
A Z-scheme NiCo2O4/S codoped 1D g-C3N4 heterojunction for solar-light-sensitive photocatalytic degradation of antibiotics in aqueous solutions exemplified by tetracycline†
Thanh-Binh
Nguyen
a,
Phung-Ngoc-Thao
Ho
a,
Chiu-Wen
Chen
b,
C. P.
Huang
c,
Ruey-an
Doong
 d and
Cheng-Di
Dong
*b
d and
Cheng-Di
Dong
*b
aInstitute of Aquatic Science and Technology, National Kaohsiung University of Science and Technology, Kaohsiung, 81157, Taiwan
bDepartment of Marine Environmental Engineering, National Kaohsiung University of Science and Technology, Kaohsiung, 81157, Taiwan. E-mail: cddong@nkust.edu.tw
cDepartment of Civil and Environmental Engineering, University of Delaware, Newark, 19716, DE, USA
dInstitute of Analytical and Environmental Sciences, National Tsing Hua University, Hsinchu, 30013, Taiwan
First published on 18th November 2021
Abstract
A NiCo2O4 nanoparticle (NP) and sulfur codoped hollow tubular g-C3N4 (NiCo–S@CN) catalyst was synthesized by a hydrothermal pyrolysis method and used to remove pharmaceuticals, exemplified by the antibiotic tetracycline (TC), from water under solar light irradiation. The synergistic effect of NiCo2O4 NPs and S@CN nanotubes in the Z-scheme heterojunction significantly enhanced the photocatalytic reactivity of the NiCo–S@CN catalyst, under solar light, toward TC degradation. Factors such as catalyst dosage, pH, TC concentration, and temperature that controlled TC degradation in synthetic and natural water media were studied. Langmuir–Hinshelwood kinetics were observed in the photocatalytic degradation of TC over NiCo–S@CN. The photocatalytic performance was the best at the catalyst dosage of 200 mg L−1. The initial effect of pH on TC photodegradation over the NiCo–S@CN/solar light system was as follows: pH 7.0 > pH 11.0 > pH 9.0 > pH 5.0 > pH 3.0. Moreover, NiCo–S@CN exhibited high TC degradation efficiency in multiple cycles while it remained chemically stable with undetectable soluble metals in the treated water. Results of electron spin resonance (EPR) and radical trapping studies indicated that holes (h+) and superoxide radicals (O2˙−) were the main reactive species for efficient TC removal in the Z-scheme/solar light system. Overall, results demonstrated that NiCo–S@CN was a novel Z-scheme photocatalyst for the remediation of pharmaceutical-contaminated water.
Environmental significanceThe presence of antibiotics in the environment has attracted increasing public attention because of potential adverse ecological and human impacts. Sunlight-active photocatalysts with high efficiency and recyclability provide friendly and sustainable solutions to the control of hazardous chemicals such as antibiotics. Herein, a NiCo2O4 nanoparticle (NP) and sulfur codoped hollow tubular g-C3N4 (NiCo–S@CN) photocatalyst was synthesized by a hydrothermal pyrolysis method and used to remove pharmaceuticals, exemplified by the antibiotic tetracycline (TC), from water under solar light irradiation. The NiCo–S@CN catalyst exhibited excellent solar-light-responsive photocatalytic activity toward TC degradation because of the synergistic effect of NiCo2O4 NPs and S@CN nanotubes in the Z-scheme heterojunction. The finding is expected to open a new avenue for fabrication of Z-scheme nanocomposites for water treatment in particular and environmental remediation in general. |
1. Introduction
Antibiotics have been widely used in the past decades to control human and animal diseases. Tetracycline (C22H24N2O8, TC) ranks second in the global production and consumption of antibiotics due to its efficacy against a variety of pathogenic bacteria.1 There has been increasing TC use recently, which leads to its increasing distribution in the aquatic environment. Furthermore, humans and animals can metabolize only a small portion of TC so that almost all TC intake is released to the environment as the parent compound or metabolites through excretions.2 The reported TC concentration in surface water, groundwater, and wastewater was in the ppt to ppb range.3 However, TC can persist in the environment for a long period of time, which contributes to the wide spread of antibiotic resistance, disruption of ecological systems, and threats to human health.4 As a result, it is critical to develop technically feasible and cost-effective methods for the elimination of TC in water.Recent years have seen a surge in interest in the creation of solar-light-active photocatalysts owing to the potential applications of such materials. Photocatalysts activated by solar light can be utilized to degrade hazardous organic pollutants in wastewater in an environmentally friendly approach under ambient settings.5 Graphitic carbon nitride (g-C3N4), a metal-free visible-light-responsive photocatalyst, has lately raised considerable interest in water splitting and environmental remediation applications because of its low cost, strong photostability, easy bulk production, and attractive optical characteristics.5 While the g-C3N4 photocatalyst has been extensively explored in recent years, it still has not been widely applied in environmental remediation because of low visible-light utilization efficiency, rapid charge carrier recombination, and small specific surface area.6 Fortuitously, after years of research efforts, there have been significant breakthroughs in enhancing the photocatalytic activity of g-C3N4, in particular by g-C3N4 heterojunctions,7 heteroatom doping,8 and dye sensitization.9 S doping has recently gained widespread interest because of the ease of synthesis, low environmental impact, and high photocatalytic activity of products. In particular, S doping effectively modifies the band structure, improves the charge carrier separation, decreases the bandgap energy, and increases the light absorption range of g-C3N4.10 Nonetheless, unfortunately, S-doped g-C3N4 (S@CN) still has limited photocatalysis reactivity without further surface modification. Therefore, there were attempts to further enhance the photocatalytic reactivity of S@CN through the synthesis of Z-scheme heterostructures to efficiently separate hole–electron pairs.11–13 The Z-scheme has gained considerable interest because of its enhanced redox capability and increased photocarrier lifetime among classic type II systems.14 Recently, Umeshbabu et al. have reported that NiCo2O4 nanoparticles (NPs) have a narrow bandgap, high electronic conductivity, tuneable valence, and low cost.15 However, NiCo2O4 NPs still suffer from limited activation efficiency because of limited dispersivity and rapid charge recombination.16,17 Hence, it has prompted us to couple NiCo2O4 NPs with g-C3N4 hollow tubes to produce efficient Z-scheme photocatalysts to enhance the photocatalytic degradation of recalcitrant chemicals, specifically antibiotics, under solar light irradiation.
The present work aimed to design and synthesize Z-scheme NiCo2O4/S-doped g-C3N4 (NiCo–S@CN) for the heterogeneous photocatalytic degradation of persistent antibiotics exemplified by TC. Scheme 1 illustrates the design and synthesis of Z-scheme NiCo–S@CN by a hydrothermal method followed by pyrolysis incorporation of NiCo2O4 NPs and doped S atoms, using L-cysteine precursor, on g-C3N4 nanotubes. Various surface analysis techniques were conducted to characterize the physicochemical and light-responsive properties of the NiCo–S@CN catalyst. A series of photocatalytic experiments were performed to investigate the feasibility of the NiCo–S@CN/solar light system for TC degradation as affected by variables such as pH, catalyst dosage, initial TC concentration, temperature, and water matrixes. Additionally, the kinetics of TC degradation over NiCo–S@CN under solar irradiation was studied. Radical scavenging experiments, EPR, and Mott–Schottky test aided in establishing the degradation mechanism. The catalytic stability of NiCo–S@CN was evaluated in six successive photocatalytic TC degradation cycles. In addition, the change in COD and TOC during the course of TC degradation was monitored to estimate the degree of mineralization in terms of the change of average carbon oxidation state (COS). To the best of our knowledge, this is the first attempt to study the photocatalytic degradation of antibiotics in aqueous solutions using the Z-scheme NiCo–S@CN heterojunction under solar-light irradiation.
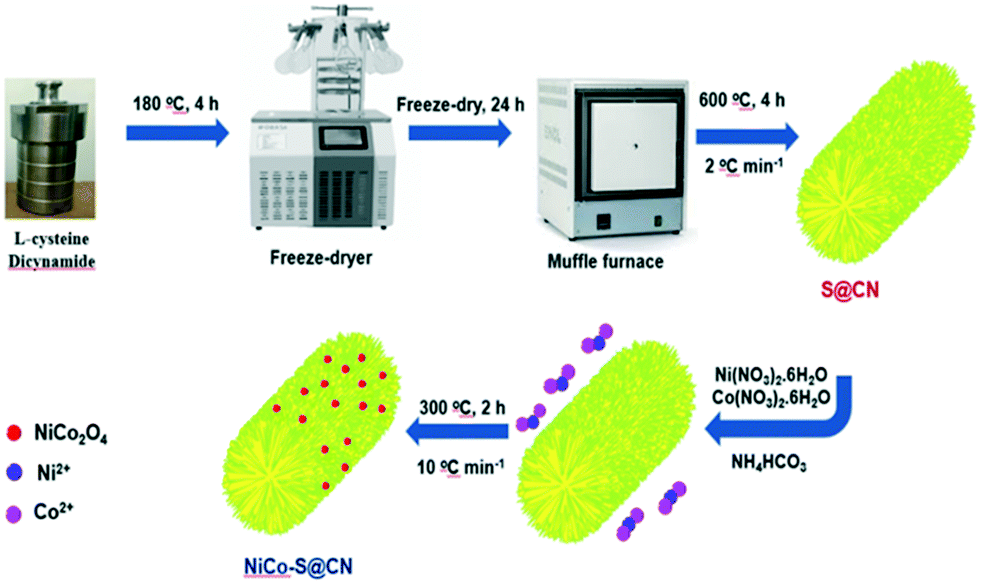 | ||
| Scheme 1 Schematic illustration of the synthesis of the NiCo–S@CN phocatalyst. | ||
2. Experimental
2.1 Chemicals
Dicyandiamide (NH2C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH)NHCN, ≥99%), L-cysteine (C3H7NO2S, 97%) and 5,5-dimethyl-1-pyrroline N-oxide (DMPO, C6H11NO, ≥97%) were supplied by Sigma-Aldrich Chemical Company (St. Louis, Missouri, USA). Tetracycline hydrochloride (≥95%) was obtained from Merck & Co., Inc. (Kenilworth, New Jersey, USA). Nickel(II) nitrate hexahydrate (Ni(NO3)2·6H2O, ≥99%), cobalt nitrate hexahydrate (Co(NO3)2·6H2O, ≥99%) and ammonium bicarbonate (NH4HCO3, 99.9%) were supplied by J.T.Baker™ (Phillipsburg, New Jersey, USA). All the chemicals were used as received without further purification.
NH)NHCN, ≥99%), L-cysteine (C3H7NO2S, 97%) and 5,5-dimethyl-1-pyrroline N-oxide (DMPO, C6H11NO, ≥97%) were supplied by Sigma-Aldrich Chemical Company (St. Louis, Missouri, USA). Tetracycline hydrochloride (≥95%) was obtained from Merck & Co., Inc. (Kenilworth, New Jersey, USA). Nickel(II) nitrate hexahydrate (Ni(NO3)2·6H2O, ≥99%), cobalt nitrate hexahydrate (Co(NO3)2·6H2O, ≥99%) and ammonium bicarbonate (NH4HCO3, 99.9%) were supplied by J.T.Baker™ (Phillipsburg, New Jersey, USA). All the chemicals were used as received without further purification.
