
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA serological aptamer-assisted proximity ligation assay for COVID-19 diagnosis and seeking neutralizing aptamers†
Ran
Liu
 a,
Lei
He
b,
Yuansheng
Hu
c,
Zhaofeng
Luo
b and
Jingjing
Zhang
*a
a,
Lei
He
b,
Yuansheng
Hu
c,
Zhaofeng
Luo
b and
Jingjing
Zhang
*a
aState Key Laboratory of Analytical Chemistry for Life Science, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, China. E-mail: jing15209791@nju.edu.cn
bHefei National Laboratory for Physical Science at the Microscale, Core Facility Center for Life Sciences, School of Life Sciences, University of Science and Technology of China, Hefei 230026, China
cThe Third Affiliated Hospital of Anhui Medical University, Binhu Hospital of Hefei City, Hefei 230022, China
First published on 12th October 2020
Abstract
Rapid and accurate diagnosis of COVID-19 plays an essential role in the current epidemic prevention and control. Despite the promise of nucleic acid and antibody tests, there is still a great challenge to reduce the misdiagnosis, especially for asymptomatic individuals. Here we report a generalizable method for highly specific and ultrasensitive detection of serum COVID-19-associated antigens based on an aptamer-assisted proximity ligation assay. The sensor is based on binding two aptamer probes to the same protein target that brings the ligation DNA region into close proximity, thereby initiating ligation-dependent qPCR amplification. Using this system, serum nucleocapsid protein has been detected quantitatively by converting protein recognition into a detectable qPCR signal using a simple, homogeneous and fast detection workflow in ∼2 hours. In addition, this system has also been transformed into a universal platform for measuring specific interactions between spike S1 and its receptor ACE2, and more importantly demonstrated the feasibility for screening and investigation of potential neutralizing aptamers. Since in vitro selection can obtain aptamers selective for many COVID-19-associated antigens, the method demonstrated here will serve as an important tool for the diagnosis and therapeutics of COVID-19.
Introduction
Corona Virus Disease 2019 (COVID-19), a highly transmittable and pathogenic viral infection caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has been spreading rapidly to more than 200 countries and challenging the global public health community.1 Therefore, rapid and accurate diagnosis of COVID-19 together with the isolation of infected individuals is one of the foremost priorities in the prevention and control of the current epidemic. To this aim, recent advances in molecular biology have produced various nucleic acid detection methods based on the conserved genome sequences of COVID-19, among which real-time quantitative polymerase chain reaction (RT-qPCR) is recommended as a gold standard for COVID-19 diagnostics.2–4 Despite the progress made, RT-qPCR tests have still been reported for false-negative results in some suspected cases with typical clinical characteristics of COVID-19 and identical specific computed tomography (CT) images.5–7 In addition, some mild or asymptomatic cases with initially negative diagnosis were confirmed with COVID-19 infection only after second, third or multiple repeated RT-qPCR tests.7,8 One of the major contribution factors to these misdiagnoses is mutations in the viral genome during genetic diversity and rapid virus evolution,2,7 which potentially facilitate COVID-19 spread through delayed patient isolation and treatment. Therefore, to improve the accuracy of COVID-19 diagnosis, serological tests for virus-induced antibodies,9 such as IgG/IgM,10 have been developed as a useful complement to nucleic acid testing. Compared with RT-qPCR, these tests have less stringent specimen requirements via uniform serum collection for indirectly detecting SARS-CoV-2.11 The majority of these systems is based on enzyme-linked immunosorbent assay (ELISA)11 or gold nanoparticle based lateral flow devices (LFDs),10 but each technique has its own drawbacks. While ELISA can provide accurate and sensitive results quantitatively, it requires multiple reagent additions, washing and separation steps, resulting in long sample-to-result time (>4 hours) that delays the onsite diagnostic decisions.11 On the other hand, although LFDs are fast (5–20 minutes) and available for point-of-care diagnostics,12 they can only provide qualitative or semi-quantitative information with a limited sensitivity.5 More importantly, these serological tests suffer from potential cross-reactivity of SARS-CoV-2 antibodies with antibodies generated against other coronaviruses due to their high genome similarities,13 leading to a high risk of false positives. Therefore, there is still an urgent need for more strategies that can be applied for rapid, accurate and sensitive diagnosis of COVID-19 patients even before an immune response can occur and more importantly for asymptomatic individuals.14,15Recent advances in structural biology have revealed that SARS-CoV-2 contains four main structural proteins, including the nucleocapsid (N), spike (S), envelope (E), and membrane (M) proteins.16 Among them, the N protein is the most abundant, and relatively conserved protein in coronaviruses, which is involved in the transcription and replication of viral RNA.17 S protein, particularly its S1 domain, is highly immunogenic and essential for host-cell infection, which is considered as a common target for neutralizing antibodies and vaccine development.18 These unique features of structural proteins have made them promising candidates for the development of new serological ELISA for COVID-19 diagnosis.19,20 Despite their importance, the detection of these structural proteins poses a considerable challenge because of their short diagnostic window in COVID-19 infection, cross-reactivity of antibodies, and extremely low abundance. Thus, it is critical to develop a rapid, sensitive, accurate, and widely available diagnostic test for the detection of serum COVID-19-associated antigens. Compared with antibodies, DNA aptamers, known as “chemical antibodies”, are a powerful class of recognition agents with several advantages of high specificity and affinity, rapid and reliable synthesis, ease of conjugation, and good feasibility to integrate with other DNA-based reactions for signal amplification.21–25 These advantages enable aptamers to be a promising receptor, with diverse applications for in vitro and in vivo diagnosis.26–39 In addition to the choice of receptor, it is also important to develop amplification methods to detect COVID-19 with high sensitivity. Toward this goal, the proximity ligation immunoassay (PLA) has been recently developed as a unique signal amplification approach,40 in which a protein target is detected by translating antibody–protein binding events into amplifiable DNA sequences for subsequent real-time polymerase chain reaction quantification.41
Inspired by this strategy, we report here a generalizable method for highly specific and ultrasensitive detection of serum COVID-19-associated antigens based on an aptamer-assisted proximity ligation assay (Apt-PLA). The method is based on binding two aptamer probes to the same antigen target that brings the ligation DNA region into close proximity, thereby initiating ligation-dependent qPCR amplification. Taking advantage of cooperative recognition by two aptamers and signal amplification via qPCR, we demonstrate that such an Apt-PLA system can provide a high degree of specificity and sensitivity for the detection of serum COVID-19-associated antigens. Furthermore, we also applied this Apt-PLA system for the investigation of the interaction of spike protein with the human ACE2 receptor, and more importantly the evaluation of potential neutralizing aptamers.
