
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing the surface chemistry of self-assembled peptide hydrogels using solution-state NMR spectroscopy†
Matthew
Wallace
*,
Jonathan A.
Iggo
and
Dave J.
Adams‡
 *
*
Department of Chemistry, University of Liverpool, Liverpool, L69 7ZD, UK. E-mail: dave.adams@glasgow.ac.uk
First published on 31st January 2017
Abstract
The surface chemistry of self-assembled hydrogel fibres – their charge, hydrophobicity and ion-binding dynamics – is recognised to play an important role in determining how the gels develop as well as their suitability for different applications. However, to date there are no established methodologies for the study of this surface chemistry. Here, we demonstrate how solution-state NMR spectroscopy can be employed to measure the surface chemical properties of the fibres in a range of hydrogels formed from N-functionalised dipeptides, an effective and versatile class of gelator that has attracted much attention. By studying the interactions with the gel fibres of a diverse range of probe molecules and ions, we can simultaneously study a number of surface chemical properties of the NMR invisible fibres in an essentially non-invasive manner. Our results yield fresh insights into the materials. Most notably, gel fibres assembled using different tiggering methods bear differing amounts of negative charge as a result of a partial deprotonation of the carboxylic acid groups of the gelators. We also demonstrate how chemical shift imaging (CSI) techniques can be applied to follow the formation of hydrogels along chemical gradients. We apply CSI to study the binding of Ca2+ and subsequent gelation of peptide assemblies at alkaline pH. Using metal ion-binding molecules as probes, we are able to detect the presence of bound Ca2+ ions on the surface of the gel fibres. We briefly explore how knowledge of the surface chemical properties of hydrogels could be used to inform their practical application in fields such as drug delivery and environmental remediation.
Introduction
Supramolecular hydrogels show great promise as new materials for a variety of applications including drug delivery and environmental remediation.1–3 These hydrogels are formed upon the self-assembly in solution of small molecules (gelators) into fibres. The fibres subsequently interact with one another to form a sample-spanning network which immobilises the water by capillary forces.4 It is important to understand the surface chemical properties of the gel fibres, including their charge, hydrophobicity/hydrophilicity and ion-binding dynamics. These properties determine not only how the gels form but also how other entities such as drug molecules or living cells interact with the gel matrices in practical applications.5–10 Nevertheless, the range of analytical methodologies commonly applied to the study of these materials cannot directly provide any information on the surface chemistry of the fibres.11 Inferences as to the surface chemistry that are based purely on the chemical structure of the gelators may be unreliable. For example, in many systems, dramatic shifts in the protonation constants of ionisable groups are known to occur upon assembly.12–14 Methods to directly probe the surface chemical properties of the fibres are thus required. Although infra-red spectroscopy can provide insight in some cases,10,15 spectra are often crowded and it can be extremely challenging to unambiguously assign the vibrations of the surface-born functional groups.16 Pioneering experimental investigations into the mobility of the gelators and solvent at the fibre-solution interface using techniques such as electron-spin paramagnetic resonance17,18 (EPR) or fast field cycling relaxometry19 (FFC) have also been reported. However, these techniques require access to specialist equipment and, in the case of EPR, labelling of the gelators, which hinders the uptake of these techniques by the broader research community.Solution-state NMR spectroscopy is commonly used to follow the formation of supramolecular hydrogels as it is a powerful, non-invasive and widely available technique.11,20 However, experimental attention is usually confined only to the mobile gelator present in solution rather than the gel networks themselves, which are usually NMR-silent.21–23 As the molecules assemble, their resonances generally broaden,21 decrease in intensity and may shift in frequency,24 thus yielding valuable insights into the molecular interactions responsible for self-assembly,25–27 and the kinetics and thermodynamics of assembly.13,28
An alternative approach is to focus not on the gelators/gel fibres themselves, but rather the mobile molecules and ions dissolved in the solvent phase of the gel that interact with the fibres.22,29–31 We recently presented a new solution-state NMR spectroscopic method to study the relative charge and hydrophobicity of self-assembled gel fibres.30 The relative affinities of different probe molecules and ions dissolved in the solvent phase of the gel for the fibres depends on the surface chemistry of those fibres. Thus, by measuring the residual quadrupolar couplings (RQCs) of two complementary probes, 14NH4+ (positive, hydrophilic) and isopropanol-d8 (neutral, hydrophobic), we were able to gain new insights into the self-assembly process of a low molecular weight dipeptide gelator (1, Fig. 1).32,33 In particular, we revealed for the first time that the gel fibres bear a significant negative charge throughout the gelation process. Here, we extend the concept outlined in that work and have developed a set of probes that includes cations, cation binders and neutral hydrophobic molecules. We apply the set to study a number of surface chemical properties across a diverse range of hydrogels formed from functionalised dipeptide gelators. By combining RQC and Saturation Transfer Difference (STD) measurements of our probe molecules, along with 23Na+T1 and T2 relaxation measurements, we can simultaneously study the charge and hydrophobicity of the fibres. We can also study the interaction with the fibres of metal ions and other species of interest.
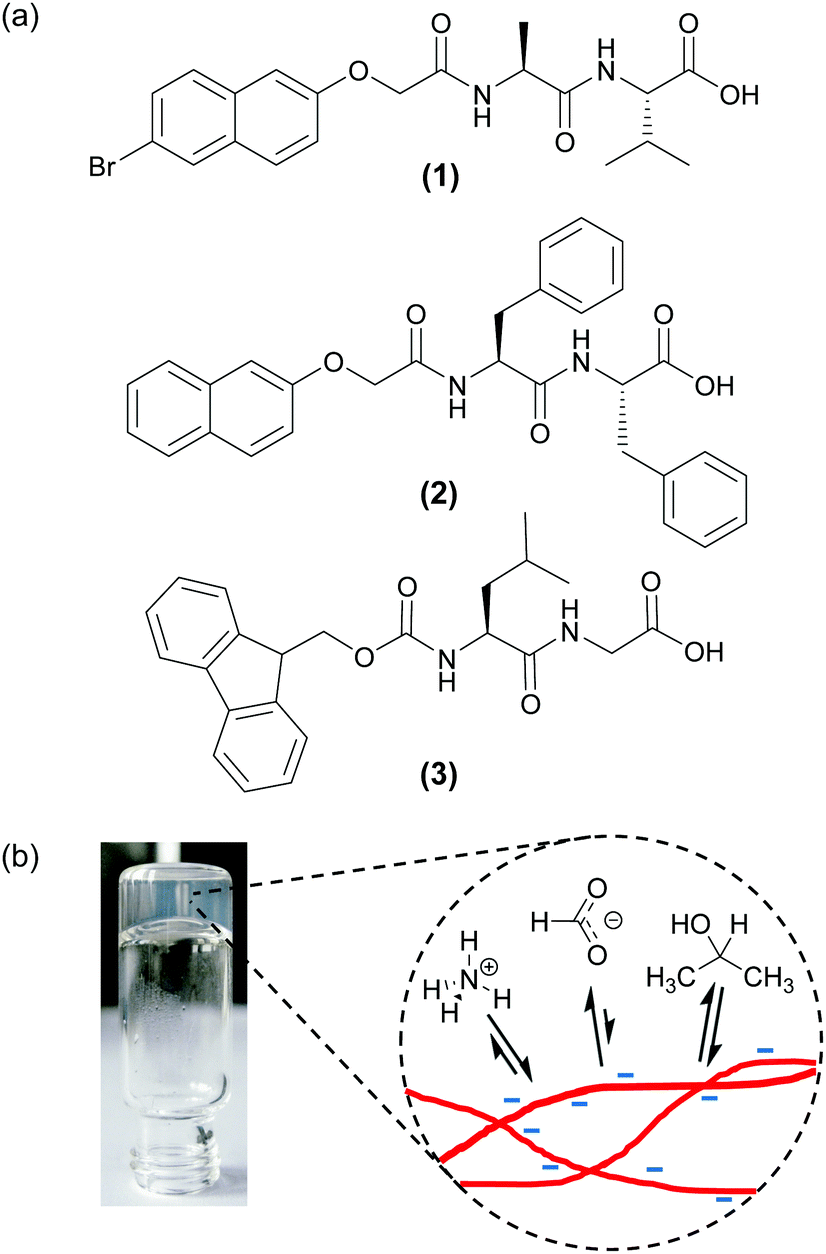 | ||
| Fig. 1 (a) Structure of the gelators used in this work. (b) Cartoon to illustrate the general concept of this work: self-supporting hydrogels can be prepared from a variety of gelators and by a variety of methods. By studying the relative affinities of a set of probe molecules and ions for the surface of the gel fibres (red) we are able to measure the surface chemical properties. | ||
N-functionalised dipeptides (see examples in Fig. 1) are an effective and versatile class of gelator.34–37 We and others have shown that some members of the class such as 1 dissolve to form free-flowing solutions when the pH of the solutions is raised above the pKa of the carboxylic acid.12,38,39 Highly reproducible gels can then be formed upon lowering the pH of the solution in a controlled manner via the hydrolysis of glucono-δ-lactone (GdL)40 or anhydrides.41 More hydrophobic gelators such as 2 are assembled at high pH into wormlike micelles, which convey significant viscosity to their solutions.16,42 The addition of salts such as CaCl2 transforms these solutions into rigid gels, attributable to a ‘cross-linking’ of the wormlike structures by the divalent metal ions.42 A third method to prepare gels involves dissolution of the gelators such as 3 in a water-miscible organic solvent, such as DMSO or acetone, followed by dilution with water. The sudden reduction in solubility induces a rapid aggregation of the gelators followed by a gradual evolution of the system into a fibrous network.43–46 Here, we demonstrate using our NMR techniques that the gels formed via these three methods possess different surface chemistries, despite the similar molecular structures of the gelators.
