
Ionic group-dependent structure of complex coacervate hydrogels formed by ABA triblock copolymers†
Seyoung
Kim‡
 a,
Jung-Min
Kim‡
a,
Kathleen
Wood
b and
Soo-Hyung
Choi
*a
a,
Jung-Min
Kim‡
a,
Kathleen
Wood
b and
Soo-Hyung
Choi
*a
aDepartment of Chemical Engineering, Hongik University, Seoul 04066, Republic of Korea. E-mail: shchoi@hongik.ac.kr
bAustralian Centre for Neutron Scattering, Australian Nuclear Science and Technology Organisation, Lucas Heights, NSW 2234, Australia
First published on 3rd May 2022
Abstract
This study investigates the nanostructure of complex coacervate core hydrogels (C3Gs) with varying compositions of cationic charged groups (i.e., ammonium and guanidinium) using small-angle X-ray/neutron scattering (SAX/NS). C3Gs were prepared by stoichiometric mixing of two oppositely charged ABA triblock copolymers in aqueous solvents, in which A end-blocks were functionalized with either sulfonate groups or a mixture of ammonium and guanidinium groups. Comprehensive small-angle X-ray/neutron scattering (SAX/NS) analysis elucidated the dependence of C3Gs structures on the fraction of guanidinium groups in the cationic end-block (x) and salt concentration (cs). As x increases, the polymer volume fraction in the cores, and interfacial tension (γcore) and salt resistance (c*) of the coacervate cores increase, which is attributed to the greater hydrophobicity and non-electrostatic association. Furthermore, we observed that the salt dependence of the interfacial tension follows γcore ∼ (1 − cs/c*)3/2 in all series of x. The results show that the variation of the ionic group provides a powerful method to control the salt-responsiveness of C3Gs as stimuli-responsive materials.
Introduction
Physical hydrogels formed by ABA triblock copolymers are fascinating due to their reversible and stimuli-responsive structures.1 The A end-blocks self-associate to construct physical junctions (i.e., the core), and the hydrophilic B mid-blocks bridge between the cores and store the stress under the applied strain.2 Among many examples of associations that are designed to produce physical junctions, the complex coacervation is particularly interesting.3 When mixed in aqueous media, two polyelectrolytes containing opposite charges form the condensed “coacervate” phase by the electrostatic interactions. This polymer-rich coacervate is inherently multi-responsive due to the susceptibility of the interactions to the constituting polyelectrolytes and their counterions.4,5 These complex coacervate core hydrogels (C3Gs) are well suited for biomedical and biological applications for several reasons: (1) the reconfigurability of the coacervate core renders the hydrogels injectable, (2) the formulation precludes the use of the organic solvent, and (3) the water-swollen core can store and release hydrophilic and/or charged compounds.6,7The nanostructure of C3Gs, including the self-assembly of the coacervate cores, is pivotal for understanding the mechanical characteristics of the C3Gs under varying physiological stimuli such as salinity, pH, and temperature. The phase behavior and the core structure of C3Gs have mostly been evaluated in relation to the length of the polyelectrolytes and salt concentration,8–10 which are perceived as universal parameters controlling the thermodynamics of coacervation.11 With longer polyelectrolyte end-blocks and lower salt concentrations, the coacervate cores are more stable and the number of associating end-blocks per core (i.e., the aggregation number) increases. In weak polyelectrolyte end-blocks, pH also controls the charge density, and thus the core stability and structure.3,12 Studies have also investigated the correlation between these thermodynamic features and the viscoelastic behavior of C3Gs: the relaxation of C3Gs is representatively slower with a longer end-block and lower salt concentration.8,13 However, less attention has been paid to the effect of ionic groups in polyelectrolyte end-blocks on both/either the structure and/or dynamics of C3Gs,14 although coacervation significantly depends on the specific types of ionic groups in polyelectrolytes.
Non-electrostatic interactions such as hydrophobic or specific interactions within polyelectrolytes influence the coacervate stability and/or the extent of hydration as found in studies on bulk coacervates,15,16 complex coacervate core micelles,17 and C3Gs.14 In particular, the interfacial tension between the coacervates and the aqueous medium, γ, depends on both the electrostatic and non-electrostatic interactions. The interfacial tension, therefore, directs the self-assembly of the block copolymers and affects the hydrogel relaxation dynamics of C3Gs.18,19 Despite the importance of interfacial tension in the structural and mechanical behavior of C3Gs, an ultralow magnitude (∼ an order of 1 mN m−1) necessitates the use of a specialized measuring technique such as atomic force microscopy20 or a microfluidic device,21 which is a hurdle in assessing interfacial tension. Nevertheless, given that self-assembled structures are coupled with the interfacial tension of the coacervate cores, we suggest that information about the interfacial tension can be retrieved from the structural characteristics of the coacervate self-assembly. Thus, a structural investigation on C3Gs would provide a facile way to assess the effect of ionic groups on the interfacial tension, along with the stability and hydration state of the coacervate cores.
In this study, we aim to evaluate the role of ionic groups on the structure of C3Gs, especially in terms of ion-specific interactions. A series of ABA triblock copolymers was prepared, where the A end-block included either the cationic or anionic groups in the repeat units and the B mid-block is hydrophilic polyethylene oxide (PEO). The cationic triblock copolymers were prepared by attaching the ammonium (A) and guanidinium (G) groups using various mixing ratios, while the anionic triblock copolymers were prepared by sulfonate (S) group functionalization. Previous works, including our efforts, have found that polyguanidines are less hydrated and result in stronger ion-pair interactions with anionic polyelectrolytes than polyamines.22–25 Therefore, as the guanidinium fraction in the cationic end-block increases, we anticipate stronger segregation of the coacervate cores and enhanced gel stability against the addition of external salt, called “salt resistance.” The nanostructures and water content in the coacervate cores as functions of guanidinium fraction and salt concentration were characterized by small-angle X-ray and neutron scattering (SAX/NS). These structural features were further utilized to assess the interfacial tension at the zero-salt limit and the salt resistance of the coacervate core based on the scaling description. Considering the current understanding of complex coacervation, the results are discussed to provide further insights into the self-assembled structures driven by the complex coacervation in block copolymer dispersions.
