proliferation and adhesion properties.38,39 Cell repellent behavior of the plasma deposited PEG rich amphiphilic PCL-co-PEG (1:4) copolymer and pDEGME homopolymer coatings were observed due to the retention of the ethylene oxide functionalities. There exist a huge number of references which explain the antifouling effect of the EO groups e.g. by providing a molecular basis for a water barrier layer which helps to reduce the protein adsorption on the surfaces. Bare (untreated) glass substrates showed some adhesion of the cell fragments but in this case most of the cells were detached from the glass surface. Optical microscopy images of cell adhesion and proliferation on plasma-polymerized coatings after incubation of 24, 48 and 72 h are shown in Fig. S8, ESI†.
In order to further confirm the cellular behavior, NIH:OVCAR-3 cells were incubated with plasma-coated glass surfaces at physiological conditions for 58 and 120 h in a polystyrene 12 well-cell culture plate. Inverted microscopic images of cell adhesion and proliferation on plasma-polymerized coatings at Peff = 1 W for different ε-CL:DEGME partial pressure ratios of the monomers were recorded after 120 h of cell culture and are shown in Fig. 5 (right side).
As expected, there was no cell adhesion observed on plasma-polymerized PEG coatings which were also stable after 120 h of cell culture. As the ε-CL:DEGME monomer ratio was gradually decreased from 100 to 25% (by varying the partial pressure of ethylene oxide monomers) the cell adhesion decreased. Polystyrene plate (PS) and soft plasma deposited PEG (pDEGME) coatings were considered as positive and negative controls respectively. The adherent cells on the surface were determined by using an inverted microscope (Nikon Instruments, Europe) and 900 μm × 600 μm size images were captured on glass cover slips by using a CCD camera to determine cell adhesion.
The adherent cells were counted by using a custom made MATLAB based image processing program (Fig. 6).
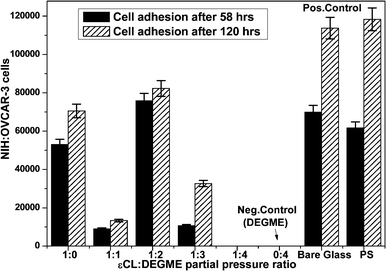 |
| | Fig. 6 Cell adhesion properties of NIH:OVCAR-3 on PCL, pDEGME (PEG) and PCL-co-PEG copolymers incubated for 58 and 120 h (error bars designate standard deviation on the mean of three measurements). | |
Cell to polymer surface interactions were examined with a SEM. At an accelerating voltage of 15 kV, images were obtained at different magnifications in the central region of each polymer surface after 24 h of cell culture. As shown in Fig. 7, the morphology of the cells seen on the SEM images were analyzed for the PCL-co-PEG (1:2) and pDEGME polymers. The magnified SEM image of the PCL-co-PEG (1:2) polymer surface showed that the cells were well proliferated and had undergone flattening on the polymer surface, which is in agreement with the fluorescent images of the cytoskeleton stain. No cell adhesion was observed on pDEGME coating, which was also in agreement with our previous cell adhesion tests.
 |
| | Fig. 7 SEM images of cell to plasma deposited polymer surface interactions: (a) PCL-co-PEG (1:2), (b) pDEGME and (c) PCL-co-PEG (1:2) (magnified image). | |
3. Experimental section
3.1 Materials
ε-CL (purity: 97%, MW: 114, empirical formula: C6H10O2) the cyclic ester monomer, DEGME (purity: 99.5%, MW: 134.17, linear formula: (CH3OCH2CH2)2O), and ethylene oxide monomers were purchased from Sigma Aldrich, France and used in this study without further purifications. For this study polished silicon (100) wafers purchased from Siltronix, France were used. They were ultrasonically cleaned in an acetone bath for 30 min and rinsed with ethanol for 30 min. The cleaned wafers were air dried and then used for XPS, WCA and ellipsometric measurements. For cell adhesion and proliferation analysis, glass cover slips (18 mm dia) were purchased from Warner Instruments, France and were used without further cleaning.