Results and discussion
Methods
We have developed a set of probe molecules and ions (Table 1) to probe simultaneously a range of surface chemistries. The probes are chosen to either interact with the gel fibres or remain in the solvent phase of the gel depending on their class (Fig. 1b). As far as possible, the probes do not bridge multiple classifications and do not significantly interfere or co-assemble with the gel fibres (ESI,† Section 1). It is, however, sometimes desirable to exclude certain probes from the samples for experimental reasons (Experimental section). In this work, we also use our in situ method for pH determination using NMR spectroscopy which has been previously described.30| Classification | Probe (abbreviation) |
|---|---|
| Positive ions | 23Na+, NH4+, MeNH3+ |
| Hydrophobic | Isopropanol (IPA), tert-butanol (tBuOH), acetone, dioxane, dimethylsulfoxide (DMSO), methanol (MeOH) |
| Ion binders | Formate, methylphosphonate (MPA), maleate |
| NMR pH indicators | Formate, acetate, MPA, glycine, methanesulfonate |
The suite of NMR techniques described here relies on the rapid exchange of the probe molecules/ions between the surface of the gel fibre and the solvent phase. When this condition applies, it can be shown that the magnitudes of RQCs, STDs and 23Na T1 and T2 relaxation times are directly proportional to the fraction of probes bound to the fibres at any instant in time (ESI,† Section 2). The principles and practices of these three techniques are discussed elsewhere. Briefly, RQCs manifest as splittings (Δ, Fig. 2) of the NMR resonances of quadrupolar nuclei such as 2H, 14N or 23Na due to the interaction of the probes with anisotropically arranged gel fibres.30,47,48 The necessary anisotropy can be induced in many systems by preparing the gels in the strong magnetic field of an NMR spectrometer.48,49 In our systems, the presence of the magnetic field during gelation does not significantly affect the surface chemical or mechanical properties of the final hydrogels (ESI,† Section 1.3). STD spectroscopy can be used to study ligand binding to a number of substrates including proteins,50 gels29,51 and solid reaction supports.52 The very broad 1H resonances of the gel fibres are saturated by a train of selective RF pulses. This saturation is then transferred via intermolecular 1H–1H dipolar couplings to probe molecules that are interacting with the gel fibres and thus exhibit lower signal intensities in the 1H NMR spectrum of the sample (Fig. 2). The T1 and T2 relaxation times of 23Na+ depend on the mobility of the Na+ ions in the sample.53,54 We and others have demonstrated that the assembly of free gelator molecules into negatively charged fibres results in a significant decrease in the mobility and relaxation times of 23Na+ as the ions bind to the negatively charged structures formed.30,55,56 A complication with 23Na relaxation measurements is the inherently biexponential relaxation of this spin-3/2 nucleus. However, in our systems, 23Na+ generally has a sufficient mobility for an effective monoexponential T1 and T2 to be determined (ESI,† Sections 2.2 and 8). Although the absolute magnitude of an STD or RQC of a probe molecule depends upon its structure and binding geometry with the gel fibres, a molecule that interacts significantly with the gel fibres is likely to exhibit RQCs and/or STDs, whereas a non-interacting molecule will exhibit neither (Fig. 2). By using our set of probe molecules and combining the three measurements discussed above, we can build a reliable picture of the surface chemistry of our gels.
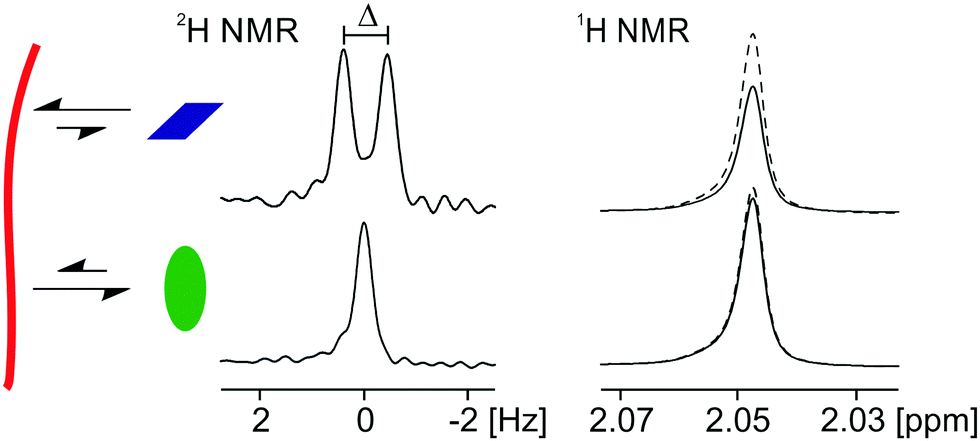 | ||
| Fig. 2 Cartoon to illustrate the use of RQC and STD measurements in this work. An interacting molecule (rhombus) will likely exhibit an RQC (Δ) on the resonance of its deuterium-labelled analogue. The 1H signal intensity will also be lower when on-resonance saturation is applied to the gel fibres (solid) than without on-resonance saturation (dashed). A weakly interacting molecule (oval) will exhibit RQCs too small to be discerned while its 1H resonance will not be significantly perturbed when on-resonance saturation is applied to the fibres. | ||
Gelation via a pH switch
We begin by discussing gels formed upon the addition of GdL to a solution of 1 at pH 9 (referred to as 1/GdL gels). A detailed analysis of the assembly process has previously been presented.30,32 Here, we briefly outline how our set of probes and combined RQC, STD and 23Na relaxation measurements can yield fresh insight into the system.The gel formation can be divided into three separate phases. In Phase I, the gelators have not yet assembled into fibres so no RQCs or STDs to the probe molecules are detectable (Fig. 3c and d). The very slight STDs plotted are within the error of the integration (≤1%). We were unable to detect NOEs from 1 to the probe molecules at pH 9 in the absence of GdL, implying minimal interaction of the probes with the unassembled gelators.
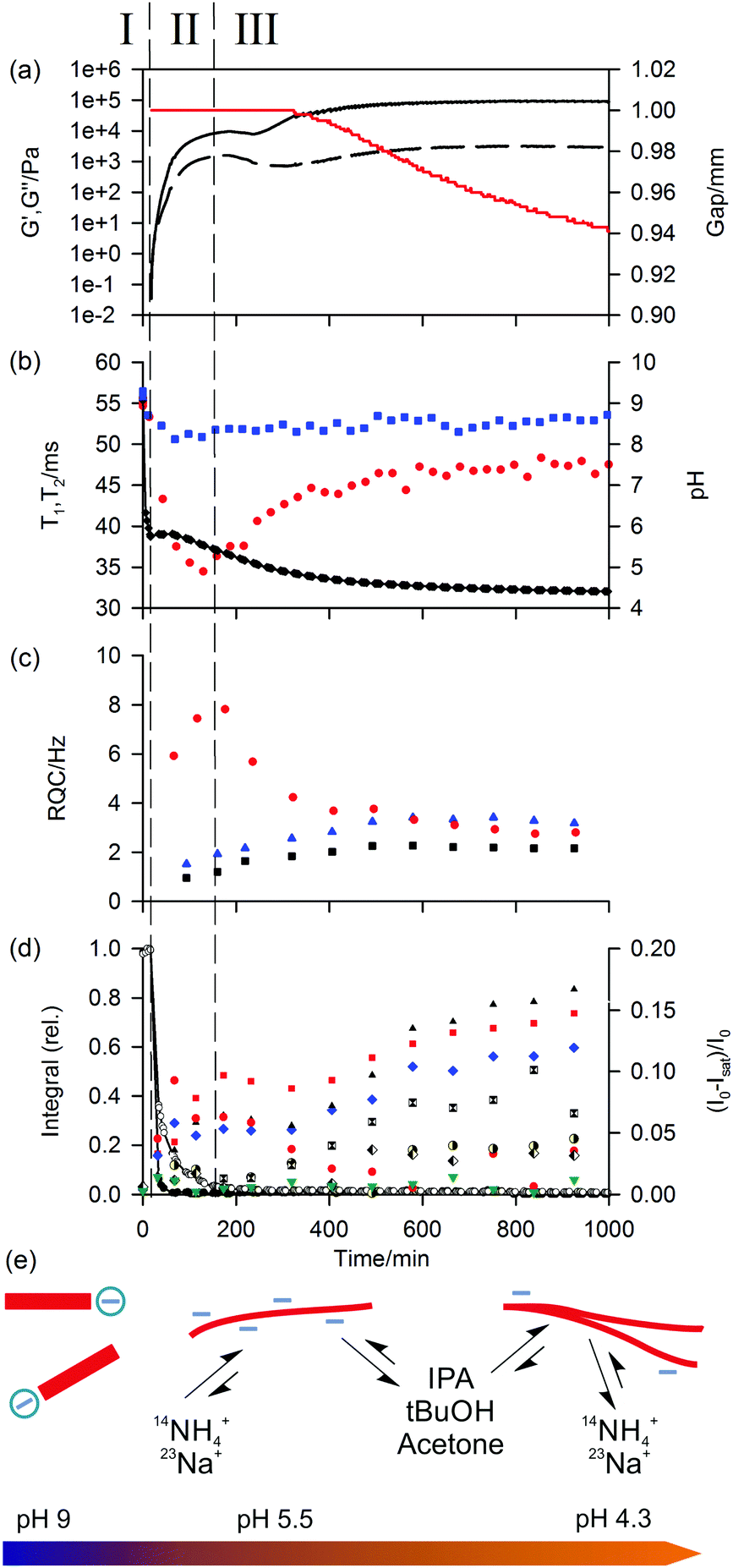 | ||
| Fig. 3 Plots of experimental observables during gelation of 1/GdL sample. Points at zero time correspond to a solution of 1 at pH 9 prior to the addition of GdL. (a) Plot of G′ (black, solid line) and G′′ (dashed) along with the gap between the rheometer plates (red). (b) Plot of 23Na+T1 (blue square) and T2 (red circle) relaxation times along with pH (black diamond). The line is a guide to the eye. (c) Plot of 14NH4+ (red circle), IPA (blue triangle) and tBuOH (black square) RQCs. (d) Plots of 1H integrals of 1: valine methyl (white circle) and aromatic protons (black circle). The lines are a guide to the eye. Plot of STDs to probe molecules, expressed in terms of the differences in the 1H signal intensities with (Isat) and without (I0) saturation applied to the gel fibres: IPA (blue diamond), acetone (black triangle), tBuOH (red square), MeNH3+ (red circle), DMSO (green down triangle), formate (half black circle), acetate (black and white square) and MeOH (black and white diamond). (e) Cartoon to illustrate the proposed assembly state of 1 during the gelation process along with the relative affinities of different probe molecules. As the pH falls, free gelator molecules (red rectangle) assemble into negatively charged fibres which subsequently lose charge while the mechanical properties of the sample increase. | ||