Experimental section
Materials
A pair of oppositely charged ABA triblock copolymers were synthesized using anionic ring-opening polymerization of poly(allyl glycidyl ether-b-ethylene oxide-b-allyl glycidyl ether) (PAGE-b-PEO-b-PAGE) and subsequent post-polymerization modification to introduce charged moieties following our established procedure (Fig. 1(a)).17,19 The allyl glycidyl ether (AGE, TCI) monomer was degassed through several freeze–pump–thaw cycles and purified with butyl magnesium chloride (Sigma-Aldrich). Tetrahydrofuran (THF, Daejung) was purified and dried using a solvent purification system (Korea Kiyon). Poly(ethylene oxide) (PEO, Mn = 20 kg mol−1, Sigma-Aldrich) was dried under vacuum and used as a macro-initiator. Under inert conditions, PEO was initiated by a potassium naphthalenide (Sigma-Aldrich) in a THF solution until a pale green color appeared. The AGE monomer was added to the reactor and polymerized at 40 °C for 24 h. Degassed methanol was added to quench the reaction, and the PAGE-b-PEO-b-PAGE block copolymer product was isolated by precipitation in n-hexane.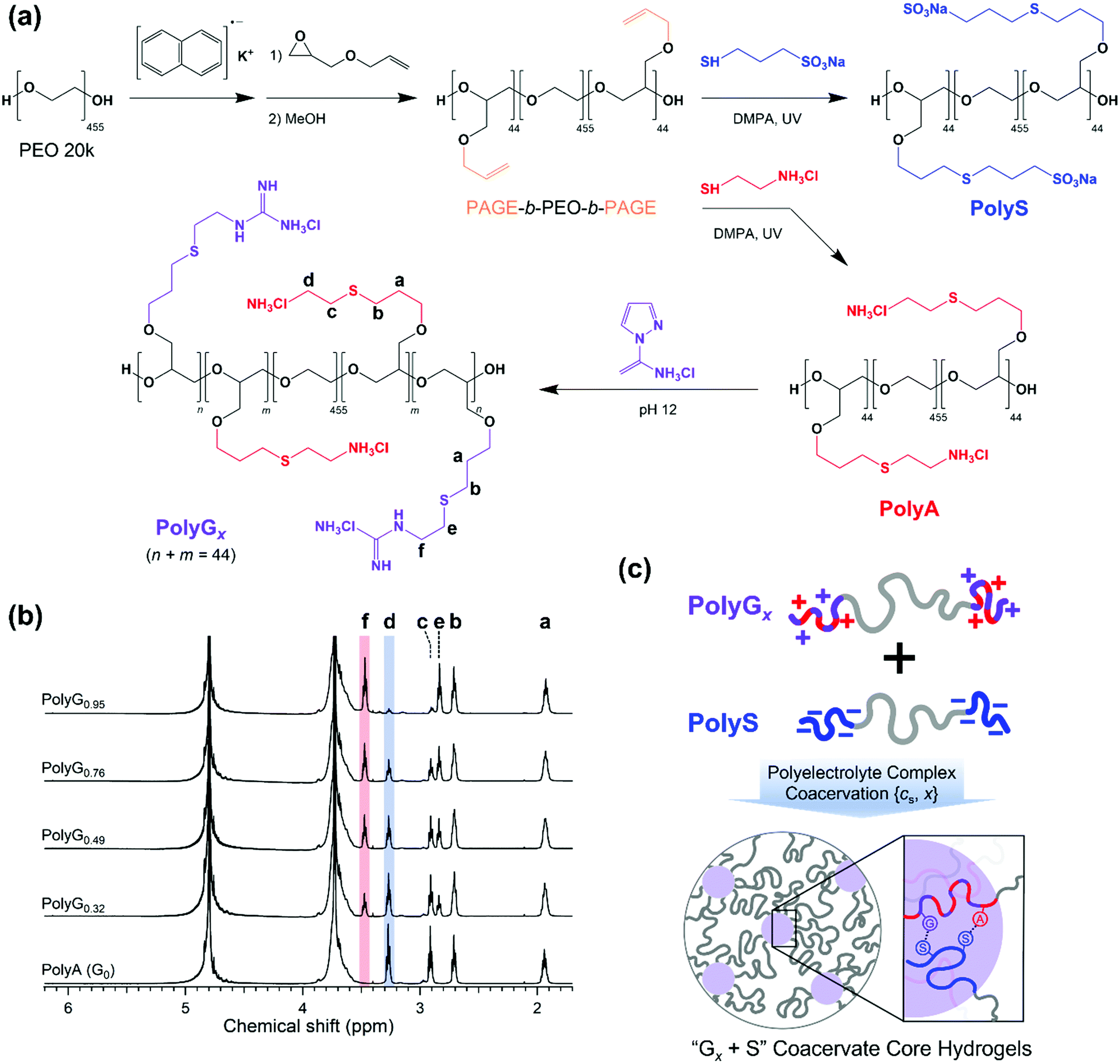 | ||
| Fig. 1 (a) Synthesis of functionalized triblock copolymers with ammonium (A), guanidinium (G), and sulfonate (S) groups. Controlled guanidination provided cationic triblock copolymers, PolyGx, of mixed A and G groups with varying compositions (x = 0.32, 0.49, 0.76, and 0.95). (b) 1H NMR spectra of the cationic triblock copolyelectrolytes with partial guanidination. The fraction of guanidinium in the cationic end-block was determined by the peak areal ratio of d and f, representing the ammonium and guanidinium groups, respectively. (c) Scheme of complex coacervate core hydrogel (C3G) preparation by mixing PolyGx and PolyS triblock copolymers in aqueous solvents. The salt concentration (cs) and guanidinium composition (x) were employed to control the C3G structures. | ||
The end-blocks of PAGE were functionalized with cationic ammonium (A) groups with chloride counterions or anionic sulfonate (S) groups with sodium counterions by thiol–ene chemistry. The PAGE-b-PEO-b-PAGE block copolymers and 2,2-dimethoxy-2-phenyl-acetophenone (DMPA, 0.05 equiv. per alkene group, Sigma-Aldrich) were dissolved in methanol, and a functional thiol reagent, either cysteamine hydrochloride (4 equiv. per alkene group, Sigma-Aldrich) or sodium 3-mercapto-1-propanesulfonate (4 equiv. per alkene group, Sigma-Aldrich), was dissolved in deionized water. The mixture of the two solutions was purged with argon, exposed to UV light for 6 h, and dialyzed with a regenerated cellulose membrane (Membrane Filtration Products, Inc., MWCO = 6–8 kDa) in pure water. Then, A- or S-functionalized triblock copolymers were recovered as a powder by lyophilization.
Guanidination of the ammonium-functionalized block copolymer was conducted using 1H-pyrazole-1-carboxamidine hydrochloride (PCA, Sigma-Aldrich) in a phosphate buffer solution (pH ∼ 12) for 3 days, followed by dialysis and lyophilization. The degree of guanidination was controlled by the amount of PCA (0.5, 0.8, 1.6, and 4 equiv. per amino group), giving cationic triblock copolymers with varying compositions of functional moieties on the end-blocks (32, 49, 76, and 95% guanidinium, respectively).
Block copolymers were characterized by a combination of 1H NMR spectroscopy (Unity-Inova 500) and SEC traces (JASCO). The degree of polymerization of the AGE end-block was calculated as Nend = 44 (Mn,end = 5.0 kg mol−1) using the molecular weight of PEO, and the overall molecular weight distribution of PAGE-b-PEO-b-PAGE was estimated as Đ = 1.06 (Fig. S1, ESI†). Nearly complete conversions in the thiol–ene reactions were confirmed by the absence of allyl group peaks at 5.8 and 5.2 ppm in the 1H NMR spectra of ammonium- and sulfonate-functionalized block copolymers (Fig. 1(b)). In addition, the degree of guanidination in partially guanidinated polymers was determined as x = 0.32, 0.49, 0.76, and 0.95 by the ratio of peak areas corresponding to the A and G groups in the 1H NMR spectra (Fig. 1(b)).
Hydrogel preparation
The anionic and cationic triblock copolymers were dissolved separately in the aqueous solution of the target NaCl concentration (cNaCl) at a polymer concentration of 8 wt%. Two solutions were mixed at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric charge ratio between the oppositely charged moieties and vortexed vigorously for 20 s at room temperature. As the copolyelectrolytes were fully neutralized by counterions prior to lyophilization, the stoichiometric mixing resulted in pH ∼ 7 without additional pH adjustment, which ensured the complete ionization of the polyelectrolyte ionic groups, i.e., ammonium, guanidinium, and sulfonate. Then, the hydrogels were left in sealed vials at 25 °C for 24 h before taking measurements. Hereafter, the hydrogels are referred to as Gx + S, where x indicates the percentage of the guanidinium groups in the cationic end-blocks; x = 0, 0.32, 0.49, 0.76, and 0.95 in this study (Fig. 1(c)).