Results and discussion
Principle of the aptamer-assisted proximity ligation assay
The construction and operation of the aptamer-assisted proximity ligation assay is illustrated in Scheme 1. The Apt-PLA system is composed of two key components: (1) two proximity ligation probes both of which consist of an aptamer region for target recognition, spacer region to minimize the structural steric hindrance during the assembly, PCR primer region, and ligation region with 5′ phosphate modification; (2) an ssDNA connector as the template to assist the ligation by hybridization with two proximity probes at the ligation region. In such a design, binding of the two aptamers to the same protein target could bring the ligation region into close proximity and thus hybridize with the connector template to form a DNA complex that is available for subsequent ligation-dependent qPCR amplification. In contrast, without the protein target, the two probes that were not in proximity produced a low ligation event, resulting in low nonspecific qPCR signals.42 The discrimination of the qPCR signal can be exhibited by a cycle threshold (Ct) value, which is defined as the PCR cycle number at which the sample's reaction curve intersects the threshold line. This value is inversely proportional to the amount of PCR DNA amplicon generated via aptamer-assisted proximity ligation reaction induced by the protein target. As a result, a higher Ct value means lower amount of ligated DNA amplicon, lower ligation efficiency, and lower concentration of the protein target accordingly. Thus, the presence and concentration of the protein target can be determined by monitoring the Ct value change in the system.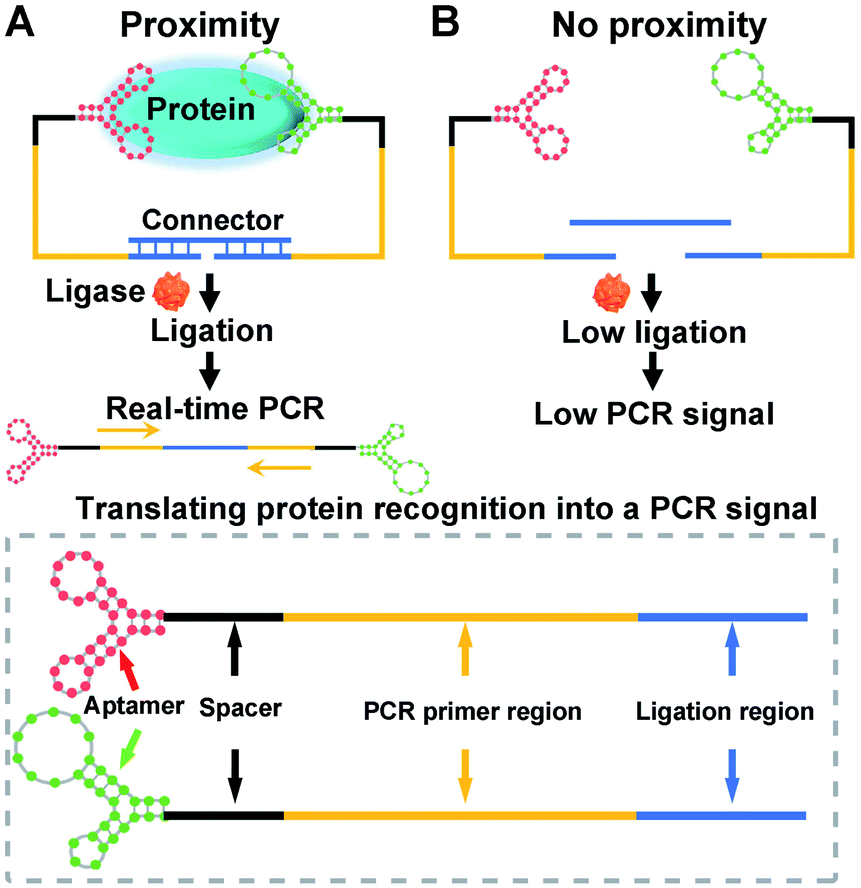 | ||
| Scheme 1 Scheme of aptamer-assisted proximity ligation assay for COVID-19 antigens. (A) Probes that bind to the same protein target are in close proximity, initiating ligation-dependent qPCR amplification. (B) Unbounded probes that are not in proximity show low ligation efficiency, resulting in low PCR signal. | ||
Feasibility study of the Apt-PLA for nucleocapsid protein
To demonstrate the above aptamer-assisted proximity ligation assay for detecting COVID-19-associated antigens, we first chose N protein as the target, and two DNA aptamers (N48 and N58) as the recognition units, which were obtained by in vitro selection.43 To achieve proximity ligation, two proximity probes (PPA and PPB) consisting of the aptamer region (N48 and N58), spacer (T5), PCR primer region (P1 and P2), and ligation region (L1 and L2) were designed (Table S1†). In addition, an ssDNA connector was designed, which assists the connection of L1 and L2 within PPA and PPB by complementary DNA hybridization, respectively. Such a design ensured that only in the presence of PPA, PPB, connector, and N protein, the subsequent ligation-dependent qPCR amplification could be initiated. The general outline of the designed Apt-PLA for N protein detection is schematically illustrated in Fig. 1A. In a typical Apt-PLA experiment, 3.5 μL of reagent A containing PPA (286 pM), PPB (286 pM), and connector (571 nM) in T4 ligase buffer was pre-incubated with 1.0 μL of N protein at 25 °C for 20 min to allow aptamer binding, followed by the addition of 0.5 μL of reagent B (T4 DNA ligase, 0.4 units) and incubated at 25 °C for 5 min to enable proximity ligation. After heat inactivation, the ligation products were amplified and analyzed by a qPCR using a One Step RT-PCR Kit, in which TB Green was utilized as the fluorescent dye for real-time detection of the PCR products.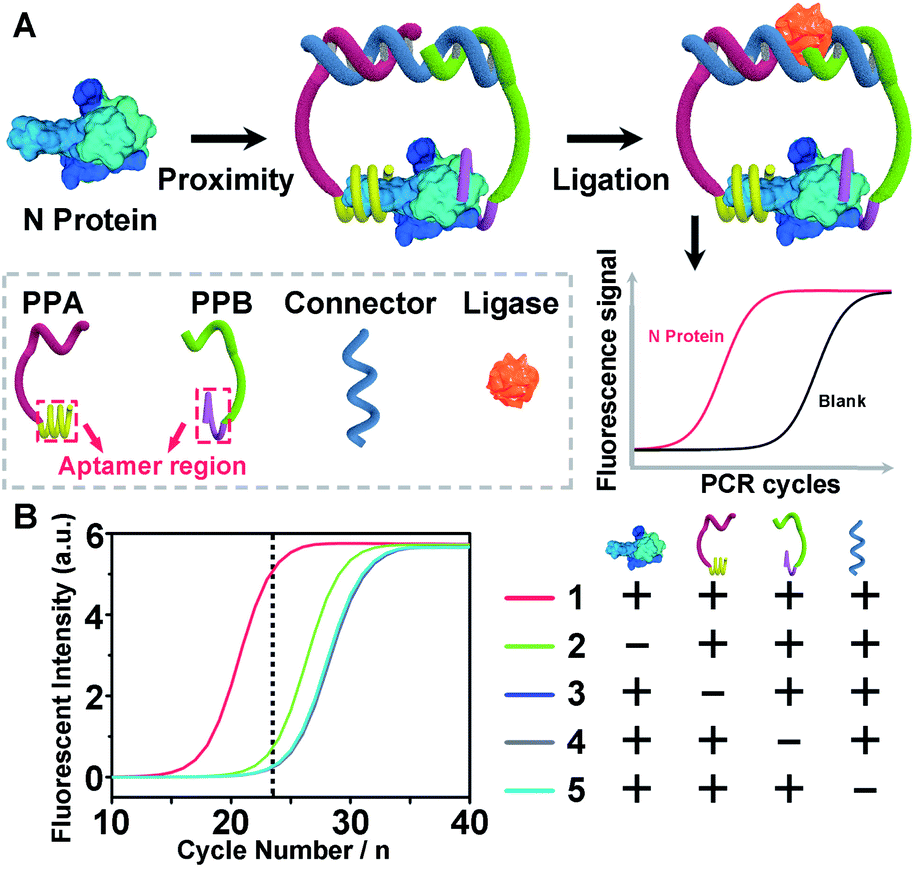 | ||
| Fig. 1 (A) Detection strategy and workflow of our aptamer-assisted proximity ligation assay for nucleocapsid protein. (B) The real-time fluorescence intensity of qPCR signals for nucleocapsid protein and control samples. | ||