2.2 Synthesis of S@g-C3N4 tubes
A mixture of 2 g of dicyandiamide and 121 mg of L-cysteine (∼1 mmol S) was dissolved in 50 mL of deionized (DI) water by ultrasonication (40 KHz model K410HTDP, RoHS, China) for 30 min in a water bath at 30 °C. Then the transparent solution was transferred to a 100 mL Teflon-lined autoclave for hydrothermal synthesis at 180 °C for 4 h in a circulating hot air oven (DK-600DT, Yihder Co. Ltd, Taiwan). After cooling to room temperature, the hydrothermally processed solution was quickly frozen to −80 °C for 2 h in a refrigerator (Glacier NU-9688, NuAire Inc., USA). The obtained icy material was lyophilized in a vacuum freeze dryer for 24 h. Finally, the slightly yellowish powder was placed in a crucible, sealed with Al foil, and further calcined in a muffle furnace (SNOL 6.7/1300 LSM01, Lithuania) at 600 °C for 4 h at a ramp rate of 2 °C min−1 in the limited presence of oxygen, followed by cooling to room temperature; the product was designated as S@CN. Pure g-C3N4 nanotubes denoted as CN were prepared following the same procedure as above except without L-cysteine, a S precursor.2.3 Synthesis of NiCo2O4–S@g-C3N4 catalysts
The S@CN tubes (240 mg) prepared above were well dispersed in 50 mL of anhydrous ethanol by ultrasonication. Then, 14.6 mg of Ni(NO3)2·6H2O and 29.7 mg of Co(NO3)2·6H2O (∼5 wt% NiCo2O4) were simultaneously dissolved in the above suspension followed by the addition of 5 mmol of NH4HCO3. After vigorous stirring for 6 h under ambient conditions, the solid product was separated by centrifugation at 4500 rpm or ∼2038g (Himac CT6E, Hitachi, Japan), then washed with DI water and absolute ethanol followed by drying in a vacuum oven at 60 °C for 6 h. Afterward, the sample was heated to 300 °C in an air atmosphere at a heating rate of 10 °C min−1 for 2 h to obtain the final product, which was denoted as NiCo–S@CN.2.4 Photocatalytic experiments
Experiments for the photocatalytic degradation of TC were carried out in a solar simulation reactor (Seton Finite Corp., Taiwan) equipped with a 300 W Xe arc lamp and a dynamic temperature control system (KISS K6, Huber USA Inc.). The initial pH of the TC stock solution was adjusted to specific values using NaOH (0.1 M) and HNO3 (0.1 M). A pH meter (model Sartorius PB-21, Germany) was used to monitor the solution pH. Prior to conducting photocatalytic experiments, a certain quantity of photocatalyst was first introduced to 50 mL of the TC aqueous solution (10 mg L−1) in a 100 mL double-walled glass beaker at 25 °C under mechanical stirring for 30 min in the dark to achieve adsorption/desorption equilibrium. Subsequently, the light was turned on to start the photocatalytic TC degradation experiments. At certain time intervals, a 2 mL aliquot of sample was withdrawn from the reactor and filtered through a 0.22 μm PVDF syringe filter (Millex-GV) to collect the filtrate for the analysis of residual TC using HPLC (Hitachi Chromaster 5110, Tokyo, Japan).3. Results and discussion
3.1 Properties of photocatalysts
Fig. 1 shows the SEM and TEM images of both pure CN and S@CN. Results demonstrated that the catalysts exhibited a well-developed tubular structure with a diameter of 1–2 μm and a longitudinal length of 3–4 μm. During hydrothermal processing, the supramolecular CN precursor was obtained via two steps: (1) hydrolysis of dicyandiamide, forming melamine, and (2) in situ formation of cyanuric acid through self-assembly, creating the supramolecular CN precursor. Thermal polymerization under air atmosphere formed the distinctive tubular structure of CN. Pure CN showed a chunky morphology assembled by stacking nanosheets of finite pores on its surface (Fig. S1a–c†). S atoms were attached onto the supramolecular structure of the CN framework during hydrothermal treatment in the presence of L-cysteine, a S precursor. Subsequent thermal polymerization of S significantly loosened the CN framework, increasing the space between nanosheets and creating a loosely hollow tubular structure with an abundance of pores (Fig. S1d–f†).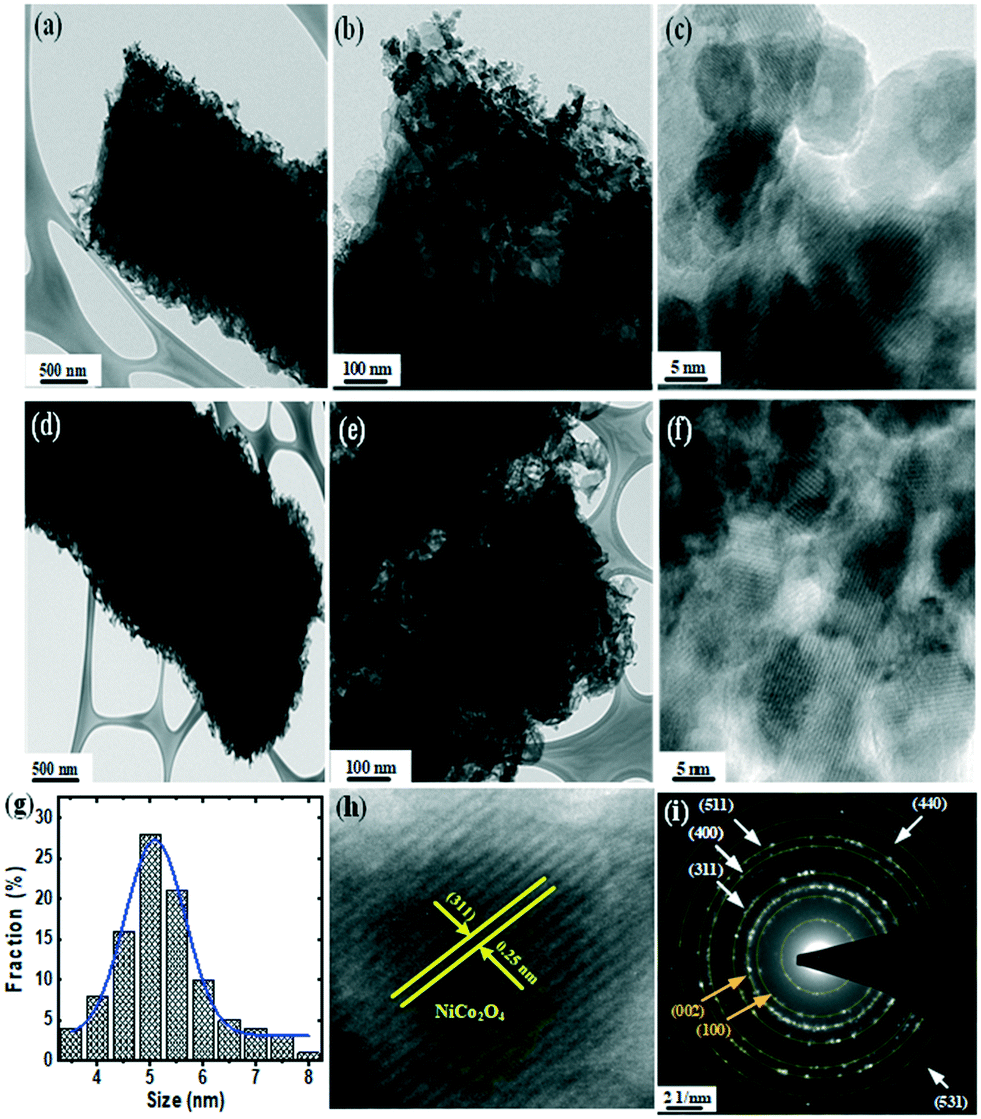 | ||
| Fig. 1 (a–f) HRTEM images and (i) SAED pattern of NiCo–S@CN. (g) Size distribution and (h) lattice spacing of NiCo2O4 NPs. | ||
EDX elemental mapping showed the presence of C, N, O, and S elements on the S@CN. All the elements were uniformly distributed on the surface of the hollow tubes (Fig. S2†). Such an intriguing structure is expected to provide sufficient surface area and reactive sites for facilitating pollutant transport and increasing light absorption.
High-resolution TEM (HRTEM) further revealed the surface structure of S@CN-doped NiCo2O4 nanoparticles (NiCo–S@CN). Incorporation of NiCo2O4 into S@CN retained its 1D tubular structure during the second synthesis stage and stabilized the NiCo2O4 nanoparticles by forming well-defined 0D/1D heterostructured NiCo–S@CN with intimate interfaces (Fig. 1a and d). Fig. 1b and e show that the electron-dense areas appear dark because of the high atomic mass and electron density of metals in contrast to non-metals, which indicated the presence of NiCo2O4 nanoparticles on the surface of 1D tubular S@CN. Obviously, NiCo2O4 nanoparticles appeared on the entire surface of the NiCo–S@CN composite (Fig. 1c and f) with a homogeneous diameter of approximately 5 nm (Fig. 2g), rendering the original surface of S@CN rougher. The result showed that S@CN contained a variety of functional groups capable of chelating metals, i.e., forming chelates and stabilizing metal-based nanoparticles on the surface.18 The corresponding high magnification of the HRTEM image obtained at the edge of hollow tubes clearly exhibited distinct lattice fringes of metal oxide particles with interplanar spacing of 0.25 nm, which was in accordance with the (311) lattice plane of NiCo2O4 (Fig. 1h). The crystallographic information based on a spotty ring pattern obtained by selected-area electron diffraction (SAED) showed that NiCo–S@CN clearly exhibits a polycrystalline structure, in which the diffraction rings of CN ({002} and {100}) and NiCo2O4 ({311}, {400}, {440}, {511} and {531}) appear, according to JCPDS#87-1526 and JCPDS#00-020-781, respectively (Fig. 1i).
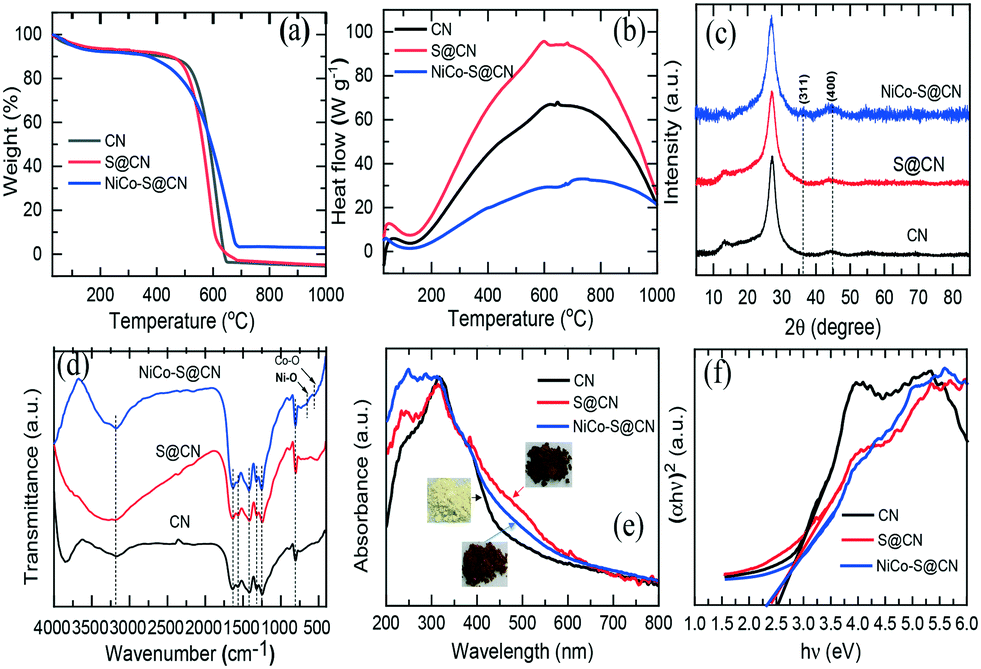 | ||
| Fig. 2 (a) TGA, (b) DSC profiles, (c) XRD patterns, (d) FTIR spectra, (e) UV-vis spectra, and (f) corresponding bandgap of CN, S@CN, and NiCo–S@CN composites. | ||
Energy-dispersive (EDX) X-ray analysis in conjunction with HRSEM revealed the elemental constituents of the NiCo–S@CN composite. The EDX spectrum showed the presence of major elements, i.e., C and N, together with minor elements, i.e., Ni, Co, and O (∼5 wt% NiCo2O4 constituent of the composite), due to the low doping level of NiCo2O4 (Fig. S3a and b†). Noticeably, the elemental composition derived from the EDX spectrum revealed the Co![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ni atomic ratio of 2:1, which agreed well with the chemical formula of NiCo2O4 and confirmed the presence of doped metal oxide nanocrystals. Note that the trace Cu was from the Cu grid. As can be seen in Fig. S3c,† the elemental mapping of the 1D tube verified the uniform dispersion of all relevant elements across the tube without apparent separation. Thus, based on the structural morphologies described above, it is clear that NiCo2O4 nanoparticles were successfully loaded on the S@CN hollow tube and displayed an intimate heterojunction between S@CN and NiCo2O4.
:Ni atomic ratio of 2:1, which agreed well with the chemical formula of NiCo2O4 and confirmed the presence of doped metal oxide nanocrystals. Note that the trace Cu was from the Cu grid. As can be seen in Fig. S3c,† the elemental mapping of the 1D tube verified the uniform dispersion of all relevant elements across the tube without apparent separation. Thus, based on the structural morphologies described above, it is clear that NiCo2O4 nanoparticles were successfully loaded on the S@CN hollow tube and displayed an intimate heterojunction between S@CN and NiCo2O4.