:1 stoichiometric charge ratio between the oppositely charged moieties and vortexed vigorously for 20 s at room temperature. As the copolyelectrolytes were fully neutralized by counterions prior to lyophilization, the stoichiometric mixing resulted in pH ∼ 7 without additional pH adjustment, which ensured the complete ionization of the polyelectrolyte ionic groups, i.e., ammonium, guanidinium, and sulfonate. Then, the hydrogels were left in sealed vials at 25 °C for 24 h before taking measurements. Hereafter, the hydrogels are referred to as Gx + S, where x indicates the percentage of the guanidinium groups in the cationic end-blocks; x = 0, 0.32, 0.49, 0.76, and 0.95 in this study (Fig. 1(c)).
Small-angle X-ray and neutron scattering (SAX/NS)
SAXS measurements were performed using the 4C SAXS-II beamline at the Pohang Accelerator Laboratory (PAL), South Korea.26 A wavelength of λ = 0.734 Å and sample-to-detector distance of 4.3 m were chosen to cover a q range of 0.007 Å−1 < q < 0.15 Å−1, where the momentum transfer, q, is defined as q = 4πλ−1sin(θ/2). The Gx + S hydrogels were loaded in 1.5 mm boron-rich capillaries and flame-sealed to prevent water evaporation, and all measurements were performed at 25 °C. The samples were exposed to X-ray for at least 3 s, and the two-dimensional scattering patterns were azimuthally averaged to provide a one-dimensional plot of intensity versus q. Background scattering obtained from the solvent was subtracted from the hydrogel scattering and the resulting data were calibrated to an absolute scale using a glassy carbon reference.
SANS measurements were performed at the Quokka beamline at the Australian National Science and Technology Organisation (ANSTO).27 The instrument configuration of a wavelength λ = 6 Å, a wavelength spread (Δλ/λ) of 0.1, and sample-to-detector distances of 8 m and 1.3 m were selected to provide a q range of 0.007 Å−1 < q < 0.4 Å−1. Two-dimensional scattering images were corrected for detector sensitivity, sample transmission, empty cell scattering, and sample thickness, and then azimuthally averaged to produce the I(q) vs. q plot following standard procedures.28 The scattering intensity was reduced to an absolute scale using the direct beam flux method, the two measurements from the two detectors were merged into a single file, and the coherent scattering intensity was obtained after subtracting solvent scattering. The Gx + S hydrogels prepared in D2O were placed in demountable quartz cells with a path length of 2 mm and exposed to the neutron beam for at least 15 min. All measurements were performed at 25 °C.
Both the SAXS and SANS profiles were fitted with a model where the ABA triblock copolymer hydrogels were treated as randomly dispersed spherical cores with polydisperse core radii and Gaussian chains attached to the core surfaces in the medium, as reported earlier.19 Knowledge about the X-ray and neutron scattering length densities (SLDs) of Gx + S coacervate cores is crucial for the fitting procedure. We estimated these values using the simultaneous fitting process of SAXS and SANS profiles with the molecular characteristics of PolyGx and PolyS. The SLD values of Gx + S cores for neutron and X-ray scattering are reported in Table 1. The fitting results indicated the core radius, the inter-core distance, the aggregation number, and the water fraction in the cores. The detailed fitting model and SLD estimation are described in the ESI.†
| Sample code | ρ N,pair [10−6 Å−2] | ρ X,pair [10−6 Å−2] |
|---|---|---|
| G0 + S | 1.31 | 12.73 |
| G0.32 + S | 1.48 | 12.72 |
| G0.49 + S | 1.58 | 12.72 |
| G0.76 + S | 1.72 | 12.71 |
| G0.95 + S | 1.83 | 12.70 |
Light scattering
Light scattering experiments were performed using a Nano Particle Analyzer (SZ-100, HORIBA) to determine the critical salt concentrations of the Gx + S hydrogels. Aqueous solutions at a polymer concentration of 2 wt% and varying NaCl concentrations were contained in the cuvettes with an optical path of 1 cm and illuminated using a laser with a wavelength of 532 nm. The intensities of the transmitted light and scattered light at a scattering angle of 90° were recorded at 25 °C. Normalized transmission was estimated as the ratio of the transmitted intensity of the sample to that of the solvent.Results
Hydrogel preparation
Complex coacervate core hydrogels (C3Gs) containing various fractions of ammonium/guanidinium groups in the cationic triblock copolymer were prepared by mixing two oppositely charged ABA triblock copolymer solutions. Complex coacervate cores were formed by two oppositely charged end-blocks and connected by neutral hydrophilic PEO blocks in aqueous solvents. Our previous study revealed that, as the polymer concentration increases, the triblock copolymer solution mixtures show a transition from a phase-separated state to a homogeneous viscoelastic fluid or gel and to an elastic solid.19 At a sufficiently high polymer concentration (i.e., ≥ 9 wt%), the spherical coacervate cores are positioned on a body-centered cubic lattice. In this study, the polymer concentration was fixed at 8 wt%, where all solution mixtures form a homogeneous viscoelastic gel regardless of the degree of guanidination, x, and the added salt concentration, cNaCl, yet the coacervate cores are still spatially disordered. Since the functionalized triblock copolymers were derived from an identical precursor triblock copolymer, only the composition of ammonium (A) and guanidinium (G) moieties (not the total number of cations) affects the cNaCl dependent structure of C3Gs. This synthetic route also minimizes the effects of the backbone length mismatch29 and the chemical differences in polymer constituents other than the ionic groups (e.g., backbone hydrophobicity)15 on the coacervation equilibrium and C3G assembly.Fig. 2 displays the viscoelastic gel-to-sol transition of C3Gs as a function of x and cNaCl. At cNaCl = 0 M, the G0 + S hydrogels containing only ammonium groups are significantly deformed within 20 s after vial inversion, which is consistent with our previous results.19 In contrast, G0.95 + S hydrogels are nearly intact upon vial inversion up to cNaCl = 1.2 M. Between these extremes, the gel-to-sol transition of the Gx + S hydrogels occurs at higher cNaCl with increasing x, indicating that the relaxation process of guanidinium-rich C3Gs becomes less salt-responsive. Since the addition of salt reduces the interaction of the intrinsic ion pairs or even breaks them and allows more water molecules to swell and lubricate the coacervate phase,30,31 the chain mobility of the coacervate phase and thus the hydrogel relaxation process are enhanced at higher salt concentrations. Consequently, the weaker salt responsiveness of guanidinium-rich C3Gs indicates that the guanidinium–sulfonate (G–S) ion pair are harder to break with salt than the ammonium–sulfonate (A–S) pair,24 leading to slower relaxation.19,22 A more detailed investigation of the ionic group effect on the relaxation kinetics will be discussed in a separate study.
 | ||
| Fig. 2 Representative photographs of 8 wt% Gx + S hydrogels at cNaCl = 0, 0.6 and 1.2 M. Vials were inverted for 20 s before the snapshot was taken. | ||
The critical salt concentration, c*, above which the coacervate cores are fully disintegrated, is also dependent on x (Fig. S2, ESI†). Light scattering experiments on diluted mixtures at 2 wt% provided c* = 0.61 and 1.05 M for G0 + S and G0.32 + S, respectively, whereas c* could not be determined for the other hydrogels of higher x because the clear solutions were not observed at higher cNaCl (up to 3 M). The larger c* for guanidinium-rich C3Gs is consistent with the weaker salt responsiveness of guanidinium-rich C3G relaxation. It is noted that hydrogel relaxation becomes faster as cNaCl approaches c*, but the C3Gs are eventually fluidized before reaching c* where the cores completely disassociate.