Fig. 1B shows the real-time fluorescence intensity of Apt-PLA for N protein and control samples. In the absence of N protein, the qPCR curve shows a high Ct value, because the two Apt-PLA probes are not in proximity and hence result in low ligation efficiency. In addition, the absence of PPA, PPB or connector produced a slightly higher Ct value than the blank sample, indicating negligible ligation efficiency. In contrast, under the same conditions, addition of N protein resulted in a significant decrease of the Ct value, which was due to the binding of PPA and PPB to the same N protein and thus yielded high ligation efficiency. Furthermore, to confirm that the qPCR signal was dependent on the ligation efficiency that generates an amplifiable PCR amplicon, we further collected the intermediate qPCR products at the 24th cycle (marked in Fig. 1B), and analyzed using 10% denatured polyacrylamide gel electrophoresis (PAGE). As shown in Fig. S1,† in the presence of PPA, PPB, connector, and N protein, a strong band at the high molecular weight (MW) region was observed (lane 1), while the absence of either of these four components produced a much weaker band at the high MW region (lanes 2–5). These results indicated that the addition of N protein could trigger the proximity ligation with high efficiency that generates a significant qPCR signal, which is consistent with the qPCR results (Fig. 1B). Moreover, to compare the rate of strand hybridization with and without the N protein, we further performed a fluorescence test by mixing PPA, PPB, ssDNA, T4 DNA ligase, and N protein (0 or 5 ng mL−1) with SYBR Green I (Fig. S2A†), which is a green fluorescent cyanine dye that has high affinity for double-stranded DNA. As shown in Fig. S2B,† in the presence of 5 ng mL−1 N−1 protein, the fluorescence intensity ratio (F/F0) increases with the increase of reaction time up to 600 s, indicating the formation of double-stranded DNA. In contrast, in the absence of N protein, the fluorescence intensity ratio (F/F0) shows a much less increase and reaches a plateau after ∼60 s. These results indicate that the presence of N protein could induce the formation of a more stable DNA ternary complex, resulting in a high ligation event and thus a high qPCR signal. Taken together, these results demonstrate the effective recognition of N protein using dual Apt-PLA probes, forming the basis for a new Apt-PLA system for COVID-19-associated antigens that can be recognized by aptamers.
Detection performance of Apt-PLA for nucleocapsid protein
The detection mechanism of Apt-PLA relies on the simultaneous binding of N protein by a pair of aptamer affinity probes which bring L1 and L2 in proximity to process the ligation with the assistance of ssDNA connector and T4 DNA ligase. Thus, to achieve good analytical performance for N protein detection, the number of complementary bases between ssDNA connector and L1/L2, the incubation time between aptamer probes and N protein, and the ligation time should be firstly optimized. Fig. S3A† shows the qPCR signals and backgrounds of the Apt-PLA when using the connector with 16, 18, 20, and 22 complementary bases (Con-16, Con-18, Con-20, Con-22) to L1/L2 for proximity hybridization. As shown in Fig. S3B,† in the presence of 2 ng mL−1 of N protein, the Ct value decreased with increasing number of complementary bases from 16 to 22. However, a larger number of complementary bases also caused a larger background due to the increased target-independent ligation events. Therefore, Con-18 was chosen for the subsequent experiments based on the maximum signal-to-background ratio (ΔCt). Similarly, according to the maximum ΔCt, the optimal incubation and ligation time for our Apt-PLA system were obtained as 20 min (Fig. S4†) and 5 min (Fig. S5†), respectively.Under the optimal conditions, we then investigate if the aptamer-assisted proximity ligation assay could detect N protein quantitatively. Fig. 2A shows qPCR curves of different concentrations of N protein added in DPBS buffer. The obtained Ct value decreased with increasing N protein concentrations from 0 to 5000 pg mL−1, indicating that two Apt-PLA probes were in proximity via binding to N protein and thus could be ligated to serve as the template for the subsequent qPCR experiment. In addition, a linear relationship was observed between the ΔCt value and the logarithm of the N protein concentration in the range of 50 pg mL−1 to 5000 pg mL−1 (Fig. 2B), and a limit of detection (LOD) of 37.5 pg mL−1 was obtained based on a 3σb/slope, where σb is the standard deviation of four blank samples. This performance is comparable to that of the commercial ELISA kit with an LOD of approximately 50 pg mL−1, and much better than that of a recent half-strip lateral flow assay with an LOD of 0.65 ng mL−1.44
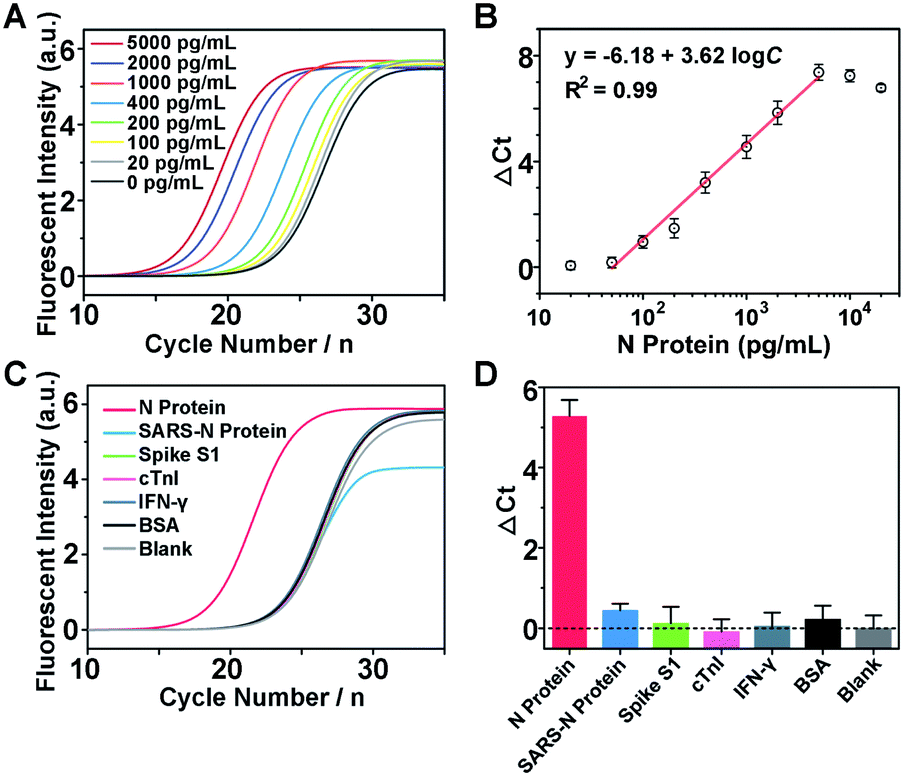 | ||