TGA/DSC technique was used to examine the thermal behaviour of catalysts prepared at temperatures in the range of 30 to 1000 °C under a static air atmosphere. Fig. 2a shows that CN was stable in air, weight loss began at approximately 480 °C, and CN completely decomposed into CO2 and NO2 at 650 °C. Interestingly, S@CN had a lower decomposition temperature (approximately 450 °C) than that of pristine CN. S-doping of the CN graphite-like structure through thermo-condensation of L-cysteine and dicyanamide in the solid phase altered the local packing motifs in the tri-s-triazine ring and weakened the van der Waals forces of the stacked layer structure, thus strengthening the thermal stability of the composite.19 In the case of NiCo–S@CN, the decomposition temperature began at 350 °C. The weight loss remained unchanged in the temperature range between 685 and 1000 °C. The results indicated the presence of metal oxides in the S@CN framework and became catalysts that facilitated the thermal decomposition of the composite and weakened the cross-linked rings of CN. Based on the high thermal stability of NiCo2O4, the estimated oxide content in the NiCo–S@CN composite was 4.8 wt%, which was close to the theoretical doping level.
The DSC thermogram clearly shows that all three materials, i.e., CN, S@CN, and NiCo–S@CN, exhibit the same decomposition reaction feature, as demonstrated by the appearance of wide exothermic peaks in the temperature range of 130 to 1000 °C (Fig. 2b). The exothermic decomposition heat increased from 67 to 94 W g−1 when CN was functionalized with sulfur owing to the enhanced decomposition of S@CN accompanied by the ready release of carbon-, nitrogen-, and sulfur-containing gases with high heating value. The exothermic peak of the NiCo–S@CN composite dropped to 33 W g−1, possibly because of NiCo2O4 doping that enhanced the crystallinity of the composite and restricted the decomposition of S@CN into volatile molecules.20
The crystallographic structure of the catalysts was characterized by XRD measurements (Fig. 2c). The XRD pattern of the CN catalyst exhibited two distinct diffraction peaks at 2θ 13.1° and 27.3°, which were well indexed as the in-plane structural packing motif of tri-s-triazine {100} and interlayer periodic stacking of the graphitic structure {002}, respectively (JCPDS#87-1526). The signal intensity of the two characteristic peaks of the S@CN catalyst were lower than that of the pristine CN due to lattice distortion by the loading of relatively large S atoms in the graphitic structure of CN and subsequent increase in the spacing between graphitic layers and decrease in layer thickness.21 Meanwhile, two additional peaks at 2θ 36° and 44.6° of NiCo–S@CN were assigned to the {311} and {400} planes of NiCo2O4 (JCPDS#00-020-781), which further demonstrated the successful loading of NiCo2O4 on S@CN. However, the peak intensity corresponding to NiCo2O4 was particularly weak owing to the low loading level of Ni and Co precursors with respect to other elements.
FT-IR spectra in the wavenumber range of 400 to 4000 cm−1 revealed the surface chemical composition of the catalysts (Fig. 2d). All three catalysts, i.e., CN, S@CN and Ni–Co–S@CN, exhibited identical IR absorption bands, implying that S and NiCo2O4 doping did not alter the π-conjugation of the CN skeleton. A sharp peak at 806 cm−1 represented the characteristic breathing vibration of tri-s-triazine rings of heterocycle-grafted CN, whilst the typical peaks in the wavenumber range of 1200 to1650 cm−1 were widely known as the stretching vibration of the aromatic C–N heterocycle.22 In addition, the broad peaks from 2900 to 3630 cm−1 were ascribed to the stretching mode of O–H and N–H bonds, possibly associated with the uncondensed amine groups and surface-bound water molecules. Note that S@CN exhibited no discernible variation and peaks of S-related groups because of low S content or overlapping of vibrational spectrum by the C–N bond. There was only a N–H peak shift from 3197 cm−1 to the higher wavenumber of 3286 cm−1, indicating the replacement of N element by S in the CN graphitic structure.21 However, NiCo2O4 loading greatly decreased the peak intensity between 2900 and 3630 cm−1, implying (i) the successful connection of NiCo2O4 NPs to the CN heterocyclic substrate via strong surface complexation with functional groups (–OH and N–H) as binding sites and (ii) homogeneous distribution of tiny 0-D metal oxide, i.e., NiCo2O4, on the surface of S@CN that limited the interaction between IR and multifunctional groups, as demonstrated in HRTEM images. Notably, the stretching vibration bonds of Co–O and Ni–O in the composite occurred at 543 and 643 cm−1, respectively, indicating the presence of metal oxide as previously reported.23 Thus, the FTIR results further confirmed the formation of a heterojunction between NiCo2O4 NPs and S@CN tubes.
The optical properties and electronic bandgap of the catalysts were characterized by UV-vis diffuse reflectance. Fig. 2e shows that the original UCN displays an absorption edge at ca. 488 nm. In contrast to pristine CN, the absorption edge of CN after S doping, i.e., S@CN, exhibited an obvious redshift of the absorption edge to approximately 518 nm, which demonstrated that S doping the CN framework could greatly broaden the spectral response range. Therefore, the colour of CN turned from yellow to light brown after S doping. Chen et al.24 have reported that doping of S on CN significantly enhances the visible light-harvesting capability, photogenerated charge separation, and surface charge transfer efficacy because of multiple reflections within the porous structure and high spin density of the catalyst. Note that NiCo–S@CN also exhibited a systematic redshift to longer visible light of 532 nm because of the broad absorption spectrum of NiCo2O4 in the UV-to-visible light region.17,25 Consequently, modification of NiCo2O4 remarkably enhanced photon absorption in the visible light range and the separation of electron–hole pairs. The macroscopic colour that shifted from light brown to dark brown also demonstrated the beneficial effect of visible light harvesting. Additionally, the bandgap (Eg) of CN, S@CN, and NiCo–S@CN, obtained by the Tauc method, was 2.54, 2.40, and 2.32 eV, respectively (Fig. 2f). Results clearly demonstrated the enhancement of the visible-light-harvesting capability of the NiCo2O4 and S@CN heterojunction.
XPS technique further characterized the chemical state and bonding of the NiCo–S@CN composite. Fig. S4† reveals the presence of C, N, S, Ni, Co and O elements on the composite. The XPS spectra of C 1s exhibited three different peaks at 285, 288.5 and 294.1 eV, corresponding to CC, C–NC and sp2 carbon, respectively (Fig. 3a).26 There were two major peaks in the S 2p orbital (Fig. 3b). The first peak at 169.2 eV was attributed to the C–S–C bond, while the low peak at 170.2 eV was formed by substituting C in the CN graphitic network (S–N bonds).10Fig. 3c shows the three deconvoluted peaks of the N 1s orbital, which were assigned to the sp2 hybridized nitrogen C–NC (399.1 eV) in the triazine ring, tertiary nitrogen in N–C3 groups (400.4 eV) connecting the C6N7 motif, and C–NH (403 eV) as a result of non-condensation of amino groups, respectively.27Fig. 3d shows the O 1s orbital of NiCo–S@CN in which the fitted peaks at 529.4 and 532.7 eV were assigned to the oxygen lattice in the bimetallic oxide (Olatt) and surface-adsorbed oxygen (Oads), respectively. The Olatt species were formed by metal oxygen bonds (M–O–M), whereas the Oads species belonged to the surface-adsorbed hydroxyl-like group (M–OH) or oxygen vacancies.28 The Ni 2p was convoluted to Ni 2p3/2 and Ni 2p3/2 at 856.6 and 875.1 eV, respectively (Fig. 3e). Additionally, two corresponding shake-up satellite peaks of spin–orbit doublets, at approximately 863.7 and 881.4 eV, respectively, appeared. The fitted peaks at 856.4 and 873.2 eV were indexed to Ni3+, whereas other fitted peaks at 857.6and 875 eV were attributed to Ni2+.29 Similarly, as shown in Fig. 3f, two distinct Co species and two shake-up satellites were identified based on the Co 2p spectrum. The fitted peaks at 781.3 and 797 eV were ascribed to Co3+, whilst the other two fitted peaks at 784.3 and 798.1 eV belonged to Co2+.30 Note that the atomic ratio of Ni to Co was 1:2, which agreed well with the stoichiometric ratio of NiCo2O4.The above results confirmed that S atoms and NiCo2O4 nanoparticles were successfully incorporated onto the surface of CN tubes at a high degree of dispersion.
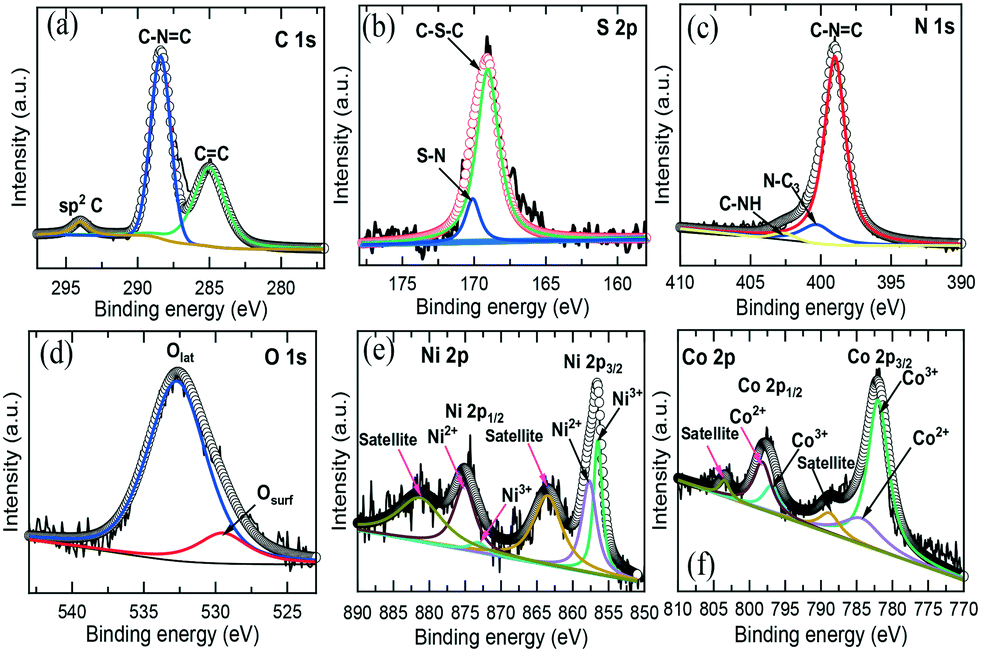 | ||
| Fig. 3 High-resolution XPS spectra of (a) C 1s, (b) N 1s, (c) S 2p, (d) O 1s, (e) Ni 2p and (f) Co 2p of the NiCo–S@CN. | ||
3.2 Photocatalytic degradation of TC
The photocatalytic performance of the catalysts was studied using TC as the model compound in a simulated solar light reactor. Fig. 4a shows that TC was hardly degraded under direct solar light irradiation (∼4% in 60 min), implying the recalcitrant nature of TC and the absence of self-photolysis. The pristine CN exhibited a slight degree of photocatalytic TC degradation (∼47% removal in 60 min). In contrast, S@CN showed significant enhancement in photocatalytic efficiency at 74% TC removal due in part to the high specific surface area and porous structure of the modified CN. It was noted that immobilization of NiCo2O4 NPs on the surface of porous hollow tubes of S@CN significantly improved TC degradation over NiCo–S@CN at 99% removal in 60 min. The above results confirmed the synergistic effect of the heterojunction between S@CN and NiCo2O4 that led to efficient solar photocatalytic degradation of antibiotics.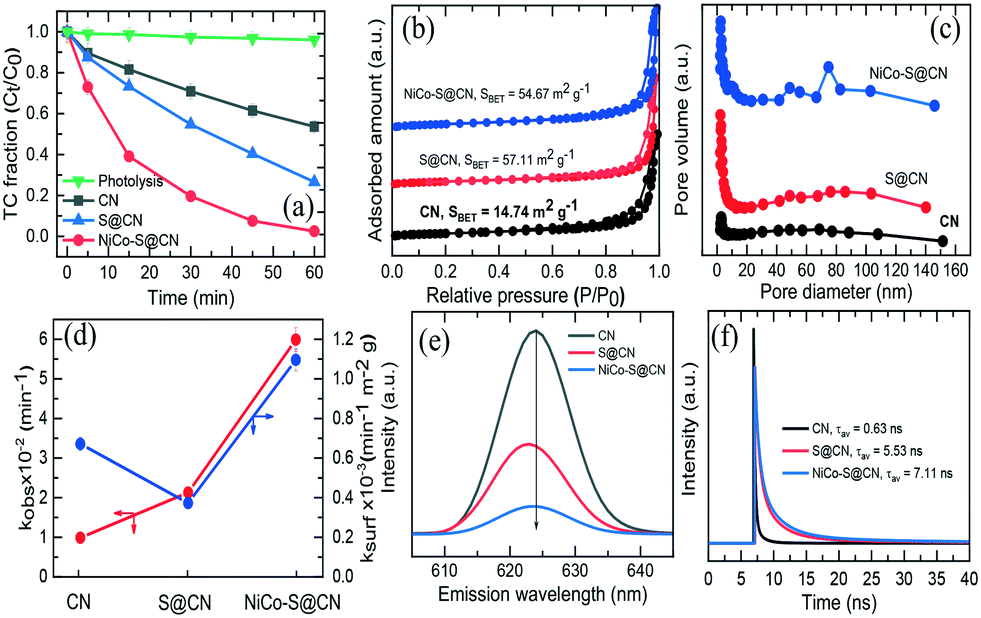 | ||
| Fig. 4 (a) Photodegradation efficiency and (d) corresponding rate constant of TC on as-prepared photocatalysts. (b) BET adsorption–desorption isotherm, (c) BJH pore size distribution, (e) photoluminescence spectra, and (f) time-resolved photoluminescence spectra of as-prepared photocatalysts. | ||
In order to elucidate the effect of specific surface area on the photocatalytic activity of catalytic materials, N2 physical adsorption/desorption was performed to determine the texture parameters, including BET specific surface area (SBET), pore volume, and pore distribution. The N2 adsorption–desorption isotherm of the catalysts (Fig. 4b) showed a type IV isotherm with an H3-type distinct inflection at P/P0 between 0.7 and 1.0, indicating the presence of a well-defined hierarchical porous structure with high abundance of various mesopores in the hollow tube.31 The SBET of S@CN (57.11 m2 g−1) and NiCo–S@CN (54.67 m2 g−1) was 3.7–3.9 times greater than that of the pristine CN (14.74 m2 g−1). Furthermore, Barrett–Joyner–Halenda (BJH) profiles (Fig. 4c) showed the presence of mesopores and macropores in the size range of 2 to 140 nm. It is noteworthy that the higher pore size distribution profiles of S@CN and NiCo–S@CN indicated the presence of a more highly porous nature of these two materials than CN. The pore volume of CN, S@CN, and NiCo–S@CN was 0.10, 0.42, and 0.37 cm3 g−1, respectively. The pore diameter of pristine CN is mainly centered at a narrow size of 2.8 nm initially. S doping widened the pore size distribution in the 2–140 nm region of the mesoscale, due mainly to spontaneous pore formation triggered by gaseous release through thermal polymerization.32 Interestingly, due to the homogeneous distribution of NiCo2O4 NPs on the S@CN surface, the NiCo–S@CN catalyst exhibited bimodal pore structures with peaks at approximately 48.7 and 74.8 nm, individually, which facilitated faster diffusion of pollutants into the catalyst pores and increased the photocatalytic reactivity.30 In conclusion, the larger surface area and pore volume of NiCO–S@CN contributed greater adsorption and external/internal diffusion of TC molecules to the photocatalyst.