Based on the observed behavior of C3Gs, we expect that the assembly of the coacervate cores and their structural features are significantly affected by x as well as cNaCl. To verify such impacts of the ionic group on the C3Gs structure, small-angle X-ray/neutron scattering measurements were conducted on the C3Gs prepared at various guanidinium fractions and salt concentrations.
Small-angle X-ray/neutron scattering (SAX/NS)
Fig. 3 shows the representative SAXS and SANS profiles obtained from 8 wt% Gx + S hydrogels at cNaCl = 0, 0.2, and 0.4 M. The liquid-like structure peak observed near q* = 0.02–0.03 Å−1 is attributed to the inter-particle correlation between the coacervate cores, and the peak position of SAXS and SANS is nearly identical for each Gx + S hydrogel. In addition, the intensity fluctuations with the minima near 0.05 Å−1 < qmin < 0.07 Å−1, corresponding to the intra-particle scattering of the coacervate cores, are clearly observed in the guanidinium-rich C3Gs. At constant cNaCl, both q* and qmin shift to lower values with increasing x, indicating a larger inter-core distance and core dimension. With increasing cNaCl, q* and qmin shift to higher values and the scattering profiles are gradually smeared (see Fig. S4–S8, ESI†). While the SAX/NS profiles at cNaCl = 0 M are nearly independent of x, the significant differences in the SAX/NS profiles obtained at higher cNaCl exhibit a pronounced effect on the guanidinium fraction. For ammonium-rich C3Gs, the minima become less distinguishable at higher cNaCl, indicating less well-defined core structures. The salt-induced structural changes become less pronounced at higher x, implying that the nanostructure in guanidinium-rich C3Gs is more robust against the salt.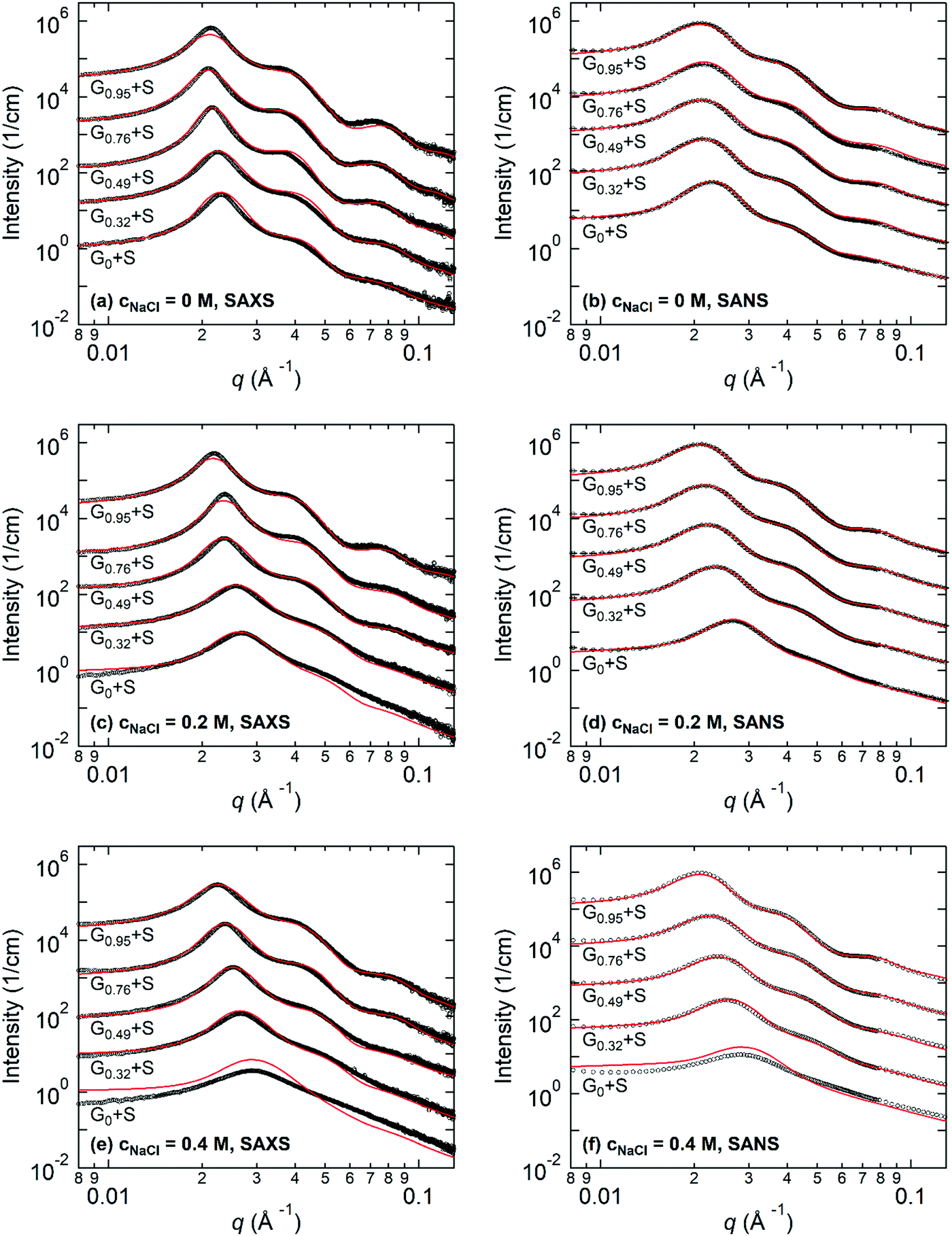 | ||
| Fig. 3 SAXS and SANS profiles for 8 wt% Gx + S hydrogels at cNaCl = 0, 0.2, and 0.4 M, where x denotes the molar percentage of the guanidinium moiety over total cations in the end-block of the triblock copolymer. Solid curves represent the best fit to the fitting model. Data and curves are vertically shifted for clarity. | ||
For the structural investigation of hydrogels, SAX/NS intensity profiles were fit using the model function of spherical cores with corona chains emanating from the core, as described in the ESI.† To determine the X-ray and neutron scattering length densities (SLDs) of the coacervates, we sampled the molecular volumes of the ion pairs (i.e., the physical basic unit of coacervates). We also performed fitting routines for the SAXS and SANS data, simultaneously, using X-ray and neutron SLDs corresponding to the sampled ion pair volumes (see the ESI† for the detailed formalism). Then, we selected the ion pair volume and the corresponding SLDs of the ion pairs for which both SAXS and SANS fitting results showed the best agreement. The resulting X-ray and neutron SLDs of the coacervate phases are summarized in Table 1. The estimated neutron SLDs for the guanidinium–sulfonate (G–S) ion pair show good agreement with the result of Ortony et al.,32 verifying the reliability of our SLD evaluation method.