| Fig. 2 Performance of the aptamer-assisted proximity ligation assay for N protein detection. (A) qPCR curves of different concentrations of N protein added in DPBS buffer. (B) Dose-dependent relationship between the ΔCt value and the N protein concentration from 20 to 5000 pg mL−1. (C) qPCR curves for the selectivity test. (D) ΔCt values for 2 ng mL−1 of N protein of SARS-CoV-2, N protein of SARS-CoV, and other competing proteins. ΔCt is the difference between the Ct value of a protein sample and a blank sample. Error bars represent the standard deviations of three parallel tests. | ||
To demonstrate the selectivity of the aptamer-assisted proximity ligation assay, we performed the PLA-qPCR using different competing proteins and the real-time fluorescence intensity of qPCR was recorded (Fig. 2C), including the N protein of SARS-CoV, spike S1, cardiac troponin I (cTnI), interferon-gamma (IFN-γ), and bovine serum albumin (BSA). As shown in Fig. 2D, compared with the blank samples, no significant difference in ΔCt was observed for competing proteins at a concentration of 2 ng mL−1 (p > 0.05), while the ΔCt value in response to 2 ng mL−1 of the N protein of SARS-CoV-2 showed a more than 20-fold increase (p < 0.001), suggesting that the high selectivity of the two N protein aptamers was maintained for the aptamer-assisted proximity ligation assay. In addition, to further explain the specificity of our Apt-PLA system, three mutated DNA aptamers were designed to replace the aptamer regions in PPA or PPB (Fig. S6A and B†). As shown in Fig. S6C,† compared with unmutated DNA aptamers, the mutated aptamers result in much higher Ct values in response to 5 ng mL−1 N protein, indicating lower ligation efficiencies due to their decreased binding affinities to N protein after mutations. In addition, the normalized signal responses to N protein using MPPA1/PPB, MPPA2/PPB, MPPA2/MPPB1, and MPPA2/MPPB1 decreased to (39.9 ± 16.2)%, (16.5 ± 4.0)%, (32.6 ± 6.6)%, and (16.6 ± 1.7)%, respectively (Fig. S6D†). These results demonstrated that the high specificity of our Apt-PLA system was due to the specific recognition of N protein by unmutated N48 and N58 aptamers.
Analysis in complex biological samples
To investigate whether the aptamer-assisted proximity ligation assay can be applied in biological samples, we further explored the detection of N protein in 100% human serum. The qPCR amplification curves for a series of human serum samples spiked with different concentrations of N protein are shown in Fig. 3A, and the Ct value decreased with increasing N protein concentrations from 0 to 5000 pg mL−1. In addition, we observed a quantitative relationship between ΔCt and the logarithm of the N protein concentration spiked in human serum in the range of 50–5000 pg mL−1 (inset of Fig. 3A), with an LOD of 30.9 pg mL−1 based on 3σb/slope. Since these results are similar to those in DPBS buffer, they suggest that other components in the serum did not interfere significantly with the Apt-PLA performance, and therefore this Apt-PLA can be used in complex biological fluids.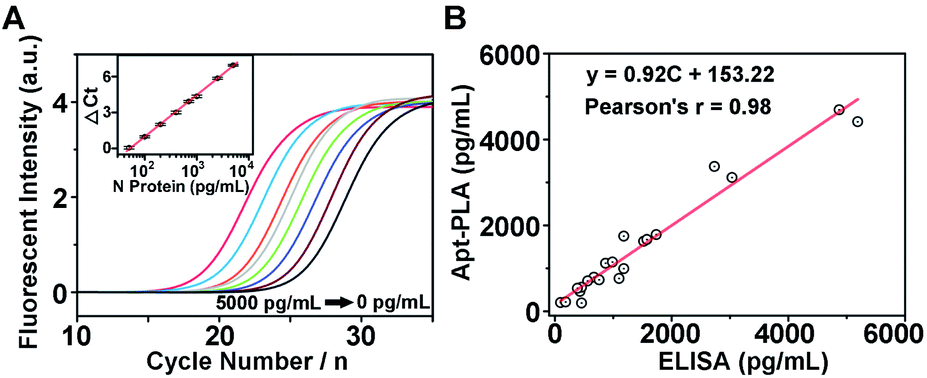 | ||
| Fig. 3 Performance of the aptamer-assisted proximity ligation assay for N protein detection in human serum. (A) qPCR curves of different concentrations of N protein spiked in 100% human serum. Inset: Linear relationship of ΔCt values and protein concentrations. (B) Comparison of the aptamer-assisted proximity ligation assay with a commercial ELISA kit for N protein detection. | ||
To verify the accuracy and reliability of the aptamer-assisted proximity ligation assay, we further compared it with a commercial ELISA kit for N protein detection by analyzing human serum samples spiked with different concentrations of N protein. Based on the calibration curves from our Apt-PLA (Fig. 3A) and the ELISA kit (Fig. S7†), the N protein concentration in each serum sample was calculated, and a total of 21 samples in human serum were evaluated (Fig. S8†). As shown in Fig. 3B, a strong positive correlation between these two methods was found, with a slope of 0.92 ± 0.04 and a correlation coefficient of 0.98, thus demonstrating that the results from the two methods were consistent to within experimental error. These results confirm that the accuracy of our Apt-PLA method is as good as that of the commercial ELISA kit but possesses remarkable merits of short sample-to-result time (∼2 hours) in a homogeneous format. Since in vitro selection can obtain aptamers selective for many COVID-19-related associated antigens, such as the recently reported aptamers targeting SARS-CoV-2 RBD,45 the method demonstrated here can expand this powerful Apt-PLA system significantly to many other targets and thus provide a new toolbox for the diagnosis of COVID-19.
