
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRedox and photocatalytic properties of a NiII complex with a macrocyclic biquinazoline (Mabiq) ligand†‡
Michael
Grübel
,
Irene
Bosque
,
Philipp J.
Altmann
,
Thorsten
Bach
 * and
Corinna R.
Hess
*
* and
Corinna R.
Hess
*
Department of Chemistry, Catalysis Research Center (CRC), Technische Universität München, 85747 Garching, Germany. E-mail: thorsten.bach@ch.tum.de; corinna.hess@ch.tum.de
First published on 23rd February 2018
Abstract
We present a late, first row transition metal photosensitizer that promotes photocatalytic C–C bond formation. The title compound, [Ni(Mabiq)]OTf, as well as its one-electron reduced form, Ni(Mabiq), were synthesized and molecular structures of both were obtained. The electronic structure of the reduced complex additionally was characterized by spectroscopic and DFT computational methods. Notably, [NiII(Mabiq)]OTf is photoactive: reduction of the compound was achieved photochemically upon irradiation at λ = 457 nm and reductive quenching by NEt3. The performance of [Ni(Mabiq)]OTf as a photoredox catalyst was examined in the cyclization of a bromoalkyl-substituted indole. In this reaction, the first-row transition metal compound is comparable if not superior to [Ru(bpy)3]2+ in terms of efficiency (turnover number) and chemoselectivity. Studies using a series of sacrificial donor amines indicate that the excited state redox potential of [Ni(Mabiq)]+* is ≥1.25 V vs. SCE. This value is similar to the excited state potential of commonly employed noble metal based photocatalysts. The Ni-Mabiq compound thus provides a rare example of an earth-abundant photoredox catalyst.
Introduction
Photoredox catalysis offers nascent opportunities to shift conventional chemical production methods to light-driven processes.1 The design of new photoactive metal compounds is key to the development of new catalytic transformations. Chromophores can effect a wide array of functional group transformations and C–C bond formation reactions via the generation of active radical species, originating from excited state electron transfer processes (Scheme 1).2 With very few exceptions, photoredox catalysis relies on the use of noble metal containing photosensitizers, mainly Ru- or Ir-polypyridyl complexes;3 other heavy metal complexes (e.g. OsII, ReI, Mo0, W0) also have occasionally been employed.4,5 [Ru(bpy)3]2+ – with its long lived, charge separated excited state ([RuIII(bpy˙−)(bpy)2]2+*) – is the classic and universal photocatalyst for a plethora of applications.2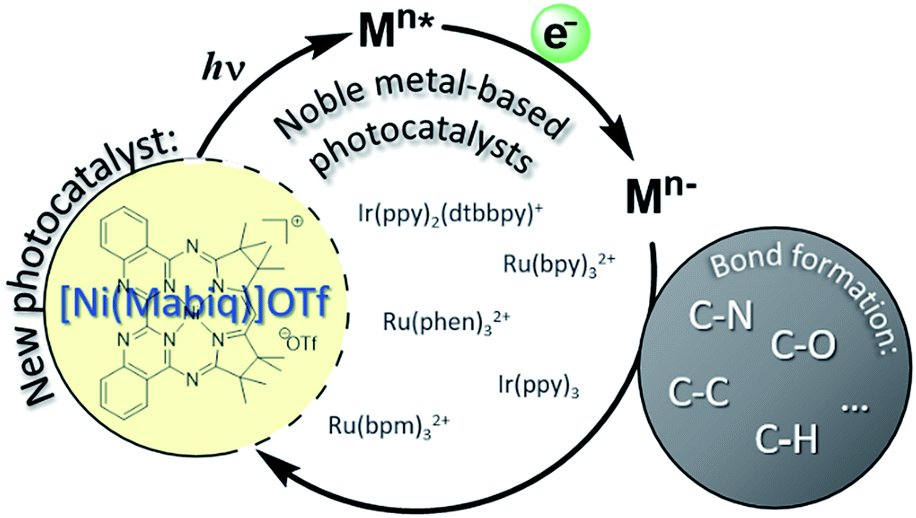 | ||
| Scheme 1 Photoredox catalysis commonly relies on noble metal complexes for organic transformations, initiated by single electron transfer upon excitation of Ru or Ir compounds. The [Ni(Mabiq)]OTf catalyst offers an earth-abundant photosensitizer. | ||
The use of less expensive, more abundant, late first row transition metal alternatives is generally precluded by their inherently short excited-state lifetimes. In the first row, nickel complexes have been used in photocatalytic cross coupling reactions,6 though the use of an added photosensitizer (commonly Ir) still is required in all of these tandem systems. A class of NiII ligand-to-ligand charge transfer complexes recently were shown to possess advantageous properties as photosensitizers, but applications have not yet been demonstrated.7 In fact, reports describing catalytic applications or reactivity of systems using only non-noble metal photosensitizers are exceedingly scarce.8–10
We now report a NiII complex with a macrocyclic biquinazoline (Mabiq) ligand,11,12 [Ni(Mabiq)]OTf (1), providing a rare example of a non-noble metal based photosensitizer. The divalent complex 1 can be photochemically reduced and the electronic structure of the product, [Ni(Mabiq)] (2), is described herein. Using a series of synthesized sacrificial donor molecules, the photo-excited state redox potential of 1 was assessed and compared to common noble-metal photocatalysts. We demonstrate the photoredox catalytic ability of 1 in a radical-based cyclization of a bromoalkyl-substituted indole. The reaction relies solely on the Ni-Mabiq photocatalyst, without the need for an additional noble metal photosensitizer.
