DOI:
10.1039/D2PY00425A
(Paper)
Polym. Chem., 2022,
13, 3489-3497
Core–shell microgels having zwitterionic hydrogel core and temperature-responsive shell prepared via inverse miniemulsion RAFT polymerization†
Received
4th April 2022
, Accepted 17th May 2022
First published on 18th May 2022
Abstract
Stimuli-responsive core–shell microgels are of significant interest because of their fascinating applications due to the different swelling/shrinkage properties of their core and shell networks. Because such stimuli-responsive core–shell microgels are conventionally prepared by precipitation polymerization, hydrophilic and biological molecules are difficult to incorporate into stimuli-responsive core–shell microgels. We have focused on the preparation of stimuli-responsive core–shell microgels with zwitterionic hydrogel core by inverse miniemulsion RAFT polymerization method because it enables the facile incorporation of hydrophilic and biological molecules maintaining their functions. This study describes the preparation of core–shell microgels composed of zwitterionic poly(methacryloyloxyethyl phosphorylcholine) (PMPC) hydrogel core and temperature-responsive poly[oligo(ethylene glycol)methacrylate-co-2-(2′-methoxyethoxy)ethyl methacrylate] (P(OEGMA-co-MEO2MA)) shell. A water-soluble block copolymer emulsifier composed of a hydrophilic/lipophilic P(OEGMA-co-MEO2MA) block and a hydrophilic PMPC block was synthesized via reversible addition fragmentation chain transfer (RAFT) polymerization. The water-in-oil (W/O) emulsions were successfully formed in a water–chloroform two-phase system in the presence of the resulting P(OEGMA-co-MEO2MA)-b-PMPC, because it stabilized the interface between water and chloroform by the distribution of PMPC and P(OEGMA-co-MEO2MA) blocks in the water and chloroform phases, respectively. The inverse miniemulsion RAFT copolymerization of MPC and N,N′-methylenebisacrylamide proceeded from the P(OEGMA-co-MEO2MA)-b-PMPC emulsifier stabilizing a water droplet of W/O emulsion, resulting in core–shell-structured microgels comprising PMPC core and P(OEGMA-co-MEO2MA) shell. The resulting PMPC core–shell microgels dispersed stably in both chloroform and water without a thorough washing process. The transmittance of the aqueous PMPC core–shell microgel dispersion decreased drastically above 38 °C. The decrease in the transmittance of the PMPC core–shell microgel dispersion was attributed to the fact that the P(OEGMA-co-MEO2MA) shell became hydrophobic above 38 °C. Because our method enables the facile encapsulation of various hydrophilic compounds into the core of temperature-responsive core–shell microgels, smart core–shell microgels have various potential applications, including smart drug delivery carriers and smart catalytic systems.
Introduction
Stimuli-responsive microgels exhibit rapid and reversible changes in size in response to external stimuli.1–4 Because of Pelton and Chibante's pioneering studies of temperature-responsive microgels,5 many researchers have synthesized various types of stimuli-responsive microgels. Particularly, stimuli-responsive core–shell microgels are of significant interest because they exhibit unique stimuli-responsive behaviour due to the different swelling/shrinkage properties of their core and shell networks.6 The stimuli-responsive core–shell microgels have various fascinating applications, such as smart nanoreactors,7,8 sensors9,10 and smart drug carriers.11,12 Conventional core–shell microgels are broadly divided into two classes: microgels composed of a hard-sphere core and a crosslinked hydrogel shell, and both core and shell crosslinked microgels.1 In the stimuli-responsive core–shell microgels comprising a hard-sphere core and hydrogel shell, the hard-sphere is usually made of polystyrene (PSt), silica nanoparticles and gold nanoparticles. The stimuli-responsive hydrogel shell is fabricated using seed emulsion polymerization and surface-initiated polymerization techniques. For example, we synthesized a bisphenol A (BPA)-responsive core–shell microgel composed of a silica nanoparticle core and β-cyclodextrin (CD)-conjugated polyacrylamide hydrogel shell via surface-initiated atom transfer radical polymerization (ATRP).10 The BPA-responsive core–shell microgels exhibited shrinkage in response to the target BPA due to the increase in the crosslinking density resulting from the formation of sandwich-like CD–BPA–CD complexes acting as dynamic crosslinks. The core–shell microgels comprising both hydrogel core and shell are conventionally synthesized via two-step precipitation polymerization.13,14 Various stimuli-responsive core–shell microgels with both hydrogel core and shell have been synthesized, such as double temperature-responsive core–shell microgels,13 pH-responsive amphoteric core–shell microgels15 and core–shell microgels consisting of a temperature-responsive hydrogel core and pH-responsive hydrogel shell.16 Such core–shell microgels are conventionally synthesized by precipitation polymerization. In precipitation polymerization, the oligomer chain produced by radical growth and chain propagation collapse itself to form the precursor particles. The precursor particles grow by aggregation with other precursors to form microgels. This means that the chemical structure of core–shell microgels with both hydrogel core and shell is limited since the polymers that do not form precursor particles during polymerization are unsuitable for the preparation of microgels. Therefore, the process of making core–shell microgels with versatile chemical structures is in high demand.
Inverse miniemulsion polymerization is a promising method for synthesizing microgels with various hydrophilic polymer networks.17 For example, polyacrylamide microgels were synthesized via inverse miniemulsion polymerization.18,19 The core–shell microgels with hydrophilic core were also synthesized using the inverse miniemulsion ATRP.20 In the inverse miniemulsion ATRP, the hydrophilic oligo(ethylene glycol)methacrylate (OEGMA) and crosslinker were copolymerized in a water droplet, followed by subsequent chain extension with St from the surface of the resulting POEGMA microgels to obtain the core–shell microgels comprising a hydrophilic POEGMA microgel core and hydrophobic PSt shell.
Controlled radical polymerization from a liquid–liquid interface of inverse miniemulsion has attracted significant attention for synthesizing polymer particles and capsules with well-defined structures.20,21 For example, the Zetterlund group synthesized various nanocapsules using reversible addition fragmentation chain transfer (RAFT) polymerization away from a droplet interface of a water-in-oil (W/O) emulsion stabilized with an amphiphilic block copolymer with a RAFT agent terminus.22–25 Water-dispersible nanocapsules were also synthesized by introducing the hydrophilic groups onto the nanocapsules via a post-polymerization reaction.26,27 However, the double hydrophilic core–shell microgels composed of both hydrogel core and shell have not been synthesized using the interfacial inverse miniemulsion polymerization technique.