Encouraged by the above successful N protein detection in spiked human serum samples, we further conducted similar tests in clinical serum samples collected from healthy people and confirmed patients to demonstrate the utility of our method for clinical diagnosis. A total of 40 serum samples from 4 healthy people and 4 patients were evaluated. As shown in Fig. S9A,† compared with the blank sample, no significant difference in Ct value was observed for healthy people, indicating the presence of a negligible amount of N protein in the serum samples of healthy people. However, the Ct values obtained from the serum of patients were much lower than that of blank samples, indicating the presence of N protein. In addition, as shown in Fig. S9C,† the average ΔCt values for the healthy people and four patients were calculated to be 0.04 ± 0.20 (n = 20), 2.27 ± 0.12 (n = 5), 0.99 ± 0.06 (n = 5), 1.04 ± 0.12 (n = 5), and 2.28 ± 0.29 (n = 5), respectively, indicating a higher level of N protein in SARS-CoV-2 infected patients. Taken together, these results demonstrated the feasibility of our Apt-PLA system for the sensitive and accurate detection of N protein in clinical samples.
Feasibility of antibody-based proximity ligation assay
To expand the capability of the proximity ligation assay, we further extended our building blocks from aptamers to antibodies and designed an antibody-based proximity ligation assay for N protein by replacing N48 and N58 aptamers with two SARS-CoV-2 Nucleoprotein antibodies. The aptamer regions in PPA and PPB were replaced by a thiolated T30 spacer, and conjugated with antibodies via the maleimide–thiol reaction. As shown in Fig. S10A,† binding of the two antibodies to the same N protein could bring the ligation region into close proximity and thus hybridize with the connector template to form a DNA ternary complex that is available for subsequent ligation-dependent qPCR amplification. In contrast, without the N protein, the two probes that were not in proximity produced a low ligation event, resulting in low nonspecific qPCR signals. Fig. S10B† shows the qPCR curves of different concentrations of N protein tested using the antibody-based proximity ligation assay. The obtained ΔCt value increased with increasing N protein concentrations from 0 to 5000 pg mL−1 (Fig. S10C†), demonstrating the feasibility of the designed antibody-based proximity ligation assay for the detection of N protein.Investigation of specific interaction between spike S1 and ACE2
Another challenge in the current COVID-19 research is to develop effective therapeutic drugs or vaccines. Previous studies have revealed that the COVID-19 virus binds to host cells via interactions with the human angiotensin converting enzyme 2 (ACE2).16,46 Therefore, neutralizing molecules targeting vulnerable sites on viral surface proteins (e.g. spike S1) are increasingly studied and have shown therapeutic blocking capabilities.16,47–51 To this aim, several methods have been developed for the screening of neutralizing antibodies or DNAs, including plaque reduction neutralization test, ELISA, and fluorescence-based high-throughput assay.49,52 Although promising, the time–to-results for these neutralization assays typically ranges from several hours to days, thereby limiting their broad applications. To address this issue, encouraged by the above successful serological antigen sensing, we sought to adapt our Apt-PLA system for the screening of potential neutralizing molecules. To do this, we first investigated the feasibility of using PLA for the measurement of interaction between spike S1 and ACE2 (Fig. 4A). Briefly, two PLA probes were prepared by replacing the aptamer region (Scheme 1) with a thiolated T30 spacer, followed by conjugating with spike S1 and ACE2 using the sulfo-SMCC cross-linking method, respectively. A control probe was also synthesized by conjugating N protein with the same DNA sequence as spike S1. Conjugation was confirmed by observing strong bands at high MW regions by 8% native PAGE (Fig. S11†). As shown in Fig. S12,† individual spike S1-PLA probe or ACE2-PLA probe produced a high Ct value of 29.8 and 23.9, respectively. In contrast, incubating spike S1-PLA probe with ACE2-PLA probe results in a much lower Ct value of 17.4 (Fig. 4B), which was due to the strong interaction between spike S1 and ACE2 that brings the two probes in close proximity for a high ligation efficiency. In addition, incubating the control N protein-PLA probe with ACE2-PLA probe generated a much higher Ct value of 22.2 (Fig. 4B), indicating a weak, non-specific interaction. These results indicated that our adapted PLA system can be used for the measurement of interaction between spike S1 and ACE2 specifically.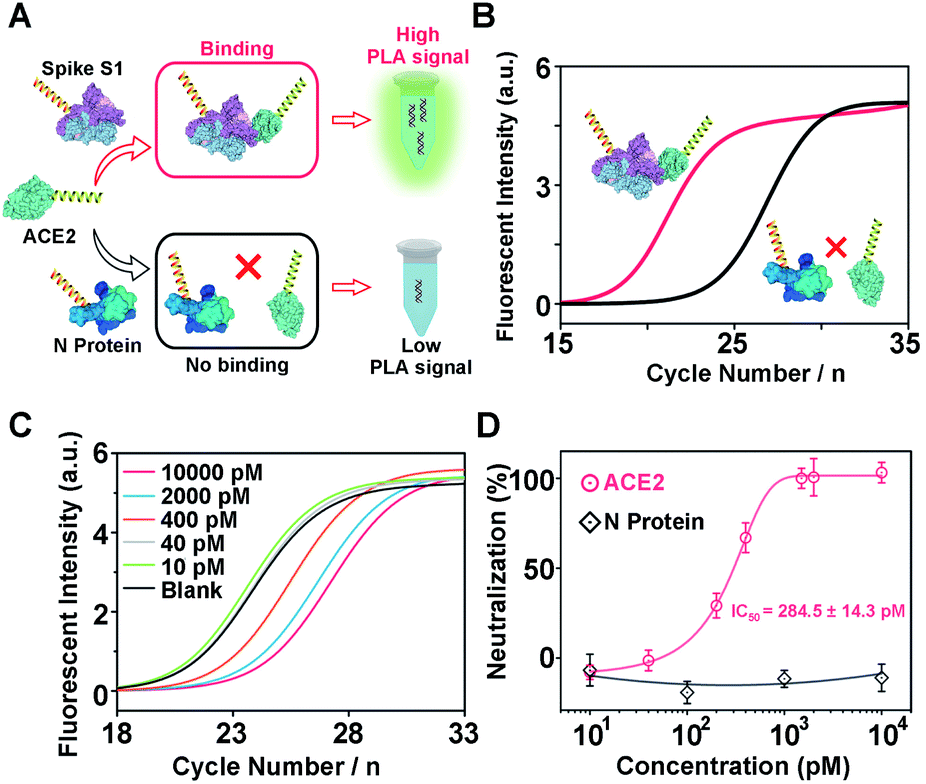 | ||
| Fig. 4 (A) Schematic illustration of our adapted proximity ligation assay for the measurement of specific interaction between spike S1 and its receptor ACE2. (B) Comparison of the qPCR curves of the specific and non-specific interactions. (C) qPCR curves of our adapted proximity ligation assay with different concentrations of ACE2. (D) Dose–response neutralization titration curves of spike S1 using free ACE2 and a control protein. The spike S1-DNA probe was pretreated with different concentrations of free ACE2 for 10 min, and then tested using our adapted proximity ligation assay. | ||
Next, to test the feasibility of the PLA system for evaluation of neutralization tests, we further used ACE2 as a model neutralizing protein, and study the binding competition to spike S1 between free ACE2 and ACE2-PLA probe. Briefly, we pre-incubated spike S1-PLA probe with different concentrations of free ACE2 at 25 °C for 10 min, followed by the addition of ACE2-PLA probe and connector and incubated for another 20 min. Then, T4 DNA ligase was added to the above mixture to initiate the ligation, and the resulting DNA sequence was analyzed using RT-PCR. As shown in Fig. 4C, the Ct value increased with increasing pre-incubated ACE2 concentrations from 40 pM to 10 nM, indicating an effective blocking of spike S1 binding to ACE2-PLA probe. The neutralization efficiency was then calculated, and a strong dose-dependent neutralization efficiency was observed for ACE2 with a half-maximal inhibitory concentration (IC50) value of 284.5 ± 14.3 pM (Fig. 4D). In contrast, compared with the blank sample, no significant difference in Ct value was observed for pre-incubation of N protein with spike S1-PLA probe even at a high concentration of 10 nM (Fig. S13†), resulting in no neutralization efficiency (Fig. 4D), which indicated that the N protein has a negligible blocking effect on the spike S1 binding to ACE2. These results demonstrate that our PLA system can be applied for neutralization tests, indicating its great potential for screening potential neutralizing molecules.