Results and discussion
The yellow diamagnetic 1 (Scheme 2) was readily prepared by complexation of the ligand with Ni(OTf)2 in ethanol solution (see ESI‡ for further details). The Ni ion adopts the expected square planar geometry in the solid state (Fig. S1‡). The electronic spectrum of 1 in DCM exhibits intense absorption bands (ε ≈ 104 M−1 cm−1) in the visible light region (λmax = 414, 435, and 457 nm; ε = 13.6, 16.4, 22.3 × 103 M−1 cm−1; Fig. 2 inset). Similar features were observed in the spectra of other M-Mabiq complexes, notably in the spectrum of Zn(Mabiq)Cl.12a The ZnII-complex displayed two strong absorption bands at 471 and 502 nm that were assigned as Mabiq π → π* transitions. However, d–d or metal-to-ligand charge transfer (MLCT) processes may additionally contribute to the corresponding absorptions of 1. The cyclic voltammogram of 1 in MeCN (Fig. S9‡) exhibits a reversible, formally NiII/I redox couple at −1.05 V vs. Fc+/0 (Fc = ferrocene; Fc+/0 = 0.4 V vs. SCE). Additional, seemingly reversible, reductive processes appear at potentials <−1.5 V.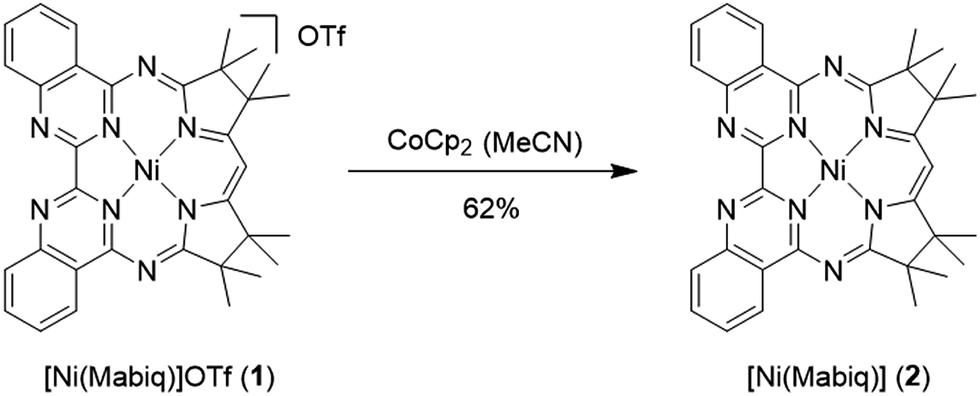 | ||
| Scheme 2 Reaction of 1 with CoCp2 (Cp = cyclopentadienyl) yields the one-electron reduced 2. | ||
The one-electron reduced Ni(Mabiq) (2) was subsequently generated from 1 using CoCp2 as the reductant (Scheme 2). The molecular structure of 2 (Fig. 1) reveals shorter Ni–N bond distances (Ni–Navg = 1.874 Å vs. Ni–Navg = 1.882 Å in 1), as well as the hallmark changes in the diketiminate C–N bonds that signify reduction of the Mabiq ligand (Table S8‡). The S = 1/2 ground state of the complex was verified by EPR spectroscopy. The spectrum is consistent with a ligand-centered radical, with giso = 1.995 (Fig. S4‡). Low energy features at λmax = 641, 711 and 801 nm (1.4, 3.5, 5.4 × 103 M−1 cm−1) are apparent in the electronic spectrum of 2 in THF (Fig. 2 inset), accounting for the vibrant green color of the complex in solution. The spectrum again closely resembles that of the one-electron reduced Zn complex, ZnII(Mabiq˙−).12a The spectroscopic data thus point to ligand-centered reduction of 1, such that the electronic structure of 2 corresponds to NiII(Mabiq˙−). Indeed, DFT calculations (B3LYP) on 2 further support this conclusion.