We synthesized reductively responsive hydrophilic gel capsules that enhanced the release of a loaded model drug under reducing environments, using the interfacial inverse miniemulsion RAFT polymerization.28 We designed a novel water-soluble block copolymer emulsifier composed of a water-soluble poly(2-methacryloyloxyethyl phosphorylcholine) (PMPC) block and a hydrophilic/lipophilic poly[oligo(ethylene glycol)methacrylate] (POEGMA) block, which is different from the conventional amphiphilic emulsifier. W/O emulsions were formed in the presence of the block copolymer (PMPC-b-POEGMA) in a water–chloroform two-phase system. The PMPC-b-POEGMA acted as an emulsifier that stabilized the water–chloroform interface because the PMPC and POEGMA blocks were distributed to the water and chloroform phases, respectively. The reductively responsive gel capsules were synthesized via the interfacial RAFT polymerization of poly(ethylene glycol)methacrylate (PEGMA) and bis(2-methacryloy)oxyethyl disulfide from the PMPC-b-POEGMA emulsifier with RAFT agent at the POEGMA block terminus. The resulting gel capsules were colloidally stable in both chloroform and water without an additional hydrophilic surface modification, because they had polyPEGMA networks. This result indicates that the interfacial miniemulsion RAFT polymerization using W/O emulsions stabilized with a water-soluble block copolymer emulsifier composed of PMPC and POEGMA blocks is a useful method for synthesizing functional hydrophilic soft nanomaterials.
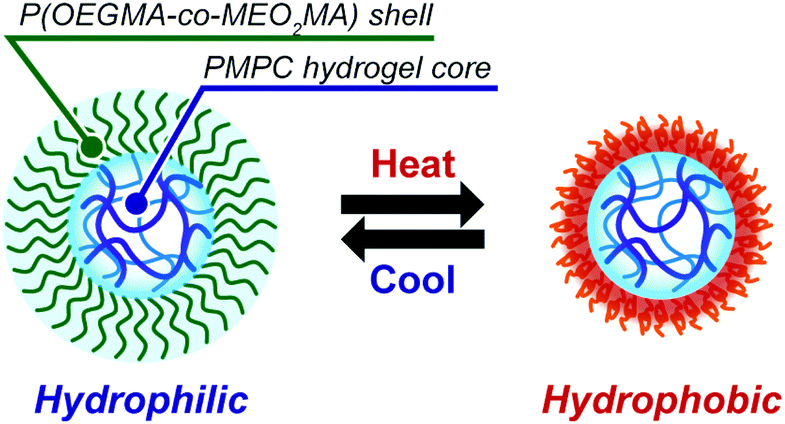
This study presents the synthesis of double hydrophilic temperature-responsive core–shell microgels using inverse miniemulsion RAFT polymerization using a water-soluble emulsifier (Fig. 1). In the inverse miniemulsion RAFT polymerization, MPC and N,N′-methylenebisacrylamide (MBAA) were copolymerized in a water-droplet from a water-soluble emulsifier with a dithiobenzoate group at the PMPC block terminus. The polymer block with the oligo(ethylene glycol) (OEG) side chain of the water-soluble emulsifier can be covalently attached to the surface of the resulting PMPC microgels as a shell because the polymerization proceeds from the water-soluble emulsifier's dithiobenzoate terminus into the water-droplet. This study also describes the temperature-responsive behaviour of resulting PMPC core–shell microgels with a side-chain OEG polymer shell that exhibits a lower critical solution temperature (LCST)-type phase transition. Only a few studies on the preparation of PMPC microgels have been conducted thus far.29–31 The PMPC microgels were synthesized via precipitation polymerization in a water/alcohol mixture and acetonitrile. Particularly, Armes and co-workers reported the synthesis of core–shell PMPC microgels with a nonionic poly(ethylene glycol) shell via precipitation polymerization.29,30 However, synthesizing PMPC microgels via precipitation polymerization restricts the encapsulation of materials such as biological macromolecules into the PMPC microgels because the precipitation polymerization should be conducted in a water/alcohol mixture. To the best of our knowledge, this is the first report of submicron-scale temperature-responsive core–shell PMPC microgels with a temperature-responsive shell. Because the biocompatible PMPC hydrogel core of the core–shell microgel can incorporate biological macromolecules without losing their functions, fundamental research on the preparation of temperature-responsive PMPC core–shell microgels will contribute significantly to creating smart drug carriers and catalytic systems.
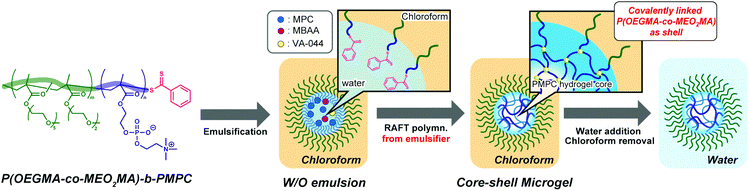 |
| | Fig. 1 Schematic illustration of the preparation of PMPC core–shell microgels by inverse miniemulsion RAFT copolymerization of MPC and MBAA using water-soluble P(OEGMA-co-MEO2MA)-b-PMPC emulsifier. | |
Experimental
Materials and methods
Chemicals.
Oligo(ethylene glycol)methacrylate (OEGMA, Mn: 300) and 4-cyano-4-(thiobenzoylthio)pentanoic acid (CTPA) were purchased from Millipore Sigma (St Louis, MO, USA). 2-(2′-Methoxyethoxy)ethyl methacrylate (MEO2MA), 4,4′-azobis(4-cyanovaleric acid) (ACVA), 2,2′-azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (VA-044) and N,N′-methylenebisacrylamide (MBAA) were purchased from FUJIFILM WAKO Pure Chemical Corporation (OSAKA, Japan). OEGMA and MEO2MA were purified using a column of basic alumina before use to remove the inhibitor. 2-Methacryloxyethyl phosphorylcholine (MPC) was kindly provided by NOF Corporation (Tokyo, Japan). All aqueous solutions were prepared with ultrapure water (Milli-Q, 18.2 MΩ cm). Other solvents were obtained from commercial sources and used without further purification.
Synthesis of P(OEGMA-co-MEO2MA).
OEGMA (1.20 g, 4.00 mmol), MEO2MA (3.01 g, 16.0 mmol), CTPA (53.5 mg, 0.20 mmol) and ACVA (11.5 mg, 6 μmol) were dissolved in 30 mL of ethanol. The solution was deoxygenated by bubbling Ar gas for 30 min, followed by stirring at 90 °C for 8 h. The product was precipitated in a 600 mL diethyl ether/hexane mixture (diethyl ether/hexane = 2/1 (v/v)) to remove the unreacted monomers. The isolated polymer product was dried under vacuum to obtain a P(OEGMA-co-MEO2MA) macroRAFT agent (2.34 g, 56% yield).
Synthesis of P(OEGMA-co-MEO2MA)-b-PMPC.