Fig. 4 illustrates the representative fitting results including the water fraction in the cores, the core radius, the aggregation number, and the stretching of the PEO corona for the Gx + S hydrogels as functions of cNaCl determined by both SAXS and SANS. The complete SAX/NS fitting results are shown in Tables S3–S10 in the ESI.† Note that the SAXS and SANS fitting results show close agreement and present qualitatively similar trends as functions of the salinity and the guanidinium fraction. At cNaCl = 0 M, the water fractions in the core, fcore,water, coincide with thermogravimetrically measured values in our previous study:24fcore,water = 0.6 and 0.4 for the A + S and G + S bulk coacervates which are analogous to G0 + S and G0.95 + S hydrogels, respectively. Furthermore, fcore,water systematically decreases as x increases at constant cNaCl. This behavior potentially indicates that the guanidinium group is more hydrophobic than the ammonium group (as discussed below).24
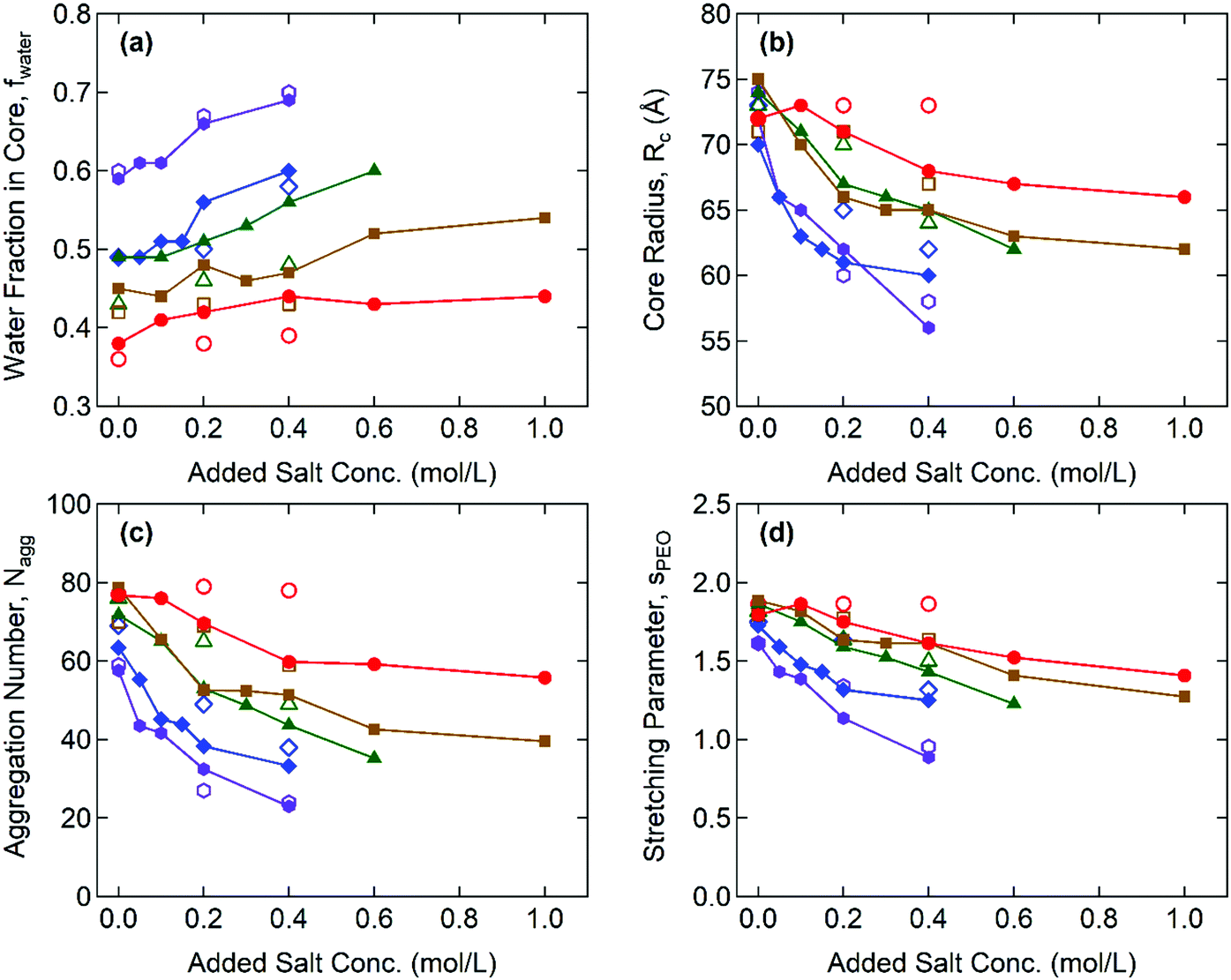 | ||
| Fig. 4 SAXS (closed symbols) and SANS (open symbols) fit results from G0 + S (purple/hexagons), G0.32 + S (blue/diamonds), G0.49 + S (green/triangles), G0.76 + S (brown/squares), and G0.95 + S (red/circles) hydrogels as functions of the added salt concentration. (a) Water fraction in the coacervate core, (b) core radius, (c) aggregation number, and (d) dimensionless parameter of the PEO chain stretching. | ||
With increasing cNaCl, the dissociation of the polyelectrolyte–polyelectrolyte ion pairs led to the enhanced hydration of coacervate phases as signified by the increase in fcore,water. This is accompanied by the simultaneous decreases in the core radius, Rcore, and the aggregation number, defined as the total number of end-blocks per core, Nagg = (1 − fcore,water)(4π/3)Rcore3/vcore, where vcore is the average molecular volume of the polyelectrolyte chains (see Fig. 4(b) and (c)). Both indicate a lower interfacial tension between the core and the aqueous medium, γcore, (i.e., milder segregation of the coacervate phases at higher salinity). Furthermore, the sensitivity of Rcore to cNaCl is weaker at higher x, reflecting that the polyelectrolyte–polyelectrolyte ion pairs become more robust against the salt as more ammonium is replaced with guanidinium. Without added salt (cNaCl = 0), the hydrogels have a similar magnitude of Rcore regardless of x. However, as cNaCl increases, Rcore rapidly decreases for the ammonium-rich hydrogels but remains nearly constant for the guanidinium-rich hydrogels. The cNaCl dependence of Nagg shows a similar trend, as Nagg is closely related to Rcore.
The extent of stretching of PEO chains in the “bridging” conformation state was evaluated by comparing the effective corona thickness, Rcorona = Rhs − Rcore, and the chain dimension of PEO in the unperturbed state. Here, Rhs is the effective hard-sphere radius determined from the SAX/NS model fitting as a measure of the overall micelle radius, i.e., the distance from the center of the core to the midpoint of the PEO corona chains bridging the cores. The dimensionless parameters of PEO chain stretching, defined as sPEO = Rcorona/Rg, decrease with increasing cNaCl, as shown in Fig. 4(d), where Rg = 4.4 nm is the radius of gyration of the PEO mid-block (20 kDa) in an aqueous solvent.33 This finding signifies that larger aggregates (and therefore a larger gap distance between the neighboring cores) are formed at lower salinity, which reduces the interfacial area per chain at the expense of a higher entropic penalty of stretching for the bridging chains.34 In addition, sPEO becomes less sensitive to cNaCl for the guanidinium-rich hydrogels, which is consistent with the weaker salt responsiveness of the C3G structure as characterized by fcore,water and Rcore. All thermodynamic and structural characteristics of Gx + S hydrogels are found to be less sensitive to the salt as the guanidinium content increases.