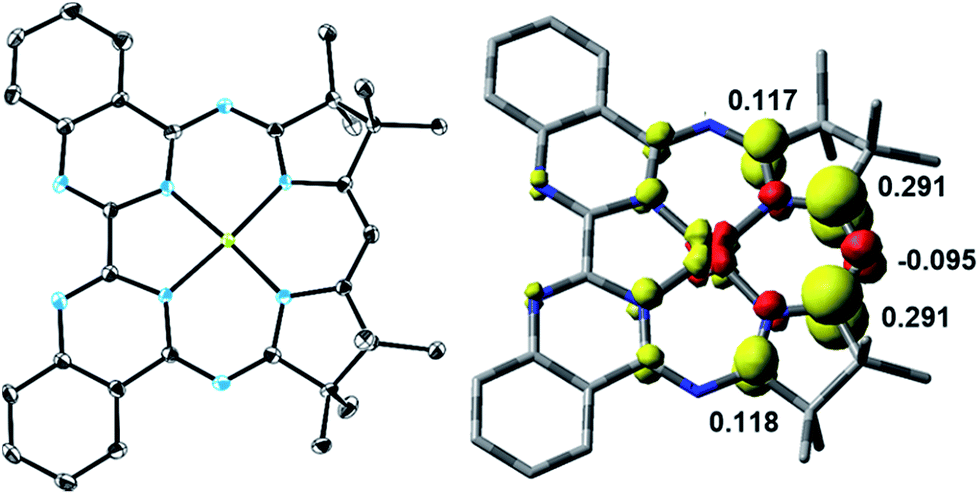 | ||
| Fig. 1 Left: Molecular structure of 2 (50% probability ellipsoids; hydrogen atoms omitted for clarity). Right: DFT-derived (B3LYP) spin density plot for 2 based on Löwdin population analysis (isosurface value = ±0.005). | ||
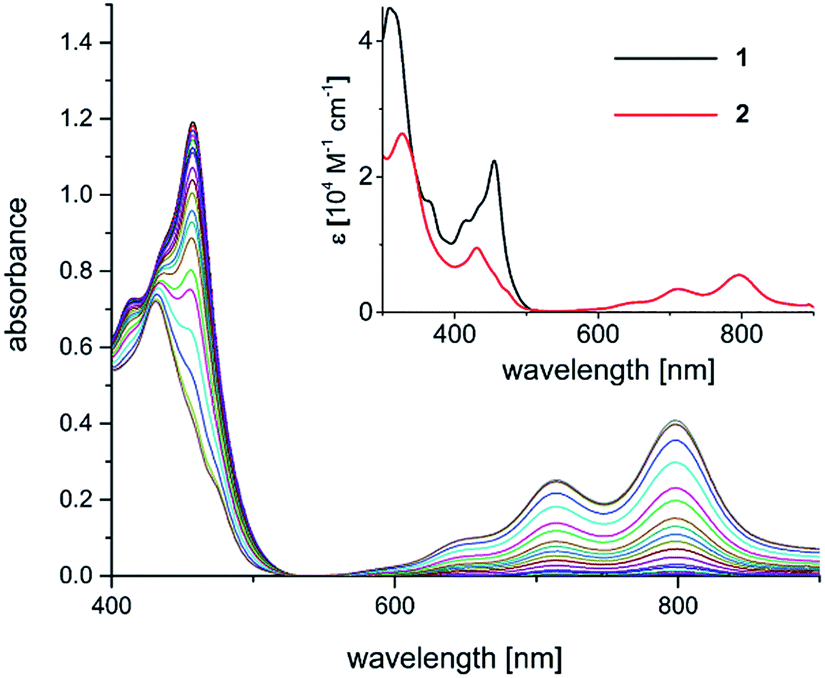 | ||
| Fig. 2 Spectral evolution during photoconversion of 1 to 2 [c (1) = 0.05 mM; c (NEt3) = 1.4 M; λ = 457 nm, DMF]. Inset: electronic spectra of 1 (black trace; CH2Cl2) and 2 (red trace; THF). | ||
The DFT-derived (B3LYP) spin density plot (Fig. 1) describes a diamagnetic d8 NiII center with an unpaired electron localized primarily on the diketiminate unit of the Mabiq ligand. Four doubly occupied d-orbitals can be identified, while the SOMO possesses only ca. 4% d-orbital character and otherwise depicts a ligand π* orbital (Fig. S5‡). The latter molecular orbital is antibonding with respect to the diketiminate C–N p-orbital interactions, which explains the lengthening of these bonds in the structure of 2. It is noteworthy that ligand-centered reduction appears to prevail across the series of metal-Mabiq compounds we have examined thus far.12
The well-behaved redox chemistry of [Ni(Mabiq)]OTf (1), its high absorbance in the visible region and its relatively high reduction potential warranted a study of its photoredox properties. As mentioned above, the compound exhibits a strong multi-structured absorption band with a maximum at λ = 457 nm (ε = 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 M−1 cm−1), which invites excitation with a visible light source and quenching studies with a suitable reductant. Gratifyingly, it was indeed found that irradiation of a DMF solution of 1 at λ = 457 nm, in the presence of NEt3 (7.5 mM to 1.4 M), leads to a color change from yellow to green and to the formation of complex 2.
300 M−1 cm−1), which invites excitation with a visible light source and quenching studies with a suitable reductant. Gratifyingly, it was indeed found that irradiation of a DMF solution of 1 at λ = 457 nm, in the presence of NEt3 (7.5 mM to 1.4 M), leads to a color change from yellow to green and to the formation of complex 2.
The formation of the reduced compound was complete after 15 minutes (using 1.4 M NEt3; c (1) = 0.05 mM), as verified spectroscopically (Fig. 2). The photoconversion of 1 to 2 occurs on a much faster timescale in MeCN/THF or DMF/THF mixtures (the solvent combination solubilizes both forms), under identical conditions. The quantum yield for the photoconversion is ≈10−4 (THF:DMF 4:1), and correlates with the Et3N concentration (14–56 mM; Table S3 and Fig. S29‡). From the data, the lifetime of the excited state form that reacts with the Et3N can be estimated as ≈1 × 10−8 s (see ESI‡ for details). Steady-state emission spectra recorded at ambient temperature and at 77 K did not reveal any luminescence. Thus, we currently cannot comment in detail on the nature of the excited state processes involved in the photo-reduction of 1. If one takes the longest wavelength absorption [λ ≅ 510 nm, E0 ≤ 235 kJ mol−1 (2.4 V)] of compound 1 to estimate the redox potential of photoexcited complex 1*, a value of ≤+1.35 V (vs. Fc+/0) is obtained.13
TDDFT (B3LYP) computational studies provide some insight into the nature of the absorptions in the visible region. The calculated transitions correlate well with the experimentally obtained absorbance spectrum of 1 (Fig. S6‡). The absorptions at 400–500 nm include a prominent LL′CT transition that corresponds to the HOMO to LUMO transition. The HOMO is localized on the bipyrimidine moiety of the Mabiq ligand, while the LUMO is a diketiminate based π* orbital (Fig. S7‡). Other, less intense, transitions in the vicinity possess d–d (Ni dz2 → Ni dx2−y2) and MLCT (Ni dz2 → Lπ*) character. These states may contribute to the unique photochemical properties of 1. However, a detailed investigation regarding the photochemistry and excited state kinetics of this compound is warranted, and will be the subject of future investigations.
We examined whether the photochemical properties of 1 might render it a suitable photoredox catalyst for C–C bond formation reactions. The radical cyclization of the N-(ω-bromoalkyl)-substituted indole 3 was chosen as a test reaction.14 The reaction had been previously studied by the Stephenson group and was found to produce mainly product 4 by C–C bond formation under optimized conditions.15 Under non-optimized conditions, hydro-de-bromination was a competing side reaction and varying product ratios of 4 and 5 were observed. Optimal conditions were reported to include the use of [Ru(bpy)3]Cl2 as the catalyst (1 mol%) and NEt3 (2 equiv.) in DMF solution and gave product 4 in 60% yield.15
Given the limited solubility of 2 in DMF, the reaction was initially attempted in a DMF/THF mixture (v/v = 1/2) with 2 mol% of catalyst 1 and 2 equiv. NEt3 as the quencher (Table 1, entry 1).16 We were pleased to find that the desired cyclization proceeded smoothly and delivered with high chemoselectivity the desired product 4. The inseparable hydro-de-brominated by-product 5 was detectable in minor quantities but the ratio of products was 95/5 in favor of cyclization product 4. When increasing the relative volume of THF in the solvent mixture both conversion and yield improved slightly (entry 2). The selectivity towards the desired reaction was high with a yield of 86% at 94% conversion, i.e. 91% yield based on conversion. For comparison, the RuII complex [Ru(bpy)3](PF6)2 was employed under identical conditions (entry 3). Although the ratio 4/5 was identical with this catalyst, the reaction suffered from a lower conversion and a lower chemoselectivity (63% yield based on conversion). Similar observations were made when the catalyst loading was further decreased to 1 mol%: while the performance of NiII catalyst 1 remained unchanged (entry 4) the reaction with the RuII catalyst was sluggish and a decrease in yield was observed (entry 5).