MPC (563 mg, 1.90 mmol), P(OEGMA-co-MEO2MA) macroRAFT agent (2.3 g, 95 μmol) and ACVA (5.6 mg, 19 μmol) were dissolved in 9.8 mL ethanol. The solution was deoxygenated by bubbling Ar gas for 30 min, followed by stirring at 90 °C for 8 h. Next, the reaction solution was poured into a seamless cellulose tubing (molecular weight cut-off: 3.5 kDa) and was dialyzed against methanol for purification. The solvent was removed under reduced pressure to obtain P(OEGMA-co-MEO2MA)-b-PMPC (1.62 g, 57% yield).
Formation of W/O emulsion using P(OEGMA-co-MEO2MA)-b-PMPC.
10 mL chloroform was added to the 0.5 mL phosphate buffered saline (PBS(−)) containing 50 mg P(OEGMA-co-MEO2MA)-b-PMPC, followed by sonication using an ultrasonic homogenizer (Sonifier SFX250, Branson Ultrasonics Corp., CT., USA) with 20% amplitude in an ice bath for 3 min.
Preparation of PMPC microgels with P(OEGMA-co-MEO2MA) shell via inverse miniemulsion polymerization.
The PMPC microgels were synthesized according to the following procedure. First, P(OEGMA-co-MEO2MA)-b-PMPC (50 mg, 1.30 μmol), MPC (132 mg, 0.45 mmol), MBAA (7.7 mg, 49 μmol) and VA-044 (0.085 mg, 0.26 μmol) were dissolved in 500 μL PBS(−). Next, 10 mL chloroform was added to the PBS(−) containing all components of inverse miniemulsion RAFT polymerization. The water–chloroform mixture was sonicated using a homogenizer (output: 20%) for 3 min in an ice bath to form a W/O emulsion. The polymerization was performed at 45 °C for 4 h under argon atmosphere. After adding 10 mL water to the miniemulsion, the chloroform was removed under reduced pressure to obtain the dispersion of microgels. The resulting microgel dispersion was purified by centrifugation (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm 15 min, 20 °C) and redispersed in deionized water.
000 rpm 15 min, 20 °C) and redispersed in deionized water.
Gel permeation chromatography (GPC) measurements.
GPC measurements were conducted using a Shimadzu Prominence HPLC system (Shimadzu corp., Kyoto, Japan) equipped with PFG 100 Å and PFG 1000 Å columns (Polymer Standards Service GmbH, Germany) at 40 °C under a flow rate of 1.0 mL min−1 using a refractive index detector. A 2,2,2-trifluoroethanol solution containing 20 mM sodium trifluoroacetate was used as an eluent. The Mn, Mw and Mw/Mn (Đ) were calculated using near-monodisperse poly(methyl methacrylate) standards.
Interfacial tension measurements.
The interfacial tension was measured using a pendant drop technique with a contact angle meter (DMo-502, Kyowa Interface Science Co., Ltd, Saitama, Japan). A pendant drop of PBS(−) containing P(OEGMA-co-MEO2MA)-b-PMPC was synthesized in chloroform at 25 °C. The interfacial tension was determined by following eqn (1).32,33where γ is the interfacial tension, Δρ is the density difference, g is the gravity acceleration, de is the maximum drop diameter, ds is the diameter measured at a distance de from the apex and 1/H is the correlation factor determined from ds/de.
Dynamic light scattering (DLS) measurements.
DLS measurements were conducted using an ELS-Z1000 spectrometer (Otsuka Electronics Co., Ltd, Osaka, Japan) equipped with a He–Ne laser (λ = 633.8 nm) and backscatter optics at 25 °C. The detection angle was fixed at 165°. The backscatter optics allows for the determination of diameter at high concentrations because it minimizes the multiple scattering effects. The diameter and polydispersity index were calculated using the cumulant method, and the size distribution was obtained via Marquart analysis.
Phase transition temperature measurements.
A UV-Vis spectrophotometer (UV-1900i, Shimadzu Co., Kyoto, Japan) is used to determine the cloud point temperature (Tcp) of an aqueous solution of P(MEO2MA-co-OEGMA)-b-PMPC and an aqueous dispersion of PMPC core–shell microgels by measuring their transmittance at 650 nm while heating the solutions and dispersions at a constant rate of 0.2 °C min−1. In this study, Tcp is defined as the temperature at which the transmittance of an aqueous solution decreases by approximately 50%.
Results and discussion