Evaluation of potential neutralizing aptamers using the PLA system
To demonstrate the generality of the PLA system for neutralization tests, we extended our methodology from proteins to aptamers for the evaluation of neutralization efficiency. Two synthetic DNA aptamers (Apt-S-79s and Apt-S-268s) targeting spike S1,53 obtained by in vitro selection, were chosen as potential neutralizing candidates, while one random DNA sequence with 16 nucleotides was used as a negative control. Then, a similar neutralization test was implemented using our PLA system (Fig. 5A), in which the spike S1-PLA probe was pre-treated with the above three DNAs at 25 °C for 10 min, respectively. The real-time fluorescence intensities of qPCR for different concentrations of Apt-S-79s, Apt-S-268s and random DNA were recorded (Fig. S14–S16†). Compared with the random DNA, a significant dose-dependent neutralization effect was observed for Apt-S-79s and Apt-S-268s, with IC50 values of 1.83 ± 0.61 μM and 12.26 ± 1.79 μM (Fig. 5B), respectively. These results indicated that Apt-S-79s and Apt-S-268s may have partially identical binding sites to the RBD region of SARS-CoV-2 spike S1 protein, revealing the potential to inhibit or block the binding of SARS-CoV-2 spike S1 protein to ACE2. This competition may be due to the binding of aptamers to several amino acid residues of RBD that are key to ACE2 binding.45 In addition, given the fact that the whole workflow could be finished within 2 hours in a homogeneous format, the proposed PLA system holds great promise for simple and rapid screening of various neutralizing candidates.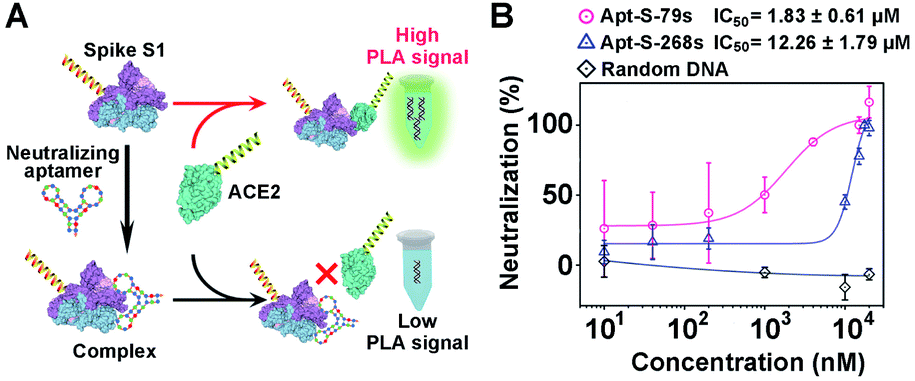 | ||
| Fig. 5 (A) Schematic illustration of our adapted proximity ligation assay for evaluation of the neutralization efficiency of potential neutralizing aptamers that bind to spike S1. (B) Dose–response neutralization titration curves of spike S1 using two aptamers targeting spike S1 and a random DNA as a negative control. The spike S1-DNA probe was pretreated with different concentrations of neutralizing aptamers for 10 min, and then tested using our adapted proximity ligation assay. | ||
Conclusion
In summary, we have demonstrated a simple and versatile aptamer-assisted proximity ligation assay that provides a target-dependent qPCR signal change, enabling sensitive and selective quantification of COVID-19-associated antigens that the aptamer recognizes. The sensor system is based on the binding of two aptamer probes to the same protein target that brings the ligation DNA region into close proximity, thereby initiating ligation-dependent qPCR amplification. Using this system, serum nucleocapsid protein has been detected quantitatively by converting protein recognition into a detectable qPCR signal using a simple, homogeneous and fast detection workflow within 2 hours. These features allow clinical laboratories to implement the nucleic acid and serological antigen test in parallel, thus improving the diagnostic accuracy of COVID-19 particularly for asymptomatic individuals. In addition, this system has also been transformed into a universal platform for the measurement of specific interactions between spike S1 and its receptor ACE2, and more importantly paves a way for further screening and investigation of potential neutralizing molecules. Since in vitro selection can obtain aptamers selective for many COVID-19-related associated antigens, the method demonstrated will serve as an important tool for the diagnosis and therapeutics of COVID-19.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We greatly acknowledge the financial support from the Fundamental Research Funds for the Central Universities (020514380196, 020514380215), Innovation Fund from Nanjing University (020514913414), and start-up funds from Nanjing University (020514912226). Zhaofeng Luo acknowledges the support of the Fundamental Research Funds for the Central Universities (YD2070002016). We thank Prof. Yi Lu for very helpful discussions and critical input.Notes and references
- R. Butowt and K. Bilinska, ACS Chem. Neurosci., 2020, 11, 1200–1203 CrossRef CAS.
- B. Udugama, P. Kadhiresan, H. N. Kozlowski, A. Malekjahani, M. Osborne, V. Y. C. Li, H. Chen, S. Mubareka, J. B. Gubbay and W. C. W. Chan, ACS Nano, 2020, 14, 3822–3835 CrossRef CAS.
- J. Wang, K. Cai, R. Zhang, X. He, X. Shen, J. Liu, J. Xu, F. Qiu, W. Lei, J. Wang, X. Li, Y. Gao, Y. Jiang, W. Xu and X. Ma, Anal. Chem., 2020, 92, 9399–9404 CrossRef CAS.
