
Synthesis of degradable double network gels using a hydrolysable cross-linker†
Takanori
Yokoi
a,
Akinori
Kuzuya
 ab,
Tasuku
Nakajima
cd,
Takayuki
Kurokawa
c,
Jian Ping
Gong
cd and
Yuichi
Ohya
*ab
ab,
Tasuku
Nakajima
cd,
Takayuki
Kurokawa
c,
Jian Ping
Gong
cd and
Yuichi
Ohya
*ab
aFaculty of Chemistry, Materials, and Bioengineering, Kansai University, 3-3-35 Yamate, Suita, Osaka 564-8680, Japan. E-mail: yohya@kansai-u.ac.jp; Fax: +81-6-633-4026; Tel: +81-6-6368-0818
bKansai University Medical Polymer Research Center (KUMP-RC), Kansai University, 3-3-35 Yamate, Suita, Osaka 564-8680, Japan
cFaculty of Advanced Life Science, Hokkaido University, Sapporo 001-0021, Japan
dInstitute for Chemical Reaction Design and Discovery (WPI-ICReDD), Hokkaido University, Sapporo 001-0021, Japan
First published on 2nd June 2022
Abstract
Double network (DN) gels have remarkably high mechanical strength and toughness and can be potentially applied in biomedical applications such as cartilage regeneration. However, most DN gels synthesised by usual radical polymerisations are non-biodegradable, and they are not desirable for replacing living tissues in the human body. In this study, we developed a DN gel with polyelectrolyte stents (St-DN gel) that exhibited high mechanical strength and biodegradability under physiological conditions. These degradable St-DN gels were prepared using poly(N,N-dimetylacrylamide) (PDMAAm) as the second network and poly(ethylene glycol) (PEG) as the first network. The PEG gel contained the molecular stent and a degradable ester bond in its network strands. To prepare the degradable PDMAAm gel, we designed a hydrolysable cross-linking agent, PEG-di(methacryloyloxyethyl succinate) (PEG-DMOS), with six ester bonds. The obtained St-DN gel showed an extremely high compressive fracture strength and strain. We also confirmed that the St-DN gel could be gradually hydrolysed under physiological conditions. Thus, this hydrolysable high-strength gel could be potentially used as an implantable biomedical material.
Introduction
Hydrogels are cross-linked three-dimensional polymer networks (NWs) that are highly water-absorbent. Hydrogels have been used in various applications owing to their softness, high water absorption, low friction, and shock absorption properties. In particular, because the physical characteristics of hydrogels are relatively close to those of soft tissues in the human body, they can be potentially used as implantable medical materials.1,2 However, the hydrogels synthesised by conventional polymerisation reactions using cross-linking agents have lower mechanical strength and are more brittle than natural tissues. This is due to the heterogeneity of the structure of conventional hydrogels.3 The wide distribution of distances between the cross-linking points leads to stress concentration during deformation; the stress is concentrated around the shortest chain, leading to the failure of the sample at a very low force.However, hydrogels with excellent mechanical properties that overcome the poor mechanical properties of conventional hydrogels have been reported in recent years, as exemplified by tetra-poly(ethylene glycol) (PEG) gels,4,5 nanocomposite (NC) gels,6,7 slide-ring (SR) gels,8,9 double network (DN) gels,10–14 and dynamic interpenetrating polymer network (IPN) systems.15,16 Tetra-PEG gels exhibit a high uniformity in their cross-linking structure. NC gels have layered clay minerals as multiple cross-linking points, and SR gel have movable cross-linking points based on rotaxane structures. Gong et al. developed a DN gel with high toughness. The DN gels comprised two independently cross-linked polymer NWs with different characteristics: densely cross-linked, rigid, and brittle first NW and loosely cross-linked, soft, and ductile second NWs, which were physically entangled with each other. There was neither any specific interaction between the two NWs nor any microphase separation between the two components. Therefore, DN gels can be categorised as IPN gels but are different from usual IPN gels. In DN gels, the first NW dissipates energy in the form of sacrificial bonds during deformation, resulting in an extremely high toughness.