The photocatalytic TC degradation over the photocatalysts followed pseudo-first-order kinetics (eqn (1)). The observed rate constant (kobs) was determined from the slope of the linear ln(Ct/C0) vs. time (t) plot, i.e.,
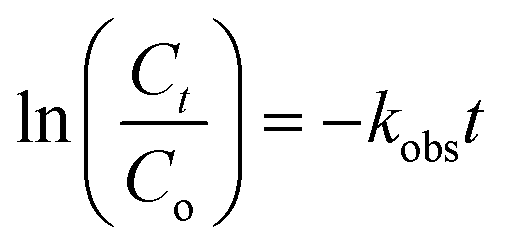 | (1) |
Photoluminescence (PL) technique well characterizes the efficacy of suppressing electron–hole pair recombination and the lifetime of charge carriers in semiconductors.22Fig. 4e shows that all three catalysts display the maximum emission peak at around 624 nm at an excitation wavelength of 310 nm and room temperature. Among all the photocatalysts, pure CN exhibited the highest PL intensity, which suggested the largest recombination rate of photoinduced charge carriers and hence limited photocatalytic activity.33 On the other hand, S@CN showed a dramatic decrease in PL emission intensity upon doping of S heteroatoms, indicating a longer lifetime of photoinduced electrons and effective separation of electron–hole pairs. Indeed, S atoms acted as trapping centres for photogenerated electrons in the mid-bandgap and thus improved the carrier mobility of pure CN.34 Noticeably, NiCo–S@CN exhibited the lowest PL peak intensity with respect to pure CN and S@CN. Coupling NiCo2O4 with S@CN promoted interfacial electron transfer efficiency while retarding the recombination of electron–hole pairs. Therefore, more charge carriers were available for the photocatalytic degradation of TC than that of pristine CN; that is, the synergistic effect of S atoms and NiCo2O4 NPs significantly enhanced the catalytic activity of CN. Furthermore, Stoke shifts on the emission wavelength of the catalyst were likely related to the change in bandgap as determined by UV-vis spectroscopy. Additionally, the lifetime of photogenerated charge carriers on all three catalysts was further investigated by time-resolved PL decay spectra (Fig. 4f). Three processes controlled the lifetime of charge carriers: non-radiative (t1), radiative (t2), and energy transfer (t3).35 The mean lifetime of NiCo–S@CN was 7.11 ns, which was significantly longer than that of S@CN (5.53 ns) and CN (0.63 ns). The PL and time-resolved PL spectra demonstrated a significant increase in lifetime of charge carriers of NiCo–S@CN (positively associated with the high efficiency of electron–hole pair separation), which was one of the major mechanisms that facilitated the photocatalytic reactions because of the unique structure of NiCo–S@CN.
3.3 Effect of catalyst dosage on TC degradation
Fig. 5a shows the TC photodegradation as affected by catalyst dosage. When the dosage was increased from 100 to 200 mg L−1, the photodegradation efficiency significantly increased from 85% to 99% (Fig. 5a), respectively, because of increase in availability of surface sites. Greater dosage tended to generate more ROSs. Nevertheless, increasing the catalyst dosage from 100 to 300 and 500 mg L−1 decreased the TC removal efficiency because of increase in solution turbidity, thereby reducing light transmittance and photon absorption of NiCo–S@CN. The TC degradation rate constant of NiCo–S@CN reached the highest value of 6 × 10−2 min−1 at the catalyst dosage of 200 mg L−1 (Fig. 5b). This dosage was selected for subsequent experiments. Table S1† shows the comparative performance in TC photodegradation by different photocatalysts. Results indicated that NiCo–S@CN exhibited complete degradation efficiency in a short time with a higher rate constant than or comparable to most reported data.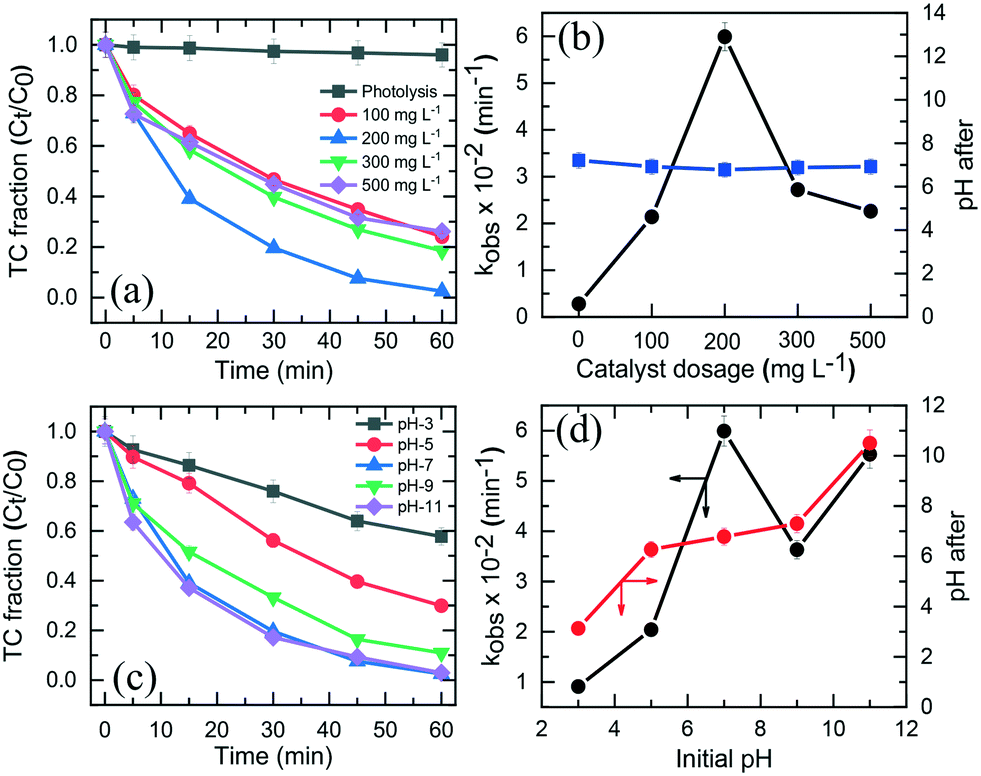 | ||
| Fig. 5 Effect of catalyst dosage and pH on (a and c) photodegradation of TC and (b and d) corresponding rate constant and pH change (reaction conditions: [TC] = 2.2 × 10−2 mM, pH 3–11, [catalyst] = 100–500 mg L−1, T = 25 °C). | ||
3.4 Effect of pH on TC degradation
The solution pH played a significant role in TC degradation because protons affected the production and distribution of oxygen reactive species and intimate interaction with the catalyst during photodegradation.19 Results (Fig. 5c) show that 70–99% of TC were removed in 60 min in the pH range of 5 to 11, but only 43% at pH 3. The kobs increased from 9.1 × 10−3 min−1 at pH 3.0 to 6 × 10−2 min−1 at pH 7.0, and slightly dropped to 3.6 × 10−2 min−1 and 5.5 × 10−2 min−1 at pH 9.0 and 11, respectively. The effect of initial pH on TC photodegradation over the NiCo–S@CN/solar light system was as follows: pH 7.0 > pH 11.0 > pH 9.0 > pH 5.0 > pH 3.0 (Fig. 5d). Note that the initial pH of 3.0, 5.0, 7.0, 9.0, and 11.0 became 3.1, 6.3, 6.8, 7.3, and 10.5, respectively (Fig. 5d). The maximum pseudo first-order degradation rate occurred at pH 7.0 with a TC degradation efficiency of 99% in 60 min.Tricarbonylamide, phenolic diketone, and dimethylamine are the three major functional groups of TC, with the acidity constants (pKa) of 3.3, 7.7, and 9.3, respectively. TC can be present in aqueous solution as cationic (TCH3+), neutral/zwitterionic (TCH2±), anionic (TC−), and dianionic (TC2−) species (Fig. S7a†). The point of zero charge (pHpzc) of NiCo–S@CN was around 3.4. Results showed that the degradation rate was slow under acidic conditions. At pH <pKa1 (3.3), a strong electrostatic repulsion between the positively charged NiCo–S@CN and TCH3+ species impeded the degradation reaction due to inefficient contact among reacting species. Likewise, electrostatic interactions hindered the adsorption of TCH− and TC2− onto the negatively charged NiCo–S@CN surface at pH >7.7. The adsorption behaviour of TC on NiCo–S@CN as a function of pH was further studied in the dark. When pH was in the range of 5 to 7, cationic TCH3+ was deprotonated to TCH2±, which resulted in increasing adsorption between the negatively charged catalysts and zwitterionic TCs.36