- G. Xue, S. Li, W. Zhang, B. Du, J. Cui, C. Yan, L. Huang, L. Chen, L. Zhao, Y. Sun, N. Li, H. Zhao, Y. Feng, S. Liu, Q. Zhang, X. Xie, D. Liu, H. Yao and J. Yuan, Anal. Chem., 2020, 92, 9699–9705 CrossRef CAS.
- N. Bhalla, Y. Pan, Z. Yang and A. F. Payam, ACS Nano, 2020, 14, 7783–7807 CrossRef CAS.
- A. Tahamtan and A. Ardebili, Expert Rev. Mol. Diagn., 2020, 20, 453–454 CrossRef CAS.
- W. Feng, A. Newbigging, C. Le, B. Pang, H. Peng, Y. Cao, J. Wu, G. Abbas, J. Song, D. B. Wang, M. Cui, J. Tao, D. L. Tyrrell, X. E. Zhang, H. Zhang and X. C. Le, Anal. Chem., 2020, 92, 10196–10209 CrossRef CAS.
- D. A. Green, J. Zucker, L. F. Westblade, S. Whittier, H. Rennert, P. Velu, A. Craney, M. Cushing, D. Liu, M. E. Sobieszczyk, A. K. Boehme and J. L. Sepulveda, J. Clin. Microbiol., 2020, 58, e00995 CrossRef CAS.
- F. Krammer and V. Simon, Science, 2020, 368, 1060–1061 CrossRef CAS.
- C. Huang, T. Wen, F. J. Shi, X. Y. Zeng and Y. J. Jiao, ACS Omega, 2020, 5, 12550–12556 CrossRef CAS.
- F. Amanat, D. Stadlbauer, S. Strohmeier, T. H. O. Nguyen, V. Chromikova, M. McMahon, K. Jiang, G. A. Arunkumar, D. Jurczyszak, J. Polanco, M. Bermudez-Gonzalez, G. Kleiner, T. Aydillo, L. Miorin, D. S. Fierer, L. A. Lugo, E. M. Kojic, J. Stoever, S. T. H. Liu, C. Cunningham-Rundles, P. L. Felgner, T. Moran, A. Garcia-Sastre, D. Caplivski, A. C. Cheng, K. Kedzierska, O. Vapalahti, J. M. Hepojoki, V. Simon and F. Krammer, Nat. Med., 2020, 26, 1033–1036 CrossRef CAS.
- Z. H. Chen, Z. G. Zhang, X. M. Zhai, Y. Y. Li, L. Lin, H. Zhao, L. Bian, P. Li, L. Yu, Y. S. Wu and G. F. Lin, Anal. Chem., 2020, 92, 7226–7231 CrossRef CAS.
- X. Ou, Y. Liu, X. Lei, P. Li, D. Mi, L. Ren, L. Guo, R. Guo, T. Chen, J. Hu, Z. Xiang, Z. Mu, X. Chen, J. Chen, K. Hu, Q. Jin, J. Wang and Z. Qian, Nat. Commun., 2020, 11, 1620 CrossRef CAS.
- Z. Huang, D. Tian, Y. Liu, Z. Lin, C. J. Lyon, W. Lai, D. Fusco, A. Drouin, X. Yin, T. Hu and B. Ning, Biosens. Bioelectron., 2020, 164, 112316 CrossRef CAS.
- J. P. Broughton, X. Deng, G. Yu, C. L. Fasching, V. Servellita, J. Singh, X. Miao, J. A. Streithorst, A. Granados, A. Sotomayor-Gonzalez, K. Zorn, A. Gopez, E. Hsu, W. Gu, S. Miller, C. Y. Pan, H. Guevara, D. A. Wadford, J. S. Chen and C. Y. Chiu, Nat. Biotechnol., 2020, 38, 870–874 CrossRef CAS.
- A. C. Walls, X. Xiong, Y. J. Park, M. A. Tortorici, J. Snijder, J. Quispe, E. Cameroni, R. Gopal, M. Dai, A. Lanzavecchia, M. Zambon, F. A. Rey, D. Corti and D. Veesler, Cell, 2019, 176, 1026–1039 CrossRef CAS.
- S. Kang, M. Yang, Z. Hong, L. Zhang, Z. Huang, X. Chen, S. He, Z. Zhou, Z. Zhou, Q. Chen, Y. Yan, C. Zhang, H. Shan and S. Chen, Acta Pharm. Sin. B, 2020, 10, 1228–1238 CrossRef.
- D. Wrapp, N. Wang, K. S. Corbett, J. A. Goldsmith, C. L. Hsieh, O. Abiona, B. S. Graham and J. S. McLellan, Science, 2020, 367, 1260–1263 CrossRef CAS.
- S. Weiss, J. Klingler, C. Hioe, F. Amanat, I. Baine, S. Arinsburg, E. M. Kojic, J. Stoever, S. T. H. Liu, D. Jurczyszak, M. Bermudez-Gonzalez, V. Simon, F. Krammer and S. Zolla-Pazner, J. Infect. Dis., 2020, 222, 1629–1634 CrossRef.
- Q. Lin, D. Wen, J. Wu, L. Liu, W. Wu, X. Fang and J. Kong, Anal. Chem., 2020, 92, 9454–9458 CrossRef CAS.
- A. Csordas, A. E. Gerdon, J. D. Adams, J. Qian, S. S. Oh, Y. Xiao and H. T. Soh, Angew. Chem., Int. Ed., 2010, 49, 355–358 CrossRef CAS.
- L. Wang, W. Li, J. Sun, S. Y. Zhang, S. Yang, J. Li, J. Li and H. H. Yang, Anal. Chem., 2018, 90, 14433–14438 CrossRef CAS.
- H. Liang, S. Chen, P. Li, L. Wang, J. Li, J. Li, H. H. Yang and W. Tan, J. Am. Chem. Soc., 2018, 140, 4186–4190 CrossRef CAS.
- E. M. McConnell, I. Cozma, D. Morrison and Y. F. Li, Anal. Chem., 2020, 92, 327–344 CrossRef CAS.
- Y. Tang, Z. Wang, X. Yang, J. Chen, L. Liu, W. Zhao, X. C. Le and F. Li, Chem. Sci., 2015, 6, 5729–5733 RSC.
- Y. Liu, W. Hou, L. Xia, C. Cui, S. Wan, Y. Jiang, Y. Yang, Q. Wu, L. Qiu and W. Tan, Chem. Sci., 2018, 9, 7505–7509 RSC.
- M. Rossetti, S. Ranallo, A. Idili, G. Palleschi, A. Porchetta and F. Ricci, Chem. Sci., 2017, 8, 914–920 RSC.
- M. Liu, A. Khan, Z. F. Wang, Y. Liu, G. J. Yang, Y. Deng and N. Y. He, Biosens. Bioelectron., 2019, 130, 174–184 CrossRef CAS.