Synthesis of P(OEGMA-co-MEO2MA)-b-PMPC
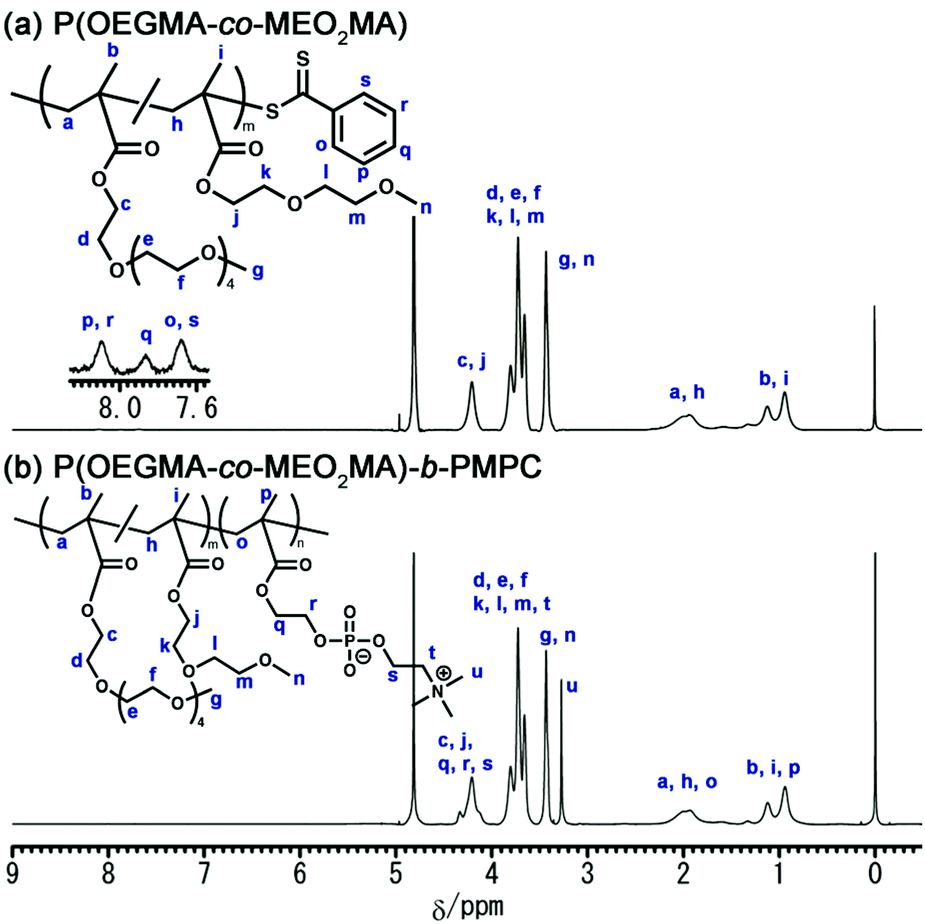
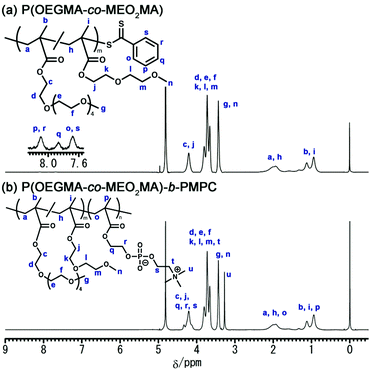
The synthesis of core–shell microgels via inverse miniemulsion RAFT polymerization requires a well-designed block copolymer emulsifier that stabilizes the water droplet of the W/O emulsion. In this study, we designed a water-soluble block copolymer emulsifier comprising a hydrophilic PMPC and hydrophilic/lipophilic POEGMA blocks. The dithiobenzoate group acting as a chain transfer agent in RAFT polymerization should be located at the terminus of the PMPC block that distributes in the water dispersed phase of W/O emulsion because inverse miniemulsion RAFT polymerization occurs in a water droplet of W/O emulsion for synthesizing core–shell microgels. The POEGMA shows LCST-type phase transition in an aqueous solution. The POEGMA with 4 to 5 ethylene oxide units as a side chain exhibits phase transition at approximately 64 °C.34 Copolymerizing OEGMA and MEO2MA can control the LCST of POEGMA.35 The LCST of P(OEGMA-co-MEO2MA) decreases as the molar fraction of OEGMA (FOEGMA) in the copolymer decreases. Considering these polymer designs and properties, the block copolymer emulsifier was synthesized via the RAFT copolymerization of OEGMA and MEO2MA, followed by the RAFT polymerization of MPC (Scheme 1). In this study, the target FOEGMA and polymerization degree of P(OEGMA-co-MEO2MA) were 0.2 and 100 because it allows the LSCT of P(OEGMA-co-MEO2MA) to be around 35 °C.36Fig. 2a shows the 1H NMR spectrum of P(OEGMA-co-MEO2MA). By comparing the overall integration of the methylene proton of both OEGMA and MEO2MA at 4.20 ppm (peak c, j) with the overall integration of the 3.59–3.95 ppm region (peaks d–f, k–m), in which 16 protons of OEGMA and 4 protons of MEO2MA are resonated, the FOEGMA of P(OEGMA-co-MEO2MA) was determined to be 0.22. The degree of polymerization (Dp), obtained by integrating the methylene resonance at 4.20 ppm (peak c, j) against the dithiobenzoate chain-end at 7.6–8.2 ppm (peak o–s), was determined to be 157. Further, the polydispersity (Đ) determined by the GPC measurement was approximately 1.1 (Table 1). The 1H NMR spectrum and GPC results demonstrate the successful synthesis of P(OEGMA-co-MEO2MA) with narrow molecular weight distribution using RAFT polymerization.
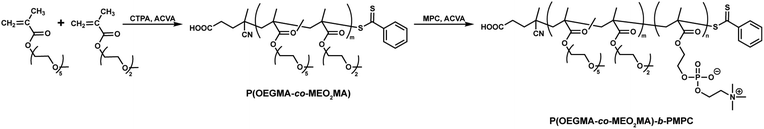 |
| | Scheme 1 Synthesis of P(OEGMA-co-MEO2MA)-b-PMPC. | |
 |
| | Fig. 2
1H NMR spectra of (a) P(OEGMA-co-MEO2MA) and (b) P(OEGMA-co-MEO2MA)-b-PMPC. | |
Table 1 Results of the synthesis of P(OEGMA-co-MEO2MA) and P(OEGMA-co-MEO2MA)-b-PMPC
| |
F
OEGMAa |
D
pa |
M
n (×104)b |
M
w (×104)b |
Đ
|
| P(OEGMA-co-MEO2MA) |
PMPC |
|
Calculated from 1H NMR.
Calculated from GPC curves using near-monodisperse poly(methyl methacrylate) calibration standards.
|
| P(OEGMA-co-MEO2MA) |
0.22 |
157 |
— |
3.32 |
3.75 |
1.13 |
| P(OEGMA-co-MEO2MA)-b-PMPC |
0.22 |
157 |
15 |
3.57 |
4.08 |
1.14 |
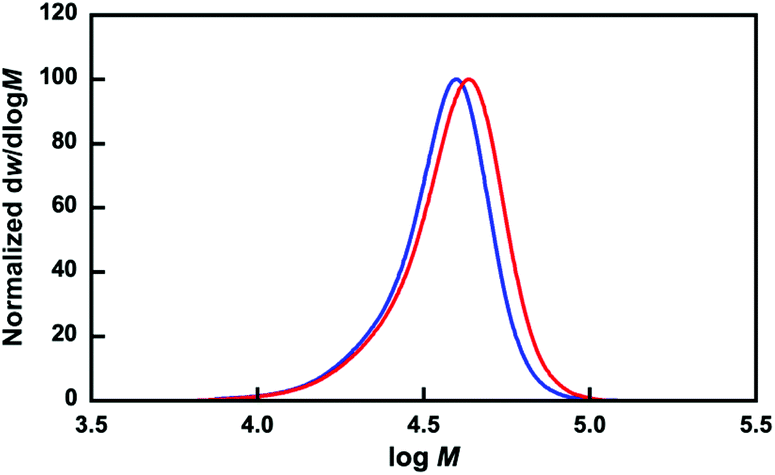
The P(OEGMA-co-MEO2MA)-b-PMPC was synthesized by the RAFT polymerization of MPC in the presence of the resulting P(OEGMA-co-MEO2MA) as a macroRAFT agent. Fig. 2b shows the 1H NMR spectrum of P(OEGMA-co-MEO2MA)-b-PMPC. Characteristic resonances for both P(OEGMA-co-MEO2MA) and PMPC blocks were observed, including signals attributed to the methoxy protons of P(OEGMA-co-MEO2MA) (3.43 ppm, peak g, n) and trimethylammonium group of PMPC (3.27 ppm, peak u). In addition, the molecular weight distribution of P(OEGMA-co-MEO2MA) shifted toward a higher molecular weight via the RAFT polymerization of PMPC using P(OEGMA-co-MEO2MA) as a macroRAFT agent (Fig. 3). The 1H NMR spectrum and GPC results indicate a successful chain extension of the PMPC block from the P(OEGMA-co-MEO2MA) macroRAFT agent. The Dp of PMPC was determined to be 15 by comparing the integration of the methyl resonance of MPC at 3.27 ppm (peak u) with the methoxy resonance of P(OEGMA-co-MEO2MA) at 3.43 ppm (peak g, n). Table 1 summarizes the P(OEGMA-co-MEO2MA) and P(OEGMA-co-MEO2MA)-b-PMPC synthetic results.