Discussion
Scattering analysis of Gx + S hydrogels clearly showed that the coacervate cores become less hydrated and the extent of hydration (fcore,water) is less sensitive to salt as more guanidinium groups are incorporated in the cationic end-blocks. Based on the scattering analysis including fcore,water, Rcore, and Nagg, we present a simple estimation of the interfacial tension of the coacervate cores, γcore, supported by classical scaling theories for general block copolymer micelles.35–37 The γcore reflects the thermodynamic cost of mixing incompatible phases and is a driving force of the self-assembly in triblock copolymer hydrogels. It is noted that the scaling theory for the hairy micelles suggested by Daoud and Cotton is used for our scaling analysis. The estimated free energy per chain due to the excluded volume effect in the corona, Fcorona, is almost twice as large as the estimated free energy per chain due to the core block stretching, Fcore, in our system35,36 (see Fig. S9, ESI†).Following the formalism for the hairy micelles,36Fcore is omitted and only Fcorona and the interfacial energy per chain, Fint, are considered to calculate the total free energy per chain involved in the hydrogels,
| Ftotal ≈ Fcorona + Fint ∼ p1/2kBT + γcoreRcore2p−1, | (1) |
| Ftotal ∼ p1/2kBT + γcorea2fcore,pol−2/3N2/3p−1/3, | (2) |
| p ∼ (γcorea2/kBT)6/5fcore,pol−4/5N4/5, | (3) |
Fig. 5 displays g as a function of the overall salt concentration, cs, at different x values based on the SAXS results, where cs represents both the added salt (i.e., cNaCl) and the counterions originally attached to the block copolymers. Previously Qin et al. proposed γ ∼ (1 − cs/c*)3/2 to illustrate the salt dependence of the coacervate interfacial tension,39 and this functional form successfully captured the experimental data over a wide range of cs.20,21 Since g is linearly proportional to γcore, the dotted lines in Fig. 5 are the best fits to the model function of g = g0(1 − cs/c*)3/2, in which the reduced interfacial tension at zero-salt limit (cs = 0), g0, and the critical salt concentration, c*, were used as fitting parameters. Since the equation proposed by Qin et al. is originally constructed for a weak-segregation limit and considers only the electrostatic interaction,39 a good agreement with our experimental data reflects that the electrostatic interaction is dominant to control the C3G structure within our experimental conditions.
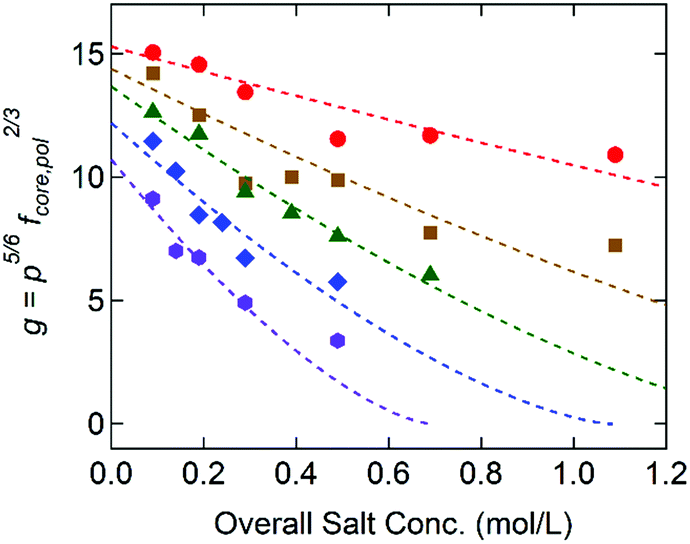 | ||
| Fig. 5 Dimensionless parameter for the interfacial tension, g = p5/6fcore,pol2/3, as a function of the overall salt concentration for G0 + S (purple/hexagons), G0.32 + S (blue/diamonds), G0.49 + S (green/triangles), G0.76 + S (brown/squares), and G0.95 + S (red/circles). The dashed lines are the best fits to the model function of g = g0(1 − cs/c*)3/2. | ||
Furthermore, the monomers containing either ammonium or guanidinium groups do not differ much in size; the molecular volumes of the A–S and G–S ion pairs were evaluated as vpair = 0.55 and 0.60 nm3, respectively. Therefore, we assume that the monomer size, a, is constant regardless of the guanidinium content, and so is the proportionality factor, defined as α = γcore/g ∼ a−2. Using the reference value of γcore = 1.4 mN m−1 for the G0 + S hydrogel at cs = 0.1 M from our previous study,19α = 0.16 mN m−1 was obtained and used to evaluate the γcore and the interfacial tension at the zero-salt limit, γcore,0 = αg0. The results, γcore,0 and c*, are displayed as functions of x in Fig. 6 and tabulated in Table S11 (ESI†).
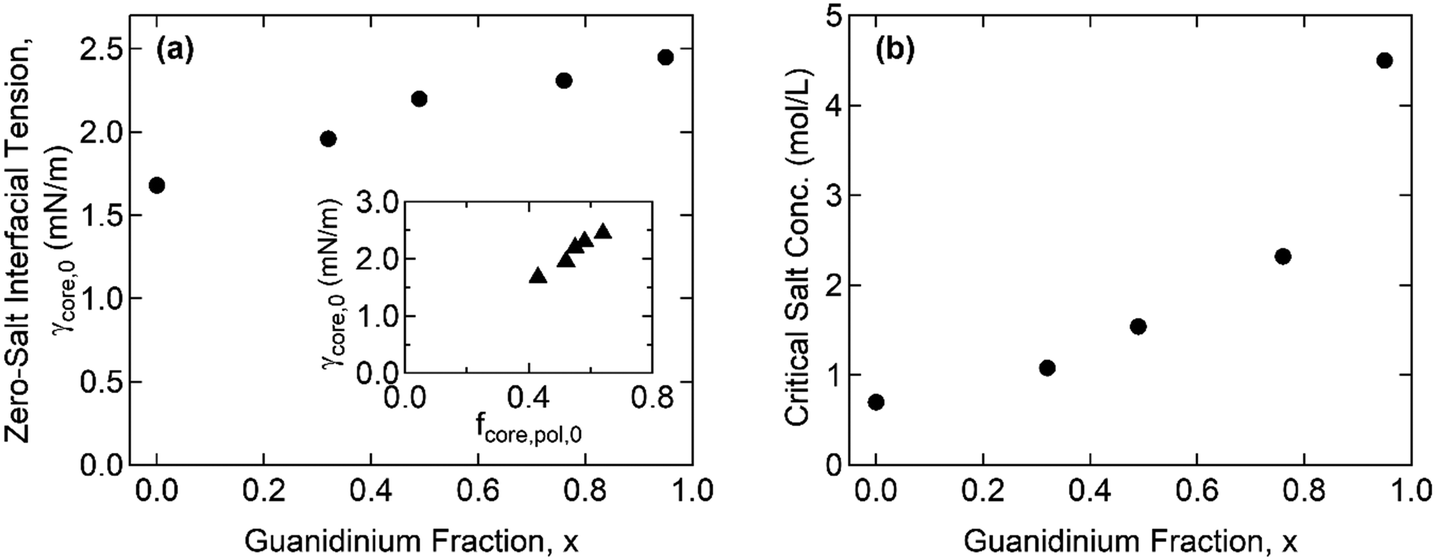 | ||
| Fig. 6 Effects of the guanidinium fraction, x, on (a) the interfacial tension at the zero-salt limit, γcore,0, and (b) the critical salt concentration, c*, of the complex coacervates. The inset in (a) compares γcore,0 with the polymer fraction in the coacervate core at the zero-salt limit, fcore,pol,0. | ||
The interfacial tension of the complex coacervate phases retrieved from the core structures gives valuable information about the ion-specificity of the coacervate cores. As shown in Fig. 6(a), γcore,0 increases with the guanidinium fraction x despite the same polyelectrolyte lengths and charge densities. This finding is attributed to the greater hydrophobicity of guanidinium than ammonium and the stronger non-electrostatic interaction between the G–S ion pair than the A–S ion pair. The guanidinium group is a poorly hydrated cation due to its broadly delocalized surface charge and exposed water-deficient hydrophobic surface resulting in the parallel stacking of like-charged ions.40,41 In addition, the formation of multiple hydrogen bonds was proposed as a possible reason for the stronger ion pair interaction involving guanidinium groups.23,24
The inset in Fig. 6(a) shows a linear correlation between γcore,0 and the polymer fraction in the coacervate core at the zero-salt limit, fcore,pol,0, which was obtained from the linear extrapolation from the data in Fig. 4(a) to cs = 0. Recently, Rumyantsev et al. reported the scaling relationships, fpol,0 ∼ u4/7σ8/7v−5/7 and γ0 ∼ u6/7σ12/7v−4/7, for the complex coacervates in salt-free good solvent (v > 0), where u = lB/a = (e2/ε0εrkBT)/a is the dimensionless Bjerrum length, σ is the charge density of polyelectrolyte, and v = B/a3 is the dimensionless second virial coefficient for the monomeric interactions.42 Since u and σ are fixed in the present system, the scaling relationships qualitatively agree with our results that both fpol,0 and γ0 increase with x through the reduction in v (i.e., the overall coacervate becomes more hydrophobic). This finding is also consistent with general observations in which a polyelectrolyte with a greater hydrophobicity led to a lower water fraction in the coacervate phases.16,43,44 Moreover, given that only v changes with x, γ0/fpol,0 ∼ u2/7σ4/7v1/7 leads to the observed linearity (i.e., γcore,0 ∼ fpol,0) since the exponent for v is negligibly small.