- R. R. Huang, L. He, Y. Y. Xia, H. P. Xu, C. Liu, H. Xie, S. Wang, L. J. Peng, Y. F. Liu, Y. Liu, N. Y. He and Z. Y. Li, Small, 2019, 15, 1900735 CrossRef.
- M. Liu, Z. F. Wang, T. Tan, Z. S. Chen, X. B. Mou, X. C. Yu, Y. Deng, G. M. Lu and N. Y. He, Theranostics, 2018, 8, 5772–5783 CrossRef CAS.
- R. R. Huang, L. He, S. Li, H. N. Liu, L. Jin, Z. Chen, Y. X. Zhao, Z. Y. Li, Y. Deng and N. Y. He, Nanoscale, 2020, 12, 2445–2451 RSC.
- X. C. Yu, L. He, M. Pentok, H. W. Yang, Y. L. Yang, Z. Y. Li, N. Y. He, Y. Deng, S. Li, T. H. Liu, X. Y. Chen and H. W. Luo, Nanoscale, 2019, 11, 15589–15595 RSC.
- D. S. Zhen, F. F. Zhong, D. Yang, Q. Y. Cai and Y. Liu, Mater. Express, 2019, 9, 319–327 CrossRef CAS.
- S. H. Yu and T. H. Kim, J. Biomed. Nanotechnol., 2019, 15, 1824–1831 CrossRef CAS.
- A. T. V. Nguyen, T. T. T. Trinh, V. T. Hoang, T. D. Dao, H. T. Tuong, H. S. Kim, H. Park and S. J. Yeo, J. Biomed. Nanotechnol., 2019, 15, 1185–1200 CrossRef CAS.
- D. L. Liu, Y. Li, R. Sun, J. Y. Xu, Y. Chen and C. Y. Sun, J. Nanosci. Nanotechnol., 2020, 20, 2114–2121 CrossRef CAS.
- M. Q. Gu, J. L. Liu, D. L. Li, M. Wang, K. N. Chi, X. Zhang, Y. Deng, Y. C. Ma, R. Hu and Y. H. Yang, Nanosci. Nanotechnol. Lett., 2019, 11, 1139–1144 CrossRef.
- H. X. Shi, W. W. Xiang, C. Liu, H. F. Shi, Y. Zhou and L. Gao, Nanosci. Nanotechnol. Lett., 2018, 10, 1707–1712 CrossRef.
- Z. J. Xi and B. Zheng, Nanosci. Nanotechnol. Lett., 2018, 10, 309–319 CrossRef.
- M. Gullberg, S. Fredriksson, M. Taussig, J. Jarvius, S. Gustafsdottir and U. Landegren, Curr. Opin. Biotechnol., 2003, 14, 82–86 CrossRef CAS.
- C. T. Tsai, P. V. Robinson, C. A. Spencer and C. R. Bertozzi, ACS Cent. Sci., 2016, 2, 139–147 CrossRef CAS.
- S. Fredriksson, M. Gullberg, J. Jarvius, C. Olsson, K. Pietras, S. M. Gustafsdottir, A. Ostman and U. Landegren, Nat. Biotechnol., 2002, 20, 473–477 CrossRef CAS.
- L. Y. Zhang, X. N. Fang, X. B. Liu, H. C. Ou, H. Y. Zhang, J. J. Wang, Q. Li, H. Y. Cheng, W. Y. Zhang and Z. F. Luo, Chem. Commun., 2020, 56, 10235–10238 RSC.
- B. D. Grant, C. E. Anderson, J. R. Williford, L. F. Alonzo, V. A. Glukhova, D. S. Boyle, B. H. Weigl and K. P. Nichols, Anal. Chem., 2020, 92, 11305–11309 CrossRef CAS.
- Y. Song, J. Song, X. Wei, M. Huang, M. Sun, L. Zhu, B. Lin, H. Shen, Z. Zhu and C. Yang, Anal. Chem., 2020, 92, 9895–9900 CrossRef CAS.
- X. Qi, B. Ke, Q. Feng, D. Yang, Q. Lian, Z. Li, L. Lu, C. Ke, Z. Liu and G. Liao, Chem. Commun., 2020, 56, 8683–8686 RSC.
- Y. Wu, F. Wang, C. Shen, W. Peng, D. Li, C. Zhao, Z. Li, S. Li, Y. Bi, Y. Yang, Y. Gong, H. Xiao, Z. Fan, S. Tan, G. Wu, W. Tan, X. Lu, C. Fan, Q. Wang, Y. Liu, C. Zhang, J. Qi, G. F. Gao, F. Gao and L. Liu, Science, 2020, 368, 1274–1278 CrossRef CAS.
- C. Wang, W. Li, D. Drabek, N. M. A. Okba, R. van Haperen, A. Osterhaus, F. J. M. van Kuppeveld, B. L. Haagmans, F. Grosveld and B. J. Bosch, Nat. Commun., 2020, 11, 2251 CrossRef CAS.
- C. M. Poh, G. Carissimo, B. Wang, S. N. Amrun, C. Y. Lee, R. S. Chee, S. W. Fong, N. K. Yeo, W. H. Lee, A. Torres-Ruesta, Y. S. Leo, M. I. Chen, S. Y. Tan, L. Y. A. Chai, S. Kalimuddin, S. S. G. Kheng, S. Y. Thien, B. E. Young, D. C. Lye, B. J. Hanson, C. I. Wang, L. Renia and L. F. P. Ng, Nat. Commun., 2020, 11, 2806 CrossRef CAS.
- A. E. Muruato, C. R. Fontes-Garfias, P. Ren, M. A. Garcia-Blanco, V. D. Menachery, X. Xie and P. Y. Shi, Nat. Commun., 2020, 11, 4059 CrossRef CAS.
- S. Jiang, C. Hillyer and L. Du, Trends Immunol., 2020, 41, 355–359 CrossRef CAS.
- Y. Cao, B. Su, X. Guo, W. Sun, Y. Deng, L. Bao, Q. Zhu, X. Zhang, Y. Zheng, C. Geng, X. Chai, R. He, X. Li, Q. Lv, H. Zhu, W. Deng, Y. Xu, Y. Wang, L. Qiao, Y. Tan, L. Song, G. Wang, X. Du, N. Gao, J. Liu, J. Xiao, X. D. Su, Z. Du, Y. Feng, C. Qin, C. Qin, R. Jin and X. S. Xie, Cell, 2020, 182, 73–84 CrossRef CAS.
- Z. F. Luo, X. N. Fang, L. Y. Zhang, J. J. Wang and L. He, CHN Pat., 202010248899.4, University of Science and Technology of China, 2020.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, additional cures, synthetic procedures, and original spectra of qPCR. See DOI: 10.1039/d0sc03920a |
| This journal is © The Royal Society of Chemistry 2020 |