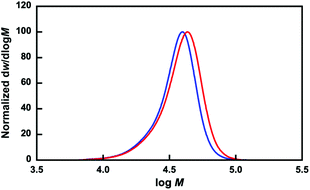 |
| | Fig. 3 Molecular weight distributions (normalized to peak height) of P(OEGMA-co-MEO2MA) (blue line) and P(OEGMA-co-MEO2MA)-b-PMPC (red line). | |
Formation of W/O emulsions using P(OEGMA-co-MEO2MA)-b-PMPC as an emulsifier
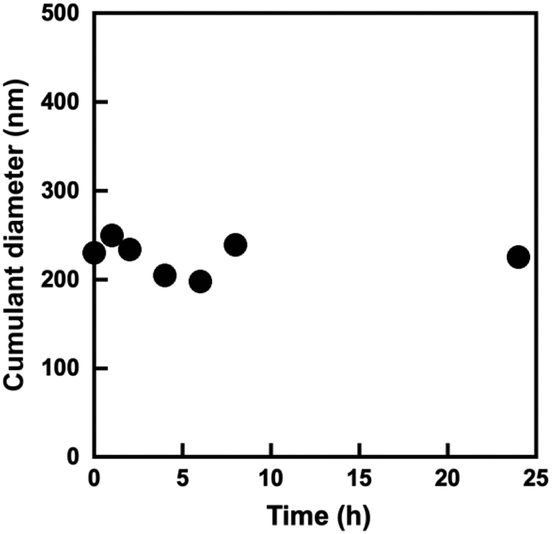
The block copolymer comprising PMPC and POEGMA blocks allows the formation of W/O emulsions in a water–chloroform two-phase system, as reported in our previous paper.28 Because the P(OEGMA-co-MEO2MA)-b-PMPC synthesized in this study had a similar structure with previously reported water-soluble PMPC-b-POEGMA emulsifier, it stabilizes the water–chloroform interface to form a W/O emulsion. The effect of P(OEGMA-co-MEO2MA)-b-PMPC concentration in a water droplet on the interfacial tension between water and chloroform was determined using the pendant drop method.33 The P(OEGMA-co-MEO2MA)-b-PMPC decreases the interfacial tension between water and chloroform as the concentration of P(OEGMA-co-MEO2MA)-b-PMPC increases, and the interfacial tension remained unchanged above 0.293 mg mL−1 (Fig. 4). This result indicates that the P(OEGMA-co-MEO2MA)-b-PMPC acts as a surfactant that stabilizes the water–chloroform interface and its critical micelle concentration (CMC) is 0.293 mg mL−1. In this study, the W/O emulsion was synthesized using 100 mg mL−1 P(OEGMA-co-MEO2MA)-b-PMPC (relative to the water dispersed phase), which was much higher than the CMC of P(OEGMA-co-MEO2MA)-b-PMPC. The water–chloroform mixture containing P(OEGMA-co-MEO2MA)-b-PMPC appeared turbid upon sonication. The microscopic observation of the resulting milky mixture revealed the successful formation of W/O emulsion (Fig. S1†). Fig. 5 shows the change in the size of water droplets of W/O emulsion as a function of time after sonication. The water droplets of the resulting W/O emulsion had a diameter of about 240 nm just after the synthesis. The diameter of the water droplets remained unchanged as the incubation time increased. No macroscopic phase separation was observed for 24 h, indicating that the W/O emulsion prepared in this study was stable. Therefore, the W/O emulsion stabilized with P(OEGMA-co-MEO2MA)-b-PMPC can provide the reaction field for producing hydrophilic microgels via inverse miniemulsion RAFT polymerization.
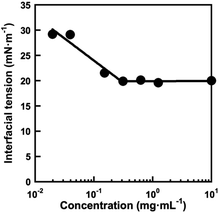 |
| | Fig. 4 Effect of P(OEGMA-co-MEO2MA)-b-PMPC concentration on the interfacial tension of a water-chloroform interface at 25 °C. The interfacial tension was measured using the pendant drop technique with a contact angle meter. | |
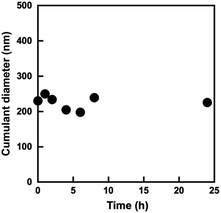 |
| | Fig. 5 Changes in cumulant diameter of water droplets of W/O emulsion in the continuous chloroform phase as a function of time after sonication for 3 min. The concentration of P(OEGMA-co-MEO2MA)-b-PMPC was 100 mg mL−1 (relative to water dispersed phase). | |
Preparation of PMPC core–shell microgels via inverse miniemulsion RAFT polymerization
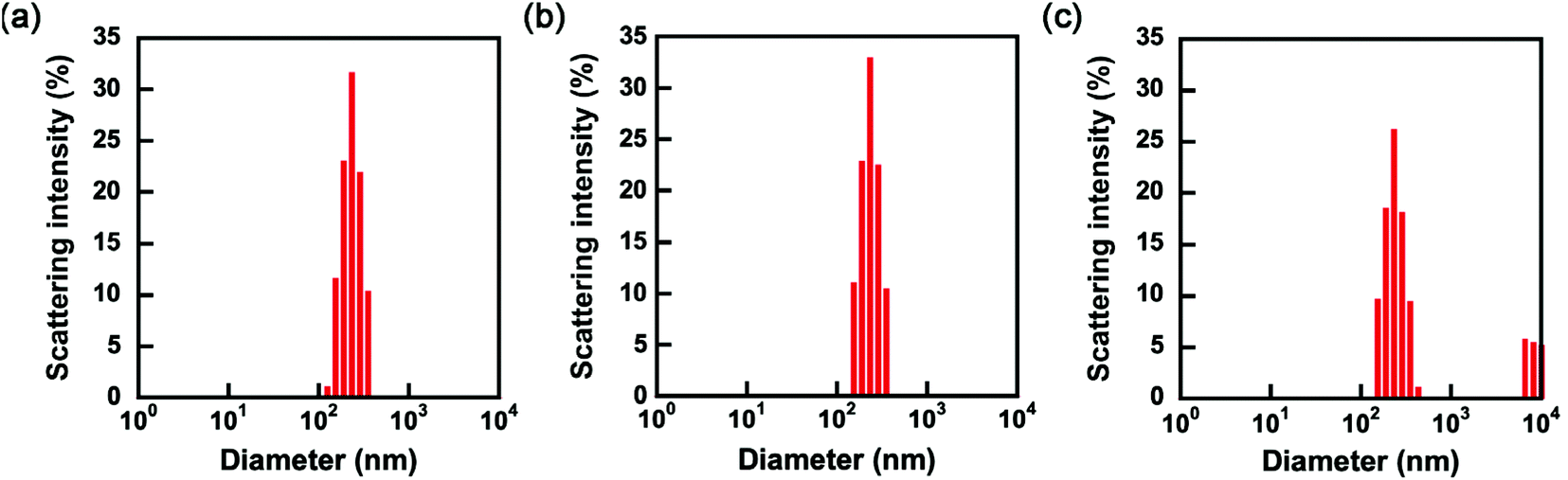
The P(OEGMA-co-MEO2MA)-b-PMPC emulsifier has a dithiobenzoate group acting as a chain transfer agent for RAFT polymerization at the terminus of the PMPC block. RAFT polymerization proceeds from P(OEGMA-co-MEO2MA)-b-PMPC emulsifier stabilizing the water–chloroform interfaces in W/O emulsion water droplet because the PMPC block with dithiobenzoate terminus distributes in a water droplet of W/O emulsion. The synthesis of microgels covalently linked with P(OEGMA-co-MEO2MA) produced from the emulsifier on their surface as a shell is possible using inverse miniemulsion RAFT polymerization of water-soluble monomers and crosslinkers. In this study, MPC and MBAA were copolymerized in a water droplet of W/O emulsion to obtain core–shell microgels composed of PMPC hydrogel core and P(OEGMA-co-MEO2MA) shell. Fig. 6a and b show the size distributions of water droplets of W/O emulsion containing monomers and initiators in chloroform before and after polymerization. The water droplets containing monomers and an initiator had monomodal size distribution, with a cumulant diameter of approximately 260 nm (Fig. 6a). We concluded that the monomers and an initiator did not affect the formation of W/O emulsion since the cumulant diameter of water droplets containing monomers and an initiator was comparable to that of water droplets without monomers and an initiator (i.e., 240 nm). The inverse miniemulsion RAFT polymerization of MPC and MBAA was initiated