Once the pH of the sample has fallen to the ‘apparent’ pKa of 1,38 the gelators assemble into a network of fibres (Phase II). The 1H integrals of the gelator resonances fall sharply as the mobile molecules assemble into large structures. The mechanical properties of the sample quickly develop into that of a gel, with G′ exceeding G′′ by approximately one order of magnitude by the end of Phase II. The formation of anisotropic structures is readily apparent from the emergence of RQCs and STDs to the probe molecules and a decrease in the 23Na T1 and T2 relaxation times as the probes interact with the newly formed gel fibres. The affinity of the positive probes, 23Na+ and 14NH4+, for the fibres quickly rises to a maximum at the end of Phase II. In Phase III, the affinity falls sharply, as the pH falls and the amount of negative charge on the fibres is gradually reduced. The increased concentration of gluconate during the experiment due to the hydrolysis of GdL will not significantly affect the 14NH4+ RQCs or 23Na+T1 and T2 relaxation times due to the low binding affinity of gluconate for these ions.57,58
In contrast to the positive probes, the affinity for the fibres of the neutral hydrophobic probes, IPA, tBuOH, acetone and dioxane, gradually increase during Phase II and Phase III, attributable to an increase in the hydrophobicity as the network develops. This assertion is supported by molecular dynamics simulations of other systems which demonstrate a clear preference of these organic solvents for hydrophobic surfaces.59,60 We attribute the absence of an observable RQC for dioxane and acetone to their binding geometry and molecular structures (ESI,† Section 2.1);61 the observation of STDs confirms a significant interaction of the molecules with the fibres. Dioxane STDs are not plotted in Fig. 3 due to spectral overlap with GdL although significant STDs are nevertheless discernible (ESI,† Section 3).
The gradual, although never complete,30 removal of charge from the fibres leads to an increase in the bulk mechanical properties of the gel, and is accompanied by a contraction of the gel (syneresis). Syneresis is readily apparent from the rheological measurements (Fig. 3a) as the sample pulls down on the upper plate of the rheometer causing the instrument to decrease the gap between the measuring plates in order to maintain a constant normal force.62,63 The syneresis is accompanied by a reduction in the magnitudes of the RQCs indicating a decrease in the anisotropy of the fibres on contraction of the gel. However, the magnitudes of the STDs to the hydrophobic solvents, which do not depend on the anisotropy of the fibres, continue to rise. This indicates an increased hydrophobicity of the syneresed network (ESI,† Section 2.4). The decrease in negative charge and increased hydrophobicity of the fibres is observed with all gelators studied, although syneresis is observed to different extents (ESI,† Section 4). As discussed in our recent work, syneresis is a consequence of both the molecular structure of the gelator and the self-assembly process followed.64
More polar or negatively charged molecules like DMSO, MPA, MeOH, formate or gluconate (ESI,† Section 3) do not show strong STDs, which is readily explained by these probes having weaker interactions with the negatively charged gel fibres. Acetate, being 75% protonated at pH 4.3, shows stronger STDs than formate possibly due to a hydrogen bonding interaction with the fibres. None of deutero-MeOH, DMSO, acetate or formate show discernible RQCs in 1/GdL gels. MeNH3+ does not show strong STDs in 1/GdL gels, even when competing positive ions are excluded (ESI,† Section 1). However, strong STDs are observed in other gel systems (see below), perhaps due to a more favourable binding geometry for saturation transfer from the gel fibres. The interaction between MeNH3+ and the gel fibres does not have to be direct and could be mediated by water molecules.65
The principles that govern the interaction of our probe molecules with 1/GdL gels can be extended to a range of larger model drug compounds (Fig. 4). The positively charged and hydrophobic D1 interacts strongly with the negatively charged gel fibres in 1/GdL gels (Fig. 4b), resulting in very large STDs and a significantly reduced magnitude of the 14NH4+ RQC due to a preferential binding of D1 (ESI,† Section 5.1). Separate resonances with different multiplicities are also observed for D1 in the gel (upfield, broad) and in the fluid exuded during syneresis (downfield, sharp), again consistent with D1 having a strong interaction with the fibres.22,66 In contrast, the negatively charged D2 and D3 exhibit weak STDs, sharp peaks and slight changes in chemical shift (0.001 ppm or less) due to the presence of the gel. Similar chemical shift changes can be observed for the organic solvents used as probe molecules (ESI,† Section 5.2). The anionic D2 and D3 thus interact only weakly with the gel fibres compared to D1.
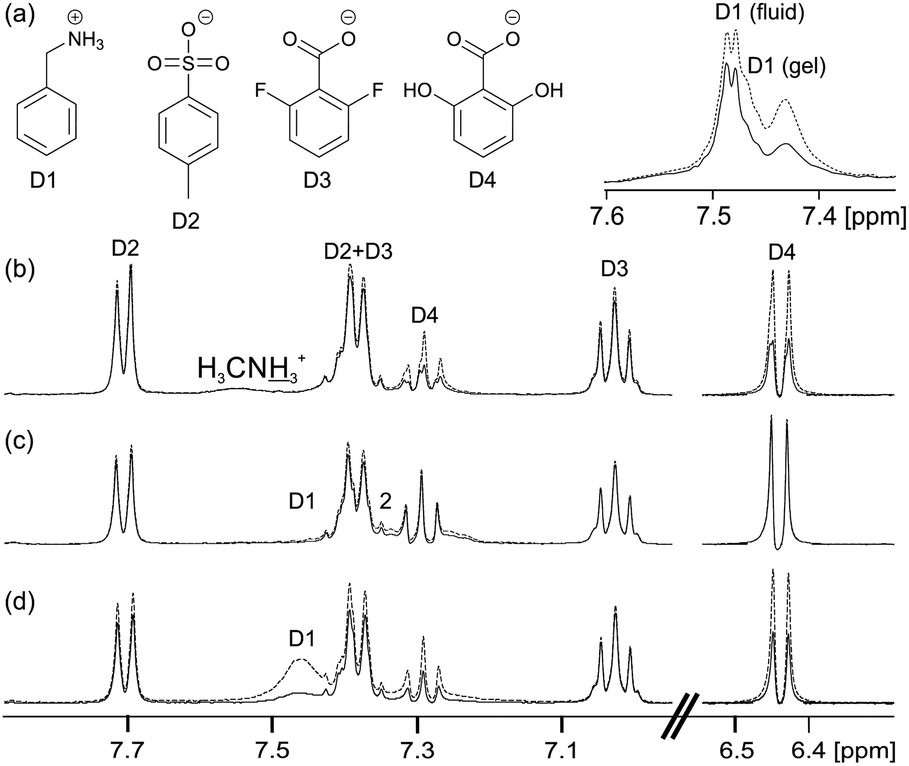 | ||
| Fig. 4 (a) Structures of model drug compounds. (b–d) NMR spectra of model drug compounds in hydrogels recorded with (solid) and without (dashed) on resonance presaturation applied to the gel fibres: (b) 1/GdL hydrogel containing D2–D4 and (inset top) hydrogel containing D1 and NH4Cl. (c) 4 mg mL−1 solution of 2 at pH 9 containing D1–D4. (d) Sample of (c) upon addition of 30 mM CaCl2. | ||
However, the negatively charged D4 interacts more strongly with the gel fibres than either D2 or D3, exhibiting large STDs and an upfield shift of 0.004 ppm. The stronger interaction of D4 with the fibres is attributable to hydrogen-bonding interactions involving the hydroxyl groups.66–68 Nevertheless, the peaks of D4 remain sharp in the gel phase while the change in chemical shift is very much smaller than that observed for D1. D1 thus shows by far the strongest interactions with the gel fibres, as would be expected on an electrostatic basis, although other non-covalent interactions also influence the interaction of these molecules with the fibres.
Salt-triggered gelation
We now discuss the gels formed upon the addition of CaCl2 to 4 mg mL−1 solutions of 2 at pH 9, referred to hereafter as 2/CaCl2 gels. The wormlike micellar structures present in solutions of 2 at pH 9 in the absence of Ca2+ bear a much greater negative charge than 1/GdL gels and thus much larger 23Na+ RQCs30 and much shorter 23Na+T1 and T2 relaxation times are observed (vide infra). The cationic D1 shows a much stronger interaction with the structures present than in 1/GdL gels and is completely invisible by 1H NMR spectroscopy (Fig. 4c). The negatively charged compounds all exhibit much weaker interactions. The addition of CaCl2 (Fig. 4d) weakens the interaction of D1 with the structures of 2 and broad NMR peaks become apparent, while enhanced STDs are observed for the other compounds. These results suggest a decrease in the effective negative charge of the structures of 2 upon the addition of CaCl2. Similar STD effects are observed when D1–D4 are diffused into a 2/CaCl2 gel after it has been formed, indicating that the presence of these molecules does not fundamentally change the gelation process (ESI,† Section 5.3).We now discuss in detail the interactions taking place upon the addition of CaCl2 and subsequent gelation of solutions of 2. In the original reports on the 2/CaCl2 system, gelation was ascribed to a ‘cross-linking’ of the wormlike structures of 2 by the Ca2+, mediated by Ca2+-carboxylate bridges.42,69 Detailed analysis with a range of analytical techniques confirms that only minimal changes to the molecular packing of the assemblies of 2 take place upon the addition of Ca2+, consistent with this hypothesis.16 However, while some reports on related systems also invoke the existence of specific interactions between the ions and the gelators,10,70–72 other reports have discussed gelation in terms of the ‘salting-in/salting-out’ effect of different ions according to the Hofmeister series73–75 or in terms of charge-screening effects.76–78 The precise role of the metal cations in promoting gelation thus remains unclear. Here, we demonstrate using our NMR methods that a strong interaction exists between the Ca2+ and the fibres of 2 upon gelation, providing firm evidence for the cross-linking hypothesis.
The gels are formed by placing a small volume of concentrated CaCl2 solution on top of a solution of the gelator. Diffusion of the CaCl2 throughout the sample forms a gel. Gelation occurs essentially instantaneously upon contact between the gelator and the Ca2+ solution and so gel formation must be studied along a concentration gradient of the Ca2+. This is possible using chemical shift imaging (CSI) techniques in which spatially resolved NMR spectra are recorded along the length of the sample (Fig. 5a).79–81 Plots of the RQCs of the various probe molecules along the Ca2+ concentration gradient are shown in Fig. 5b. Additional plots at earlier and later stages of the experiment are presented in the ESI,† Section 6.
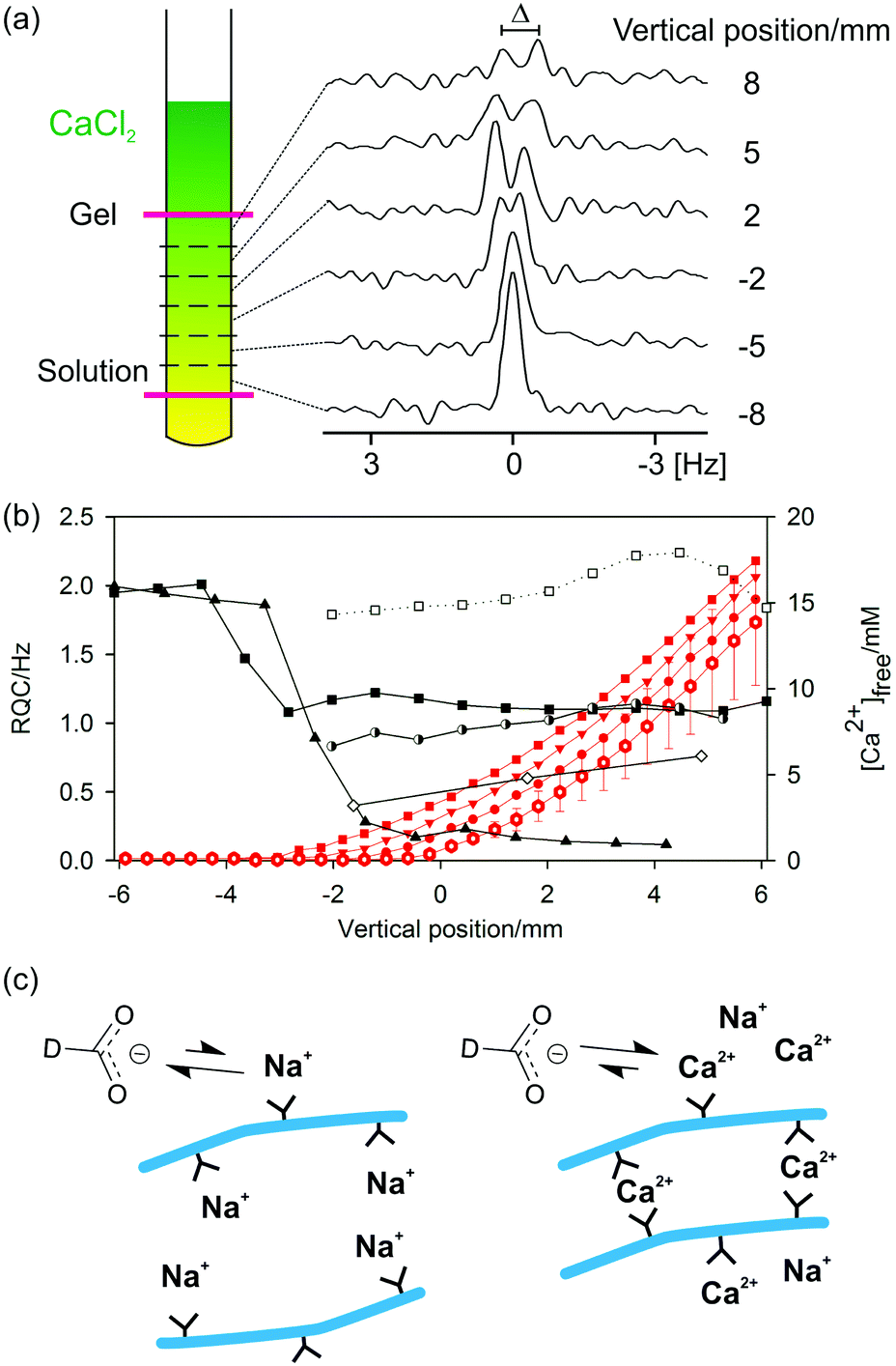 | ||
| Fig. 5 (a) Cartoon to illustrate the principle of chemical shift imaging (CSI): spatially resolved NMR spectra are recorded from slices along the length of a Ca2+ gradient. The pink lines represent the NMR-active region of the sample, centred 18 mm from the base of the NMR tube. 2H spectra of formate-d are illustrated as an example. (b) Plots of experimental observables along the length of the NMR sample. 0 mm corresponds to the centre of the NMR-active region. RQCs: 23Na+ (black triangle), formate-d (white diamond), HDO (black square), dioxane (white square) and tBuOH (half-black circle). The 23Na+ RQC has been scaled down by a factor of 500. Only resolvable RQCs are plotted. Due to experimental limitations RQCs were recorded in separate images and hence at different [Ca2+]free profiles. Red: Profiles of [Ca2+]free before all images (hollow hexagon) – error bars indicate uncertainty in [Ca2+]free measurements – after 23Na image (circle), after formate-d image (down triangle) and after 2H HDO, tBuOH and dioxane image (square). The lines are to guide the eye. The vertical position of a data point corresponds to the centre of the slice while the separation between the points corresponds to the slice width. (c) Cartoon to illustrate the proposed surface chemistry of the fibres (blue) in the absence (left) and presence (right) of Ca2+ and the interaction of the formate-d ions with the associated metal ions. Carboxylate groups of 2 are shown as black Y shapes. | ||
The concentration of free Ca2+ ions in solution can be determined from the 1H chemical shift of maleate (ESI,† Section 7) and is also plotted. In the absence of Ca2+, a very large RQC for 23Na+ is observed of 1 kHz. As the Ca2+ diffuses down the tube, the 23Na+ RQC falls precipitately as the Na+ is displaced from the assemblies of 2 by the Ca2+. Displacement of Na+ is further confirmed by 23Na relaxation measurements. The T1 relaxation time of 23Na+ increases from 41 ± 1 ms in the absence of Ca2+ to 54 ± 1 ms in the final gel. Spatially resolved 23Na relaxation measurements demonstrate how the T2 relaxation transforms from biexponential and fast in the absence of Ca2+ to slow and monoexponential when an excess of Ca2+ is present (ESI,† Fig. S8b). However, the Na+ ions are never completely displaced and remain in competition with the Ca2+; the relaxation behaviour of 23Na+ in the final gels is very different to that in an analogous solution in the absence of 2 while the 23Na+ RQC never completely vanishes to zero (ESI,† Section 8). Similar effects are observed when methylammonium chloride is included as a probe (ESI,† Section 9). Methylammonium was excluded from 2/CaCl2 gel samples as standard, owing to its very strong interaction with the structures of 2 in the absence of Ca2+.