For instance, poly(2-acrylamido-2-methylpropane sulfonic acid)/polyacrylamide (PAMPS/PAAm) DN gels have a compressive fracture strength of about 20 MPa despite containing over 90% water.10,11 Moreover, they show a very low coefficient of friction of about 0.01 between the same gels.14 The first NW in typical DN gels is charged and highly swollen to work as sacrificial bonds and produces high toughness. However, Gong et al. reported that tough DN gels could also be produced using non-ionic polymers for both first and second NWs by embedding a charged linear polymer, called molecular stent, in the first NW (St-DN gel).17–19 The introduced stent provides high osmotic pressure to the non-ionic gel and makes it highly swollen and suitable for use as the first NW of tough DN gels. Most of the abovementioned gels prepared by conventional radical polymerisation reactions, including DN gels, do not degrade under physiological conditions and are not absorbable. Therefore, they are unsuitable as implantable medical materials, where degradation and absorption are desirable.
One of the expected biomedical applications of such high-strength hydrogels is the repair of damaged cartilage tissue. The functional repair of joint (hyaline) cartilage defects in joint surgeries is still challenging. The most common strategy for repairing articular cartilage defects is to fill osteochondral defects with artificial, cell-disseminated, or tissue-engineered cartilage-like materials. For example, autologous chondrocyte transplantation,20–23 tibial osteotomy,24,25 and partial joint replacement with prosthesis (silicone)26,27 have been attempted for treating cartilage lesions on the knee joint. Artificial cartilage transplantation for cartilage defects is expected to be a potential treatment option. For example, the development of artificial cartilage using poly(vinyl alcohol) (PVA) hydrogels28–31 has been reported. However, such artificial cartilages cannot be used in clinical treatment because of their insufficient strength, toughness, and frictional properties.30 More recently, PVA/hydroxyapatite/polyamide composites,32 silk-fibroin/gelatin, and cross-linked hyaluronates33 have also been reported to replace cartilage. However, attempts to find materials that provide sufficient strength, lubrication, and good scaffolding for transplanted cells are still ongoing.
As DN gels have high mechanical strength and lubrication properties, they can be used as materials for the replacement of cartilage, which experiences repeated applications of large loads. Gong et al. realised the spontaneous regeneration of cartilage tissues (hyaline) in four weeks by implanting DN gels under the cartilage defect site in the knee meniscus of rabbits.34–36 These results overturned the medical myth that hyaline cartilage could not be regenerated in vivo.37,38 In addition, hyaline cartilage regeneration was achieved by implanting the DN gel without any chemical treatment; this process was less invasive and safer than other articular cartilage repair methods. However, because the DN gel used was non-degradable, it remained in the tissue even after the articular cartilage regenerated. Therefore, there are concerns about the long-term stability of these gels. Moreover, they have to be removed by reoperation when inflammation occurs. Despite the reported use of degradable natural polymers as DN gel components,39–43 degradable DN gels acting as tissue-regenerating materials that (i) can be prepared by conventional polymerisation reactions, (ii) decompose into soluble macromolecules, (iii) are eliminated during tissue regeneration, and (iv) are completely replaced by living tissue are challenging to develop.
Ohya et al. have studied biodegradable biomedical materials44,45 that can be absorbed by the human body. Herein, we attempted to design a DN gel that can be degraded under physiological conditions to use as an implant for cartilage regeneration. We chose poly(ethylene glycol) (PEG) for the first NW and poly(N,N-dimethylacrylamide) (PDMAAm) for the second NW because of their biocompatibility. Based on the fact that ester bonds are hydrolysable, we preliminary synthesised a PDMAAm gel using poly(ethylene glycol)diacrylate (PEG-DA) (Fig. 1) containing ester bonds as a hydrolysable cross-linking agent for the second NW of the DN gel. However, contrary to our expectations, the obtained PDMAAm/PEG-DA gel showed almost no degradability. Therefore, we newly designed PEG-di(methacryloyloxyethyl succinate) (PEG-DMOS) (Fig. 1) with four additional (total six) ester bonds as the hydrolysable cross-linking agent and synthesised a degradable PDMAAm gel. In this study, we prepared a degradable St-DN gel using cross-linked branched PEG (4-arm PEG) containing ester bonds in its main chain as the first NW. PAMPS was used as a stent, and PDMAAm cross-linked with PEG-DMOS was used in the second NW (Scheme 1). Moreover, we report the mechanical and degradation properties of the synthesised St-DN gel. Experimental details are reported in the ESI.†