by VA-044 in the water droplets of W/O emulsion stabilized with P(OEGMA-co-MEO2MA)-b-PMPC emulsifier with dithiobenzoate terminus. The P(OEGMA-co-MEO2MA) block of the emulsifier was located on the surface of the resulting PMPC microgel since the polymerization of MPC and MBAA proceeded from the RAFT terminus of the P(OEGMA-co-MEO2MA)-b-PMPC emulsifier. This implies that the resulting microgels had a core–shell structure comprising a PMPC core and a P(OEGMA-co-MEO2MA) shell. The monomodal size distribution of the W/O emulsion was maintained after polymerization (Fig. 6b). This indicates that the polymerization of MPC and MBAA occurred within the individual water droplets of the W/O emulsion without undesirable interparticle crosslinking and fusion of water droplets during polymerization. Then, the continuous chloroform phase was exchanged for water by adding more water, followed by the removal of chloroform under reduced pressure. The scattering intensity drastically decreased when the continuous chloroform phase of the W/O emulsion stabilized with P(OEGMA-co-MEO2MA)-b-PMPC emulsifier was exchanged for water, and the diameter could not be determined by DLS due to the decomposition of the W/O emulsion. Alternatively, the polymerization of MPC and MBAA in a water droplet of W/O emulsion formed stable core–shell microgels dispersed in both chloroform and water continuous phases. The size of the resulting microgels in water was determined via DLS measurement. Although the size distribution showed a slight aggregation with a large diameter, the main peak of PMPC core–shell microgels in water was comparable to that of the PMPC core–shell microgels in chloroform (Fig. 6b and c), and the cumulant diameter of PMPC core–shell microgels was approximately 270 nm in water. Further, a scanning electron microscope and transmission electron microscope images of dried PMPC core–shell microgels were used to reveal its spherical structure (Fig. S2†). The average, maximum and minimum diameters of the dried PMPC core–shell microgels obtained from the SEM image were 242, 447 and 86 nm, respectively. Notably, the PMPC core–shell microgels were stably dispersed in water without a thorough purification process. In general, the hydrophilic microgels synthesized using the conventional inverse miniemulsion polymerization method are covered with surfactants used for the emulsion preparation. The adsorbed surfactants are washed thoroughly using polar organic solvents. For example, in synthesizing polymer carboxybetaine microgels via inverse miniemulsion polymerization, the sodium bis(2-ethylhexyl) sulfosuccinate, which is a surfactant stabilizing the W/O emulsion, was removed by thorough washing with THF.37 Various biomolecules, such as proteins and enzymes, can be loaded into microgels since our system uses a water-soluble emulsifier and does not require a thorough washing process with a polar organic solvent for redispersion of microgels in water.
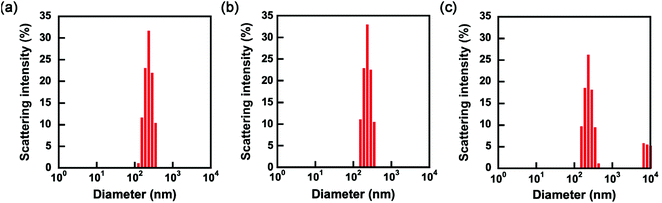 |
| | Fig. 6 Size distributions of W/O emulsions containing monomers and an initiator before (a) and after polymerization with VA-044 in chloroform continuous phase (b), and core–shell microgels with PMPC core and P(OEGMA-co-MEO2MA) shell after exchanging chloroform continuous phase for water (c). | |
Temperature-responsive behaviour of PMPC core–shell microgels
The P(OEGMA-co-MEO2MA) shows the LCST-type phase transition. The LCST of P(OEGMA-co-MEO2MA) can be controlled by the molar fraction of OEGMA (FOEGMA). First, the LCST of the P(OEGMA-co-MEO2MA)-b-PMPC emulsifier synthesized in this study was determined using the turbidity method. Upon heating, the transmittance of aqueous P(OEGMA-co-MEO2MA)-b-PMPC solution decreased drastically. For a given type of temperature-responsive polymer, Tcp depends on s concentration. The Tcp of P(OEGMA-co-MEO2MA)-b-PMPC decreased as its concentration increased, and remained unchanged above 4 mg mL−1 (Fig. S3†). Therefore, the LCST of P(OEGMA-co-MEO2MA)-b-PMPC was determined to be 38 °C. According to a previous report, LCST of P(OEGMA-co-MEO2MA) with FOEGMA of 0.21 is approximately 37 °C.36 Interestingly, the LCST of the P(OEGMA-co-MEO2MA)-b-PMPC synthesized in this study is consistent with that of P(OEGMA-co-MEO2MA) with FOEGMA of 0.21, although the latter has no PMPC block. Therefore, we conclude that the temperature-responsive phase transition of P(OEGMA-co-MEO2MA)-b-PMPC is governed by the P(OEGMA-co-MEO2MA) block.