Formate, maleate and MPA2− all have appreciable binding constants to Ca2+ and so can be used to probe the presence of the bound divalent ion.57,82,83 Concomitant with the decrease in the 23Na+ RQC is the emergence of an RQC of the formate-d ions. The observation of RQCs to formate in 2/CaCl2 gels, but not in acid-triggered gels (ESI,† Section 4), indicates the presence of bound Ca2+ ions on the surface of the fibres with which the negatively charged formate ions can interact (Fig. 5c). Residual dipolar couplings (RDCs) are also observed on the 1H resonances of MPA2− and maleate (ESI,† Section 10) which can be similarly attributed. We note that RDCs are becoming increasingly important in the structural and conformational analysis of biomolecules.84 Dipeptide hydrogels may thus prove to be valuable and hitherto unexplored media for their measurement.
Towards the top of the sample, the 23Na+ and formate-d RQCs approach steady values while the concentration of free Ca2+ ions in solution rises steeply. These observations indicate that the gel fibres in the upper part of the sample are essentially saturated with Ca2+; further additions of Ca2+ have no further effect on either the anisotropy of the fibres or their surface chemical properties (ESI,† Section 6). Accordingly, the mechanical properties of the gels are largely invariant when between 1 and 10 equivalents of Ca2+ to peptide are added, with significant decreases in the mechanical properties only observed at very high (>0.1 M) Ca2+ concentrations.42
It can also be seen in Fig. 5b that the 23Na+ RQC decreases and an RQC of formate-d and dioxane-d8 appear before a significant concentration of free Ca2+ is apparent in solution. These observations suggest a strong initial interaction between the structures of 2 and the Ca2+. In an attempt to further probe the dynamics of the Ca2+ ions, we turned to 43Ca NMR.85,86 No 43Ca resonances are apparent in the final gels whereas a sharp resonance is observed in an analogous 20 mM solution of CaCl2. The ‘free’ Ca2+ ions thus spend a significant fraction of their time (>1%) associated with the gel fibres (ESI,† Section 11). Experiments at higher spatial resolution, but with twice the concentration of (2H) probes, demonstrate that the formate-d RQC becomes observable after that of dioxane-d8 (ESI,† Fig. S1f). A slight excess of Ca2+ is thus required in order to sufficiently decrease the effective negative charge of the fibres for the anionic formate to interact and exhibit an RQC.
In contrast to GdL and acid-triggered gels (ESI,† Section 4), no significant STDs are observed to any of the probe molecules in 2/CaCl2 gels. However, that RQCs and RDCs are observed indicates that the interaction of the probe molecules with the fibres differs from those in the GdL and acid-triggered gels. For example, the bound Ca2+ may hinder the intimate contact between the molecule and the protons of the gel fibre required for an STD.87 In contrast, RQCs and RDCs arise as a result of the restricted motions of the molecules when interacting with the gel fibres so do not require intimate contact in order to be observed (ESI,† Section 2). The increase in the RQCs to the organic solvents upon the addition of CaCl2 would seem to imply an increase in the hydrophobicity of the structures. However, interpretation of the results is considerably complicated by the potential ‘salting-out’ effect of the CaCl2 on the organic solvents and a detailed interpretation of the results is beyond the scope of the present study.88 The decrease in the RQC of HDO is likely due to a change in the orientation of the bound water molecules upon coordination of Ca2+.47 RQCs of acetone and MeOH can also be observed but are not shown on Fig. 5 for brevity (ESI,† Section 6).
The ‘free’ Ca2+ can diffuse out of the gel into an external NaCl solution at pH 9 (Fig. 6). The gel remained intact during this process; no swelling or erosion of the gels was observed over the entire experiment (ESI,† Section 12) while the RQCs of HDO and dioxane, which are sensitive to both the structure and anisotropy of the gel fibres as well as the surface chemical properties, also remain constant. However, a slight decrease of the formate-d RQC is apparent along with a slight increase of the 23Na+ RQC (ESI,† Section 13) indicating that the bound Ca2+ ions are partially exchanged for Na+. Such behaviour of the gels could be of considerable interest for environmental remediation or sensing applications. For example, the gel could be placed in a much larger volume of water whereupon other species of interest could diffuse in, the excess of Ca2+ providing additional stability.1
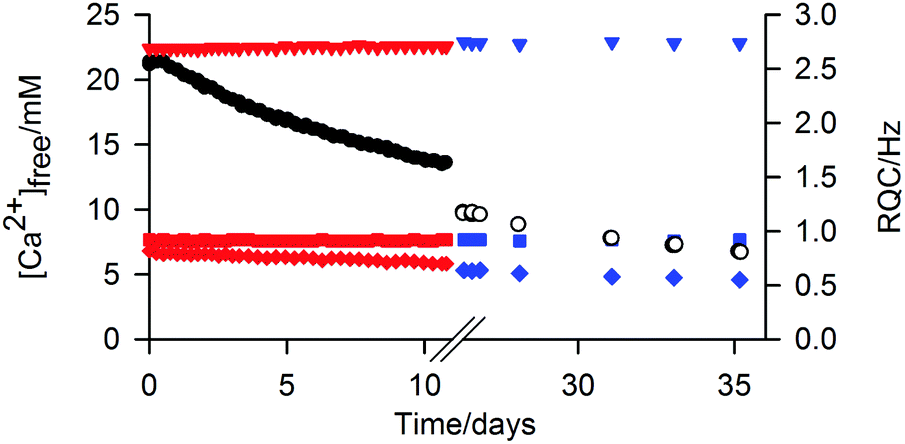 | ||
| Fig. 6 Plots of [Ca2+]free in 2/CaCl2 gel versus time after an NaCl solution was placed on top of the sample. After the first solution (black circle) had stood on top of the gel for 26 days, the supernatant solution was removed, fresh solution placed on top and the second series of measurements (white) commenced. The RQCs of HDO (square), dioxane (down triangle) and formate (diamond) are plotted for the first (red) and second (blue) series of measurements. | ||
Gelation via a solvent-switch
Finally, we discuss gels formed upon the addition of H2O to solutions of 3 in DMSO, referred to hereafter as 3/DMSO gels. A gel prepared by the addition of MilliQ water to 3 in DMSO was found to have a pH of 3.9, in agreement with measurements on related systems,44,89 implying that the gel fibres have an appreciable acidity. When sodium formate (7 mM), sodium methanesulfonate (2 mM) and methylammonium chloride (2 mM) were included as probes in the water along with neutral organic solvents, a self-supporting gel was formed with a pH of 4.5. Analysis of the STDs observed for this sample (Fig. 7) imply that the gel fibres bear a negative charge; a strong STD is observed to the positively charged methylammonium ions while negligible STDs are observed to the negative methanesulfonate and formate ions. Weak STDs are observed to the organic solvents. Furthermore, the resonance of the formate ions is shifted upfield due to protonation by an amount equivalent to inclusion of 2.4 ± 0.2 mM HCl in the absence of 3. It can thus be calculated that 20 ± 2% of the gelators are deprotonated (ESI,† Section 14). Reductions in the 23Na T1 and T2 relaxation times from 36 ± 2 and 35 ± 2 ms in an analogous solution in the absence of 3 to 31 ± 2 and 19 ± 2 ms in the gel are observed, again consistent with the fibres carrying negative charge. Less than 5% of 3 is visible in the gels by 1H NMR and we can thus be confident that the negative charge resides on the self-assembled structures rather than on unassembled gelator molecules (ESI,† Section 15). We are thus able to introduce negative charge onto the fibres by a judicious choice of additives in the H2O solution.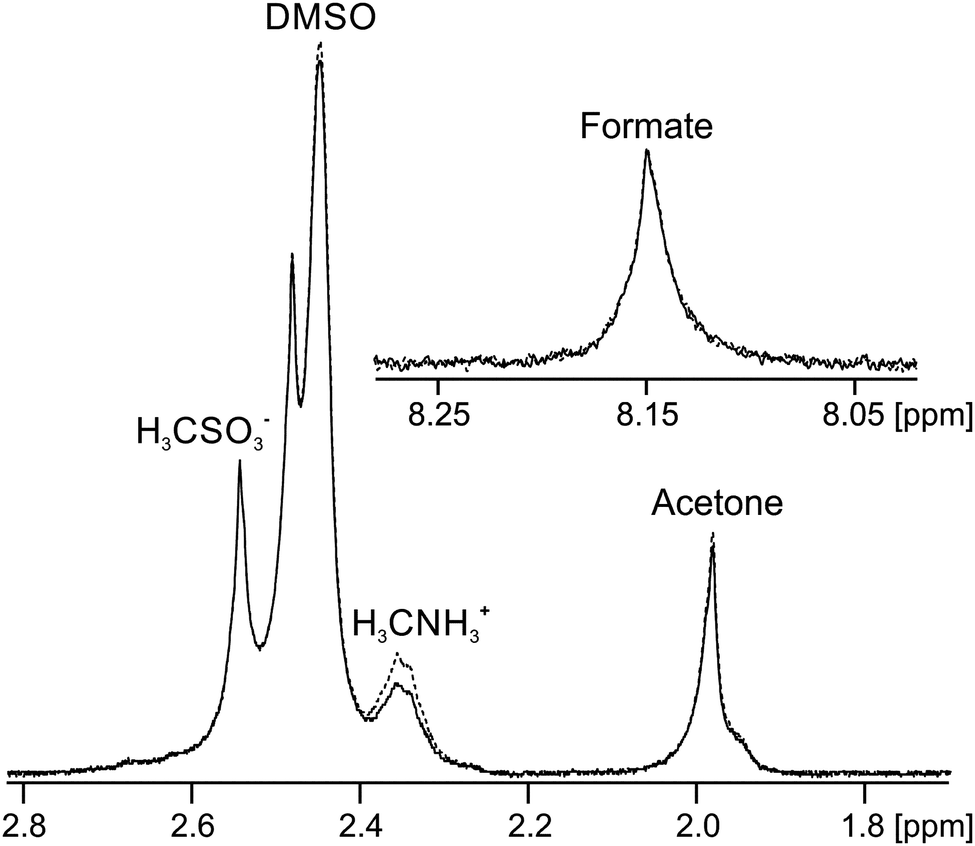 | ||
| Fig. 7 NMR spectra of probe molecules in 3/DMSO gel with (solid) and without (dashed) on resonance pre-saturation applied to the gel fibres. The downfield and upfield resonances of acetone and DMSO correspond to the totally protonated (H6) and partially deuterated (HD5) molecules respectively. The formate resonance is shown inset. | ||
The acidic nature of the fibres in 3/DMSO gels, as well as in analogous gels formed from related dipeptides, is further supported by previous reports concerning the effect of basic additives on the materials.46,89,90 Working with the related gelator, FmocFF, Raeburn et al.89 reported that the mechanical properties of gels formed upon the addition of buffer solutions to DMSO solutions of the gelator depended on the pH of the buffer used, with strong gels formed only at acidic pH. Elsewhere, Orbach et al.90 reported that gels formed from other related Fmoc-capped peptides were unstable when exposed to buffer solutions above pH 6.5. The weakly acidic nature of the gel fibres is of significance were the gels to be used in cell culturing or drug delivery applications where weakly basic organic compounds would likely be present.45,46,90
No RQCs were detected in these gels for any of the probes indicating that the fibres in 3/DMSO gels do not possess a significant degree of alignment with respect to the magnetic field (ESI,† Section 4.1). The observation of STDs to the organic solvents and reduced 23Na+T1 and T2 relaxation times confirms the interaction of these species with the fibres. RQCs are observed, however, in gels of 3 triggered by the addition of GdL to a solution of the gelator at high pH. We attribute this observation to differences in the assembly mechanisms of the two methods.33,43
Summary of methods
RQC, STD and 23Na relaxation measurements have been presented as complementary techniques to study the interaction of a range of probe molecules and ions with the gel fibres. We now summarise a general strategy for their use. In all systems presented here, 23Na relaxation measurements are a useful tool to study the interaction of Na+ with the self-assembled gel fibres. The interaction of Na+ with the negatively charged gel fibres increases the rate of its NMR relaxation. 23Na+ relaxation measurements do not require the gels to be aligned in the spectrometer field and can, in principle, be performed on the background Na+ in a sample without the need for probes. RQCs are only observed when a stringent set of conditions applies: firstly, the probe must interact significantly with the gel fibres. Secondly, the gel fibres must possess a degree of alignment relative to the magnetic field of the spectrometer. Thirdly, the probe must possess a favourable structure and interact in a suitable orientation with the gel fibres (ESI,† Section 2.1). Nevertheless, when observable, RQCs are very sensitive to the surface chemistry of the gels (Fig. 3b and 5). STDs are also sensitive to the surface chemistry of the fibres but do not require them to be aligned. The sensitivity of RQCs to the anisotropy of the fibres, but not STDs, is apparent by comparing the RQCs and STDs to IPA and tBuOH in 1/GdL gels (Fig. 3). However, qualitatively, RQCs and STDs to the same probe molecule convey the same information; an increase in hydrophobicity with time (Fig. 3 and ESI,† Section 4.2) or a loss of negative charge from the fibres (ESI,† Section 9).To summarise, when studying the interaction of a set of probe molecules with self-assembled gels, it is advisable to use two or more complementary NMR methods. The choice of method depends upon the probes used and the system under study.22,64 The absolute size of an STD or RQC should not be used alone to judge the affinity of a probe molecule for the gel fibres. Finally, we note that in very heterogeneous systems possessing cationic, anionic and hydrophobic sites, a significant interaction with the gel fibres may be detected for all our probes. In such systems, it may be fruitful to study how these interactions change as the sample conditions such as the pH are changed. We have demonstrated how this may be achieved using chemical shift imaging (CSI) methods (Fig. 5 and ESI,† Section 4.2).
Conclusions
We have presented a general analytical approach by which the surface chemical properties of the gel fibres in a range of hydrogel systems can be studied using only standard solution-state NMR equipment. We applied our techniques to study the similarities and differences in the surface properties of hydrogels formed from N-functionalised dipeptides via different preparation methods. By combining STD and RQC measurements of a range of positive ions and hydrophobic probe molecules, along with 23Na+ relaxation measurements, we have shown that all fibres bear at least a slight negative charge due to deprotonation of the terminal carboxylic acid groups. As a result, cations exhibit much stronger interactions with the gel fibres than similar anions. Particularly strong interactions are observed with the benzylammonium ion (D1) which is both cationic and hydrophobic. With gels triggered by the addition of H2O to solutions of the gelators in DMSO, additional negative charge was introduced onto the fibres by the inclusion of the weakly basic formate.By studying cation-binding probes such as formate or methylphosphonate, we are able to detect the presence of bound Ca2+ ions on the surface of the gel fibres formed from a hydrophobic dipeptide gelator at alkaline pH. Our methods reveal that the binding of Ca2+ to the gel fibres is very strong, consistent with a Ca2+ mediated cross-linking mechanism of gelation. It is also apparent that the gels can be pre-loaded with an excess of Ca2+ without affecting their integrity. Together, these properties convey a high stability to the gels when exposed to an external Ca2+-free solution at alkaline pH. Our methods may thus be of considerable use in the development of gel systems for the controlled entrapment or release of metal ions and other species of interest. Overall, we anticipate that our methods will be adopted wherever the surface chemical properties of self-assembled hydrogels are of interest.
Experimental
Materials
Gelators 1, 2 and 3 were synthesised as described previously.38,40 All other chemicals were purchased from Sigma-Aldrich and used as received.Preparation of samples
All samples were prepared in H2O to allow for analysis by 2H NMR. Unless otherwise stated, 1/GdL and 2/CaCl2 gels were transferred to the NMR spectrometer (9.4 T) within 3 minutes of preparation of the pre-gel mixtures/solutions and allowed to gel in the magnetic field for at least 14 hours. For the 3/DMSO gels, it was not feasible to transfer the samples to the spectrometer before gelation commenced (<1 minute). A stock solution of the NMR pH indicators was prepared containing sodium formate (0.4 M), glycine (0.2 M), methylphosphonic acid (0.2 M), sodium acetate (0.2 M), sodium methanesulfonate (0.1 M) and sodium hydroxide (0.6 M). Stock solutions of the organic solvents were prepared at 5 vol% (2H) or 2 vol% (1H) of each solvent. Methanol-d3 was included at 2 vol% to avoid excessive truncation artefacts from this resonance in 2H NMR experiments. MeNH3Cl (0.4 M) was also included in the 1H solvent mixture while sodium formate-d (0.5 M) was included in the 2H mixture. Stock solutions of NH4Cl and disodium maleate were prepared at 0.5 M and 0.2 M respectively.To collect the data of Fig. 6, a 2/CaCl2 gel was prepared and aged for two weeks. A solution containing 20 mM NaCl and the other probe molecules was prepared and the pH adjusted to 8.9–9.2 with the addition of 1–2 mM HCl. 1800 μL of solution was then carefully placed on top of the gel. The sample was maintained at 298 K in either an NMR spectrometer or a water bath. Periodically, the solution was removed, analysed and a fresh solution placed on top. For Fig. 4, 5 mM each of D1–D4 were included in a 4 mg mL−1 solution of 2 at pH 9.2 (Fig. 4c). CaCl2 was then added to the solution and the sample aged for two weeks before the spectra of Fig. 4d were recorded.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with the DMSO solution of 3: sodium formate (2 mM), sodium formate-d (5 mM), sodium methanesulfonate (2 mM), methylammonium chloride (2 mM), and all of the 1H and 2H solvents listed in Table 1 at 0.05 and 0.01 vol% respectively. Methanol-d3 was included at 0.02 vol%. The pH indicator probes, MPA2−, glycinate and acetate, were excluded as they do not function in 20% DMSO/80% H2O mixtures. These probes cause additional deprotonation of the gelators (ESI,† Fig. S15b) and do not convey any further information on the system. To prepare gels, a 9′′ glass pipette was placed in a 5 mm NMR tube and 140 μL of the solution of 3 in DMSO was placed in the pipette and allowed to settle at the base of the tube. 560 μL of the H2O solution was then rinsed down the pipette. Upon mixing of the two solutions, the sample immediately became turbid and white whereupon the pipette was withdrawn and the sample transferred to the spectrometer for analysis. The samples resolved into clear gels after standing for less than a minute. The NMR-observable properties of 3/DMSO samples were found to be stable, once measured, within twenty minutes of gel preparation. The NMR resonances on Fig. 7 are broad due to the formation of air bubbles in the gel on standing. Fig. 7 was recorded 20 minutes after preparation.
:1 with the DMSO solution of 3: sodium formate (2 mM), sodium formate-d (5 mM), sodium methanesulfonate (2 mM), methylammonium chloride (2 mM), and all of the 1H and 2H solvents listed in Table 1 at 0.05 and 0.01 vol% respectively. Methanol-d3 was included at 0.02 vol%. The pH indicator probes, MPA2−, glycinate and acetate, were excluded as they do not function in 20% DMSO/80% H2O mixtures. These probes cause additional deprotonation of the gelators (ESI,† Fig. S15b) and do not convey any further information on the system. To prepare gels, a 9′′ glass pipette was placed in a 5 mm NMR tube and 140 μL of the solution of 3 in DMSO was placed in the pipette and allowed to settle at the base of the tube. 560 μL of the H2O solution was then rinsed down the pipette. Upon mixing of the two solutions, the sample immediately became turbid and white whereupon the pipette was withdrawn and the sample transferred to the spectrometer for analysis. The samples resolved into clear gels after standing for less than a minute. The NMR-observable properties of 3/DMSO samples were found to be stable, once measured, within twenty minutes of gel preparation. The NMR resonances on Fig. 7 are broad due to the formation of air bubbles in the gel on standing. Fig. 7 was recorded 20 minutes after preparation.
NMR measurements
All NMR experiments were performed on a Bruker Avance II 400 MHz wide bore spectrometer operating at 400.20 MHz for 1H. The temperature of the samples was maintained at 298 ± 0.5 K, the variation in the temperature being less than 0.1 K. 1H integrals (Fig. 3d) were recorded in a single scan with presaturation (50 Hz power) applied to the H2O resonance for 5 s followed by a spoil gradient pulse (27 G cm−1, 1 ms) prior to a π/2 hard pulse and signal acquisition. 65536 data points were acquired with a sweep width of 15 ppm, giving a total acquisition time of 35 s including a 25 s delay at the start of the experiment prior to any pulses. DMSO was used as the reference for integration. The STD effects to the gelator resonances arising from presaturation of the H2O resonance were not significant (ESI,† Section 16). Integrals have been normalised to their values before the addition of GdL. Essentially all (>90%) of a theoretical 5 mg mL−1 of 1 was visible in the solution at pH 9 when integrated against an internal standard. Very accurate quantification by NMR is extremely challenging in H2O;91 presaturation can affect the gelator resonances by saturation-transfer effects from the H2O while WATERGATE sequences give distorted lineshapes due to J-modulation. Very similar integrals are observed when D2O is used instead of H2O.92 Methanesulfonate (2.815 ppm) was used as a chemical shift reference for 1H NMR in all H2O samples.
1H STD experiments were performed using the double-echo WATERGATE sequence of Liu et al.93 (Bruker library ZGGPW5) with presaturation applied during the relaxation delay (8.1 s) and with a 2 s signal acquisition time. The delay between successive hard pulses in the selective pulse train was set at 250 μs corresponding to a 4000 Hz separation between the null points. Presaturation was applied using a train of 157 Gaussian pulses of 50 ms in duration and separated by 1 ms. The peak pulse power was 380 Hz. Saturation was applied at −5 ppm (on resonance) or −250 ppm (off resonance). On and off resonance spectra were acquired in alternate scans to minimise the effect of any change in the sample over the time-course of the STD acquisition. The spectra of Fig. 3 and 7 were acquired using 16 dummy scans prior to signal acquisition (8 scans on and off resonance) in order to attain steady-state conditions, giving a total acquisition time of 5 minutes 20 s. Omitting these dummy scans resulted in very slight (≤3%) apparent STDs, even in the absence of a gel phase, owing to the first off-resonance scan being acquired before the first on-resonance. Measurement of the STDs as a function of presaturation time confirmed that a point measurement at 8 s presaturation provides an adequate indication of the strength of the probe-fibre interaction (ESI,† Section 2.3). Very similar STDs were observed in 1/GdL gels using our standard protocol and when a very long (40 s) relaxation delay was elapsed to allow complete relaxation to thermal equilibrium between scans (ESI,† Fig. S2b). The spectra of Fig. 4 were acquired with no dummy scans in order for STD build up curves to be constructed. The omission of dummy scans gave a very slight relaxation artefact of ≤1% for D1–D3 and ≤2% for D4. All spectra on Fig. 4 were processed with an exponential line broadening factor of 1 Hz. On Fig. 3, STDs and RQCs were recorded on the same sample while 1H integrals, 23Na+ relaxation times and the pH were recorded on a separate sample. As in our previous work,30 the pH and kinetics of gel formation are highly reproducible and so the datasets are directly comparable.
2H spectra were recorded via the lock channel with 4422 data points, a 200 μs pulse (70°) and a sweep width of 12 ppm. Between 256 and 1024 scans were acquired in 12 to 46 minutes respectively, depending on the magnitude of the IPA RQC and the extent of syneresis of 1/GdL samples. 14N{1H} spectra were acquired with the aring sequence (π/2–τ–π/2–τ–π/2-acquire) in order to suppress acoustic ringing effects in our probe. A 46 μs π/2 pulse was used with a τ of 4 μs and a relaxation time of 0.1 s. CPD was applied (Waltz 16) during the pulses and signal acquisition in order to remove the effect of 14N–1H coupling. Spectra were acquired with 578 data points and a sweep width of 10 ppm in either 256 or 1024 scans depending on the magnitude of the 14NH4+ RQC. The time plotted for 2H and 14N spectra corresponds to the time halfway through the acquisition.
23Na T1 and T2 were measured using the inversion-recovery and CPMG pulse sequences respectively. For T1, the inversion recovery time, t, was varied between 1 and 300 ms in 8 steps. For T2, the spacing between the π pulses was fixed at 1 ms and the number of pulses varied between 2 and 256 in 8 steps. 32 scans were performed with 6144 points, a 100 ppm sweep width and a relaxation delay of 0.1 s, giving acquisition times of 2 minutes for both T1 and T2 measurements. No de-gassing of samples was attempted prior to relaxation measurements owing to the insignificant effect of oxygen on the relaxation of 23Na, the dominant relaxation mechanism of this nucleus being the quadrupolar mechanism. The 23Na T1 of 10 mM NaCl in H2O was measured as 59 ± 1 ms after preparation and 60 ± 1 ms after de-gassing with argon.
CSI experiments were performed using the gradient phase encoding sequence of Trigo-Mouriño et al.79 (π/2–τ1–g–τ2-acquire) where g is a gradient pulse and τ1 and τ2 are delays of 10 and 200 μs respectively. g was a minimum of 100 μs in duration and was varied between a maximum of −50 and 50 G cm−1, depending on the nucleus and the number of gradient steps acquired. 2H CSI experiments were acquired with either 8 gradient steps and 128 scans (formate-d) or 32 steps and 32 scans (other probes). 3684 points were acquired with a sweep width of 12 ppm. The theoretical spatial resolution was 0.81 mm for 32 step images or 3.3 mm for 8 step images. For 23Na, the sequence was modified to include a solid-echo (π/2–τ–π/2–τ1–g–τ2-acquire) to refocus the evolution of the quadrupolar coupling during the gradient pulse and delays. 32 steps were acquired with 256 scans and 2118 points. The theoretical spatial resolution was 0.94 mm. All 2H and 23Na CSI experiments were 45 minutes in duration. For 1H, the π/2 pulse was replaced with a double-echo WATERGATE sequence to suppress the H2O resonance. A spoil gradient (27 G cm−1) was also employed at the end of the signal acquisition period (1 s) to destroy any transverse magnetisation. 64 steps were acquired with 8 scans giving a total acquisition time of 10 minutes and a theoretical spatial resolution of 0.41 mm.