 | ||
| Fig. 1 Structures of the cross-linkers used in this study: PEG-DA and PEG-DMOS. | ||
 | ||
| Scheme 1 Synthesis of St-DN gel comprising 4-arm PEG/PEG-DA as the first NW, linear PAMPS as the stent, and PDMAAm/PEG-DMOS as the second NW. | ||
Results and discussion
Degradation of PDMAAm/PEG-DMOS gel (PDMAAm gel) (second NW gel)
PEG-DA contains ester bonds, which should be relatively easily hydrolysed. However, at the preliminary stage of this study, we noticed that gels obtained by polymerisation using PEG-DA as the cross-linking agent were difficult to hydrolyse. It is well known that ester bonds in the main chain of aliphatic polyesters, such as polylactic acid and poly(ε-caprolactone), are hydrolysed under physiological conditions. However, ester bonds on the side-chains of polymethacrylates and polyacrylates, such as poly(methylmethacrylate) (PMMA) and poly(2-hydroxyethylmethacrylate) (PHEMA), are difficult to hydrolyse even under acidic or alkaline conditions. As described later, the ester bonds of PEG-DA can be hydrolysed when it becomes the main chain after the thiol–ene chain extension reaction. Based on these facts, we hypothesised that ester bonds adjacent to the polymer main chain would be difficult to hydrolyse. To verify this hypothesis, we synthesised a cross-linker with additional ester bonds in places away from the terminal double bonds (PEG-DMOS) (molecular weight (MW) of starting PEG = 600 g mol−1, total MW of PEG-DMOS = 1100 g mol−1). Subsequently, we synthesised a PDMAAm gel using PEG-DMOS as the cross-linking agent, which became the second NW of the DN gel, and the hydrolysis behaviour of the resultant gel was investigated.Synthetic details for PEG-DMOS and the PDMAAm/PEG-DMOS gel (20 wt%) are described in the ESI.†Fig. 2 shows the results of the hydrolysis behaviour by monitoring the swelling ratios of the PDMAAm/PEG-DMOS and PDMAAm/PEG-DA gels, which is a control synthesised using PEG-DA (MW = 700 g mol−1) as the cross-linking agent. Photographs of the gels before and after 95 d of degradation are shown in the ESI (Fig. S4†).
 | ||
| Fig. 2 Time course of the swelling ratio during the degradation test for the PDMAAm/PEG-DMOS and PDMAAm/PEG-DA gels in PBS (pH = 7.4) at 37 °C. | ||
The PDMAAm/PEG-DMOS gel gradually swelled during the degradation test, indicating the cleavage of the cross-links by hydrolysis. On the 95th day, it swelled 5–6 times the initial swelling ratio (equilibrium swelling ratio at t = 0 d), softened, and then completely dissolved on the 98th day. In contrast, the PDMAAm/PEG-DA gel showed almost no change in the swelling ratio even after 95 days, indicating almost no degradability. This supports our hypothesis that the ester bonds adjacent to the polymer main chains are difficult to hydrolyse. This cross-linking agent, PEG-DMOS, is useful for preparing gels that decompose via hydrolysis under physiological conditions. These degradable gels can be synthesised by radical copolymerisation with methacrylate, acrylate or other vinyl monomers.
Optimisation of the preparation of the first NW gel (PEG gel) and its physical properties
In this study, considering degradability and biocompatibility, a PEG-based gel containing ester bonds was chosen as the first NW gel. The thiol–ene reaction of a 4-arm PEG derivative with terminal thiol groups (4-arm PEG-SH) (MW = 5000 or 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) and PEG-DA (MW = 700 g mol−1) was performed. The first NW PEG gels were synthesised by thiol–ene polyaddition under various conditions, and optimisation was carried out by measuring the mechanical properties of the PEG gels. The total concentration of the prepolymers (4-arm PEG-SH + PEG-DA) was set to 5–20 wt%, while the amount of the acryl and thiol groups was equivalent. Subsequently, PEG-DA containing acryl groups was reacted with 4-arm PEG-SH to produce the PEG gels (Fig. S7†).
000 g mol−1) and PEG-DA (MW = 700 g mol−1) was performed. The first NW PEG gels were synthesised by thiol–ene polyaddition under various conditions, and optimisation was carried out by measuring the mechanical properties of the PEG gels. The total concentration of the prepolymers (4-arm PEG-SH + PEG-DA) was set to 5–20 wt%, while the amount of the acryl and thiol groups was equivalent. Subsequently, PEG-DA containing acryl groups was reacted with 4-arm PEG-SH to produce the PEG gels (Fig. S7†).
Fig. 3 shows the compressive fracture strength and strain of the PEG5k and PEG10k gels prepared using 4-arm PEG-SH with MWs of 5000 and 10000 g mol−1, respectively. The compressive fracture strength was maximal at a concentration of 15 wt% in both cases. These results are in good agreement with those reported by Sakai et al., where the mechanical strength of the tetra-PEG gel increases as the NW becomes more uniform at a concentration of 160 mg mL−1.46 Furthermore, the compressive fracture strength of the PEG5k gel was higher than that of the PEG10k gel, reflecting the larger cross-linking density of the PEG5k gel. In all the cases, the compressive fracture strain values were almost the same. Based on these results, the concentration of the first NW in the subsequent experiments was set at 15 wt%.
 | ||
| Fig. 3 Effect of total macromonomer concentration (4-arm PEG-SH + PEG-DA) on compressive fracture strength and strain for the (A) PEG5k and (B) PEG10k gels. | ||
Preparation of the St-first NW gel (PEG gel containing PAMPS) and its physical properties
Since the first NW used in this study is a non-ionic PEG-based gel, the introduction of a stent in the first NW is required to produce the high toughness of the DN gel.17–19 In this study, we employed 2-acrylamido-2-methyl-1-propanesulfonic acid (AMPS), which has already been used as a precursor for a stent in various DN gel studies.17–19 To optimise the stent concentration, we synthesised the St-first NW gels (St-PEG5k and St-PEG10k gels) using AMPS at various concentrations and evaluated their elastic moduli and swelling ratios. As shown in Fig. S7,† the volume after equilibrium swelling increased when the stent was introduced. In both the St-PEG5k and St-PEG10k gels, the elastic moduli decreased when the stent was introduced. In contrast, the swelling ratio increased to a stent concentration of 1.0 M but hardly changed at higher concentrations (Fig. 4). These results indicated that the mesh of the first NW was almost fully extended at a stent concentration of 1.0 M, which is suitable for sacrificial bonds. Therefore, in the subsequent synthesis of the St-DN gel, the concentration of AMPS was set to 1.0 M. | ||
| Fig. 4 Effect of stent concentration on the swelling ratios and Young's moduli of the (A) St-PEG5k and (B) St-PEG10k gels. | ||
Physical properties and degradation behaviour of St-DN gel
Under the preparative conditions determined above (first NW concentration = 15 wt%, and stent (AMPS) concentration = 1.0 M), the St-DN5k and St-DN10k gels were synthesised from the St-PEG5k and St-PEG10k gels, respectively, by changing the concentration of DMAAm and the feeding ratio of the cross-linking agent (PEG-DMOS/DMAAm (mol%)). Highly transparent gels were obtained for both St-DN5k and St-DN10k. Photographs of the gels are shown in Fig. S8.†Fig. 5A shows the results of the compression tests for the St-DN5k gels prepared by changing the concentration of DMAAm with the feeding ratio of the cross-linking agent to the monomer fixed at 0.01 mol%. Moreover, the St-DN5k gel was prepared by changing the monomer concentration. Consequently, the gel exhibited the highest compressive fracture strength of 20 MPa when it was prepared at a DMAAm concentration of 2.0 M; the compressive fracture strain was close to 100%. Subsequently, St-DN5k gels were synthesised by changing the feeding ratio of the cross-linking agent while maintaining the DMAAm concentration at 2.0 M (Fig. 5B). The compressive fracture strength and strain were the highest when the DMAAm concentration was 0.01 mol%. Fig. 5C shows the stress–strain (S–s) curve obtained from the compression tests of the gels prepared under these conditions. Moreover, the first and second NW gels separately synthesised under the same conditions as the St-DN5k gel were used as controls. The tensile test results are shown in Fig. S9,† and the movies for the compression test and the gels being stretched by hand are also included in the ESI (Movies S1 and S2).† This gel did not break even when compressed at 99% and exhibited a high compressive fracture strength of 20 MPa. The elongation at break and breaking stress in the tensile test were 1600% and 0.53 MPa, respectively. DN gels and St-DN gels prepared by Gong et al. exhibited a compressive fracture strength of 17–40 MPa,10,19 while their breaking stress and elongation at break in tensile tests were determined as 1.0–2.0 MPa and 1200–2000%, respectively.17–19 The mechanical strengths of our gels are further compared with those of other DN gels in Table S1.† Thus, the values obtained for the St-DN5k gel are comparable to those of previously reported DN and St-DN gels and exceed the general load on knee cartilage (3–18 MPa).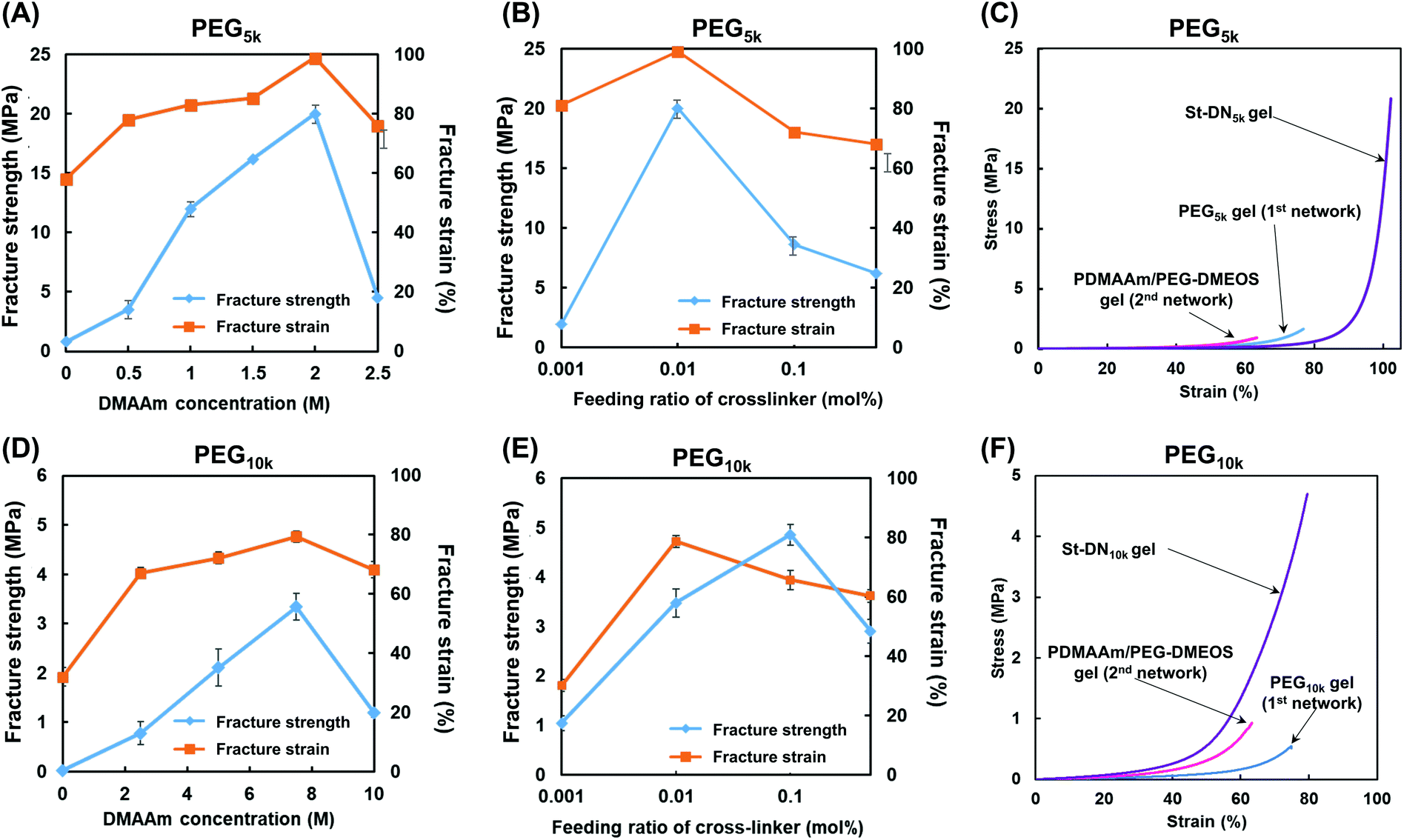 | ||
| Fig. 5 Mechanical properties of St-DN5k (A)–(C) and St-DN10k (E)–(F) gels. Effect of DMAAm (second NW monomer) concentration on the compressive fracture strength and strain of (A) St-DN5k and (D) St-DN10k gels. Effect of the feeding ratio of the cross-linker for the second NW (PEG-DMOS/DMAAm (%)) on the compressive fracture strength and strain of (B) St-DN5k and (E) St-DN10k gels. Stress–strain curves of (C) St-DN5k and (F) St-DN10k gels recorded during compression tests. St-DN5k/St-DN10k gels were prepared at [DMAAm] = 2.0/7.5 M and cross-linker feeding ratio = 0.01/0.1 mol%. Data were expressed as means ± standard deviations (n = 3). | ||
The results for the St-DN10k gel are shown in Fig. 5D–F. The feeding ratio of the cross-linking agent was fixed at 0.1 mol%, and the monomer concentration was varied. In these cases, the maximum breaking strength was 3.2 MPa when the monomer concentration was 7.5 M. Moreover, when the monomer concentration was fixed at 7.5 M, and the feeding ratio of the cross-linking agent was changed, the maximum breaking strength was 4.7 MPa at 0.1 mol%. The fracture strain was 80%, with a maximum at 0.01 mol%. However, these values were lower than those of the St-DN5k gel, and the performance of the St-DN gel was not optimal. This may have occurred because of the insufficiently low cross-linking density of the original PEG10k gel, which did not function as effectively as the sacrificial bonds.
Finally, the hydrolysis behaviour of the St-DN5k gel and PEG5k gel used for the first NW was investigated under physiological conditions (Fig. 6A). The PEG5k gel contained PEG-DA as a bifunctional chain extender for the thiol–ene polyaddition reaction; however, it gradually swelled and completely dissolved on the 37th day. These results indicated that the ester bonds in PEG-DA were hydrolysed under physiological conditions when used in a polymer backbone. This result supports our hypothesis that an ester group is difficult to hydrolyse when located adjacent to the polymer backbone but easy to hydrolyse in other cases.
 | ||
| Fig. 6 (A) Time course of swelling ratio during degradation test for 4-arm-PEG/PEG-DA (1st NW) gel and St-DN gel in PBS (pH = 7.4) at 37 °C. (B) and (C): photographs for St-DN gel before hydrolysis test and after 60 days, respectively. | ||
However, the St-DN5k gel exhibited gradual swelling, suggesting that the hydrolysis continuously occurred until day 65. The duration of the study was set as 65 days due to limited resources. A longer degradation study for this gel may be carried out in a future study on St-DN gels, where the general degradation properties of the DN gels may be improved. Photographs of the gel before and after hydrolysis are shown in Fig. 6B and C, respectively. These figures show that the shape of the gel changed significantly due to the swelling and degradation. Based on these results, we assume that if a longer-term degradation test is carried out, the gel will be completely hydrolysed. The first (PEG5k gel) and second NW gels (PDMAAm/PEG-DMOS gel, Fig. 2) reached complete dissolution after 98 and 37 days, respectively. The second NW gel (PDMAAm/PEG-DMOS gel) did not completely dissolve in 65 days. Moreover, the hydrolytic rate for the St-DN5k gel tends to be slower than that of the component gels. This trend might be natural because the polymer chains cannot be dissolved unless both NWs are dissociated; however, the hydrolysis may have been suppressed owing to the entanglement of the first and second NW and the stent in the St-DN gel.
Conclusions
We synthesised a bifunctional PEG methacrylate derivative (PEG-DMOS) with additional ester bonds as a degradable cross-linking agent. We then prepared a degradable hydrogel (PDMAAm/PEG-DMOS gel) using PEG-DMOS as a cross-linking agent. Furthermore, we successfully synthesised the St-DN gels by combining PDMAAm/PEG-DMOS as the second NW and PEG gel (prepared from 4-arm PEG-SH and PEG-DA) as the first NW. PAMPS was used as the stent. When the synthetic conditions of the St-DN gel were optimised using a 4-arm PEG-SH with a molecular weight of 5000, the obtained St-DN5k gel showed a fracture strength of 20 MPa with very high toughness. The St-DN5k gel slowly hydrolysed under physiological conditions. We successfully synthesised a biodegradable DN gel that exhibits high mechanical strength under physiological conditions. It would be interesting to apply this gel to cartilage regeneration. However, the hydrolysis rate of the St-DN5k gel may be too slow (the ideal degradation period for cartilage regeneration is 2–3 months). Moreover, the remarkable increase in the swelling ratio and volume of the gel during degradation may be an issue for in vivo cartilage regeneration. Therefore, the immediate application of the St-DN gels for cartilage regeneration may be difficult. In addition, the toxicity and absorbability of the degradation products should be investigated. However, because the preparation of tough and degradable hydrogels is now possible, we expect that they will be used in biomedical applications after they are improved further.Author contributions
Y. O. and J. P. G. conceived the idea of designing the experiments. T. Y. performed all the experiments. A. K., T. N, and T. K. performed some parts of the measurements and analysed the data. Y. O. prepared the manuscript. All authors discussed the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the Private University Research Branding Project: Matching Fund Subsidy from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) Japan (2016–2020) and Grants-in-Aid for Scientific Research (16H01854, 20K21228 and 20H00670) from the Japan Society for the Promotion of Science (JSPS). The authors thank NOF Co. for providing 4-arm PEG10k-SH.References
- A. S. Hoffman, Adv. Drug Delivery Rev., 2012, 64, 18 CrossRef.
- J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337 CrossRef CAS.
- G. Hild, Prog. Polym. Sci., 1998, 23, 1019 CrossRef CAS.
- T. Sakai, T. Matsunaga, Y. Yamamoto, C. Ito, R. Yoshida, S. Suzuki, N. Sasaki, M. Shibayama and U. Chung, Macromolecules, 2008, 41, 5379 CrossRef CAS.
- M. Shibayama, X. Li and T. Sakai, Colloid Polym. Sci., 2019, 297, 1 CrossRef CAS.
- K. Haraguchi and T. Takehisa, Adv. Mater., 2002, 14, 1120 CrossRef CAS.
- K. Haraguchi, Polym. J., 2011, 43, 223 CrossRef CAS.
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485 CrossRef CAS.
- K. Ito, Polym. J., 2007, 39, 489 CrossRef CAS.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155 CrossRef CAS.
- J. P. Gong, Soft Matter, 2010, 6, 2583 RSC.
- M. A. Haque, T. Kurokawa and J. P. Gong, Polymer, 2012, 53, 1805 CrossRef CAS.
- C. Azuma, K. Yasuda, Y. Tanabe, H. Taniguro, F. Kanaya, A. Nakayama, Y. M. Chen, J. P. Gong and Y. Osada, J. Biomed. Mater. Res., Part A, 2007, 81, 373 CrossRef.
- D. Kaneko, T. Tada, T. Kurokawa, J. P. Gong and Y. Osada, Adv. Mater., 2005, 17, 535 CrossRef CAS.
- B. Zhang, J. Ke, J. R. Vakil, S. C. Cummings, Z. A. Digby, J. L. Sparks, Z. Ye, M. B. Zanjani and D. Konkolewicz, Polym. Chem., 2019, 10, 6290 RSC.
- S. Monemian and L. T. J. Korley, Macromolecules, 2015, 48, 7146 CrossRef CAS.
- T. Nakajima, H. Sato, Y. Zhao, S. Kawahara, T. Kurokawa, K. Sugahara and J. P. Gong, Adv. Funct. Mater., 2012, 22, 4426 CrossRef CAS.
- T. Nakajima, Y. Fukuda, T. Kurokawa, T. Sakai, U.-I. Chung and J. P. Gong, ACS Macro Lett., 2013, 2, 518 CrossRef CAS PubMed.
- T. Matsuda, T. Nakajima, Y. Fukuda, W. Hong, T. Sakai, T. Kurokawa, U.-I. Chung and J. P. Gong, Macromolecules, 2016, 49, 1865 CrossRef CAS.
- M. Brittberg, A. Lindahl, A. Nilsson, C. Ohlsson, O. Isaksson and L. Peterson, N. Engl. J. Med., 1994, 331, 889 CrossRef CAS PubMed.
- J. E. Browne, A. F. Anderson, R. Arciero, B. Mandelbaum, J. B. Moseley, L. J. Micheli, F. Fu and C. Erggelet, Clin. Orthop. Relat. Res., 2005, 436, 237 CrossRef PubMed.
- B. Mandelbaum, J. E. Browne, F. Fu, L. J. Micheli, J. B. Moseley, C. Erggelet and A. F. Anderson, Am. J. Sports Med., 2007, 35, 915 CrossRef PubMed.
- K. Zaslav, B. Cole, R. Brewster, T. Deberardino, J. Farr, P. Fowler and C. Nissen, Am. J. Sports Med., 2009, 37, 42 CrossRef PubMed.
- H. Bergenudd, O. Johnell, I. Redlund-Johnell and L. S. Lohmander, Acta Orthop. Scand., 1992, 63, 413 CrossRef CAS PubMed.
- K. Yasuda, T. Majima, T. Tsuchida and K. Kaneda, Clin. Orthop. Relat. Res., 1992, 282, 186 CrossRef.
- R. H. Emerson and L. L. Higgins, J. Bone Jt. Surg., Am. Vol., 2008, 90, 118 CrossRef.
- D. Naudie, J. Guerin, D. A. Parker, R. B. Bourne and C. H. Rorabeck, J. Bone Jt. Surg., Am. Vol., 2004, 86, 1931 CrossRef PubMed.
- Z. Q. Gu, J. M. Xiao and X. H. Zhang, J. Biomed. Mater. Eng., 1998, 8, 75 CAS.
- M. Kobayashi and M. Oka, Artif. Organs, 2004, 28, 734 CrossRef CAS PubMed.
- M. Kobayashi and M. Oka, J. Biomater. Sci., Polym. Ed., 2004, 15, 741 CrossRef CAS.
- T. Gui, X. Tian, B. Li, T. Yang and Y. Li, J. Orthop. Surg. Res., 2017, 12, 176 CrossRef.
- W. Shi, M. Sun, X. Hu, B. Ren, J. Cheng, C. Li, X. Duan, X. Fu, J. Zhang, H. Chen and Y. Ao, Adv. Mater., 2017, 29, 2701089 Search PubMed.
- S. P. Xiao, L. S. Tang, J. Y. Chen, Z. T. Li, G. H. Cheng, Q. Q. Chen, S. H. Liu and W. G. Liu, Orthop. Surg., 2019, 11, 379 Search PubMed.
- K. Arakaki, N. Kitamura, H. Fujiki, T. Kurokawa, M. Iwamoto, M. Ueno, F. Kanaya, Y. Osada and J. P. Gong, J. Biomed. Mater. Res., Part A, 2010, 93, 1160 Search PubMed.
- K. Yasuda, N. Kitamura, J. P. Gong, K. Arakaki, H. J. Kwon, S. Onodera, Y. M. Chen, T. Kurokawa, F. Kanaya, Y. Ohmiya and Y. Osada, Macromol. Biosci., 2009, 9, 307 CrossRef CAS.
- K. Arakaki, N. Kitamura, T. Kurokawa, S. Onodera, F. Kanaya, J. P. Gong and K. Yasuda, J. Mater. Sci.: Mater. Med., 2011, 22, 417 CrossRef CAS PubMed.
- B. R. Mandelbaum, J. E. Browne, F. Fu, L. Micheli, J. B. Mosely Jr., C. Erggelet, T. Minas and L. Peterson, Am. J. Sports Med., 1998, 26, 853 CrossRef CAS PubMed.
- J. A. Buckwalter and H. A. Mankin, Arthritis Rheumatol., 1998, 41, 1331 CrossRef CAS.
- A. Nakayama, A. Kakugo, J. P. Gong, Y. Osada, M. Takai, T. Erata and S. Kawano, Adv. Funct. Mater., 2004, 14, 1124 CrossRef CAS.
- M. A. Haque, T. Kurokawa and J. P. Gong, Polymer, 2012, 53, 1805 CrossRef CAS.
- Y. Zhang, M. Chen, J. Tian, P. Gu, H. Cao, X. Fan and W. Zhang, Biomater. Sci., 2019, 7, 3266 RSC.
- Z. Fang, K. Qiao, Y. Wang, Y. Zheng, W. He, Y. Xie and H. Yang, Polym. Test., 2022, 106, 107452 CrossRef CAS.
- Z. Li, D. Chu, G. Chen, L. Shi, L. Jin, X. Zhang and J. Li, J. Biomed. Nanotechnol., 2019, 15, 2209 CrossRef CAS.
- Y. Ohya, A. Takahashi and K. Nagahama, Adv. Polym. Sci., 2012, 247, 65 CrossRef CAS.
- T. Ouchi and Y. Ohya, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 453 CrossRef CAS.
- T. Sakai, Y. Akagi, T. Matsunaga, M. Kurakazu, U.-I. Chung and M. Shibayama, Macromol. Rapid Commun., 2010, 31, 1954 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py00360k |
| This journal is © The Royal Society of Chemistry 2022 |