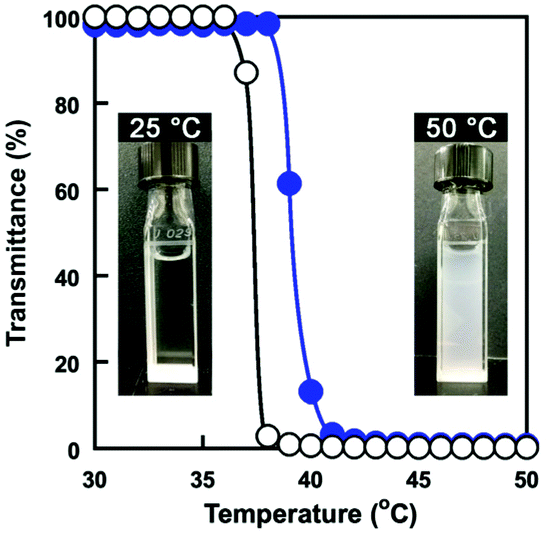
The PMPC core–shell microgels had the temperature-responsive P(OEGMA-co-MEO2MA) chains on their surface as the shell. The temperature-responsive behaviour of PMPC core–shell microgels was evaluated via turbidity measurement. Fig. 7 shows the changes in transmittance of an aqueous PMPC core–shell microgel dispersion and an aqueous P(OEGMA-co-MEO2MA) solution as a function of temperature. The transmittance of PMPC core–shell microgel dispersion decreased drastically above 38 °C and appeared turbid. We also investigated the temperature-responsive behaviour of P(OEGMA-co-MEO2MA) having the same composition as the P(OEGMA-co-MEO2MA) shell of PMPC core–shell microgels (i.e., FOEGMA: 0.22; Dp: 157). The aqueous P(OEGMA-co-MEO2MA) solution became turbid upon heating owing to the hydrophilic-to-hydrophobic transition and its Tcp was approximately 37 °C (Fig. 7). These results indicate that the temperature-responsive changes in transmittance of PMPC core–shell microgels were due to the hydrophilic-to-hydrophobic transition of P(OEGMA-co-MEO2MA) chains as the shell. The P(OEGMA-co-MEO2MA) shell of the PMPC core–shell microgel changed from hydrophilic-to-hydrophobic above 38 °C. The P(OEGMA-co-MEO2MA) shell's hydrophilic-to-hydrophobic transition of core–shell microgels changes their reflactive index, resulting in light scattering. In addition, the hydrophobically changed PMPC microgel surfaces undergo interparticle aggregation. Thus, the transmittance of PMPC core–shell microgel dispersion decreased drastically upon heating. The Tcp of an aqueous P(OEGMA-co-MEO2MA) solution is subject to the ionic strength due to the weak salting-out effect. The Tcp of P(OEGMA-co-MEO2MA) in PBS(−) (I = 0.16 M) is approximately 4 °C lower than that in water.36 In fact, the Tcp of PMPC core–shell microgel dispersion decreased in a PBS(−) (Fig. S4†). This result also indicates that the temperature-responsive behaviour of PMPC core–shell microgels was governed by the P(OEGMA-co-MEO2MA) shell.
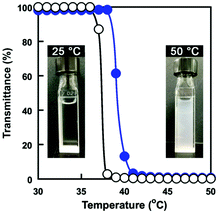 |
| | Fig. 7 Changes in transmittance (650 nm) of a PMPC core–shell microgel dispersion in water (●) and an aqueous P(OEGMA-co-MEO2MA) solution (○) as a function of temperature. (Inserted photos: PMPC core–shell microgel dispersion at 25 °C and 50 °C.) | |
We conclude from these results that the microgels synthesized by the inverse miniemulsion RAFT copolymerization of MPC and MBAA from the P(OEGMA-co-MEO2MA)-b-PMPC emulsifier stabilizing a water droplet have well-defined core–shell structures comprising a PMPC hydrogel core and a P(OEGMA-co-MEO2MA) shell that exhibits temperature-responsive phase transition (Fig. 8). Because the temperature-responsive P(OEGMA-co-MEO2MA) shell regulates molecule permeability via temperature-responsive hydrophilic-to-hydrophobic transition, temperature-responsive core–shell microgels will be used to construct smart drug delivery system carriers and catalytic systems.
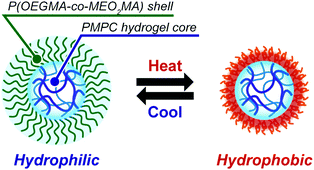 |
| | Fig. 8 Schematic illustration of the morphology of PMPC core–shell microgels and their temperature-responsive behaviour. | |
Conclusions
This study proposes a novel synthetic method of core–shell microgels with a temperature-responsive shell via inverse miniemulsion RAFT polymerization using a water-soluble emulsifier. The water-soluble P(OEGMA-co-MEO2MA)-b-PMPC possessing a dithiobenzoate group at the PMPC block terminus was synthesized via RAFT copolymerization. The resulting P(OEGMA-co-MEO2MA)-b-PMPC stabilized a water droplet containing MPC, MBAA and VA-044 in a continuous chloroform phase. Upon heating the W/O emulsion, MPC and MBAA were copolymerized in the water droplet of W/O emulsion from the dithiobenzoate terminus of P(OEGMA-co-MEO2MA)-b-PMPC emulsifier stabilizing the water droplet of W/O emulsion. The resulting PMPC core–shell microgels were colloidally stable in both chloroform and water continuous phases. The resulting PMPC core–shell microgels had a diameter of approximately 270 nm in water with a well-defined core–shell structure comprising a PMPC hydrogel core and a temperature-responsive P(OEGMA-co-MEO2MA) shell. Because the P(OEGMA-co-MEO2MA) was dissolved in both water and chloroform, the resulting PMPC core–shell microgels with a P(OEGMA-co-MEO2MA) shell on their surface exhibited good colloidal stability in both water and chloroform continuous phases. Upon heating, the PMPC core–shell microgel dispersion appeared turbid above 38 °C due to the hydrophilic-to-hydrophobic transition of the P(OEGMA-co-MEO2MA) shell in response to temperature. In our method for synthesizing core–shell microgels, various water-soluble materials, such as low molecular weight drugs, proteins, enzymes and metal nanoparticles, can be loaded into the core of microgels by simply dissolving in water droplets of W/O emulsion before inverse miniemulsion RAFT polymerization. Microgels with a temperature-responsive shell can regulate molecule permeability in response to temperature, making them a viable platform for developing smart catalytic systems and drug delivery systems.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
This work was partly supported by JSPS KAKENHI (Grant No. JP21K05202) from the Japan Society for the Promotion of Science, Iketani Science and Technology Foundation (Grant No. 0321032-A) and MEXT-Supported Program for Private University Research Branding Project.