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Catalysta | mol% | DMF/THF [v/v] | Conv.b [%] | Yieldc [%] | 4/5d |
| a All reactions were performed on a scale of 0.08 mmol (c = 25 mM) with a LED lamp (3 W power output) as light source. Irradiation time: 13 h. b The conversion was calculated from recovered starting material. c Total yield of isolated products 4 and 5. d Ratio of cyclized to hydro-de-brominated product as determined by 1H-NMR. e Attempted reaction without irradiation. f No catalyst was added. g No NEt3 was added. n.d. = not detected. | ||||||
| 1 | 1 | 2 | 1/2 | 93 | 84 | 95/5 |
| 2 | 1 | 2 | 1/4 | 94 | 86 | 95/5 |
| 3 | [Ru(bpy3)(PF6)2 | 2 | 1/4 | 73 | 46 | 95/5 |
| 4 | 1 | 1 | 1/4 | 95 | 84 | 95/5 |
| 5 | [Ru(bpy3)(PF6)2 | 1 | 1/4 | 59 | 23 | 95/5 |
| 6 | 1 | 2 | 1/4 | n.d. | — | — |
| 7 | —f | — | 1/4 | 12 | 12 | 55/45 |
| 8 | 1 | 2 | 1/4 | <5 | <5 | — |
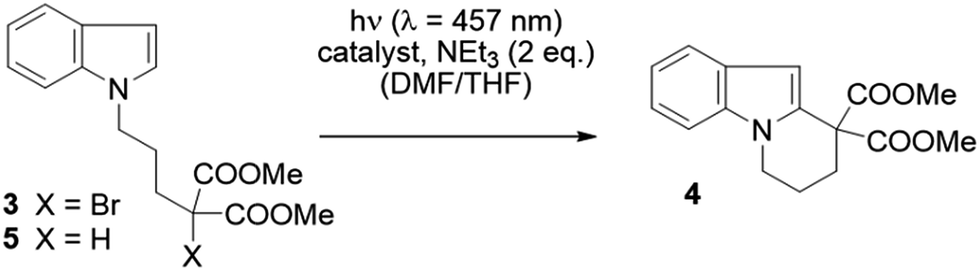
The above reaction is induced by visible light as no conversion occurs without irradiation (entry 6). In the absence of the NiII catalyst,17 only 12% of a product mixture was obtained, which was composed of the cyclized product 4 and the reduced product 5 in a 55/45 ratio (entry 7). In the absence of the reductant, no reaction was observed (entry 8). The free HMabiq ligand is not photocatalytically active. The quantum yield for the [Ni(Mabiq)]OTf catalysed cyclization reaction was determined to be Φ = 0.006.
Mechanistically, it is suggested that the photoreduction of complex 1 by NEt3 (reductive quenching cycle)1 generates complex 2, which may engage in SET to the bromide 3 (Scheme 3). Complex 2 was shown to be competent to reduce 3. We note that modification of the Ni-Mabiq complex during the cyclization reaction was not observed, as verified by ESI-MS and 1H-NMR (Fig. S8 and S41‡).
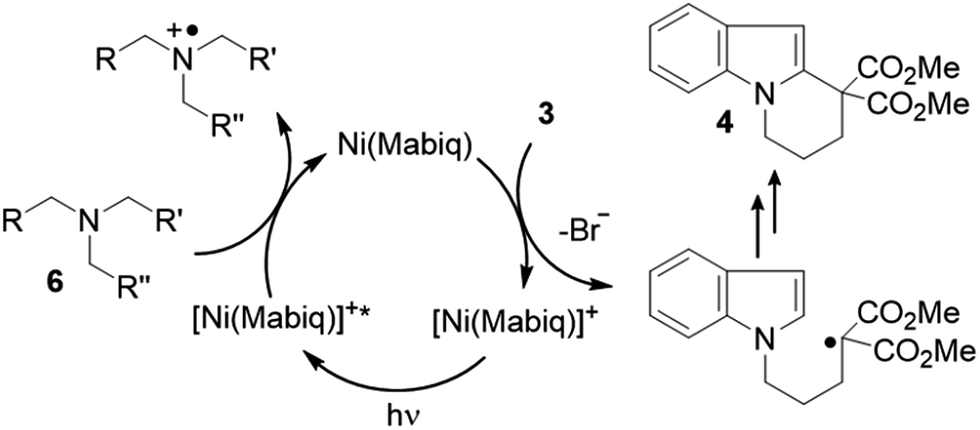 | ||
| Scheme 3 Proposed catalytic cycle for the cyclization of 3 to give 4. | ||
To experimentally verify the estimated photoexcited state redox potential of 1*, we synthesized a series of amines with oxidation potentials in the range of 0.78–1.59 V (vs. SCE; Tables 2 and S1‡), as determined by CV. The amines were employed as sacrificial donors in the cyclization reaction. Excellent yields of 4 were obtained using the donor molecules with oxidation potentials up to 1.25 V (Table 2, amines 6a–6c), whereas a drastic decrease in yield was observed using those with higher redox potentials. Only 40% and 20% product yields were obtained with 6d (Eox = 1.41 V vs. SCE) and 6e (Eox = 1.59 V vs. SCE), respectively. The product yield in the control reaction using 6e in the absence of photocatalyst 1 was 7%, a result that is comparable to that obtained using NEt3 (Table 1, entry 7). The results confirm that the excited state redox potential of 1* is at least 1.25 V (vs. SCE). Thus, our new Ni-Mabiq complex is a more powerful oxidant than [Ru(bpy)3]2+ (E1/2 [Ru2+*/Ru+] 0.78 V vs. SCE),18 and comparable to [Ir(dF(CF3)ppy)2(dtbbpy)]+ (E1/2 [Ir3+*/Ir2+] 1.21 V vs. SCE).19
|
|
||
|---|---|---|
| Aminea | E ox (vs. SCE) | Yieldb [%] |
|
a All reactions were performed on a scale of 0.08 mmol (c = 25 mM) with a 457 nm LED lamp (3 W power output) as the light source. Irradiation time: 13 h.
b Total yield of isolated products 4 and 5, with a 95:5 ratio of cyclized to hydro-de-brominated product as determined by 1H-NMR.
c Average of two runs. Fc+/0 = 0.4 V vs. SCE. Cy = cyclohexyl.
|
||
| Et3N (6a) | 0.83 | 84 |
| 6b | 1.05 | 97 |
| 6c | 1.25 | 95 |
| 6d | 1.41 | 40 |
| 6e | 1.59 | 20c |
| 6f | 0.78 | 84 |
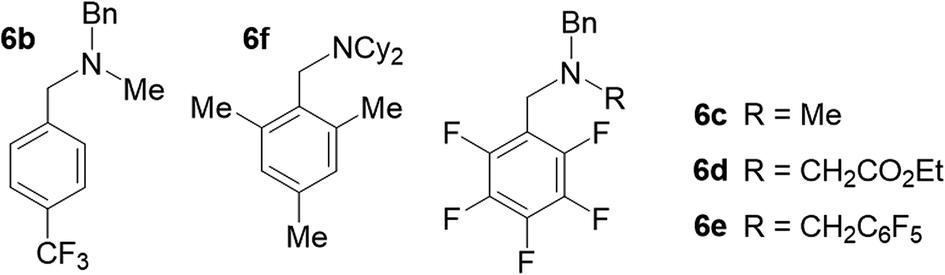
We additionally generated a sterically hindered amine, N(CH2Mes)Cy2 (6f), to assess whether coordination of the sacrificial donor molecules influences the reactivity of 1. In contrast to the bpy- and phen-based photosensitizers, the Ni-Mabiq complexes are coordinatively unsaturated and intramolecular electron transfer from a coordinated amine to the divalent metal center could potentially occur. The molecular structure of 6f (Fig. S3‡) suggests that coordination of this amine group to the Ni center is unlikely; the cyclohexyl and mesityl groups encapsulate the nitrogen atom rendering the lone pair inaccessible. Whereas noticeable changes in the absorption spectrum of 1 were observed upon addition of Et3N (400 equiv.) to a solution of the complex in THF/DMF, the addition of 6f has no effect (Fig. S30 and S31‡). However, the yield of the catalytic reaction using 6f as sacrificial donor was found to be 84%, a value that is comparable to the one obtained using Et3N (6a) and 6b–6e as sacrificial donors.
Conclusions
In summary, we have discovered a new photoredox catalyst, [Ni(Mabiq)]OTf (1) that is based on the earth-abundant metal nickel. The diamagnetic, bench-stable compound was readily prepared, its redox properties were studied and the one-electron reduced form Ni(Mabiq) (2) likewise was isolated. Further studies to elucidate the detailed photophysical properties of 1* are warranted. However, we have already demonstrated that the photoexcited complex is a strong oxidant, with the capacity to induce C–C bond formation in an initial test reaction. The Ni-Mabiq compound may offer an alternative to noble metal photosensitizers for other synthetic transformations in organic photoredox chemistry, as well as for energy conversion processes. The Mabiq ligand also features a second metal binding site that could be exploited for tandem catalysis. Thus, the macrocycle represents a new type of platform for the development of photoactive systems. With evidence of the ability of 1 to act as a photosensitizer and photoredox catalyst, the broader photocatalytic applications of our system subsequently will be investigated.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Alex Pöthig for contributions to the crystallography, and Dr Andreas Bauer for assistance with fluorescence and quantum yield measurements. We are grateful to Prof. Klaus Köhler for the use of his EPR instrument, and to Dr Carmen Haeßner and Manuel Kaspar for technical assistance. I. B. acknowledges the Ramón Areces Foundation (Becas para Estudios Postdoctorales) and the Alexander von Humboldt Foundation for a postdoctoral fellowship. M. G acknowledges the TUM Graduate School for financial support.Notes and references
- For seminal studies, see: (a) M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef CAS PubMed; (b) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed; (c) J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef CAS PubMed.
- Reviews: (a) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 985 CrossRef CAS PubMed; (b) Y. Xi, H. Yi and A. Lei, Org. Biomol. Chem., 2013, 11, 2387–2403 RSC; (c) M. Reckenthäler and A. G. Griesbeck, Adv. Synth. Catal., 2013, 355, 2727–2744 CrossRef; (d) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (e) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (f) K. Zeitler, Angew. Chem., Int. Ed., 2009, 48, 9785–9789 CrossRef CAS PubMed.
- D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803–5820 RSC.
- (a) L. A. Büldt and O. S. Wenger, Angew. Chem., Int. Ed., 2017, 56, 5676–5682 CrossRef PubMed; (b) L. A. Büldt, X. Guo, A. Prescimone and O. S. Wenger, Angew. Chem., Int. Ed., 2016, 55, 11247–11250 CrossRef PubMed; (c) J.-P. Sauvage, J.-P. Collin, J.-C. Chambron, S. Guillerez and C. Coudret, Chem. Rev., 1994, 94, 993–1019 CrossRef CAS; (d) D. J. Stufkensa and A. Vlcek Jr, Coord. Chem. Rev., 1998, 177, 127–179 CrossRef; (e) K. S. Schanze, D. B. MacQueen, T. A. Perkins and L. A. Cabana, Coord. Chem. Rev., 1993, 122, 63–89 CrossRef CAS; (f) S. Meister, R. O. Reithmeier, M. Tschurl, U. Heiz and B. Rieger, ChemCatChem, 2015, 7, 690–697 CrossRef CAS.
- For the use of organic compounds as photoredox catalysts, see: N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–11166 CrossRef CAS PubMed.
- (a) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437–440 CrossRef CAS PubMed; (b) D. T. Ahneman and A. G. Doyle, Chem. Sci., 2016, 7, 7002–7006 RSC; (c) M. Jouffroy, D. N. Primer and G. A. Molander, J. Am. Chem. Soc., 2016, 138, 475–478 CrossRef CAS PubMed; (d) B. P. Woods, M. Orlandi, C.-Y. Huang, M. S. Sigman and A. G. Doyle, J. Am. Chem. Soc., 2017, 139, 5688–5691 CrossRef CAS PubMed; (e) M. K. Nielsen, B. J. Shields, J. Liu, M. J. Williams, M. J. Zacuto and A. G. Doyle, Angew. Chem., Int. Ed., 2017, 56, 7191–7194 CrossRef CAS PubMed; (f) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 1–18 CrossRef; (g) E. R. Welin, C. Le, D. M. Arias-Rotondo, J. K. McCusker and D. W. C. MacMillan, Science, 2017, 355, 380–385 CrossRef CAS PubMed.
- L. A. Cameron, J. W. Ziller and A. F. Heyduk, Chem. Sci., 2016, 7, 1807–1814 RSC.