Fig. S7b† shows the Langmuir adsorption isotherm of TC on NiCo–S@CN as a function of pH. The data fitted well with the Langmuir adsorption isotherm (r2 > 0.99) (Fig. S7c†), demonstrating the monolayer nature of TC adsorption on the homogeneous adsorbent surface. The maximum TC adsorption density (qm) at different pH values, calculated from the Langmuir adsorption isotherm, followed the order pH 3.0 (5.1 × 10−2 mmol g−1) < pH 11.0 (6.3 × 10−2 mmol g−1) < pH 9.0 (9.3 × 10−2 mmol g−1) < pH 5.0 (10.2 mmol g−1) < pH 7.0 (15.9 × 10−2 mmol g−1) (Table S2 and Fig. S7d†). The KL (log) value, an indication of the strength of the TC bond with the catalyst, was relatively larger in the pH range of 5 and 11 than that of 3 due to strong interactions between the NiCo–S@CN surface and TC species (TCH2±, TCH− and TC2−) (Fig. S7d†). The result showed that neutral pH favoured TC adsorption onto the surface of NiCo–S@CN, thus enhancing photocatalytic degradation. Furthermore, the abundance of hydroxide ions improved the photocatalytic degradation of TC because the increasing number of anions in solution under basic conditions (pH 11.0) drastically accelerated the production of ˙OH radicals via reaction with photogenerated holes (h+).37
| OH− + h+ → ˙ OH | (2) |
3.5 Effect of initial TC concentration on TC degradation
Fig. 6a shows the effect of initial TC concentration (1.1 × 10−2–6.7 × 10−2 mM) on the degradation efficiency of TC over NiCo–S@CN under solar light irradiation at 25 °C and pH 7. The photodegradation efficiency and rate decreased when the initial TC concentration was increased from 1.1 × 10−2 to 6.7 × 10−2 M. The NiCo–S@CN/solar light system almost completely (∼100%) photodegraded TC at concentrations of 1.1 × 10−2 and 2.2 × 10−2 mM in 60 min under solar light irradiation (>99%). Nevertheless, the efficiency significantly decreased to 95–88% when the TC concentration was increased to 4.5 × 10−2–6.7 × 10−2 mmol L−1. Moreover, the kobs of TC photodegradation decreased from 6.6 × 10−2 to 3.3 × 10−2 min−1 when TC concentration was increased from 1.1 × 10−2 to 6.7 × 10−2 mmol L−1 (Fig. S8†).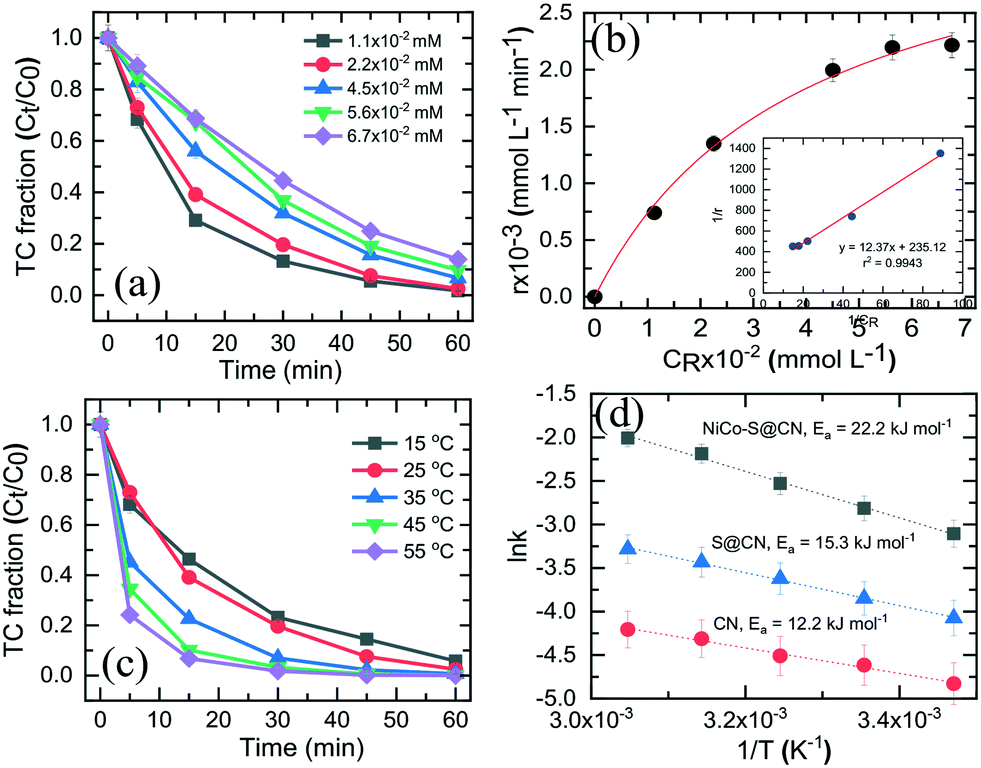 | ||
| Fig. 6 (a) Photocatalytic degradation efficiency and (b) initial rate of TC over NiCo–S@CN as a function of initial TC concentration at pH 7 (the inset of Fig. 6b is the linear regression of the Langmuir–Hinshelwood model). (c) Effect of temperature on TC degradation over NiCo–S@CN and (d) Arrhenius plot for the photocatalytic degradation of TC. | ||
It was assumed that the photocatalytic TC degradation was a surface-mediated reaction and strongly reliant on the catalytic sites on the photocatalyst surface.22,38 A limited number of active sites on the NiCo–S@CN limited the rapid adsorption of TC onto the photocatalyst surface, which significantly decreased the reaction rate constant. Langmuir–Hinshelwood (LH) kinetics was used to describe heterogeneous catalytic reactions as follows:
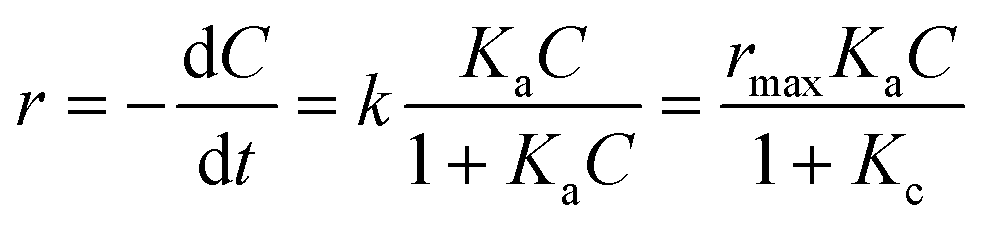 | (3) |
3.6 Effect of temperature on TC degradation
The reaction kinetics as a function of temperature was further studied. Fig. 6c shows the reaction rate increase from 4.4 × 10−2 to 1.3 × 10−1 min−1 as temperature increased from 15 to 55 °C. Barka et al.39 suggested that the increase in temperature increased the collision frequency of reacting chemical species with the photocatalyst surface due to elevated kinetic energy, thereby enabling competitive reactions over hole–e− recombination. The apparent activation energy was estimated by the Arrhenius equation as follows:40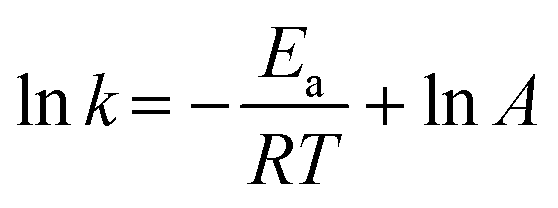 | (4) |
k versus 1/T with r2 >99%. The activation energy, Ea, was approximately 12.2, 15.3, and 22.2 kJ mol−1 for CN, S@CN and NiCo–S@CN, respectively (Fig. 6d). The findings suggested that the photocatalytic TC degradation rate over NiCo–S@CN was more sensitive to temperature than the other catalysts.
Furthermore, the Eyring equation was used to determine the enthalpy (ΔH) and entropy (ΔS) for the photocatalytic TC degradation process by the following equation:
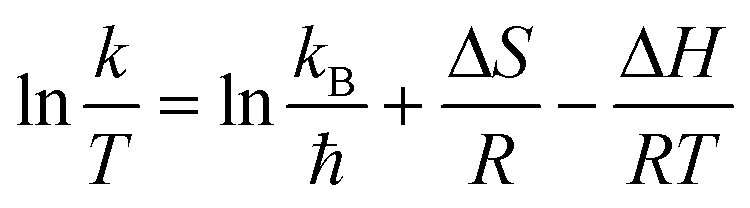 | (5) |
3.7 Reusability, stability, and applicability of NiCo–S@CN
To assess the reusability and chemical stability of NiCo–S@CN, the catalyst was collected using a high-speed centrifuge after each cycle of TC degradation experiment, washed with DI water to remove organic residues, and dried at 70 °C in a vacuum oven. The TC degradation test was repeated with the spent catalyst and the same reaction conditions in five successive runs at a catalyst dosage of 200 mg L−1, TC concentration of 0.05 mM, T of 25 °C, and pH 7. Fig. 7a shows nearly complete TC degradation at ∼99% in 60 min after 6 cycles, while the TC degradation rate constant was slightly decreased from 6 × 10−2 to 5.6 × 10−2 min−1. The degree of TC mineralization, measured in terms of TOC conversion, was also high, ranging between 78.5% and 83% after six consecutive runs (Fig. 7b). Furthermore, the leaching concentration of Ni and Co was insignificant during the five-cycle operation, less than the permissible water quality standard of Co (<50 μg L−1) and Ni (<100 μg L−1) (Fig. 7b). Finally, the pH of treated water varied in the normal range of natural water from 6.81 to 7.27. Therefore, there was no need to adjust the pH of treated water in order to meet source water quality standard.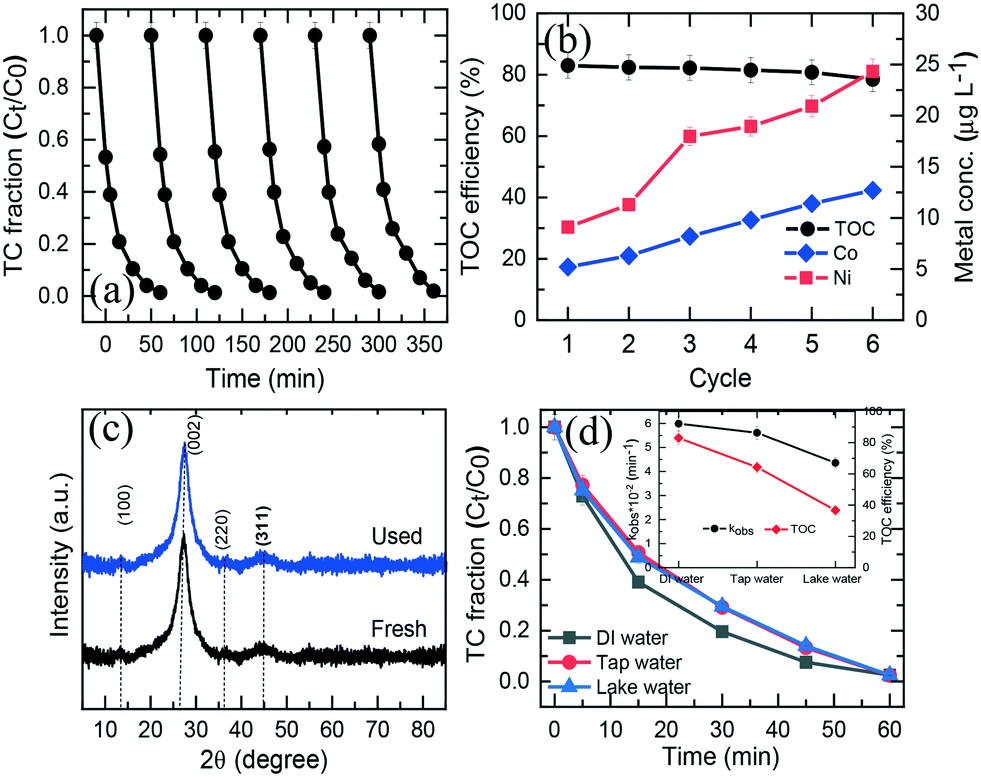 | ||
| Fig. 7 (a) Recyclability of the NiCo–S@CN catalyst in consecutive cycles, (b) TOC removal efficiency and metal leaching during 6 consecutive cycles, (c) XRD spectra of NiCo–S@CN before and after use, and (d) photocatalytic performance of NiCo–S@CN in the presence of real water. | ||
The crystal phase and chemical valence of the spent NiCo–S@CN catalyst were evaluated using XPS and XRD methods. Results showed that the XRD spectrum remained unchanged with no appearance of any new crystalline orientation after six consecutive cycles (Fig. 7c). Additionally, the chemical composition of spent CoMn2O4 exhibited an oxidation state of Ni and Co compatible with that of fresh NiCo–S@CN (Fig. S10a and b†). The molar ratio of Ni2+/Ni3+ and Co2+/Co3+ were only slightly different from that of the fresh catalyst, implying the preservation of the oxidation state of Ni and Co in the solar light activation reaction with respect to electron transfer. Above all, the results demonstrated that the NiCo–S@CN4 catalyst was highly durable, reactive, and capable of heterogeneous photocatalytic activation toward the decontamination of antibiotics from water.