References
- S. Nayak and L. A. Lyon, Angew. Chem., Int. Ed., 2005, 44, 7686–7708 CrossRef CAS PubMed
 .
.
- G. Agrawal and R. Agrawal, Small, 2018, 14, e1801724 CrossRef PubMed .
- A. Kawamura, Y. Hata, T. Miyata and T. Uragami, Colloids Surf., B, 2012, 99, 74–81 CrossRef CAS PubMed .
- A. Kawamura, A. Harada, S. Ueno and T. Miyata, Langmuir, 2021, 37, 11484–11492 CrossRef CAS PubMed .
- R. H. Pelton and P. Chibante, Colloids Surf., 1986, 20, 247–256 CrossRef CAS .
- W. Richtering and A. Pich, Soft Matter, 2012, 8, 11423–11430 RSC .
- Y. Lu and M. Ballauff, Prog. Polym. Sci., 2011, 36, 767–792 CrossRef CAS .
- V. Sabadasch, L. Wiehemeier, T. Kottke and T. Hellweg, Soft Matter, 2020, 16, 5422–5430 RSC .
- W. Wu, J. Shen, P. Banerjee and S. Zhou, Biomaterials, 2010, 31, 7555–7566 CrossRef CAS PubMed .
- A. Kawamura, T. Katoh, T. Uragami and T. Miyata, Polym. J., 2015, 47, 206–211 CrossRef CAS .
- N. Welsch, A. L. Becker, J. Dzubiella and M. Ballauff, Soft Matter, 2012, 8, 1428–1436 RSC .
- L. K. Meena, H. A. Rather and R. Vasita, ACS Appl. Polym. Mater., 2020, 2, 4902–4913 CrossRef CAS .
- C. D. Jones and L. A. Lyon, Macromolecules, 2000, 33, 8301–8306 CrossRef CAS .
- T. Brandel, V. Sabadasch, Y. Hannappel and T. Hellweg, ACS Omega, 2019, 4, 4636–4649 CrossRef PubMed .
- K. E. Christodoulakis and M. Vamvakaki, Langmuir, 2010, 26, 639–647 CrossRef CAS PubMed .
- Y. Kim, L. T. Thuy, Y. Kim, M. Seong, W. K. Cho, J. S. Choi and S. M. Kang, Langmuir, 2022, 38, 1550–1559 CrossRef CAS PubMed .
- I. Capek, Adv. Colloid Interface Sci., 2010, 156, 35–61 CrossRef CAS PubMed .
- E. A. Doherty, C. W. Kan and A. E. Barron, Electrophoresis, 2003, 24, 4170–4180 CrossRef CAS PubMed .
- E. A. Doherty, C. W. Kan, B. M. Paegel, S. H. Yeung, S. Cao, R. A. Mathies and A. E. Barron, Anal. Chem., 2004, 76, 5249–5256 CrossRef CAS PubMed .
- J. K. Oh, C. Tang, H. Gao, N. V. Tsarevsky and K. Matyjaszewski, J. Am. Chem. Soc., 2006, 128, 5578–5584 CrossRef CAS PubMed .
- A. V. Fuchs and K. J. Thurecht, Macromol. Chem. Phys., 2015, 216, 1271–1281 CrossRef CAS .
- R. H. Utama, Y. Guo, P. B. Zetterlund and M. H. Stenzel, Chem. Commun., 2012, 48, 11103–11105 RSC .
- R. H. Utama, M. H. Stenzel and P. B. Zetterlund, Macromolecules, 2013, 46, 2118–2127 CrossRef CAS .
- F. Ishizuka, R. H. Utama, S. Kim, M. H. Stenzel and P. B. Zetterlund, Eur. Polym. J., 2015, 73, 324–334 CrossRef CAS .
- F. Ishizuka, R. P. Kuchel, H. Lu, M. H. Stenzel and P. B. Zetterlund, Polym. Chem., 2016, 7, 7047–7051 RSC .
- R. H. Utama, M. Drechsler, S. Förster, P. B. Zetterlund and M. H. Stenzel, ACS Macro Lett., 2014, 3, 935–939 CrossRef CAS PubMed .
- R. H. Utama, Y. Jiang, P. B. Zetterlund and M. H. Stenzel, Biomacromolecules, 2015, 16, 2144–2156 CrossRef CAS PubMed .
- H. Nakaura, A. Kawamura and T. Miyata, Langmuir, 2019, 35, 1413–1420 CrossRef CAS PubMed .
- H. Ahmad, D. Dupin, S. P. Armes and A. L. Lewis, Langmuir, 2009, 25, 11442–11449 CrossRef CAS PubMed .
- S. Sugihara, K. Sugihara, S. P. Armes, H. Ahmad and A. L. Lewis, Macromolecules, 2010, 43, 6321–6329 CrossRef CAS .
- Y. F. Tian, M. Lei, L. K. Yan and F. F. An, Polym. Chem., 2020, 11, 2360–2369 RSC .
- J. M. Andreas, E. A. Hauser and W. B. Tucker, J. Phys. Chem., 1938, 42, 1001–1019 CrossRef CAS .
- J. D. Berry, M. J. Neeson, R. R. Dagastine, D. Y. Chan and R. F. Tabor, J. Colloid Interface Sci., 2015, 454, 226–237 CrossRef CAS PubMed .
- J.-F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS .
- J.-F. Lutz and A. Hoth, Macromolecules, 2006, 39, 893–896 CrossRef CAS .
- J.-F. Lutz, A. Hoth and K. Schade, Des. Monomers Polym., 2012, 12, 343–353 CrossRef .
- B. Li, Z. Yuan, P. Zhang, A. Sinclair, P. Jain, K. Wu, C. Tsao, J. Xie, H. C. Hung, X. Lin, T. Bai and S. Jiang, Adv. Mater., 2018, 30, e1705728 CrossRef PubMed .
Footnote |
| † Electronic supplementary information (ESI) available: Microscope images of W/O emulsions stabilized with P(OEGMA-co-MEO2MA)-b-PMPC emulsifier; SEM and TEM image of PMPC core–shell microgels; determination of the LCST of P(OEGMA-co-MEO2MA)-b-PMPC; turbidimetry of PMPC core–shell microgels in phosphate buffered saline. See DOI: https://doi.org/10.1039/d2py00425a |
|
| This journal is © The Royal Society of Chemistry 2022 |

 *ab and
Takashi
Miyata
*ab and
Takashi
Miyata