Fig. 6(b) shows that c* increases with x, which is consistent with previous reports in which the greater salt resistance of coacervates at a higher guanidinium content is attributed to the hydrophobic nature of guanidinium groups.22,24 Although the difference in the pKa of ammonium and guanidinium (pKa = 9.2 and 13.6 for ammonium and guanidinium chloride, respectively) was previously pointed out to account for the different salt resistance,14 the pH ∼ 7 in the present study ensures a negligible difference in the degree of ionization in both the ammonium and guanidinium groups. We note that increasing c* with enhanced hydrophobicity in homologous polyelectrolytes was also reported by others, where the hydrophobicity was controlled by the length of alkyl chains per charged repeat unit43 or copolymerization of hydrophobic moieties with ionic monomers.44
The critical salt concentrations obtained from the fit, c*, are consistent with the light scattering measurements of c* = 0.61 M and 1.05 M for G0 + S and for G0.32 + S, respectively, within experimental errors. However, the light scattering failed to detect c* at a higher x. Specifically, the complete disintegration of the coacervate cores into weakly scattering single chains was not found below cs < 3 M. We attribute this phenomenon to the self-associative interactions of guanidinium-rich polyelectrolyte blocks, such as hydrophobicity and π–π interactions,45,46 which drives phase separation at high salinity where the electrostatic self-repulsion is screened out.24 This “salting-out” behavior is believed to occur simultaneously with the dissociation of complex coacervation, thereby interfering with the determination of c* using light scattering. This observation suggests a promising future avenue to investigate the behavior of polyelectrolytes containing guanidinium.
Conclusion
The effect of the ionic group on the complex coacervate core hydrogel (C3Gs) structure was investigated using small-angle X-ray/neutron scattering (SAX/NS) experiments using a detailed fitting model. A series of ABA triblock copolyelectrolytes, with various cationic compositions of the ammonium and guanidinium groups or anionic sulfonate group, was prepared by anionic ring-opening polymerization and post-polymerization modification. We achieved consistency between the SAXS and SANS results obtained from all C3G samples by adjusting the partial molecular volumes of the ion pairs evaluated. As the salt concentration increases and the guanidinium fraction decreases, the core size and the aggregation number decrease, and the water fraction in the coacervate cores increases. This is mainly attributed to the weaker ion-pair interaction and thus the reduced interfacial tensions of the complex coacervate cores, γcore. Based on the structural parameters obtained from SAX/NS analyses, we estimated the γcore with the help of a classical scaling model for the block copolymer micelles and observed that the scaling of γcore ∼ γ0(1 − cs/c*)3/2 shows good agreement with our experimental data. In addition, γ0 and the critical salt concentrations, c*, increase with an increasing guanidinium fraction, which is attributed to the more hydrophobic nature and stronger hydrogen bonding in the guanidinium group than in the ammonium group. Overall, the systematic investigation of C3Gs with varying guanidinium compositions provides insights into the effect of an ionic group on the structures of C3Gs and analogous coacervate-driven self-assemblies.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Research Foundation (NRF) Grant funded by the Korea Government (No. NRF-2021R1A2C2011164, NRF-2018R1A5A1024127, and NRF-2020K1A3A7A09077997). QUOKKA data were collected during proposal DB9220.References
- B. Jeong, S. W. Kim and Y. H. Bae, Adv. Drug Delivery Rev., 2012, 64, 154–162 CrossRef.
- T. Annable, R. Buscall, R. Ettelaie and D. Whittlestone, J. Rheol., 1993, 37, 695–726 CrossRef CAS.
- M. Lemmers, J. Sprakel, I. K. Voets, J. van der Gucht and M. A. Cohen Stuart, Angew. Chem., Int. Ed., 2010, 49, 708–711 CrossRef CAS PubMed.
- S. Srivastava and M. V. Tirrell, Adv. Chem. Phys., 2016, 161, 499–544 CrossRef CAS.
- C. E. Sing and S. L. Perry, Soft Matter, 2020, 16, 2885–2914 RSC.
- K. M. Jin and Y. H. Kim, J. Controlled Release, 2008, 127, 249–256 CrossRef CAS PubMed.
- F. Vecchies, P. Sacco, E. Decleva, R. Menegazzi, D. Porrelli, I. Donati, G. Turco, S. Paoletti and E. Marsich, Biomacromolecules, 2018, 19, 3936–3944 CrossRef CAS PubMed.
- D. V. Krogstad, N. A. Lynd, S. H. Choi, J. M. Spruell, C. J. Hawker, E. J. Kramer and M. V. Tirrell, Macromolecules, 2013, 46, 1512–1518 CrossRef CAS.
- D. V. Krogstad, N. A. Lynd, D. Miyajima, J. Gopez, C. J. Hawker, E. J. Kramer and M. V. Tirrell, Macromolecules, 2014, 47, 8026–8032 CrossRef CAS.