The effect of water chemistry, commonly encountered in field scenarios, on the catalytic performance of the NiCo–S@CN/solar light system toward TC removal was studied using real water media including tap water and lake water. Fig. 7d shows that TC was completely degraded in all three water media in 30 min following the rate constant of lake water (4.36 × 10−2 min−1) < tap water (5.61 × 10−2 min−1) < DI water (6 × 10−2 min−1) (inset of Fig. 7d). It is interesting to note that the conversion of TC in terms of TOC was significantly higher in DI water (78%) than in tap water (64.3%) and lake water (36.7%). As a result, the degree of TC mineralization appeared to be inhibited in tap and lake water. The decrease in TC mineralization was ascribed to competition for reactive oxidation species (ROSs) between the main constituents of TOC (i.e., hydrophobic, transphilic and hydrophilic DOCs) and TC (Table S3†).6 Regardless of the possible presence of inhibitory natural organic matter in tap and lake water, the photocatalytic TC degradation efficiency over NiCo–S@CN remained high, indicating that the present NiCo–S@CN /solar light system was able to generate highly selective radicals in abundance for TC oxidation in real contaminated waters.
3.8 Identification of main reactive oxidation species and possible photocatalytic mechanism
Radical scavenging experiments (indirect method) were performed in the presence of appropriate scavengers to determine the reactive species and electron transfer mechanism involved in the photocatalytic degradation of TC over NiCo–S@CN. It is well known that sodium dihydrogen EDTA (EDTA-2Na), tert-butanol (tBuOH), and p-benzoquinone (pBQ) effectively scavenged holes (h+), hydroxyl radicals (˙OH), and superoxide anion radicals (O2˙−), respectively.38,41Fig. 8a shows that the addition of 10 mM pBQ, tBuOH, or EDTA-2Na remarkably hindered the degradation efficiency of TC by 47%, 33%, and 69%, respectively. The TC degradation rate constant in the presence of a scavenger followed the ascending order: kEDTA-2Na (6.2 × 10−3 min−1) < tBuOH (1.3 × 10−2 min−1) < kpBQ (1.6 × 10−2 min−1) (Fig. 8b). The radical scavenging results clearly demonstrated that the three reactive species, namely, h+, ˙OH and O2˙−, were produced in the photocatalytic system and contributed to TC degradation.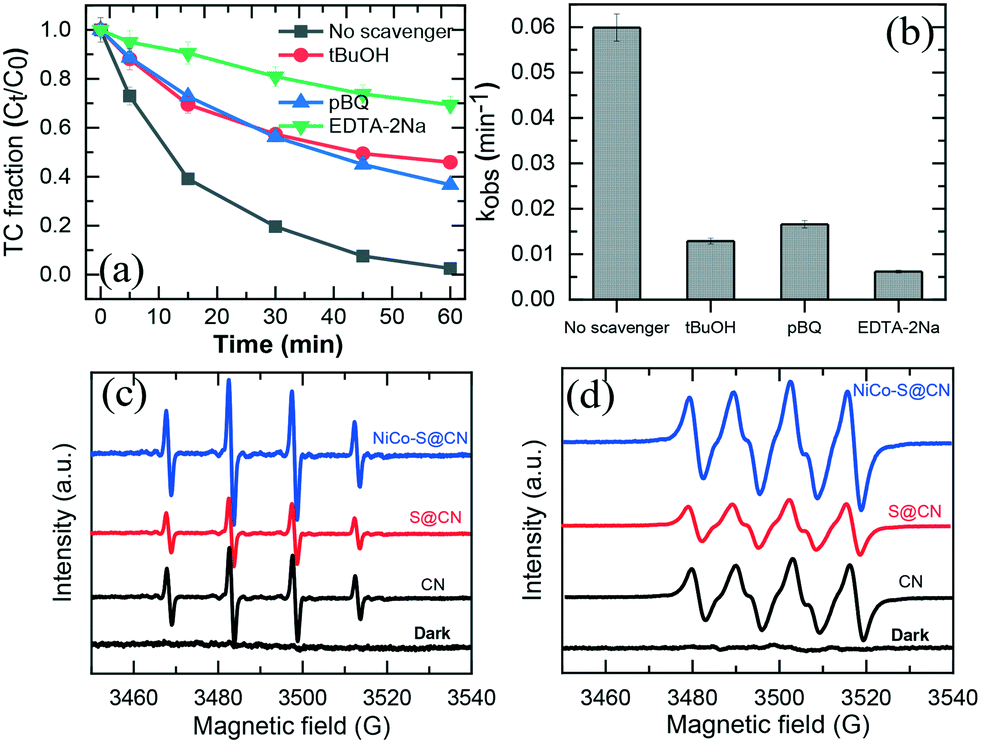 | ||
| Fig. 8 (a) and (b) Effect of different quenchers for TC degradation in the NiCo–S@CN/solar light system. EPR spectra of (c) ˙OH and (d) O2˙− radicals generated in the NiCo–S@CN/solar light system. | ||
Results confirmed that h+ played a major role in the photocatalytic TC degradation at 69%, whereas O2˙− and OH˙ radicals were secondary ROSs contributing to TC photodegradation at 34% and 47%, respectively.
Electron spin resonance (EPR) in the presence of 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as spin trapping reagent directly revealed the production of ROSs in the NiCo–S@CN/solar light system. Fig. 8c shows the DMPO–HO˙ signal with 1:2:2:1 intensity distribution in EPR spectra for all three catalysts under simulated solar light irradiation except for the control run under dark conditions.19Fig. 8d shows that there was no O2˙− signal in the dark; however, the quartet-line EPR signals (1:1:1:1) could be obviously found after all catalysts were excited by the simulated solar light.42 Furthermore, both the DMPO–HO˙ and DMPO–O2˙− signal intensities of NiCo–S@CN were stronger than that of CN and S@CN, implying more OH˙ and O2˙− production in the Z-scheme heterojunction between NiCo2O4 and S@CN. The result of radical scavenging assay and EPR analysis confirmed the formation of all three reactive species (h+, OH˙ and O2˙−) in the NiCo–S@CN/solar light system, and all played a role in TC degradation in the photocatalytic process.
To gain insight into the photocatalytic mechanism, a Mott–Schottky plot was constructed to establish the CB potential (ECB) of the catalysts, being created at a set frequency as a function of the applied DC potential, E. Since ECB is nearly identical to the flat band potential (Efb), it may be determined according to the Mott–Schottky relation,43,44i.e.,:
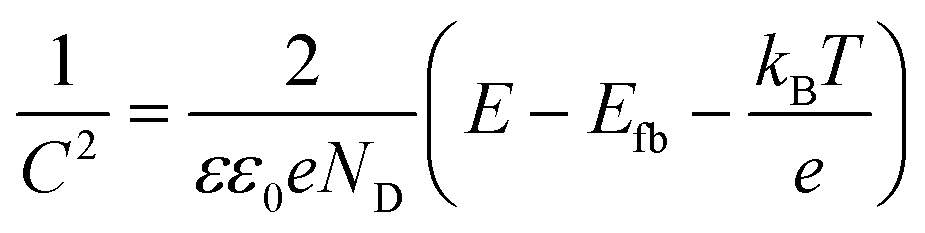 | (6) |
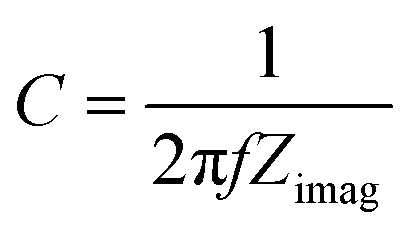 | (7) |
Fig. S11† shows that the positive slope was a typical characteristic of an n-type semiconductor of the as-prepared catalysts.43 The flat band potential of CN, S@CN and NiCo2O4 was −1.11, −0.97, and −1.3 V vs. Ag/AgCl, which was further converted to −0.91, −0.77 and −1.10 V vs. normal hydrogen electrode (NHE), respectively. For many n-type semiconductors, the ECB is about 0.1 V more negative than its flat-band potential. Therefore, the ECB of CN, S@CN and NiCo2O4 was roughly −1.01, −0.87 and −1.2 V (vs. NHE), respectively. The valence band potential (EVB) can be determined by the empirical equation, EVB = ECB + Eg. Consequently, the EVB of CN, S@CN and NiCo2O4 was 1.53, 1.53 and 0.33 V (vs. NHE), respectively.
Fig. 9 illustrates the reaction mechanism based on the direct Z-scheme heterojunction during the heterogeneous photocatalytic TC degradation on the reaction surface site of NiCo–S@CN catalysts. Confirmed by UV-vis reflectance spectra and Mott–Schottky results, in situ S doping changed the electronic structure and induced surface states near the bottom of the conduction band of CN, leading to a narrower bandgap (2.40 eV) and a downshift of the valence band from −1.2 eV to −0.87 eV. The result was in good agreement with EPR measurement that the signal intensity of HO˙ and O2˙− radicals of S@CN was lower than that of pure CN due to its lower reduction potential. Solar light irradiation of S@CN and NiCo2O4 photogenerated e− and h+ pairs on the CB and VB, respectively. The photoexcited e− on the CB of S@CN (−0.87 eV) migrated favourably, via the built-in electric field of the Z-scheme heterojunction between S@CN and NiCo2O4, to the VB of NiCo2O4 (+0.33 eV, NHE) because of a favourable redox potential difference.21,45 As a result, the strongly reducing e− on the CB of NiCo2O4 (−1.29 eV) and oxidizing h+ on the VB of S@CN were retained for subsequent reactions. In the conventional heterojunction system, if h+ from the VB of S@CN (+1.53 eV) migrated to the VB of NiCo2O4 (+0.33 eV), the oxidation potential of the accumulated h+ was not strong enough to directly oxidize TC species, which contradicted with the results of radical scavenging experiments.14,24,43 Therefore, the photocarrier transfer on NiCo–S@CN should essentially follow the Z-scheme mechanism, which reduced the probability of e−–h+ recombination.
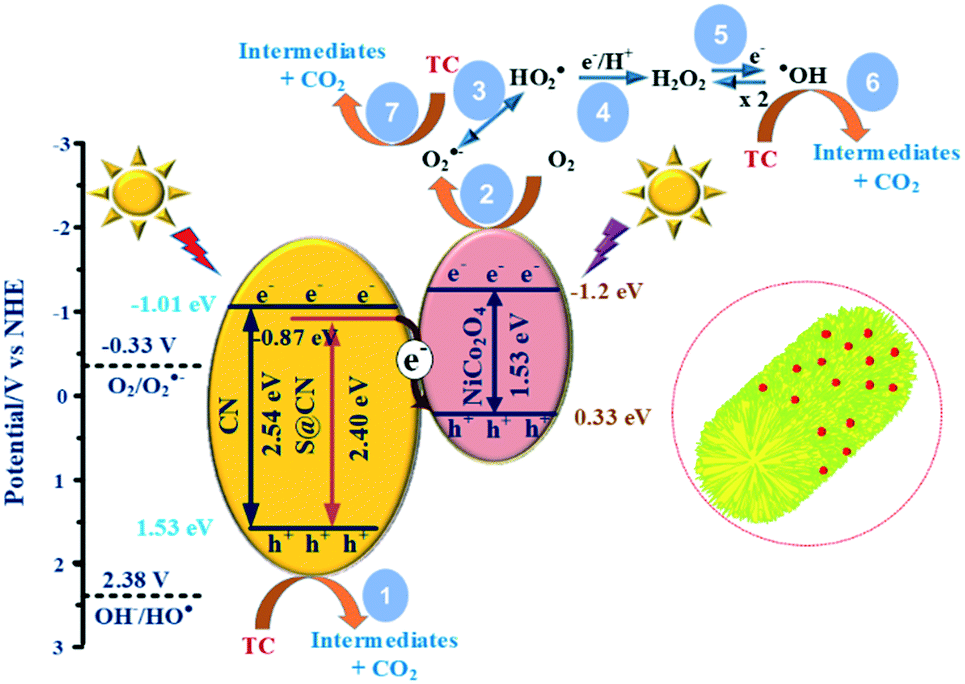 | ||
| Fig. 9 Proposed photocatalytic mechanism of the NiCo–S@CN catalyst under solar light irradiation. | ||
The holes created at the VB of S@CN took part in the direct oxidation of TC, yielding by-product CO2 (pathway 1), while the electrons at the CB of NiCo2O4 were trapped by dissolved oxygen to produce superoxide radicals [E0(O2/O2˙−) = −0.33 V] (pathway 2).41 Nevertheless, holes lacked sufficient redox capability to create hydroxyl radicals [E0(OH/HO˙) = +2.38 V].33,41 As a result, hydroxyl radicals were generated indirectly. The superoxide radicals could be reduced to peroxyl (HO2˙) radicals [E0(O2/HO2˙) = −0.05 V] (pathways 3),5 and then further reactions of peroxyl radicals via pathways 4 and 5 resulted in the formation of more potent ROSs, such as highly reactive HO˙. Meanwhile, HO˙ and O2˙− radicals contributed directly to the oxidation of TC and further mineralized the degradation intermediates during the photocatalytic process (pathways 6 and 7).