3.2 Plasma copolymerization set up for PCL-co-PEG coatings
Plasma copolymerization of ε-CL and DEGME was carried out in a low-pressure inductively excited radio frequency tubular quartz plasma reactor system at soft plasma conditions (Fig. S1, ESI†). The tubular plasma reactor had dimensions of 4 cm in diameter and 50 cm in length with an externally wound copper coil for RF power coupling. The power was generated by a Dressler Cesar RF generator which was delivered through the L–C matching network. The substrate is placed 9.0 cm below the coil. The plasma reactor was connected to the single stage rotary pump (Pfeiffer vacuum) via a chemical filter trap (Edwards high vacuum, Britain). The base pressure and operating pressure were 0.03 and 0.5 mbar, respectively. V1 and V2 are multi-turn manual valves used to introduce precursor vapour in the plasma reactor. The partial pressure ratio of monomer feed was controlled by flow rate of carrier gas (i.e. Ar), which was regulated and measured by electronic mass flow controllers (MKS instruments). The partial pressure of ε-CL and DEGME exhibited linear correlations with the flow rate of argon gas and were comparable with each other (Fig. S2, ESI†). Prior to each experimental run, the reactor was scrubbed and cleaned with detergent, organic solvents and dried using compressed air. The plasma reactor was reassembled and cleaned further with 30 W argon plasma discharge at 0.5 mbar pressure for 30 min. PCL-co-PEG coatings were deposited on (100) silicon wafers by simultaneously introducing ε-CL and DEGME monomer vapour. In the present work copolymer coatings at 20 W CW (continuous wave) and 50 W CW plasma were deposited and compared to pulsed plasma deposited coatings at 0.25 W PW (pulse wave) and 1 W PW. For the pulsed plasma discharge, the peak power (Ppk) was 25 W and duty cycle (DC = (ton/(ton + toff), where ton and toff were the “plasma ON” and “plasma OFF” times respectively) varied from 1 to 4% to obtain the effective plasma power (Peff) of 0.25 W PW (ton = 1 ms, toff = 99 ms) and 1 W PW (ton = 4 ms, toff = 96 ms), respectively. All the coatings were deposited for 20 min. After polymer deposition, the reactor was again evacuated to base pressure before the plasma-polymerization system was vented to atmospheric pressure with air.
3.3 Precursor flow rates and sample nomenclature
The flow rate of carrier gas varies from 5 to 20 sccm with the increment of 5 sccm which was denoted by “1” for 5 sccm and “4” for 20 sccm, respectively. Total flow rate was varied from 5 sccm to 25 sccm by keeping the same operating pressure.
The bulk chemical properties of plasma copolymerized coatings were analyzed by recording FT-IR spectra of PCL-co-PEG coatings using a Bruker Tensor 27 FT-IR spectrophotometer in the attenuated total reflectance (ATR) mode, using Si wafers as coating substrates. The FT-IR ATR spectra were recorded with a resolution of 4 cm−1 by using germanium as an ATR crystal. Baseline correction was performed by OPUS 6.5 software after 64 scans of each sample.
Surface chemical analysis of the PCL-co-PEG coatings was obtained by using XPS. A Thermo Scientific ESCALAB 250 spectrometer was used equipped with a microspot monochromated Al-Kα source and a flood gun combined with an argon gun for compensation of sample electrostatic charging. The Al-Kα line (1486.6 eV) was used throughout the work and the base pressure in the analytical chamber was ∼5 × 10−10 mbar. The emitted photoelectrons were detected by a multichannel detector. Survey scans (pass energy = 100 eV) and detailed spectra (pass energy = 20 eV) were recorded for each sample. A photoelectron take-off angle of 90° relative to the surface was used, which corresponds to a sampling depth of approximately 5–8 nm for the C1s peak. The values of binding energies (BE, eV) were taken relative to the binding energy (285.0 eV) of the C1s electrons of hydrocarbon present on the sample surface. XPS peaks were fitted using the Avantage software package, which consists of a non-linear least-square fitting program. The surface composition was determined using the instrument manufacturer's sensitivity factors. The curve fitting process was done by imposing the peak full width at half maximum (FWHM) of the most resolved peak (C1s at 285.0 eV) to the other peaks.
3.6 Ellipsometry
Coating thickness measurements were obtained with a UVISEL spectroscopic ellipsometer (Horiba Jobin Vyon) at a fixed angle of incidence of 69.8°. Spectroscopic data was acquired over the range 300–800 nm with a 1 nm increment. Three measurements were carried out on each coating and resulting values were averaged. A three-layer model (Si + SiO2 + polymer) was used to simulate experimental data.
3.7 Surface wettability measurements
Sessile drop contact angle values were measured using a video capture apparatus (Digidrop GBX-3S system, France). For each measurement, 6 μL of DI water droplets were dispensed onto the coating surface and images of water droplets were captured continuously to measure the time resolved water contact angle values. Time varying water contact angle measurement was performed at room temperature. Four measurements were carried out on each coating and resulting values were averaged.
3.8 Proton nuclear magnetic resonance (1H NMR)
Proton nuclear magnetic resonance (1H NMR) spectra of the plasma copolymerized coatings were recorded in CDCl3 by using a Bruker Avance 400 MHz spectrometer. Chemical shifts (δ) were given in ppm using tetramethylsilane (TMS) as internal standard.
3.9 Matrix-assisted laser desorption/ionization time of flight (MALDI ToF)
Mass spectrometry measurements were performed using the MALDI-ToF method working in linear mode (Applied Biosystems Voyager System 4130). The MALDI, ionization was triggered by a beam of an N2 laser at λ = 337 nm. The plasma copolymerized coatings were dissolved in α-cyano-4-hydroxycinnamic acid (alpha-cyano or alpha-matrix) and/or 2,5-dihydroxybenzoic acid (DHB) for the laser ionization. All the samples were prepared in acetonitrile (ACN). The calibration was made with polyethylene oxide standards.
3.10 Cell adhesion and proliferation
Cellular adhesion tests have been performed using human bone marrow endothelial cells (HBMEC), human ovarian carcinoma (NIH:OVCAR-3) and embryologic fibroblast which were cultured in Dulbecco's Modified Eagle's Medium (DMEM). The culture medium was supplemented with 1% (v/v) antibiotics (10000 U/ml penicillin-G sodium, 10 mg/ml streptomycin), 2 mM L-glutamine (Fisher scientific, France) and 10% fetal bovine serum (FBS, Sigma Aldrich, France). Cells were expanded by routine cell culture techniques in 25 cm2 cell culture flasks containing 5 ml of 10% serum supplemented medium and were incubated in a humidified atmosphere of 95% air–5% CO2 (Napco, Model 5410, France) at 37 °C for 24 h.
3.11 Cytoskeleton observation on plasma deposited coatings
After 24, 48 and 72 h of HBMEC culture, the glass surfaces were washed in 1X PBS for three times, and fixed with 2.5% glutaraldehyde (Sigma, France) at room temperature for 15 min. The samples were treated with 5% Triton PBS solution for 10 min and washed in 1× PBS three times. All the plasma deposited glass surfaces were stained with phalloidin for 30 min and given a final wash. Then they were mounted on glass slides by glycerol PBS solution. All the samples were examined by a fluorescence microscope (Nikon, Japan) and imaging was done by CCD camera.
3.12 Image processing for adherent cell counting
The adherent cells on the plasma-polymerized coatings were determined by using an inverted microscope (Nikon Instruments, Europe) and images of 900 μm × 600 μm were captured on glass cover slips by using the CCD camera to determine cell adhesion. The adherent cells were counted by using a custom script developed using the Image Processing Toolbox in the MATLAB software package. The Image J image analysis software was used for further processing. The microscopic images were converted into gray scale ones and then histogram equalization operation was performed. It was observed that cell nucleus showed high saturation and was above 0.5, i.e. threshold value. Grayscale images were converted into binary images by applying the above threshold value. For the image processing, 10–20 microscopic images were carried out on each coating and resulting values of adherent cells were averaged. Polystyrene cell culture plate and bare glass substrate were considered as positive control and plasma-polymerized PEG (pDEGME) coatings were taken as negative control.
The morphologies of the embryologic fibroblast cells on the different plasma deposited glass substrate after seeding for 24 h was observed by SEM. Plasma-polymerized coatings were washed with 0.1 M PBS (pH 7.4) to remove the unattached cells. The cells were fixed with 2.5% glutaraldehyde solution for 1 h at 4 °C. Then the samples were sequentially dehydrated in 75 and 95% aqueous ethanol solution, each for 15 min. The samples were put in a pressure chamber with liquid CO2, and the CO2 was vaporized at a temperature above its critical point. The dried samples were coated with gold (Au) by a sputter coater and examined by scanning electron microscopy (SEM).