- S. Srivastava, A. E. Levi, D. J. Goldfeld and M. V. Tirrell, Macromolecules, 2020, 53, 5763–5774 CrossRef CAS.
- J. T.-G. Overbeek and M. J. Voorn, J. Cell. Comp. Physiol., 1957, 49, 7–26 CrossRef CAS.
- C. Papadakis and C. Tsitsilianis, Gels, 2017, 3, 3 CrossRef PubMed.
- H. Jung, S. E. Gang, J. M. Kim, T. Y. Heo, S. Lee, E. Shin, B. S. Kim and S. H. Choi, Macromolecules, 2020, 53, 10339–10348 CrossRef CAS.
- J. N. Hunt, K. E. Feldman, N. A. Lynd, J. Deek, L. M. Campos, J. M. Spruell, B. M. Hernandez, E. J. Kramer and C. J. Hawker, Adv. Mater., 2011, 23, 2327–2331 CrossRef CAS PubMed.
- J. C. Fu, H. M. Fares and J. B. Schlenoff, Macromolecules, 2017, 50, 1066–1074 CrossRef CAS.
- J. Z. Lou, S. Friedowitz, J. Qin and Y. Xia, ACS Cent. Sci., 2019, 5, 549–557 CrossRef CAS PubMed.
- T.-Y. Heo, I. Kim, L. Chen, E. Lee, S. Lee and S.-H. Choi, Polymers, 2019, 11, 455 CrossRef PubMed.
- E. B. Zhulina and O. V. Borisov, Macromolecules, 2012, 45, 4429–4440 CrossRef CAS.
- J.-M. Kim, T.-Y. Heo and S.-H. Choi, Macromolecules, 2020, 53, 9234–9243 CrossRef CAS.
- E. Spruijt, J. Sprakel, M. A.-C. Stuart and J. van der Gucht, Soft Matter, 2010, 6, 172–178 RSC.
- S. Ali and V. M. Prabhu, Macromolecules, 2019, 52, 7495–7502 CrossRef CAS PubMed.
- K. Sadman, Q. F. Wang and K. R. Shull, ACS Macro Lett., 2019, 8, 117–122 CrossRef CAS.
- V. Petrauskas, E. Maximowitsch and D. Matulis, J. Phys. Chem. B, 2015, 119, 12164–12171 CrossRef CAS PubMed.
- S. Kim, M. Lee, W. B. Lee and S.-H. Choi, Macromolecules, 2021, 54, 7572–7581 CrossRef CAS.
- T.-Y. Heo, S. Kim, L. Chen, A. Sokolova, S. Lee and S.-H. Choi, ACS Macro Lett., 2021, 10, 1138–1144 CrossRef CAS PubMed.
- K.-W. Kim, J. Kim, Y. D. Yun, H. Ahn, B. Min, N. H. Kim, S. Rah, H.-Y. Kim, C.-S. Lee, I. D. Seo, W.-W. Lee, H. J. Choi and K. S. Jin, Biodesign, 2017, 5, 24–29 Search PubMed.
- K. Wood, J. P. Mata, C. J. Garvey, C. M. Wu, W. A. Hamilton, P. Abbeywick, D. Bartlett, F. Bartsch, P. Baxter, N. Booth, W. Brown, J. Christoforidis, D. Clowes, T. d’Adam, F. Darmann, M. Deura, S. Harrison, N. Hauser, G. Horton, D. Federici, F. Franceschini, P. Hanson, E. Imamovic, P. Imperia, M. Jones, S. Kennedy, S. Kim, T. Lam, W. T. Lee, M. Lesha, D. Mannicke, T. Noakes, S. R. Olsen, J. C. Osborn, D. Penny, M. Perry, S. A. Pullen, R. A. Robinson, J. C. Schulz, N. Xiong and E. P. Gilbert, J. Appl. Crystallogr., 2018, 51, 294–314 CrossRef CAS.
- S. R. Kline, J. Appl. Crystallogr., 2006, 39, 895–900 CrossRef CAS.
- A. Harada and K. Kataoka, Science, 1999, 283, 65–67 CrossRef CAS PubMed.
- M. Yang, J. Shi and J. B. Schlenoff, Macromolecules, 2019, 52, 1930–1941 CrossRef CAS.
- F. J. Morin, M. L. Puppo and J. E. Laaser, Soft Matter, 2021, 17, 1223–1231 RSC.
- J. H. Ortony, S.-H. Choi, J. M. Spruell, J. N. Hunt, N. A. Lynd, D. V. Krogstad, V. S. Urban, C. J. Hawker, E. J. Kramer and S. Han, Chem. Sci., 2014, 5, 58–67 RSC.
- N. Zeibacz, S. A. Wieczorek, T. Kalwarczyk, M. Fiałkowski and R. Hołyst, Soft Matter, 2011, 7, 7181–7186 RSC.
- J. J. Shin, E. J. Kim, K. H. Ku, Y. J. Lee, C. J. Hawker and B. J. Kim, ACS Macro Lett., 2020, 9, 306–317 CrossRef CAS.
- P.-G. De Gennes, Solid State Phys., Suppl., 1978, 14, 1–18 CAS.
- M. Daoud and J. P. Cotton, J. Phys., 1982, 43, 531–538 CrossRef CAS.
- A. Halperin, Macromolecules, 1987, 20, 2943–2946 CrossRef CAS.
- A. M. Rumyantsev, E. B. Zhulina and O. V. Borisov, ACS Macro Lett., 2018, 7, 811–816 CrossRef CAS.
- J. Qin, D. Priftis, R. Farina, S. L. Perry, L. Leon, J. Whitmer, K. Hoffmann, M. Tirrell and J. J. de Pablo, ACS Macro Lett., 2014, 3, 565–568 CrossRef CAS.
- P. E. Mason, G. W. Neilson, C. E. Dempsey, A. C. Barnes and J. M. Cruickshank, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4557–4561 CrossRef CAS PubMed.
- P. E. Mason, G. W. Neilson, J. E. Enderby, M. L. Saboungi, C. E. Dempsey, A. D. MacKerell and J. W. Brady, J. Am. Chem. Soc., 2004, 126, 11462–11470 CrossRef CAS PubMed.
- A. M. Rumyantsev, E. B. Zhulina and O. V. Borisov, Macromolecules, 2018, 51, 3788–3801 CrossRef CAS.
- K. Sadman, Q. F. Wang, Y. Y. Chen, B. Keshavarz, Z. Jiang and K. R. Shull, Macromolecules, 2017, 50, 9417–9426 CrossRef CAS.
- J. Huang and J. E. Laaser, ACS Macro Lett., 2021, 10, 1029–1034 CrossRef CAS PubMed.
- M. Vazdar, F. Uhlig and P. Jungwirth, J. Phys. Chem. Lett., 2012, 3, 2021–2024 CrossRef CAS.
- N. Zydziak, M. H. Iqbal, A. Chaumont, A. Combes, E. Wasielewski, M. Legros, L. Jierry, P. Lavalle, F. Boulmedais and D. Chan-Seng, Eur. Polym. J., 2020, 125, 109528 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: The molecular characterizations, critical salt concentration measurements, SAX/NS analysis model and results, scaling analyses for the free energies, and the interfacial tension table. See DOI: https://doi.org/10.1039/d2sm00255h |
| ‡ The authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |