
Chiral graphene-based supramolecular hydrogels toward tumor therapy†
Xueqian
Wang
,
Beibei
Wu
,
Yaqian
Zhang
 and
Chuanliang
Feng
*
and
Chuanliang
Feng
*
State Key Lab of Metal Matrix Composites, Shanghai Key Laboratory for Molecular Engineering of Chiral Drugs, School of Materials Science and Engineering, Shanghai Jiao Tong University, Shanghai 200240, China. E-mail: clfeng@sjtu.edu.cn
First published on 17th February 2022
Abstract
Drugs with a chiral property are playing very important roles in the precise treatment of diseases (especially antitumor drugs); however, the enantioselective delivery of chiral anticancer drugs is still a challenge. Herein, a chiral hydrogel system is constructed by the co-assembly of the D-phenylalanine derivative gelator (DPFEG) with graphene oxide (GO). The chiral property can be regulated by near-infrared light irradiation, which makes it an ideal material for the on-demand control of chiral anticancer drug (oxaliplatin) release. In the absence of infrared light, the drug molecules can hardly leak out of the system for up to seven hours and the drugs can be released upon near-infrared light irradiation. Meanwhile, the light-heat conversion characteristics of GO enable the material to be capable of photothermal therapy. Consequently, the DPFEG-GO material shows a good suppression effect on T47D breast cancer cells. This exquisitely designed supramolecular system provides a simple and effective method for the absorption and precise release of chiral drugs, which has potential for the combination of chemotherapy and photothermal-therapy in the treatment of cancer.
Introduction
Chiral helical nanostructures are ubiquitous in nature and play a significant role in the field of medicine.1–5 Most of the available drugs possess chiral centers, and their enantiomers generally exhibit differences in pharmacodynamic or pharmacokinetic activities, thus leading to different influence on their therapeutic effect.6–10 To date, chemotherapy remains the predominant means of cancer treatment. Nevertheless, conventional chemotherapy is confronted with a certain hindrance, for instance uncontrolled drug release, non-specific distribution and serious side effects, which vastly limit their therapeutic efficiency and might cause damage to normal tissues.11 The emergence of drug delivery system (DDS) made of nanoparticles, liposomes, hydrogels and other materials can effectively carry and release drugs, thereby improving the treatment efficiency and reducing the toxicity of drugs.12–14 Although many notable achievements have been made, most research concentrated on the triggering release of achiral anticancer drugs, and concerns about enantioselective loading and the release of chiral drugs in an accurately controlled way remain a main difficulty in biomedicine. In this regard, there is an urgent need to design functional systems with an enantioselective recognition capability for tumor therapy.In recent years, with the tremendous development of biomimetic helical structures, functional materials with an enantioselective recognition or separation ability have appeared.15–23 For example, Yan et al.10 reported a photo-responsive M-helix based supramolecular vesicle that was capable of the selective release of chiral drug enantiomers. In addition, Chen and co-workers24 synthesized a short peptide carrier for the controlled capture and release of enantiomers. Therefore, supramolecular hydrogels with a specific helical structure formed by non-covalent bonds have potential applications in chiral drug delivery systems. The dynamic properties of non-covalent interactions involved in arranged assemblies endow the supramolecular system with splendid adjustability and responsiveness, thus making them easy to be affected by external stimuli.25–28 Among various environmental factors (temperature,29–33 light,34–40 pH,41–44 solvents45–47 and magnetic fields), photoirradiation attracts extensive attention since it is non-invasive, convenient and controllable, especially for infrared light, which shows a deeper tissue penetration capability and less damage to normal cells.48–50 However, photo-responsive functional groups, such as azobenzene, benzidine, fulgide and spirooxazine, are needed to achieve reversible helical switching, which makes the design procedure more complicated. In this respect, incorporating light-responsive components into multicomponent systems through co-assembly is a noteworthy strategy. Remarkably, graphene oxide (GO), an important derivative of graphene, has emerged as a new category of stimuli-responsive material with an excellent performance. On the one hand, the affluent oxygenated functional groups enable it to interact with diverse molecules through host–guest interaction, π–π stacking interaction and electrostatic interaction. On the other hand, GO exhibits biocompatible and photo-responsive characteristics, which makes it an excellent drug carrier and candidate material for tumor treatment. Moreover, GO has been extensively used in photothermal therapy (PTT) due to its significant photothermal conversion performance.51–59
In this paper, a D-phenylalanine derivative (DPFEG)-based supramolecular hydrogel for the selective delivery of chiral cancer drugs was synthesized, in which GO was introduced to endow the system with stimulating responsive characteristics (Scheme 1). Under near infrared light (NIR) irradiation, DPFEG can realize a helical transformation from left-handed (M) nanostructures to a right-handed (P) helix. A chiral anticancer drug (oxaliplatin) was selected and the R-isomer displayed a specific loading property in the hydrogel. The drug molecules can be quickly released from the system under infrared irradiation. Combining the chemotherapy and photothermal therapy effects, the DPFEG-GO material exhibits a good inhibitory activity toward breast cancer cells. This kind of supramolecular system provides an efficient and facile method for the release of enantioselective drugs and is expected to be applied in the biomedicine field.
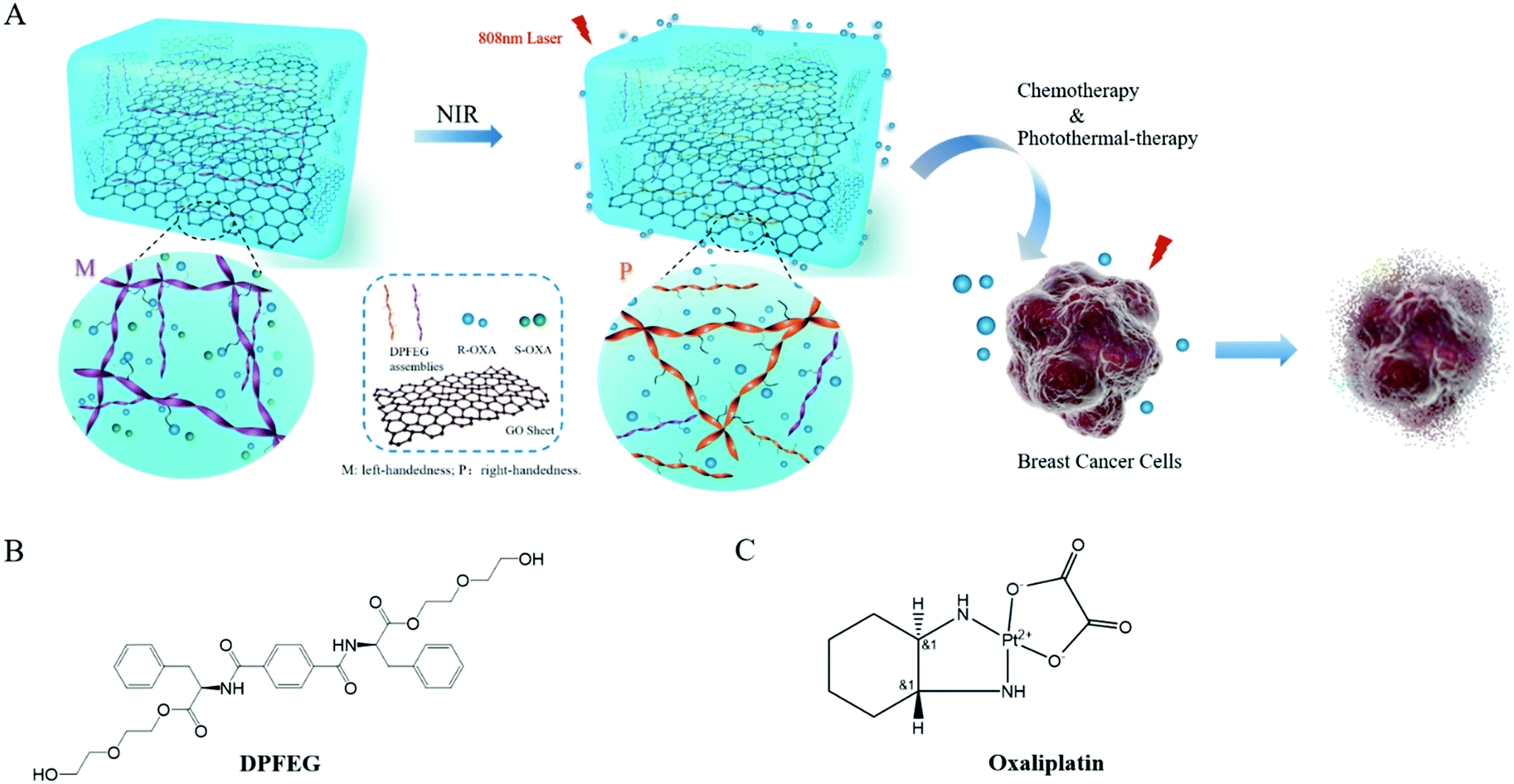 | ||
| Scheme 1 (A) Schematic diagram of phenylalanine-based hydrogel co-assembled with GO and its handedness reversal induced selective delivery of chiral anticancer drugs under IR irradiation. The chemical structures of (B) DPFEG and (C) oxaliplatin. | ||
Experimental section
Materials
All the reagents utilized in this work are described explicitly in the ESI.†Preparation of D/LPFEG gelator
The D/LPFEG gelator was prepared according to the reported literature.60 The specific steps of the synthesis are shown in the ESI.† All of the chemical compounds are identified with 1H NMR and EI-MS (ESI Fig. S18†).Preparation of GO
Graphene oxide (GO) was synthesized by a modified Hummers’ method from graphite powder according to previous reports.61,62 All steps of the synthesis are shown in the ESI.†Preparation of DPFEG-GO composite hydrogels
Graphene oxide (GO) was first dispersed in deionized water under sonication to obtain a uniform dispersion with different concentrations varying from 0.015 to 0.12 mg mL−1. Then 3 mg of DPFEG was added into the GO suspensions (1 mL) at various mass ratios of DPFEG/GO, 1/0.5, 1/1, 1/2, 1/3 and 1/4, in a 5 mL glass vial and heated. The operation was halted until the solid fully disappeared, and then the solution was set aside and cooled to room temperature to form a hydrogel. The final hydrogels were termed as DPFEG-GO 0.5%, DPFEG-GO 1%, DPFEG-GO 2%, DPFEG-GO 3% and DPFEG-GO 4%. Pure DPFEG hydrogel was produced in the same way for comparison and termed DPFEG-GO 0%.Instruments and characterization
The morphology of the D/LPFEG hydrogels and composite hydrogels was studied by applying a field emission scanning electron microscope (FM-SEM, FEI Quanta 250) under a 10 kV acceleration voltage. The specimens were diluted and dripped onto the silicon wafer before the measurement. After drying, Au plating was sprayed on the surface of the sample. The morphologies of graphite oxide and hydrogels were presented on a transmission electron microscope (TEM, Tecnai G2 spirit BioTwin) with a working voltage of 200 kV. The surface structure information of GO was imaged using atomic force microscopy (AFM, MFP-3D). X-ray photoelectron spectroscopy (XPS) was accomplished with an AXIS ULTRA DLD instrument (Kratos, Japan). The infrared spectrum of the composite hydrogels before and after irradiation was obtained by using a Fourier transform spectrometer (FTIR, Bruck EQUINOX55) after mixing freeze-dried gel powder and KBr with a measurement wavelength range from 4000 to 400 cm−1. Information on molecular vibration can be obtained by Raman spectroscopy. The Raman spectra of the hydrogels were produced with a confocal Raman micro-spectrometer (Renishaw inVia Qontor) equipped with a CCD camera. The samples were excited by a 532 nm laser source with a measurement range of 400–3500 cm−1. Circular dichroism spectroscopy was utilized to reflect the optical activity and secondary structure of the substances. On a JASCO J-1500 CD spectrometer, the experiment was carried out in the ultraviolet range of 190–600 nm using a quartz colorimetric dish (optical diameter, 0.2 mm) in a N2 atmosphere with a bandwidth of 1.0 nm. An ultraviolet-visible spectrophotometer (Evolution 201 Thermo Fisher Scientific, U.S.A) was operated to obtain the absorption spectrum of the chiral drugs. Ultraperformance liquid chromatography (UPLC) analyses were performed by an Acquity UPLC I-class/VION IMS QTOF system. A microplate reader (TECAN Infinite M200 Pro) was used to read the absorbance values of the cells at 450 nm.Photothermal effect of the DPFEG-GO composite hydrogels
DPFEG-GO hydrogels (1.0 mL) with various concentrations of GO were added into 96 plates and irradiated with an 808 nm NIR laser (Changchun Leishi Photoelectric Technology Co., Ltd, China) for 5 min at 2.0 W cm−2. The temperature was monitored using an infrared thermal imaging camera (FLIR E98, USA). The photothermal stability of DPFEG-GO was also studied by conducting three cycles of first irradiation followed by cooling to room temperature.Rheological measurement of the DPFEG-GO composite hydrogels
The rheological measurements of DPFEG-GO composite hydrogels were conducted on a rheometer (AR G2 rheometer, TA Instruments) with a 20 mm diameter parallel-plate. The trials were carried out by using a dynamic frequency sweep measurement within a frequency range from 0.1 to 10 Hz at 25 °C and applying a sinusoidal shear strain with a 0.1% strain to record the data of modulus G′ and G′′.Loading and release of chiral anticancer drugs
In vitro drug release was investigated. Firstly, a certain amount of chiral anticancer drugs was dissolved in 15 mL of deionized water by ultrasonication, and put aside for further use. Afterwards, DPFEG-GO mixtures of a certain proportion were heated and cooled to form hydrogels. The racemic drug solution was then added to the hydrogel vials for co-culture, and the resulting mixture was diffused by gentle stirring for half an hour and remained stationary for 24 hours under the environmental conditions. Through this treatment, the hydrogel collapsed and bound with specific chiral drug molecules, which were then collected by centrifugation. For the drug release studies, 5 mL of the phosphate buffered saline (PBS, pH 7.4) was added into the system, using the NIR laser to irradiate. During this period, the sample was placed in an ice water bath to maintain the temperature, and then the PBS was taken out at set intervals to monitor the drug release. The concentration of chiral drug released from the hydrogel could be determined directly by UV-vis measurements. The spectra of chiral drugs were recorded in the range of 200–500 nm with a 0.1 mm quartz cuvette. Besides, the standard calibration curves of oxaliplatin and its enantiomers were established. The drug release experiments were performed three times to obtain valid values.Cell culture
Hs27 human skin fibroblasts and T47D Human Breast Cancer Cell lines were selected for the experiment. The Hs27 cells were cultured on 6-well plates inside a 37 °C incubator under a humidified atmosphere of 5% CO2 in DMEM/high glucose (GIBCO) containing 10% FBS and 1% penicillin–streptomycin. The T47D cells were cultured in the same way.In vitro biocompatibility test
To examine the biocompatibility of DPFEG-GO composite hydrogels in vitro, a CCK (Dojindo's cell counting kit-8) assay was used to perform on the Hs27 cells. The Hs27 cells were seeded in 96-well plates at a density of 1 × 104 cells per well and first cultivated in DMEM for 24 h. Afterwards, the solution in 96-well plates was substituted with a mixture of DPFEG-GO hydrogel and culture medium (200 μL in total) and co-cultured with cells for 3 days. After that, according to the manufacturer's instructions, the medium was removed from the 96-well plate, and then 20 μL of CCK-8 solution was added to each well and incubated for 4 hours. After 4 hours of incubation, the absorbance at 450 nm was measured with a microplate reader. The relative cell viability was calculated in accord with ESI eqn (1).†In vitro cancer cytotoxicity test
In vitro cancer cytotoxicity tests were conducted to evaluate the anticancer effect of the samples, monitoring the T47D cancer cell line on the first, second, and third days of culture. We first seeded the T47D cells in 96 well plates at a density of 2 × 104 cells per well and allowed the attachment of cells to the substrate for 24 h, and then the hydrogel was added. For the sake of verifying the synergistic effect of PTT and chemotherapy on cells, the trial was divided into five groups: control group, DPFEG-GO group, DPFEG-GO/drug group, DPFEG-GO + NIR group and DPFEG-GO/drug + NIR group. In order to eliminate the interference of the environment during the NIR irradiation treatment, the cells with or without the NIR irradiation were separately seeded on different 96 well plates. The T47D cells were irradiated for 5 min every day. After 1 day, 2 day and 3 days of incubation, the viability of the cells was determined by CCK-8 assay separately. The relative cell viability was calculated referring to the above formula in the ESI.†Live/dead fluorescence staining
For the live/dead fluorescence staining studies, the T47D cells were washed with Dulbecco's PBS after 3 days of incubation. Then, the cells were cultivated in a mixture of 2 μM AM/4 μM PI and Dulbecco's PBS solution for 40 min. The results of live/dead fluorescence staining were observed by confocal laser scanning microscopy. Calcein-AM residing in the living cells revealed a green fluorescence and the presence of propidium iodide in dead cells showed a red fluorescence.Cell morphology
To evaluate the morphologies of the nucleus and cytoskeleton, cells were first co-cultured with different groups of hydrogels on 12-well plates, then chemically fixed with 4% pfa for 30 min, followed by staining with Phalloidin YF and Hoechst 33258, respectively, according to the instructions. The morphology of cells was evaluated under a fluorescence microscope. (Olympus LDP72).Results and discussion
Preparation of DPFEG-GO composite hydrogels
A C2-symmetric phenylalanine chiral gelator (DPFEG) was synthesized according to the reported method, and its synthesis procedure and characterization are specifically described in the ESI.†60 In order to obtain the expected performance of external field stimulation, GO was introduced into the system to form a composite hydrogel. Herein, GO was obtained by a modified Hummer's technique and measured as a single atomic layer with a thickness of about 1.1 nm (ESI Fig. S1 and S2†). Due to the introduction of a large number of oxygen-containing functional groups on the surface and edge of the plates, GO could be well dispersed in an aqueous solution and a polar solvent (ESI Fig. S3 and S4†). Through heating–cooling, the DPFEG was found to form uniform and translucent hydrogels in water with a critical gelation concentration of 0.5% (w/v), showing a right-handed helical fibrillar microstructure with a width of about 55–65 nm and a helical spacing of 300–350 nm (ESI Fig. S5a†). By contrast, its enantiomer LPFEG showed the opposite homogeneous left-handed helical nanofibers (ESI Fig. S5b†). When the ultrasonic dispersion of GO was mixed with DPFEG, brown semi-transparent and homogeneous composite hydrogels were obtained by simply heating and cooling, which could stably exist at room temperature for at least two weeks (ESI Fig. S5c†).Mechanical properties of DPFEG-GO composite hydrogels
To investigate the influence of GO content on the mechanical properties of the hydrogels, five kinds of DPFEG-GO composite hydrogels with weight ratios of 0.5, 1, 2, 3 and 4 wt% were prepared under the conditions of a fixed DPFEG at 3 mg mL−1 (ESI Fig. S6†). The dynamic rheological research (Fig. 1A and B) shows that the storage modulus (G′) of hydrogels was greater than the loss modulus (G′′), revealing that hydrogel was formed. For the DPFEG-GO composite hydrogels, the maximum values of G′ and G′′ reached up to about 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Pa and 5400 Pa, respectively, nearly three times larger than that of pure hydrogels. Remarkably, with the increase of GO content, the difference between G′ and G′′ became more significant, which demonstrated that the introduction of GO could increase the mechanical properties of the hydrogels. However, the DPFEG-GO hydrogels showed an apparent decrease in mechanical strength when the amount of GO was more than 3%. The possible reason is that a high GO concentration is helpful for agglomeration, which may reduce the interaction area between the gelator and GO, thus weakening the binding force between them and leading to a decrease in the mechanical strength. Therefore, appropriate GO doping can improve the mechanical strength of composite hydrogels.
000 Pa and 5400 Pa, respectively, nearly three times larger than that of pure hydrogels. Remarkably, with the increase of GO content, the difference between G′ and G′′ became more significant, which demonstrated that the introduction of GO could increase the mechanical properties of the hydrogels. However, the DPFEG-GO hydrogels showed an apparent decrease in mechanical strength when the amount of GO was more than 3%. The possible reason is that a high GO concentration is helpful for agglomeration, which may reduce the interaction area between the gelator and GO, thus weakening the binding force between them and leading to a decrease in the mechanical strength. Therefore, appropriate GO doping can improve the mechanical strength of composite hydrogels.
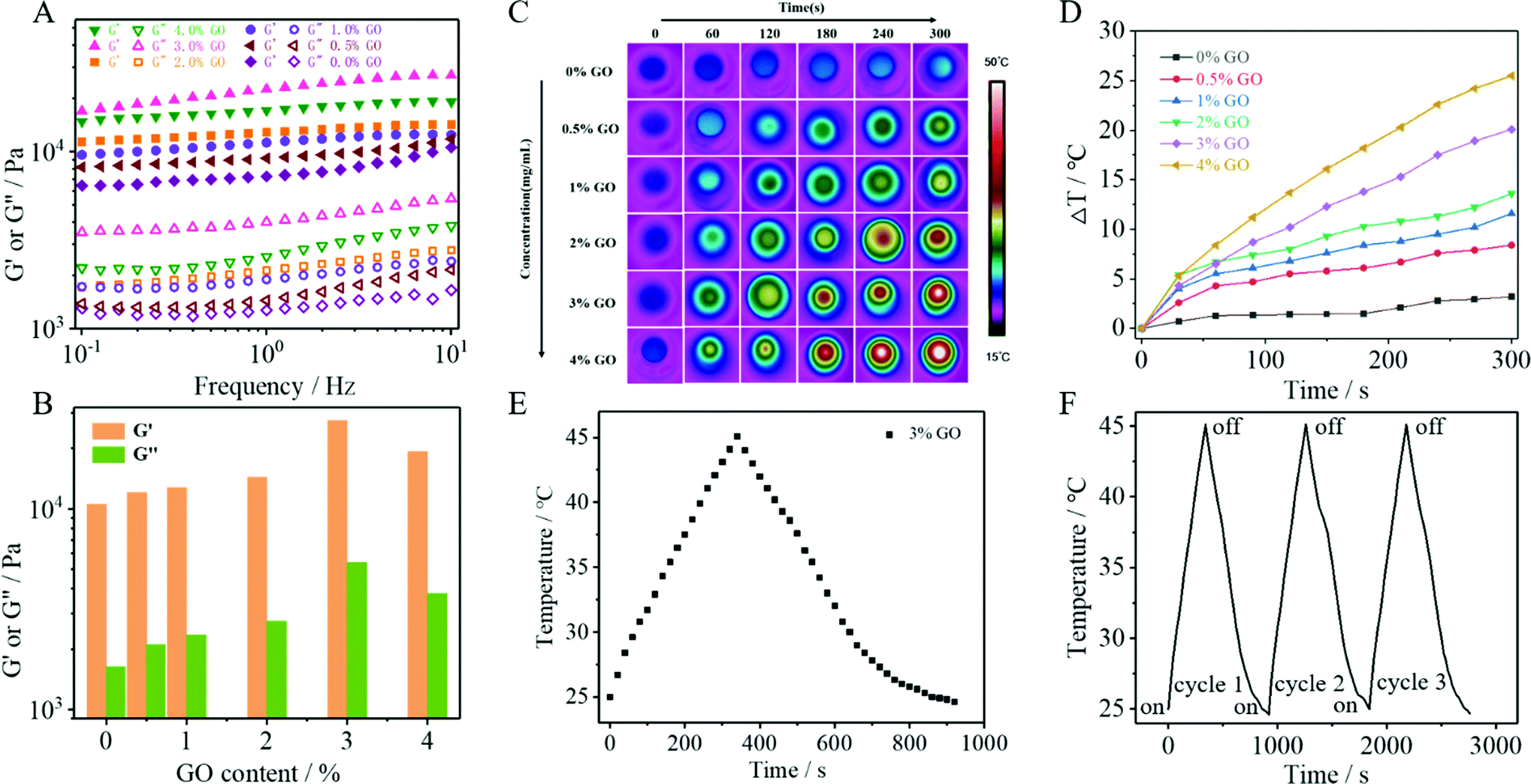 | ||
| Fig. 1 (A) Rheological properties of DPFEG-GO hydrogels with different concentrations of GO. (B) Comparison of the storage modulus (G′) and loss modulus (G′′) for the DPFEG-GO hydrogels. (C) Thermal infrared imaging and (D) photothermal effects (the initial temperature was 25 °C) of DPFEG-GO hydrogels with different concentrations of GO over 5 min. (E) Temperature rise and fall curve with or without 808 nm irradiation, (F) cycle curve of heating and cooling three times toward DPFEG-GO 3% hydrogels. | ||
Photothermal effect of DPFEG-GO composite hydrogels
The photothermal effect of DPFEG-GO composite hydrogels containing different concentrations of GO was measured by 808 nm NIR laser irradiation with a power of 2.0 W cm−2 for 5 min. The IR thermogram exhibited a temperature change for 5 min for the DPFEG-GO hydrogel (Fig. 1C). As shown in Fig. 1D, the pure hydrogel (DPFEG-GO 0%) showed a negligible temperature increment compared to those of the composite hydrogels with the same irradiation time. Meanwhile, the temperature elevated significantly along with the increment of GO concentration, and the surface temperature of the composite hydrogels could rise to about 50 °C (the initial temperature was 25 °C). These results indicate that the DPFEG-GO composite hydrogels have a great photothermal therapeutic potential. Thus, combined with the results of the rheological tests, we chose the DPFEG hydrogel containing 3 wt% GO for subsequent study.The heating/cooling curve of the DPFEG-GO 3% composite hydrogel is presented in Fig. 1F. The temperature rose rapidly up to 45 °C under infrared laser irradiation, and then naturally cooled to room temperature. During the cooling period, the temperature dropped slowly. Furthermore, the temperature rising and falling process could be repeated three times without an obvious change, demonstrating its good photothermal stability.
NIR irradiation induced chiral inversion
Spectroscopic characterization of the hydrogels was first performed. The pure DPFEG hydrogel presented a positive CD signal peak at about 227 nm and a negative one at 278 nm (ESI Fig. S7†). Whilst the CD pattern of the DPFEG-GO 3% hydrogel showed a mirror symmetry relationship, a negative Cotton effect peak and a positive one at around 217 nm and 275 nm were observed (ESI Fig. S8†). Based on the preceding literature analysis, the dichroism signals at 227 nm and 278 nm are generated by an amide group and 1,4-benzenedicarbonamide chromophore.60 The negative signal peak at 227 nm was probably due to the jumping of an amido linkage or terminal phenyl to a central aryl group, while the positive one at 278 nm was attributed to the intermolecular π–π* transition of a central aryl group.60 It turns out that the introduction of foreign building blocks disturbs the intermolecular interaction, and the transformations of intramolecular chirality can induce supramolecular self-assembly to form a helical structure with a preferred chirality, inducing the chiral transition from a molecular level to a macroscopic level.When DPFEG-GO 3% composite hydrogels were exposed to infrared light irradiation for a period of time, the chiral optical properties and color changed (ESI Fig. S9†). The intensity of the CD signal in the aryl absorption region (Fig. 2A) decreased significantly and showed a complete reversal with a further increase of irradiation time. The Cotton effect in the peptide bond absorption region was basically the same, suggesting that the chirality could be regulated by non-covalent bonding between the central benzene ring and phenylalanine residues. Instead, the pure hydrogels did not produce a chiral inversion phenomenon with the extension of irradiation (ESI Fig. S10 and S11†). The possible mechanism may be due to the extra formation of intermolecular hydrogen bonds between gelators and GO. Along with the change of GO response to infrared light at different times,63–66 molecular rearrangement of DPFEG took place in the co-assembled system. Thus, the stacking model of intermolecular non-covalent interactions in the dynamic self-assembly process may be changed, and ultimately the supramolecular self-assembly behavior at the macroscopic level was affected.
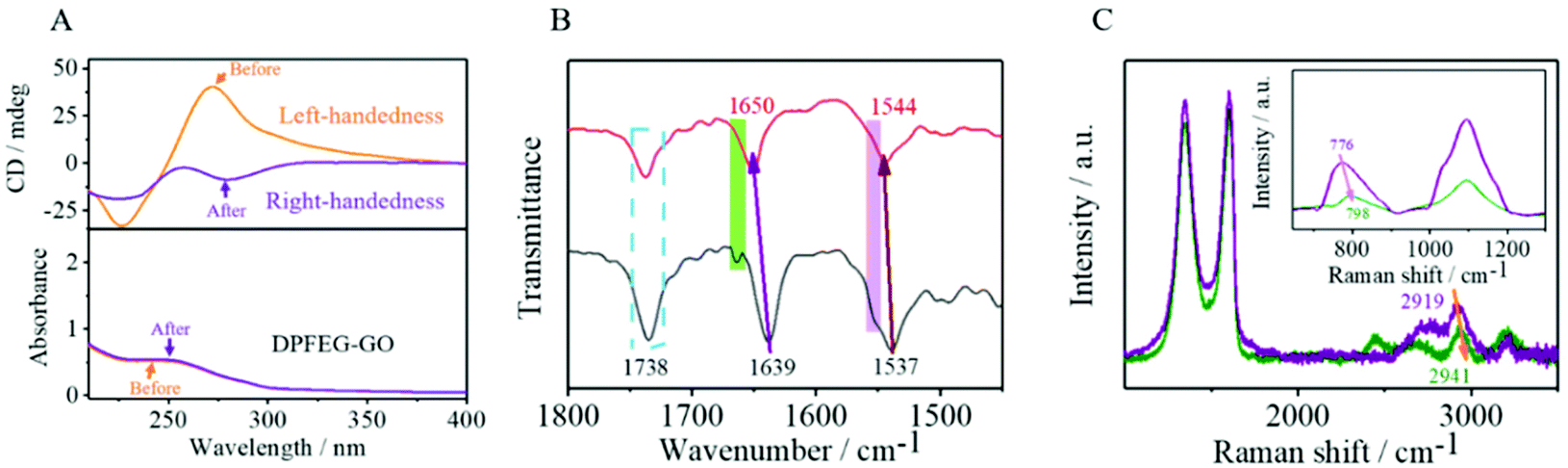 | ||
| Fig. 2 CD and corresponding UV-vis spectra of (A) DPFEG-GO 3% before and after IR irradiation. (B) FTIR and (C) Raman of DPFEG-GO 3% hydrogels. | ||
Scanning electron microscope (SEM) was used to visualize the morphology changes of the hydrogel and the results showed that they all exhibited a microscale helical fiber network structure. The pure DPFEG gel fibers showed a unique right helix; nevertheless, the DPFEG-GO 3% hydrogels formed continuous uniform left-handed spiral fibers with a diameter of hundreds of nanometers (ESI Fig. S12†). As shown in Fig. 3A, after NIR irradiation (6 h), a large number of right-handed nanofibers could be found among the twisted left-handed fibers (ESI Fig. S13a†). With the extension of illumination time, the assemblies tend to arrange into right-handed helix and the helix structure could be maintained when the irradiation was removed. As a contrast, the bare DPFEG hydrogel did not exhibit chiral inversion under NIR illumination (ESI Fig. S13b† and Fig. 3B).
 | ||
| Fig. 3 SEM images of (A) DPFEG hydrogels and (B) DPFEG-GO 3% before and after IR irradiation (scale bar: 1 μm). | ||
Fourier transform infrared spectroscopy (FTIR) was used to further investigate the mechanism of the co-assemblies. In Fig. 2B, the amide I and amide II bands appeared at 1639 cm−1 and 1537 cm−1, respectively; in addition, a stretching vibration band of carboxyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 1738 cm−1 co-existed for the composite hydrogels, suggesting a strong hydrogen bond in the composite. A blue-shift occurred in the amide I band from 1639 cm−1 to 1650 cm−1 after NIR irradiation, and the rift of the amide I band at 1664 cm−1 disappeared. The same phenomenon emerged for amide II band, with a shift from 1537 cm−1 to 1544 cm−1, and a vanish of the rift at 1556 cm−1. These consequences demonstrated that the oxygen groups of GO were gradually eliminated after NIR irradiation, and the existing hydrogen bond with the amide band was steadily decreased, stimulating the rearrangement of hydrogen bonds between the amides.
O at 1738 cm−1 co-existed for the composite hydrogels, suggesting a strong hydrogen bond in the composite. A blue-shift occurred in the amide I band from 1639 cm−1 to 1650 cm−1 after NIR irradiation, and the rift of the amide I band at 1664 cm−1 disappeared. The same phenomenon emerged for amide II band, with a shift from 1537 cm−1 to 1544 cm−1, and a vanish of the rift at 1556 cm−1. These consequences demonstrated that the oxygen groups of GO were gradually eliminated after NIR irradiation, and the existing hydrogen bond with the amide band was steadily decreased, stimulating the rearrangement of hydrogen bonds between the amides.
These results were further confirmed by Raman spectroscopy. In Fig. 2C, the peak of the DPFEG-GO 3% gel at 2919 cm−1(O–H tensile vibration) blue-shifted to 2941 cm−1, and the peak value simultaneously decreased after NIR irradiation, suggesting that the reduction of GO resulted in the weakening of hydrogen bonds in the composite hydrogel. In Fig. 2D, the vibrational band shifted from 776 to 798 cm−1, and the characteristic band at 1091 cm−1 was broadened, accompanied by a significant decline in intensity. Therefore, it can be speculated that the introduction of photo-responsive groups into a supramolecular system will help to establish a hydrogen bond network and modify the supramolecular configurations.
Controlled delivery of chiral anticancer drug
Chiral oxaliplatin (R-OXA) and its enantiomers (S-OXA) were selected as a drug model to investigate the drug loading and controlled release capability of DPFEG-GO 3% hydrogels. As aforementioned, DPFEG-GO 3% presented a negative Cotton effect peak and a positive one at about 217 nm and 275 nm, respectively, similar to that of oxaliplatin (R-OXA). In this case, it could be reasonably inferred that DPFEG-GO aggregates are more inclined to interact with oxaliplatin (R-OXA) because the homochiral structures are more attractive to each other than the heterochiral structures with the known literature.7,10 For verification, an R/S-oxaliplatin solution (0.4 mg mL−1) was added into the DPFEG-GO 3% hydrogel separately and diffused by gentle stirring for half an hour followed by standing still for 24 h under ambient conditions. Ultimately, the mixture was collected by centrifugation and rinsed with water again (ESI Fig. S14†). The changes of the UV-vis spectra of the supernatant before and after culture showed that the absorption intensity of R-OXA decreased significantly whilst the ultraviolet absorption of S-OXA remained unchanged (Fig. 4A). These results confirmed that the DPFEG-GO 3% composite hydrogel is selective to adsorb R-OXA via the preferred supramolecular interactions.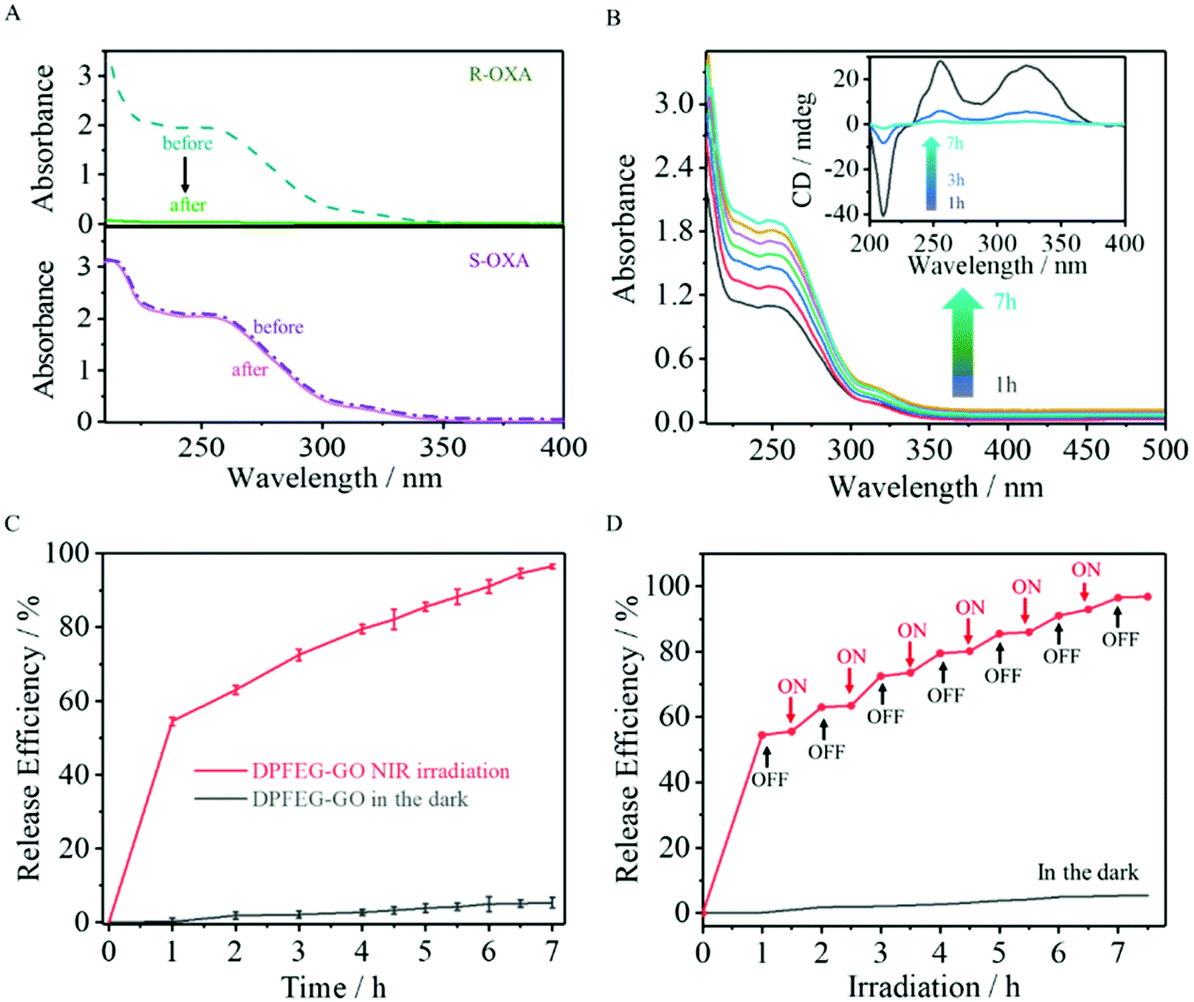 | ||
| Fig. 4 (A) UV-vis spectra of R-OXA (top) and S-OXA (bottom) solutions before and after absorption employing DPFEG-GO hydrogels. (B) UV-vis spectra of R-OXA released from the DPFEG-GO hydrogel at specified irradiation intervals. The inset is the corresponding CD spectra of the release medium. (C) R-OXA release profiles from the DPFEG-GO hydrogel at different times under irradiation and in the dark. (D) The on–off switching release curve of R-OXA released from the DPFEG-GO hydrogel. | ||
UV-vis spectrophotometry was operated to assess the radiation induced release characteristics of DPFEG-GO 3% hydrogels based on the standard curves (ESI Fig. S15†). During the irradiation, the samples were placed in an ice water bath to maintain the temperature. The supernatant was gathered through centrifugation at each prescribed time interval and the detection outcome showed that there was an almost negligible drug molecule leak from the gels in the absence of NIR radiation (placed in the dark to avoid other influences, as shown in Fig. 4B). However, the absorption signals of R-OXA were obviously observed in samples after NIR irradiation and its release efficiency gradually increased with the prolongation of the irradiation time, reaching about 95% in 7 hours (Fig. 4B, C and S16†). The on–off switch experiment could better monitor the responsive release under NIR light stimulation (Fig. 4D). With NIR light, the drug loaded aggregates could commendably release R-OXA molecules owing to the weaker binding force between the drug molecules and complex. After stopping irradiation, this process was inhibited, and the samples treated by irradiation again would show a continuous delivery. Apparently, the drug release process could be well modulated by switching on and off the NIR light. Before irradiation, the gelator assembled into left-handed nanofibers, which have strong interaction with R-OXA. Irradiation weakened the interaction force between gelators and drug molecules due to a chiral transformation from left- to right-handedness. Its enantioselective ability can also be well demonstrated by UPLC-TOM-MSe by using a racemic mixture solution of R-OXA and S-OXA (ESI Fig. S17†).
In vitro biocompatibility of composite hydrogels
To investigate the biocompatibility of the compound hydrogels we carried out a CCK-8 method and AM/PI staining using HS27 cells for a 3-day cytotoxicity test. After the days of cultivation, no obvious cytotoxicity was monitored. As shown in Fig. 5A, cell viabilities of the DPFEG hydrogel and DPFEG-GO 3% hydrogel were both beyond 90%, which is not affected by the toxicity of GO. Although some surveys have revealed that GO influences cells to a certain extent, it can be applied in the biological field by controlling the concentration in a reasonable range.67 The selected GO concentration in the experiment could maintain a cell survival rate of more than 80%. Samples embracing the drug were also characterized and found to have little effect on cell growth. The morphology of cells cultured with various hydrogel groups could also be observed by a fluorescence microscope (ESI Fig. S18†). It was fond that the cells were fibroblasts and showed good growth. Consequently, this kind of mixed hydrogel with a pleasant biocompatibility can maintain the normal growth of cells, promising for biological applications. | ||
| Fig. 5 (A) In vitro biocompatibility test of different hydrogels. (B) Cell viability of T47D Human Breast Cancer Cells treated with A: control, B: DPFEG-GO, C: DPFEG-GO/R-OXA, D: DPFEG-GO + NIR and E: DPFEG-GO/R-OXA + NIR. (C) Live/Dead staining images of T47D cells with various treatment (scale bar: 50 μm). | ||
In vitro antitumor synergistic effect
To further assess the effect of DPFEG-GO 3% hydrogels on cancer cells, materials combined with PTT and chemotherapeutic drugs were performed on T47D cancer cells in vitro via CCK-8 assay. Among them, the groups that needed infrared light were exposed to an 808 nm near infrared laser at 2 W cm−2 for 3 min on the first, second and third days. As illustrated in Fig. 5B, compared with the control group, the DPFEG-GO hydrogels without irradiation exhibited almost a negligible effect on the T47D cells. But in the DPFEG-GO 3% hydrogels with NIR, cell growth was inhibited, dropping to about 34% over time after three days. The cell survival rate was slightly affected in DPFEG-GO drug-containing hydrogels. However, the viability of cells in the combination of drugs and NIR changed most obviously for 3 days, declining to approximately 20%. This is probably because less drug exuded from the hydrogel in the absence of infrared light, resulting in little influence on cell growth. When subjected to near-infrared irradiation, drug molecules could be released quickly, and the photothermal effect also hinders the growth of the cells. The outcomes evidently manifest that the near-infrared irradiation promotes the release of drugs, and thus an outstanding therapeutic effect can be attained by integrating chemotherapy and photothermal therapy into the same stand. These consequences can be further elucidated intuitively by live/death staining (Fig. 5C). Compared with NIR irradiation alone, the drug-containing group exposed to NIR showed a stronger red fluorescence due to cell death, which was consistent with the previous cell viability results. Collectively, such biocompatible supramolecular hydrogels can efficiently kill T47D cancer cells through combination therapy, which is expected to develop into a candidate material for tumor therapy.The staining of the cytoskeleton and nucleus can make further efforts to explore the reasons for the decreased survival rate of T47D cells (Fig. 6). In the case of a combined treatment, many cells showed a crumpled structure while the cytoskeleton shape of some cells was destroyed compared with those in the control group, which suggests that the cell death was caused by apoptosis and the damage was irrevocable.
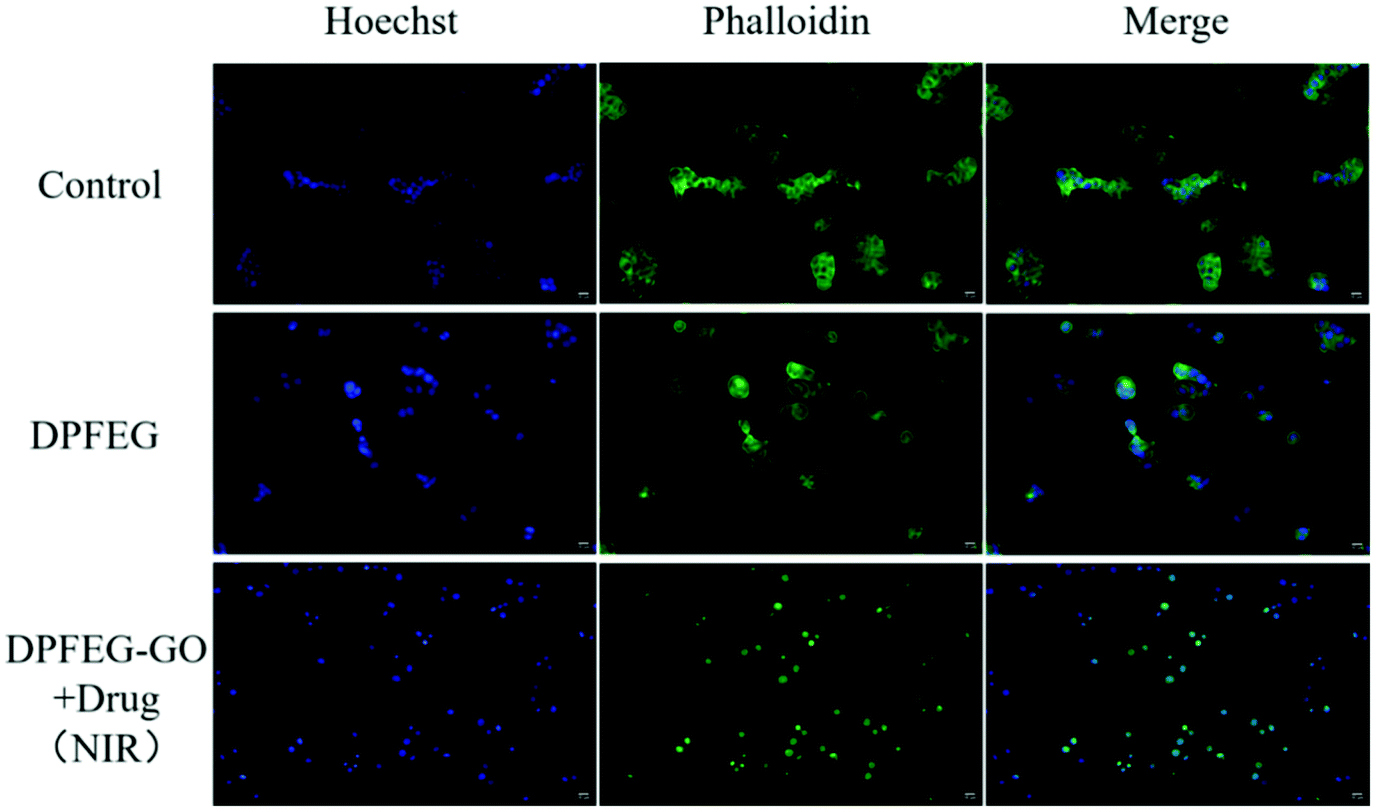 | ||
| Fig. 6 Fluorescence staining images of the nucleus with Hoechst (blue fluorescence) and the cytoskeleton with Phalloidin (green fluorescence) in vitro (scale bar: 10 μm). | ||
Conclusions
In summary, a chiral hydrogel system was constructed by the co-assembly of D-phenylalanine derivative gelator (DPFEG) with graphene oxide (GO). The chiral properties can be regulated by near-infrared light irradiation, which makes it an ideal material for the on-demand control of chiral anticancer drug (oxaliplatin) release. The potential therapeutic effect of the excellent enantioselective delivery ability combined with the photothermal effect was confirmed by significant tumor growth inhibition and conspicuous cell morphological changes. The study renders a theoretical basis for the specific loading of chiral drug molecules, and is expected to be an advanced carrier in the field of chiral drug delivery. Moreover, remote irradiation-mediated technology contributes convenience and controllability for cancer therapy. It broadens the way for the application of chiral multifunctional materials in the treatment of other cancers.Author contributions
Xueqian Wang: methodology, investigation, data curation, formal analysis, writing – original draft, visualization. Beibei Wu: formal analysis, review and editing. Yaqian Zhang: review and editing. Chuanliang Feng: conceptualization, resources, review and editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Nature Science Foundation of China (51833006), the Innovation Program of Shanghai Municipal Education Commission (201701070002E00061), the Science and Technology Commission of Shanghai Municipality (20S31904600, 19ZR1425400), and SJTU Trans-Med Awards Research (WF540162603).Notes and references
- M. H. Liu, L. Zhang and T. Y. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed.
- J. Yan, Y. Yao, S. Q. Yan, R. Q. Gao, W. Y. Lu and W. X. He, Nano Lett., 2020, 20, 5844–5852 CrossRef CAS PubMed.
- P. Y. Xing and Y. L. Zhao, Acc. Chem. Res., 2018, 51, 2324–2334 CrossRef CAS PubMed.
- X. Q. Dou, N. Mehwish, C. L. Zhao, J. Y. Liu and C. L. Feng, Acc. Chem. Res., 2020, 53, 852–862 CrossRef CAS PubMed.
- R. A. Scanga and J. F. Reuther, Polym. Chem., 2021, 12, 1857–1897 RSC.
- Y. H. Chen, W. H. Cheng, L. J. Teng, J. Min and B. H. Lu, Macromol. Mater. Eng., 2018, 303, 1700660 CrossRef.
- H. Noguchi, M. Takafuji, V. Maurizot, I. Huc and H. Ihara, J. Chromatogr., A, 2016, 1437, 88–94 CrossRef CAS PubMed.
- K. Shimomura, T. Ikai, S. Kanoh, E. Yashima and K. Maeda, Nat. Chem., 2014, 6, 429–434 CrossRef CAS PubMed.
- X. J. Zhang, S. H. Qi, C. Y. Liu and X. L. Zhao, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1063, 31–35 CrossRef CAS PubMed.
- T. F. Yan, F. Li, S. W. Qi, J. Tian, R. Z. Tian, J. X. Hou, Q. Luo, Z. Y. Dong, J. Y. Xu and J. Q. Liu, Chem. Commun., 2019, 56, 149–152 RSC.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- Q. P. Duan, Y. Cao, Y. Li, X. Y. Hu, T. X. Xiao, C. Lin, Y. Pan and L. Y. Wang, J. Am. Chem. Soc., 2013, 135, 10542–10549 CrossRef CAS PubMed.
- J. N. Liu, W. B. Bu, L. M. Pan and J. L. Shi, Angew. Chem., Int. Ed., 2013, 52, 4375–4379 CrossRef CAS PubMed.
- Y. W. Sun, Y. F. Yan, M. Q. Wang, C. X. Chen, H. Xu and J. R. Lu, ACS Appl. Mater. Interfaces, 2013, 5, 6232–6236 CrossRef CAS PubMed.
- G. M. Whitesides and M. Boncheva, Proc. Natl. Acad. Sci. U. S. A., 2002, 16, 4769–4774 CrossRef PubMed.
- F. Wang, H. B. Qiu and C. L. Feng, Adv. Funct. Mater., 2020, 30, 2002936 CrossRef CAS.
- D. P. Wang, H. Chen, B. L. Song, T. T. Yan, Z. L. Zhai, X. M. Pei and Z. G. Cui, J. Agric. Food Chem., 2020, 68, 10056–10062 CrossRef CAS PubMed.
- M. Kumar, P. Brocorens, C. Tonnelé, D. Beljonne, M. Surin and S. J. George, Nat. Commun., 2014, 5, 5793 CrossRef CAS PubMed.
- G. F. Liu and Y. L. Zhao, Adv. Sci., 2017, 4, 1700021 CrossRef PubMed.
- G. F. Liu, D. Zhang and C. L. Feng, Angew. Chem., Int. Ed., 2014, 53, 7789–7793 CrossRef CAS PubMed.
- H. JÈ©drzejewska, M. Kwit and A. Szumna, Chem. Commun., 2015, 51, 13799–13801 RSC.
- L. Chen, J. Xiang, Y. Zhao and Q. Yan, J. Am. Chem. Soc., 2018, 140, 7079–7082 CrossRef CAS PubMed.
- N. Mehwish, X. Q. Dou, C. L. Zhao, C. L. Feng and Q. Fu, Adv. Fiber Mater., 2020, 2, 204–211 CrossRef CAS.
- X. Chen, Y. He, Y. J. Kim and M. Lee, J. Am. Chem. Soc., 2016, 138, 5773–5776 CrossRef CAS PubMed.
- Q. D. Hu, G. P. Tang and P. K. Chu, Acc. Chem. Res., 2014, 47, 2017–2025 CrossRef CAS PubMed.
- J. Zhou, G. C. Yu and F. H. Huang, Chem. Soc. Rev., 2017, 46, 7021–7053 RSC.
- Y. C. Chang, Y. Jiao, H. E. Symons, J. F. Xu, C. F. J. Faul and X. Zhang, Chem. Soc. Rev., 2019, 48, 989–1003 RSC.