3.9 Mineralization of tetracycline and toxicity evaluation
The degradation and mineralization of TC were further studied by COD and TOC analysis. According to the above result, 99% of TC were rapidly eliminated in the NiCo–S@CN/solar light system in 60 min, while 83% of TOC was removed in the same period. Based on TOC and COD measurements, it is possible to estimate the average oxidation number (or state) of carbon (COS) (eqn (8)).46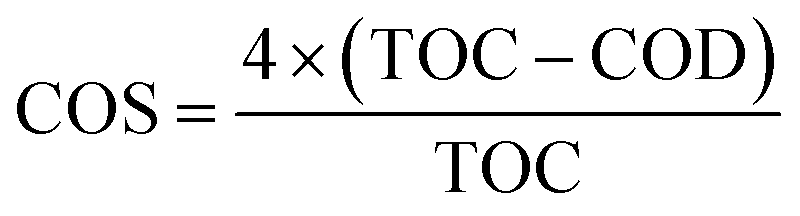 | (8) |
Results showed that COS increased from −0.52 to 1.3 as a function of oxidation time (Fig. 10a), which clearly indicated significant TC mineralization on NiCo–S@CN during the photocatalytic oxidation reaction.
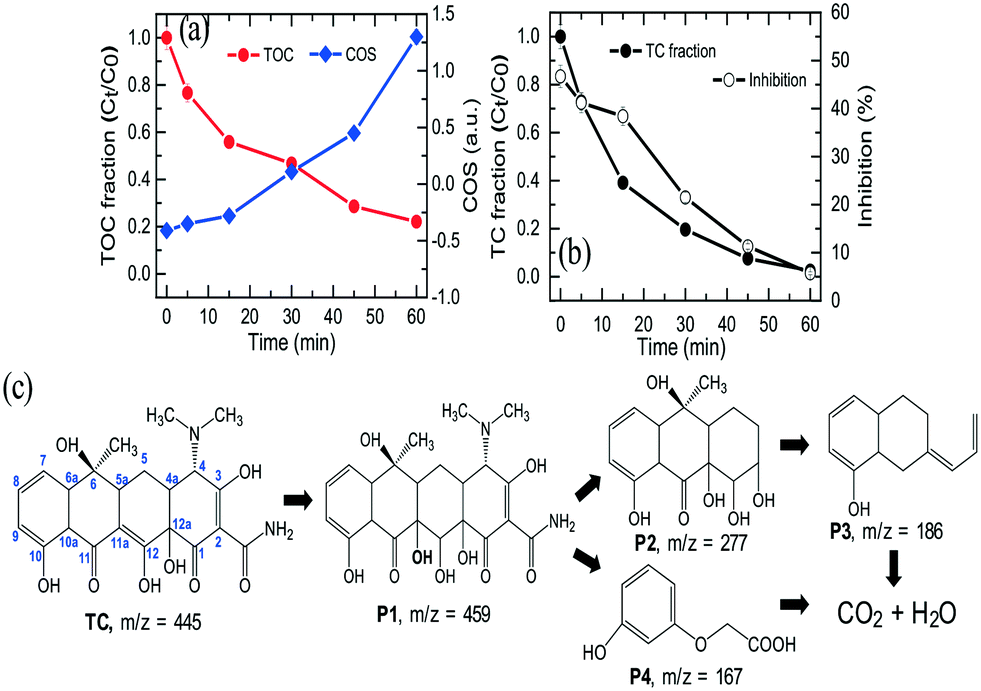 | ||
| Fig. 10 (a) TOC decrease and course of average carbon oxidation state (COS), (b) inhibition change during photocatalytic degradation of TC, and (c) photodegradation pathway of TC over the NiCo–S@CN/solar light system. | ||
Although complete degradation into small organic fragments and CO2 will decrease the toxicity of the parent compound,47 TC degradation intermediates can be more toxic than their parent compound.48 The toxicity change of aqueous solution was assessed with Microtox using luminescent bacteria (Vibrio fischeri) during 60 min of TC oxidation process. Fig. 10b shows that TC, at the original concentration of 10 mg L−1, exhibited a certain degree of biotoxicity to V. fischeri with a bioluminescence inhibition ratio of 52.3%. The inhibition ratio gradually decreased to 46.7% after 30 min, indicating the possible formation and accumulation of some highly toxic intermediates. Furthermore, the inhibition effect was significantly decreased to 5.7% after 60 min of photocatalytic treatment, which was much lower than that of the original TC. The results implied that although highly toxic intermediates were produced in the beginning period of the reaction, they could be further degraded into non-toxic chemicals and ultimately converted to relatively non-harmful CO2, HNO3, and H2O by ROSs generated during solar light activation of Z-scheme NiCo–S@CN.
To gain a better understanding of the TC degradation pathway in the NiCo–S@CN/solar light system, ultra-performance liquid chromatography coupled with time-of-flight mass spectrometry detector (UPLC/ToF MS) analysis was employed to identify TC oxidation intermediates. Fig. S12† shows the MS spectra and the possible molecular structure of intermediates from TC degradation (Fig. 10c). The photogenerated ˙OH favourably attacked the C11a position of TC because of high electron density at this position and generated the primary P1 product (m/z = 459). Further radical oxidation and rupture of the ring structures yielded P2 (m/z = 277), P3 (m/z = 186), and P4 (m/z = 167). These intermediates were subsequently converted to carbon dioxide, water, and other mineral substances. The results revealed that NiCo–S@CN was an effective solar-light-sensitive photocatalyst for the degradation of antibiotics such as TC.
4. Conclusion
A Z-scheme NiCo–S@CN photocatalyst was successfully synthesized by a hydrothermal process. The NiCo–S@CN catalyst exhibited excellent solar-light-responsive photocatalytic activity with respect to TC degradation because of the synergistic effect of NiCo2O4 NPs and S@CN nanotubes in the Z-scheme heterojunction. The photocatalytic performance was the best at the catalyst dosage of 200 mg L−1. The initial pH effect on TC photodegradation over the NiCo–S@CN/solar light system was as follows: pH 7.0 > pH 11.0 > pH 9.0 > pH 5.0 > pH 3.0. The photocatalytic TC degradation over NiCo–S@CN followed the Langmuir–Hinshelwood kinetics. Increase in temperature enhanced the photocatalytic degradation rate of TC over NiCo–S@CN. The NiCo–S@CN catalyst exhibited significant stability and reusability with a consistently high degree of TC degradation in multiple runs. The NiCo–S@CN/solar light system was capable of degrading TC in real water media. Results of EPR and radical trapping experiments were used to describe the reaction mechanism of TC degradation, in which holes (h+) and superoxide radicals (O2˙−) were the main reactive species in the Z-scheme/solar light system. Additionally, the structure and composition of NiCo–S@CN remained relatively unaltered after consecutive TC photodegradation runs. The leaching concentration of metal ions in the treated water was lower than that of the permissible drinking water standard. NiCo–S@CN will be a promising and effective heterogeneous photocatalyst for the degradation of recalcitrant contaminants, such as TC. NiCo–S@CN has high reactivity, stable structure, and reusability. The findings of this study have opened a new venue for the synthesis of Z-scheme heterojunction-based nanocomposites for water treatment and general remediation of contaminated environments.Author contributions
Thanh-Binh Nguyen: conceptualization, methodology, data curation, investigation, writing – review and editing. Phung-Ngoc-Thao Ho: investigation, data curation, writing – original draft, validation. Chiu-Wen Chen: resources. C. P. Huang: data curation, visualization, writing – review and editing. Ruey-an Doong: conceptualization, resources. Cheng-Di Dong: supervision, validation, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors highly acknowledge funding support from MOST, Taiwan, under grant number MOST 109-2222-E-992-002.References
- S. Adhikari, H. H. Lee and D. H. Kim, Efficient visible-light induced electron-transfer in z-scheme MoO3/Ag/C3N4 for excellent photocatalytic removal of antibiotics of both ofloxacin and tetracycline, Chem. Eng. J., 2020, 391, 13 CrossRef.
- I. Chopra and M. Roberts, Tetracycline antibiotics: Mode of action, applications, molecular biology, and epidemiology of bacterial resistance, Microbiol. Mol. Biol. Rev., 2001, 65(2), 232–260 CrossRef CAS PubMed.
- V. Choudhary, K. Vellingiri, M. I. Thayyil and L. Philip, Removal of antibiotics from aqueous solutions by electrocatalytic degradation, Environ. Sci.: Nano, 2021, 8(5), 1133–1176 RSC.
- A. H. Malik and P. K. Iyer, Conjugated Polyelectrolyte Based Sensitive Detection and Removal of Antibiotics Tetracycline from Water, ACS Appl. Mater. Interfaces, 2017, 9(5), 4433–4439 CrossRef CAS PubMed.
- P. Suyana, P. Ganguly, B. N. Nair, A. P. Mohamed, K. G. K. Warrier and U. S. Hareesh, Co3O4-C3N4 p-n nano-heterojunction for the simultaneous degradation of a mixture of pollutants under solar irradiation, Environ. Sci.: Nano, 2017, 4(1), 212–221 RSC.
- J. S. Xu, T. J. K. Brenner, Z. P. Chen, D. Neher, M. Antonietti and M. Shalom, Upconversion-Agent Induced Improvement of g-C3N4 Photocatalyst under Visible Light, ACS Appl. Mater. Interfaces, 2014, 6(19), 16481–16486 CrossRef CAS PubMed.
- S. Tonda, S. Kumar, M. Bhardwaj, P. Yadav and S. Ogale, g-C3N4/NiAl-LDH 2D/2D Hybrid Heterojunction for High-Performance Photocatalytic Reduction of CO2 into Renewable Fuels, ACS Appl. Mater. Interfaces, 2018, 10(3), 2667–2678 CrossRef CAS PubMed.
- L. K. B. Paragas, M. D. G. de Luna and R. A. Doong, Rapid removal of sulfamethoxazole from simulated water matrix by visible-light responsive iodine and potassium co-doped graphitic carbon nitride photocatalysts, Chemosphere, 2018, 210, 1099–1107 CrossRef CAS PubMed.
- A. Naseri, M. Samadi, A. Pourjavadi, A. Z. Moshfegh and S. Ramakrishna, Graphitic carbon nitride (g-C3N4)-based photocatalysts for solar hydrogen generation: recent advances and future development directions, J. Mater. Chem. A, 2017, 5(45), 23406–23433 RSC.
- M. H. Vu, M. Sakar, C. C. Nguyen and T. O. Do, Chemically Bonded Ni Cocatalyst onto the S Doped g-C3N4 Nanosheets and Their Synergistic Enhancement in H2 Production under Sunlight Irradiation, ACS Sustainable Chem. Eng., 2018, 6(3), 4194–4203 CrossRef CAS.
- L. Acharya, S. Nayak, S. P. Pattnaik, R. Acharya and K. Parida, Resurrection of boron nitride in p-n type-II boron nitride/B-doped-g-C3N4 nanocomposite during solid-state Z-scheme charge transfer path for the degradation of tetracycline hydrochloride, J. Colloid Interface Sci., 2020, 566, 211–223 CrossRef CAS PubMed.