4. Conclusions
In summary, a catalyst-free strategy to obtain nanosized biocompatible PCL-PEG copolymer coatings was successfully developed by using a low-pressure soft pulsed plasma copolymerization. The latter being a dry process, as compared to the conventional multistep wet chemical techniques, therefore overcome the disadvantage of incomplete removal of catalysts used in the wet processes. Polymerization mechanisms were investigated and could be monitored by following the chemical functionality on the plasma deposited surfaces, the chemical structure analysis and the distribution of the molar masses of the polymer fragments of plasma-polymerized PCL-co-PEG coatings. The elemental analysis of PCL-co-PEG coatings confirmed the incorporation of hydrophobic and hydrophilic groups in the copolymers. Coating stability test showed that the plasma-polymerized coatings, deposited under 1 W plasma power conditions, were stable after soaking in water. The chemical linkage of PCL and PEG segments and molecular weight distribution of the fragments of plasma copolymerized coatings were successfully performed by using FTIR-ATR, 1H NMR and MALDI ToF analyses. The present studies reveal that MALDI can allow one to understand the mechanism and distributions of polymer fragments of plasma deposited coatings. Based on MALDI-ToF analysis, one can conclude that the average copolymer formula is approximately (PCL)4(PEG)6. This observation is what can be expected from a low-pressure plasma process. Indeed, in the gas phase, the mixing of the two monomers is quite homogenous at the molecular level and thus the number of repeating unit should be very low. Therefore, we can conclude that we have obtained very homogenous copolymers at the molecular level by this soft plasma-polymerization process, and as a consequence, that it is not possible to obtain high molecular weight copolymers or block copolymers. Furthermore our copolymer coatings were characterized by a good retention of monomer functionalities and PCL/PEG mixing that is close to molecular. Finally, cell adherent or repellent PCL-PEG copolymer coatings can be obtained by varying the monomer ratio in the plasma process. Our results have demonstrated that plasma-copolymerized PCL-PEG coatings can be tailored in such a way to be cell adherent, convenient for biomedical implants such as artificial skin substrates, or cell repellent, which can be used as antibiofouling surfaces for urethral catheters, cardiac stents etc. In the present study, we have also shown that the plasma processes are promising tools to develop nanothick polymer coatings for tailorable 2D cultures which can therefore; in the near future be used to modify 3D biocompatible (nano) scaffolds for cell culture and further clinical studies.
Acknowledgements
The authors gratefully acknowledge University Pierre & Marie Curie (UPMC), France for offering the PhD financial support and the Institut de Biologie Intégrative (IFR 83, UPMC/INSERM) for SEM analysis.
References
- M. A. Woodruff and D. W. Hutmacher, Prog. Polym. Sci., 2010, 35, 1217–1256 CrossRef CAS.
- X. Wei, C. Gong, M. Gou, S. Fu, Q. Guo, S. Shi, F. Luo, G. Guo, L. Qiu and Z. Qian, Int. J. Pharm., 2009, 381, 1–18 CrossRef CAS.
-
J. M. Harris and S. Zaplipsky, Poly(ethylene glycol): Chemistry and Biological Applications, ACS Symposium Series, Washington, DC, 1997 Search PubMed.
- M. J. Mondrinos, R. Dembzynski, L. Lu, V. K. Byrapogu, D. M. Wootton, P. I. Lelkes and J. Zhou, Biomaterials, 2006, 27, 4399–4408 CrossRef CAS.
- V. V. Seregin and J. L. Coffer, Biomaterials, 2006, 27, 4745–4754 CrossRef CAS.
- M. F. Koenig and S. J. Huang, Polymer, 1995, 36, 1877–1882 CrossRef CAS.
- D. R. Chen, J. Z. Bei and S. G. Wang, Polym. Degrad. Stab., 2000, 67, 455–459 CrossRef CAS.
- H. T. Moon, Y. K. Lee, J. K. Han and Y. J. Byun, J. Biomater. Sci., Polym. Ed., 2002, 13, 817–828 CrossRef CAS.
- S. B. Zhou, X. M. Deng and H. Yang, Biomaterials, 2003, 24, 3563–3570 CrossRef CAS.
- B. Bogdanov, A. Vidts, V. D. Bulcke, R. Verbeeck and E. Schacht, Polymer, 1998, 39, 1631–1636 CrossRef CAS.
- S. Cometa, F. Chiellini, I. Bartolozzi, E. Chiellini, E. DeGiglio and L. Sabbatini, Macromol. Biosci., 2010, 10, 317–327 CrossRef CAS.
- K. C. Remant Bahadur, B. Shanta Raj, A. Santosh, B. Narayan, M. L. Byoung and Y. K. Hak, Polym. Int., 2007, 56, 518–524 CrossRef.
- H. L. Guan, Z. G. Xie, P. B. Zhang, X. Wang, X. S. Chen and X. H. Wang, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4771–4780 CrossRef CAS.
- L. H. Piao, Z. L. Dai, X. M. Deng, X. S. Chen and X. B. Jing, Polymer, 2003, 44, 2025–2031 CrossRef CAS.
- J. Yang, L. Jia, L. Yin, J. Yu, Z. Shi, Q. Fang and A. Cao, Macromol. Biosci., 2004, 4, 1092–1104 CrossRef CAS.
- Z. Weipu, X. Weihui, T. Xiaowei and S. Zhiquan, Eur. Polym. J., 2007, 43, 3522–3530 CrossRef.
- X. M. Deng, C. D. Xiong, L. M. Cheng and R. P. Xu, J. Polym. Sci. Polym. Lett., 1990, 28, 411–416 CAS.
-
C. G. Pitt and A. Schindler, in Long-Acting Contraceptive Delivery Systems, ed. G. L. Zatuchni, A. Goldsmith, J. D. Shelton and J. J. Sciarra, Harper and Row Publishers, Philadelphia, PA, 1984, pp. 48–63 Search PubMed.
- H. Feng and C. M. Dong, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5353–5361 CrossRef CAS.
- M. Myers, E. F. Connor, T. Glauser, A. Mock, G. Nyce and J. L. Hedrick, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 844 CrossRef CAS.
- J. Casas, P. V. Persson, T. Iversen and A. Cordova, Adv. Synth. Catal., 2004, 346, 1087 CrossRef CAS.
- S. Y. Kim, Y. M. Lee, H. J. Shin and J. S. Kang, Biomaterials, 2001, 22, 2049 CrossRef CAS.
- S. Y. Kim and Y. M. Lee, Biomaterials, 2001, 22, 1697 CrossRef CAS.
- Y. Shibasaki, F. Sanda and T. Endo, Macromol. Rapid Commun., 1999, 20, 532–535 CrossRef CAS.
- Y. Shibasaki, H. Sanada, M. Yokoi, F. Sanda and T. Endo, Macromolecules, 2000, 33, 4316–4320 CrossRef CAS.
- M. S. Kim, K. S. Seo, G. Khang and H. B. Lee, Macromol. Rapid Commun., 2005, 26, 643–648 CrossRef CAS.
-
H. Yasuda, Plasma Polymerization, Academic Press, Inc, Orlando, FL, 1985, 112 Search PubMed.
- P. A. Tamirisa, K. C. Liddell, P. D. Pedrow and M. A. Osman, J. Appl. Polym. Sci., 2004, 93, 1317–1325 CrossRef CAS.
- J. Zhang, W. V. Ooij, P. France, S. Datta, A. Radomyselskiy and H. Xie, Thin Solid Films, 2001, 390, 123–129 CrossRef CAS.
- J. Friedrich, Plasma Processes Polym., 2011, 8, 783–802 CrossRef CAS.
- Y. J. Wu, R. B. Timmons, J. S. Jen and F. E. Molock, Colloids Surf., B, 2000, 18, 235–248 CrossRef CAS.
- F. Brétagnol, M. Lejeune, A. P. Bouraoui, M. Hasiwa, H. Rauscher, G. Ceccone, P. Colpo and F. Rossi, Acta Biomater., 2006, 2, 165–172 CrossRef.
- D. Briggs and G. Beamson, Anal. Chem., 1992, 64, 1729–1736 CrossRef CAS.
- P. Viville, R. Lazzaroni, E. Pollet, M. Alexandre, P. Dubois, G. Borcia and J. J. Pireaux, Langmuir, 2003, 19, 9425–9433 CrossRef CAS.
- V. Kumar, J. Pulpytel, G. Giudetti, H. Rauscher, F. Rossi and F. Arefi-Khonsari, Plasma Processes Polym., 2011, 8, 373–385 CrossRef CAS.
- M. Chausson, A. S. Fluchere, E. Landreau, Y. Aguni, Y. Chevalier, T. Hamaide, N. A. Malak and I. Bonnet, Int. J. Pharm., 2008, 362, 153–162 CrossRef CAS.
- L. Peng and G. R. Kinsel, Langmuir, 2010, 26, 17477–17481 CrossRef CAS.
- B. Dalton, C. McFarland, T. Gengenbach, H. Griesser and J. Steele, J. Biomater. Sci., Polym. Ed., 1998, 9, 781 CrossRef CAS.
- J. Steele, G. Johnson, K. McLean, G. Beumer and H. Griesser, J. Biomed. Mater. Res., 2000, 50, 475 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S10. See DOI: 10.1039/c2ra21211k/ |
|
| This journal is © The Royal Society of Chemistry 2012 |