- G. F. Liu, J. H. Sheng, H. W. Wu, C. L. Yang, G. B. Yang, Y. X. Li, R. Ganguly, L. L. Zhu and Y. L. Zhao, J. Am. Chem. Soc., 2018, 140, 6467–6473 CrossRef CAS PubMed.
- M. Smulders, T. Buffeteau, D. Cavagnat, M. Wolffs, A. Schenning and E. W. Meijer, Chirality, 2008, 20, 1016–1022 CrossRef CAS PubMed.
- S. Cantekin, D. W. R. Balkenende, M. M. J. Smulders, A. R. A. Palmans and E. W. Meijer, Nat. Chem., 2011, 3, 42–46 CrossRef CAS PubMed.
- Y. Nakano, T. Hirose, P. J. M. Stals, E. W. Meijer and A. R. A. Palmans, Chem. Sci., 2012, 3, 148–155 RSC.
- J. J. Van Gorp, J. A. Vekemans and E. W. Meijer, J. Am. Chem. Soc., 2002, 124, 14759–14769 CrossRef CAS PubMed.
- P. F. Duan, H. Cao, L. Zhang and M. H. Liu, Soft Matter, 2014, 10, 5428–5448 RSC.
- J. D. Barrio, R. M. Tejedor, L. S. Chinelatto, C. Sanchez, M. PinOl and L. Oriol, Chem. Mater., 2010, 22, 1714–1723 CrossRef.
- K. Takaishi, M. Kawamoto, K. Tsubaki, T. Furuyama, A. Muranaka and M. Uchiyama, Chemistry, 2011, 17, 1778–1782 Search PubMed.
- R. M. Tejedor, J. L. Serrano and L. Oriol, Eur. Polym. J., 2009, 45, 2564–2571 CrossRef CAS.
- F. Vera, J. L. Serrano, M. P. D. Santo, R. Barberi, M. B. Ros and T. Sierra, J. Mater. Chem., 2012, 22, 18025–18032 RSC.
- Q. C. Hu, Y. Y. Wang, J. Jia, C. S. Wang, L. Feng, R. H. Dong, X. Sun and J. C. Hao, Soft Matter, 2012, 8, 11492–11498 RSC.
- A. Gopal, M. Hifsudheen, S. Furumi, M. Takeuchi and A. Ajayaghosh, Angew. Chem., Int. Ed., 2012, 51, 10505–10509 CrossRef CAS PubMed.
- T. Peng, A. Y. Dang-I, J. Y. Liu and C. L. Feng, Adv. Fiber Mater., 2019, 1, 241–247 CrossRef.
- P. F. Duan, Y. G. Li, L. C. Li, J. G. Deng and M. H. Liu, J. Phys. Chem. B, 2011, 115, 3322–3329 CrossRef CAS PubMed.
- R. Lin, H. Zhang, S. H. Li, L. Q. Chen, W. G. Zhang, T. Bin, W. H. Zhang and H. P. Xia, Chemistry, 2011, 17, 2420–2427 CrossRef CAS PubMed.
- D. Kurouski, X. F. Lu, L. Popova, W. Wan, M. Shanmugasundaram, G. Stubbs, R. K. Dukor, I. K. Lednev and L. A. Nafie, J. Am. Chem. Soc., 2014, 136, 2302–2312 CrossRef CAS PubMed.
- D. Kurouski, R. K. Dukor, X. F. Lu, L. A. Nafie and I. K. Lednev, Chem. Commun., 2012, 48, 2837–2839 RSC.
- C. X. Liu, Q. X. Jin, K. Lv, L. Zhang and M. H. Liu, Chem. Commun., 2014, 50, 3702–3705 RSC.
- Y. Y. Mao, K. Y. Liu, L. Y. Meng, C. Liang and Y. Tao, Soft Matter, 2014, 10, 7615–7622 RSC.
- Y. W. Huang, J. C. Hu, W. F. Kuang, Z. X. Wei and C. F. J. Faul, Chem. Commun., 2011, 47, 5554–5556 RSC.
- X. Y. Wang, C. P. Wang, Q. Zhang and Y. Y. Cheng, Chem. Commun., 2016, 52, 978–981 RSC.
- Y. S. Cai, Z. Q. Guo, J. M. Chen, W. L. Li, L. B. Zhong, Y. Gao, L. Jiang, L. F. Chi, H. Tian and W. H. Zhu, J. Am. Chem. Soc., 2016, 138, 2219–2224 CrossRef CAS PubMed.
- H. J. Jiang, Y. Q. Jiang, J. L. Han, L. Zhang and M. H. Liu, Angew. Chem., Int. Ed., 2019, 58, 785 CrossRef CAS PubMed.
- D. Iglesias, M. Melle-Franco, M. Kurbasic, M. Melchionna, M. Abrami, M. Grassi, M. Prato and S. Marchesan, ACS Nano, 2018, 12, 5530–5538 CrossRef CAS PubMed.
- S. Merino, C. Martín, K. Kostarelos, M. Prato and E. Vázquez, ACS Nano, 2015, 9, 4686–4697 CrossRef CAS PubMed.
- C. Y. Cha, S. R. Shin, N. Annabi, M. R. Dokmeci and A. Khademhosseini, ACS Nano, 2013, 7, 2891–2897 CrossRef CAS PubMed.
- G. Cirillo, G. S. Umile, M. Curcio, S. Hampel, O. Vittorio, D. Restuccia, N. Picci and F. Iemma, Mini Rev. Med. Chem., 2016, 16, 658–667 CrossRef CAS PubMed.
- X. Y. Yang, X. Y. Zhang, Z. F. Liu, Y. F. Ma, Y. S. Huang and J. Phys, Chem. C, 2008, 112, 17554–17558 CAS.
- J. W. Tian, Y. P. Luo, L. W. Huang, Y. Q. Feng, H. X. Ju and B. Y. Yu, Biosens. Bioelectron., 2016, 80, 519–524 CrossRef CAS PubMed.
- A. Deb, N. G. Andrews and V. Raghavan, Int. J. Biol. Macromol., 2018, 113, 515–525 CrossRef CAS PubMed.
- M. Ma, L. Cheng, A. Y. Zhao, H. L. Zhang and A. P. Zhang, Photodiagn. Photodyn. Ther., 2020, 29, 101640 CrossRef CAS PubMed.
- B. Adhikari and A. Banerjee, Soft Matter, 2011, 7, 9259–9266 RSC.
- G. F. Liu, X. Li, J. H. Sheng, P. Z. Li, W. K. Ong, S. Z. Phua, H. Agren, L. L. Zhu and Y. L. Zhao, ACS Nano, 2017, 11, 11880–11889 CrossRef CAS PubMed.
- K. Narasimharao, G. Venkata Ramana, D. Sreedhar and V. Vasudevarao, J. Mater. Sci. Eng., 2016, 5, 1000284 Search PubMed.
- S. N. Alam, N. Sharma and L. Kumar, Graphene, 2017, 06, 1–18 CrossRef CAS.
- R. Maiti, A. Midya, C. Narayana and S. K. Ray, Nanotechnology, 2014, 25, 495704 CrossRef CAS PubMed.
- S. Bhattachraya, R. Maiti, A. C. Das, S. Saha, S. Mondal, S. K. Ray, S. N. B. Bhaktha and P. K. Datta, J. Appl. Phys., 2016, 120, 3677–3694 CrossRef.
- H. L. Guo, M. Peng, Z. M. Zhu and L. N. Sun, Nanoscale, 2013, 5, 9040–9048 RSC.
- N. Habibi, N. Kamaly, A. Memic and H. Shafiee, Nano Today, 2016, 11, 41–60 CrossRef CAS PubMed.
- O. Akhavan and E. Ghaderi, ACS Nano, 2010, 4, 5731–5736 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py01724a |
| This journal is © The Royal Society of Chemistry 2022 |