- S. S. M. Bhat, S. A. Lee, T. H. Lee, C. Kim, J. Park, T. W. Lee, S. Y. Kim and H. W. Jang, All-Solution-Processed BiVO4/TiO2 Photoanode with NiCo2O4 Nanofiber Cocatalyst for Enhanced Solar Water Oxidation, ACS Appl. Energy Mater., 2020, 3(6), 5646–5656 CrossRef CAS.
- B. Palanivel and A. Mani, Conversion of a Type-II to a Z-Scheme Heterojunction by Intercalation of a 0D Electron Mediator between the Integrative NiFe2O4/g-C3N4 Composite Nanoparticles: Boosting the Radical Production for Photo-Fenton Degradation, ACS Omega, 2020, 5(31), 19747–19759 CrossRef CAS PubMed.
- X. Zeng, S. Lan and I. M. Lo, Rapid disinfection of E. coli by aternary BiVO4/Ag/g-C3N4 composite under visible light: photocatalytic mechanism and performance investigation in authentic sewage, Environ. Sci.: Nano, 2019, 6(2), 610–623 RSC.
- E. Umeshbabu, P. H. K. Charan, P. Justin and G. R. Rao, Hierarchically organized NiCo2O4 microflowers anchored on multiwalled carbon nanotubes: efficient bifunctional electrocatalysts for oxygen and hydrogen evolution reactions, ChemPlusChem, 2020, 85(1), 183–194 CrossRef CAS.
- J. J. Jiang, X. Y. Wang, C. L. Yue, S. D. Liu, Y. H. Lin, T. F. Xie and S. S. Dong, Efficient photoactivation of peroxymonosulfate by Z-scheme nitrogen-defect-rich NiCo2O4/g-C3N4 for rapid emerging pollutants degradation, J. Hazard. Mater., 2021, 414, 14 CrossRef PubMed.
- A. Shawky, M. Alhaddad, R. M. Mohamed, N. S. Awwad and H. A. Ibrahium, Magnetically separable and visible light-active Ag/NiCo2O4 nanorods prepared by a simple route for superior photodegradation of atrazine in water, Prog. Nat. Sci.: Mater. Int., 2020, 30(2), 160–167 CrossRef CAS.
- T. B. Nguyen, R. A. Doong, C. P. Huang, C. W. Chen and C. D. Dong, Activation of persulfate by CoO nanoparticles loaded on 3D mesoporous carbon nitride (CoO@meso-CN) for the degradation of methylene blue (MB), Sci. Total Environ., 2019, 675, 531–541 CrossRef CAS PubMed.
- T. B. Nguyen, C. P. Huang, R. A. Doong, C. W. Chen and C. D. Dong, Visible-light photodegradation of sulfamethoxazole (SMX) over Ag-P-codoped g-C3N4 (Ag-P@UCN) photocatalyst in water, Chem. Eng. J., 2020, 384, 13 CrossRef.
- S. Elbasuney and M. Yehia, Thermal decomposition of ammonium perchlorate catalyzed with CuO nanoparticles, Def. Technol., 2019, 15(6), 868–874 CrossRef.
- M. Jourshabani, Z. Shariatinia and A. Badiei, Synthesis and characterization of novel Sm2O3/S-doped g-C3N4 nanocomposites with enhanced photocatalytic activities under visible light irradiation, Appl. Surf. Sci., 2018, 427, 375–387 CrossRef CAS.
- T. B. Nguyen, C. P. Huang and R. A. Doong, Enhanced catalytic reduction of nitrophenols by sodium borohydride over highly recyclable Au@graphitic carbon nitride nanocomposites, Appl. Catal., B, 2019, 240, 337–347 CrossRef CAS.
- S. Karmakar, S. Varma and D. Behera, Investigation of structural and electrical transport properties of nano-flower shaped NiCo2O4 supercapacitor electrode materials, J. Alloys and Compd., 2018, 757, 49–59 CrossRef CAS.
- L. Chen, D. Y. Zhu, J. T. Li, X. X. Wang, J. F. Zhu, P. S. Francis and Y. H. Zheng, Sulfur and potassium co-doped graphitic carbon nitride for highly enhanced photocatalytic hydrogen evolution, Appl. Catal., B, 2020, 273, 9 CrossRef.
- P. C. Nagajyothi, M. Pandurangan, S. V. P. Vattikuti, C. O. Tettey, T. V. M. Sreekanth and J. Shim, Enhanced photocatalytic activity of Ag/g-C3N4 composite, Sep. Purif. Technol., 2017, 188, 228–237 CrossRef CAS.
- R. S. Sahu and R. A. Doong, Functionalized Fe/Ni@g-C3N4 nanostructures for enhanced trichloroethylene dechlorination and successive oxygen reduction reation activity, Environ. Sci.: Nano, 2020, 7(11), 3469–3481 RSC.
- I. Papailias, N. Todorova, T. Giannakopoulou, N. Ioannidis, N. Boukos, C. P. Athanasekou, D. Dimotikali and C. Trapalis, Chemical vs thermal exfoliation of g-C3N4 for NOx removal under visible light irradiation, Appl. Catal., B, 2018, 239, 16–26 CrossRef CAS.
- J. Di, M. Z. Zhu, R. Jamakanga, X. K. Gai, Y. Li and R. Q. Yang, Electrochemical activation combined with advanced oxidation on NiCo2O4 nanoarray electrode for decomposition of Rhodamine B, J. Water Process. Eng., 2020, 37, 9 Search PubMed.
- B. Naresh, T. N. V. Krishna, S. S. Rao and H. J. Kim, Reagent induced morphological changes in NiCo2O4 electrode material for flexible supercapacitor, Mater. Lett., 2019, 248, 218–221 CrossRef CAS.
- T. B. Nguyen, C. P. Huang, R. A. Doong, C. W. Chen and C. D. Dong, CoO-3D ordered mesoporous carbon nitride (CoO@mpgCN) composite as peroxymonosulfate activator for the degradation of sulfamethoxazole in water, J. Hazard. Mater., 2021, 401, 11 Search PubMed.
- I. Papailias, T. Giannakopoulou, N. Todorova, D. Demotikali, T. Vaimakis and C. Trapalis, Effect of processing temperature on structure and photocatalytic properties of g-C3N4, Appl. Surf. Sci., 2015, 358, 278–286 CrossRef CAS.
- V. Hasija, P. Raizada, A. Sudhaik, K. Sharma, A. Kumar, P. Singh, S. B. Jonnalagadda and V. K. Thakur, Recent advances in noble metal free doped graphitic carbon nitride based nanohybrids for photocatalysis of organic contaminants in water: A review, Appl. Mater. Today, 2019, 15, 494–524 CrossRef.
- C. C. Hu, Y. H. Lin, M. Yoshida and S. Ashimura, Influence of Phosphorus Doping on Triazole-Based g-C3N5 Nanosheets for Enhanced Photoelectrochemical and Photocatalytic Performance, ACS Appl. Mater. Interfaces, 2021, 13(21), 24907–24915 CrossRef CAS PubMed.
- F. Raziq, A. Hayat, M. Humayun, S. K. B. Mane, M. B. Faheem, A. Ali, Y. Zhao, S. B. Han, C. Cai, W. Li, D. C. Qi, J. B. Yi, X. J. Yu, M. B. H. Breese, F. Hassan, F. Ali, A. Mavlonov, K. Dhanabalan, X. Xiang, X. T. Zu, S. Li and L. Qiao, Photocatalytic solar fuel production and environmental remediation through experimental and DFT based research on CdSe-QDs-coupled P-doped-g-C3N4 composites, Appl. Catal., B, 2020, 270, 12 Search PubMed.
- D. M. Ruan, S. Kim, M. Fujitsuka and T. Majima, Defects rich g-C3N4 with mesoporous structure for efficient photocatalytic H2 production under visible light irradiation, Appl. Catal., B, 2018, 238, 638–646 CrossRef CAS.
- S. Y. Zheng, L. J. Kong, J. L. Meng, N. Song, Y. N. Jiang, J. L. Guo, T. W. Mu Guo and M. H. Huang, MIL-88A grown in-situ on graphitic carbon nitride (g-C3N4) as a novel sorbent: Synthesis, characterization, and high-performance of tetracycline removal and mechanism, Adv. Powder Technol., 2020, 31(10), 4344–4353 CrossRef CAS.
- T. S. Bui, P. Bansal, B. K. Lee, T. Mahvelati-Shamsabadi and T. Soltani, Facile fabrication of novel Ba-doped g-C3N4 photocatalyst with remarkably enhanced photocatalytic activity towards tetracycline elimination under visible-light irradiation, Appl. Surf. Sci., 2020, 506, 12 CrossRef.
- T. B. Nguyen and R. A. Doong, Heterostructured ZnFe2O4/TiO2 nanocomposites with a highly recyclable visible-light-response for bisphenol A degradation, RSC Adv., 2017, 7(79), 50006–50016 RSC.
- N. Barka, S. Qourzal, A. Assabbane, A. Nounah and Y. Ait-Ichou, Factors influencing the photocatalytic degradation of Rhodamine B by TiO2-coated non-woven paper, J. Photochem. Photobiol., A, 2008, 195(2–3), 346–351 CrossRef CAS.
- Z. Ghasemi, H. Younesi and A. A. Zinatizadeh, Kinetics and thermodynamics of photocatalytic degradation of organic pollutants in petroleum refinery wastewater over nano-TiO2 supported on Fe-ZSM-5, J. Taiwan Inst. Chem. Eng., 2016, 65, 357–366 CrossRef CAS.
- J. Z. Y. Tan, N. M. Nursam, F. Xia, M. A. Sani, W. Li, X. D. Wang and R. A. Caruso, High-Performance Coral Reef-like Carbon Nitrides: Synthesis and Application in Photocatalysis and Heavy Metal Ion Adsorption, ACS Appl. Mater. Interfaces, 2017, 9(5), 4540–4547 CrossRef CAS PubMed.
- F. Guo, W. Shi, H. Wang, M. Han, W. Guan, H. Huang, Y. Liu and Z. Kang, Study on highly enhanced photocatalytic tetracycline degradation of type II AgI/CuBi2O4 and Z-scheme AgBr/CuBi2O4 heterojunction photocatalysts, J. Hazard. Mater., 2018, 349, 111–118 CrossRef CAS PubMed.
- T. Giannakopoulou, I. Papailias, N. Todorova, N. Boukos, Y. Liu, J. G. Yu and C. Trapalis, Tailoring the energy band gap and edges' potentials of g-C3N4/TiO2 composite photocatalysts for NOx removal, Chem. Eng. J., 2017, 310, 571–580 CrossRef CAS.
- C. Baumanis and D. W. Bahnemann, TiO2 Thin Film Electrodes: Correlation between Photocatalytic Activity and Electrochemical Properties, J. Phys. Chem. C, 2008, 112(48), 19097–19101 CrossRef CAS.
- S. Chakrabarty, A. Mukherjee and S. Basu, RGO-MoS2 Supported NiCo2O4 Catalyst toward Solar Water Splitting and Dye Degradation, ACS Sustainable Chem. Eng., 2018, 6(4), 5238–5247 CrossRef CAS.
- W. Stumm and J. J. Morgan, Aquatic Chemistry. Chemical Equilibrim and Rates in Natural Waters, John & Wiley, New York, NY, 3rd edn, 1996 Search PubMed.
- S. Yahiat, F. Fourcade, S. Brosillon and A. Amrane, Removal of antibiotics by an integrated process coupling photocatalysis and biological treatment - Case of tetracycline and tylosin, Int. Biodeterior. Biodegrad., 2011, 65(7), 997–1003 CrossRef CAS.
- R. R. Solís, Ö. Dinc, G. Fang, M. N. Nadagouda and D. D. Dionysiou, Activation of inorganic peroxides with magnetic graphene for the removal of antibiotics from wastewater, Environ. Sci.: Nano, 2021, 8(4), 960–977 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: SEM/TEM images of S@CN, XPS survey spectra of as-prepared catalysts, kinetic curve, TOC removal efficiency, TC speciation and zeta potential as a function of pH, XPS spectra of NiCo–S@CN before and after photocatalytic degradation, Mott–Schottky plot, and MS spectra. See DOI: 10.1039/d1en00888a |
| This journal is © The Royal Society of Chemistry 2022 |