- For a recent review of earth-abundant photosensitizers see: C. B. Larsen and O. S. Wenger, Chem.–Eur. J., 2018, 24, 2039–2058 CrossRef CAS PubMed.
- (a) F. J. Sarabia and E. M. Ferreira, Org. Lett., 2017, 19, 2865–2868 CrossRef CAS PubMed; (b) S. M. Stevenson, M. P. Shores and E. M. Ferreira, Angew. Chem., Int. Ed., 2015, 54, 6506–6510 CrossRef CAS PubMed; (c) A. Gualandi, M. Marchini, L. Mengozzi, M. Natali, M. Lucarini, P. Ceroni and P. G. Cozzi, ACS Catal., 2015, 5, 5927–5931 CrossRef CAS; (d) P. Zimmer, P. Müller, L. Burkhardt, R. Schepper, A. Neuba, J. Steube, F. Dietrich, U. Flörke, S. Mangold, M. Gerhards and M. Bauer, Eur. J. Inorg. Chem., 2017, 1504–1509 CrossRef CAS; (e) S. Otto, A. M. Nauth, E. Ermilov, N. Scholz, A. Friedrich, U. Resch-Genger, S. Lochbrunner, T. Opatz and K. Heinze, ChemPhotoChem, 2017, 1, 344–349 CrossRef CAS.
- S. J. Hwang, D. C. Powers, A. G. Maher, B. L. Anderson, R. G. Hadt, S.-L. Zheng, Y.-S. Chen and D. G. Nocera, J. Am. Chem. Soc., 2015, 137, 6472–6475 CrossRef CAS PubMed.
- E. Müller, G. Bernardinelli and A. von Zelewsky, Inorg. Chem., 1988, 27, 4645–4651 CrossRef.
- (a) P. Banerjee, A. Company, T. Weyermüller, E. Bill and C. R. Hess, Inorg. Chem., 2009, 48, 2944–2955 CrossRef CAS PubMed; (b) E. V. Puttock, P. Banerjee, M. Kaspar, L. Drennan, D. S. Yufit, E. Bill, S. Sproules and C. R. Hess, Inorg. Chem., 2015, 54, 5864–5873 CrossRef CAS PubMed; (c) M. Kaspar, P. J. Altmann, A. Pöthig, S. Sproules and C. R. Hess, Chem. Commun., 2017, 53, 7282–7285 RSC.
- The redox potential of 1*/2 was calculated from the redox potential of 1/2 (−1.05 V vs. Fc+/0) by adding the energy of the excited state E0 (see ref. 3).
- For recent work on photoredox-induced alkylation reactions at the indole core, see: D. Alpers, M. Gallhof, J. Witt, F. Hoffmann and M. Brasholz, Angew. Chem., Int. Ed., 2017, 56, 1402–1406 CrossRef CAS PubMed and refs cited therein.
- J. W. Tucker, J. M. R. Narayanam, S. W. Krabbe and C. R. J. Stephenson, Org. Lett., 2010, 12, 368–371 CrossRef CAS PubMed.
- For the reaction set-up, see: (a) D. Rackl, V. Kais, P. Kreitmeier and O. Reiser, Beilstein J. Org. Chem., 2014, 10, 2157–2165 CrossRef CAS PubMed; (b) D. Lenhart, A. Pöthig and T. Bach, Chem.–Eur. J., 2016, 22, 6519–6523 CrossRef CAS PubMed.
- Radical reactions of amines can be initiated in the absence of a photoredox catalyst if an alkyl halide is present: (a) J. F. Franz, W. B. Kraus and K. Zeitler, Chem. Commun., 2015, 51, 8280–8283 RSC; (b) A. Böhm and T. Bach, Chem.–Eur. J., 2016, 22, 15821–15928 CrossRef PubMed; (c) A. M. Nauth, J. C. O. Pacheco, S. Pusch and T. Opatz, Eur. J. Org. Chem., 2017, 6966–6974 CrossRef CAS.
- A. Juris, V. Balzani, P. Belser and A. von Zelewsky, Helv. Chim. Acta, 1981, 64, 2175–2182 CrossRef CAS.
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal Jr, G. G. Mailliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712–5719 CrossRef CAS.
Footnotes |
| † Dedicated to the memory of Inge Grübel. |
| ‡ Electronic supplementary information (ESI) available: CIF files, synthetic procedures, spectroscopic, electrochemical and DFT computational data. CCDC 1538675, 1538676 and 1586948. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc05320g |
| This journal is © The Royal Society of Chemistry 2018 |