Data processing
All NMR data was processed in Bruker Topspin 3.2. 23Na+ and 2H RQCs were obtained by Lorentzian deconvolution of the spectra while 14NH4+ RQCs were obtained by peak picking between the peak maxima. pH values were extracted from 1H spectra following the procedure described in our previous work.30 [Ca2+]free values were obtained from the chemical shift of maleate using the procedure described in the ESI,† Section 7. CSI images were processed and phase corrected following the procedure of Trigo-Mouriño et al.79 Beyond ±7 mm from the sample centre, the sensitivity of the CSI spectra falls markedly as the limits of the NMR coils are exceeded. Data is thus only plotted to ±6 mm on Fig. 5. 23Na+T1 and T2 values were extracted by fitting the data to eqn (1) and (2) respectively using a non-linear regression method developed for Microsoft Excel:94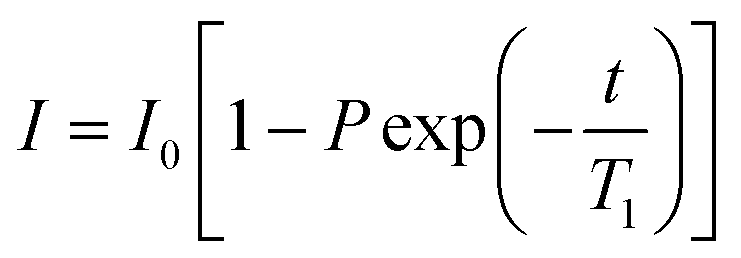 | (1) |
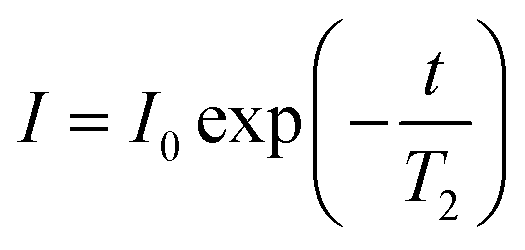 | (2) |
Rheology
The time series of Fig. 3 was recorded on an Anton Paar Physica MCR301 Rheometer. A solution of 1 with the standard set of probes was mixed with 5 mg mL−1 GdL and 2 mLs transferred to the stationary (silicon rubber) lower plate whereupon the upper plate (50 mm, sandblasted) was lowered onto the sample to a gap of 1 mm. Low viscosity mineral oil was placed around the edge of the sample to prevent drying artefacts. G′ and G′′ were recorded at a frequency of 10 rad s−1 and 0.5% strain, a data point being recorded every 15 s for 15 hours. The gap between the measuring plates was adjusted automatically in order to maintain a constant normal force on the upper measuring plate. The temperature of the sample was maintained at 25 °C.Acknowledgements
We thank Unilever for a Case Award (MW) and the EPSRC for funding a DTA (MW). We thank the EPSRC for funding (EP/C005643/1 and EP/K039687/1). DA thanks the EPSRC for a Fellowship (EP/L021978/1).Notes and references
- B. O. Okesola and D. K. Smith, Chem. Soc. Rev., 2016, 45, 4226–4251 RSC.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef CAS PubMed.
- N. Zweep and J. H. van Esch, in Functional Molecular Gels, ed. B. Escuder and J. F. Miravet, The Royal Society of Chemistry, 2014, pp. 1–29 10.1039/9781849737371-00001.
- S. Boothroyd, A. F. Miller and A. Saiani, Faraday Discuss., 2013, 166, 195–207 RSC.
- D. Roberts, C. Rochas, A. Saiani and A. F. Miller, Langmuir, 2012, 28, 16196–16206 CrossRef CAS PubMed.
- K. Nagy-Smith, Y. Yamada and J. P. Schneider, J. Mater. Chem. B, 2016, 4, 1999–2007 RSC.
- C. J. Newcomb, S. Sur, J. H. Ortony, O. S. Lee, J. B. Matson, J. Boekhoven, J. M. Yu, G. C. Schatz and S. I. Stupp, Nat. Commun., 2014, 5, 3321 Search PubMed.
- V. Jayawarna, S. M. Richardson, A. R. Hirst, N. W. Hodson, A. Saiani, J. E. Gough and R. V. Ulijn, Acta Biomater., 2009, 5, 934–943 CrossRef CAS PubMed.
- E. V. Alakpa, V. Jayawarna, A. Lampel, K. V. Burgess, C. C. West, S. C. J. Bakker, S. Roy, N. Javid, S. Fleming, D. A. Lamprou, J. Yang, A. Miller, A. J. Urquhart, P. W. J. M. Frederix, N. T. Hunt, B. Péault, R. V. Ulijn and M. J. Dalby, Chem, 2016, 1, 298–319 CAS.
- G. Yu, X. Yan, C. Han and F. Huang, Chem. Soc. Rev., 2013, 42, 6697–6722 RSC.
- C. Tang, R. V. Ulijn and A. Saiani, Langmuir, 2011, 27, 14438–14449 CrossRef CAS PubMed.
- M. Tena-Solsona, B. Escuder and J. F. Miravet, Chem. Mater., 2015, 27, 3358–3365 CrossRef CAS.
- C. Tang, A. M. Smith, R. F. Collins, R. V. Ulijn and A. Saiani, Langmuir, 2009, 25, 9447–9453 CrossRef CAS PubMed.
- S. L. Zhou, S. Matsumoto, H. D. Tian, H. Yamane, A. Ojida, S. Kiyonaka and I. Hamachi, Chem. – Eur. J., 2005, 11, 1130–1136 CrossRef CAS PubMed.
- A. Z. Cardoso, L. L. E. Mears, B. N. Cattoz, P. C. Griffiths, R. Schweins and D. J. Adams, Soft Matter, 2016, 12, 3612–3621 RSC.
- A. Caragheorgheopol, W. Edwards, J. G. Hardy, D. K. Smith and V. Chechik, Langmuir, 2014, 30, 9210–9218 CrossRef CAS PubMed.
- J. H. Ortony, C. J. Newcomb, J. B. Matson, L. C. Palmer, P. E. Doan, B. M. Hoffman and S. I. Stupp, Nat. Mater., 2014, 13, 812–816 CrossRef CAS PubMed.
- J. Tritt-Goc, A. Rachocki and M. Bielejewski, Soft Matter, 2014, 10, 7810–7818 RSC.
- V. J. Nebot and D. K. Smith, in Functional Molecular Gels, ed. B. Escuder and J. F. Miravet, The Royal Society of Chemistry, 2014, pp. 30–66 10.1039/9781849737371-00030.
- D. C. Duncan and D. G. Whitten, Langmuir, 2000, 16, 6445–6452 CrossRef CAS.
- B. Escuder, M. Llusar and J. F. Miravet, J. Org. Chem., 2006, 71, 7747–7752 CrossRef CAS PubMed.
- S. Bouguet-Bonnet, M. Yemloul and D. Canet, J. Am. Chem. Soc., 2012, 134, 10621–10627 CrossRef CAS PubMed.
- A. Reddy, A. Sharma and A. Srivastava, Chem. – Eur. J., 2012, 18, 7575–7581 CrossRef CAS PubMed.
- F. Piana, D. H. Case, S. M. Ramalhete, G. Pileio, M. Facciotti, G. M. Day, Y. Z. Khimyak, J. Angulo, R. C. D. Brown and P. A. Gale, Soft Matter, 2016, 12, 4034–4043 RSC.
- Nonappa, D. Šaman and E. Kolehmainen, Magn. Reson. Chem., 2015, 53, 256–260 CrossRef CAS PubMed.
- M. Suzuki, M. Yumoto, M. Kimura, H. Shirai and K. Hanabusa, Chem. – Eur. J., 2003, 9, 348–354 CrossRef CAS PubMed.
- C. A. Angulo-Pachon, C. Gasco-Catalan, J. J. Ojeda-Flores and J. F. Miravet, ChemPhysChem, 2016, 17, 2008–2012 CrossRef CAS PubMed.
- M. D. Segarra-Maset, B. Escuder and J. F. Miravet, Chem. – Eur. J., 2015, 21, 13925–13929 CrossRef CAS PubMed.
- M. Wallace, J. A. Iggo and D. J. Adams, Soft Matter, 2015, 11, 7739–7747 RSC.
- M. Wallace, D. J. Adams and J. A. Iggo, Soft Matter, 2013, 9, 5483–5491 RSC.
- L. Chen, K. Morris, A. Laybourn, D. Elias, M. R. Hicks, A. Rodger, L. Serpell and D. J. Adams, Langmuir, 2010, 26, 5232–5242 CrossRef CAS PubMed.
- A. Z. Cardoso, A. E. Alvarez Alvarez, B. N. Cattoz, P. C. Griffiths, S. M. King, W. J. Frith and D. J. Adams, Faraday Discuss., 2013, 166, 101–116 RSC.
- W. J. Frith, Philos. Trans. R. Soc., A, 2016, 374, 20150135 CrossRef PubMed.
- D. J. Adams and P. D. Topham, Soft Matter, 2010, 6, 3707–3721 RSC.
- S. Awhida, E. R. Draper, T. O. McDonald and D. J. Adams, J. Colloid Interface Sci., 2015, 455, 24–31 CrossRef CAS PubMed.
- N. Singh, M. Kumar, J. F. Miravet, R. V. Ulijn and B. Escuder, Chem. – Eur. J., 2017, 23, 981–993 CrossRef CAS PubMed.
- L. Chen, S. Revel, K. Morris, L. C. Serpell and D. J. Adams, Langmuir, 2010, 26, 13466–13471 CrossRef CAS PubMed.
- Z. Yang, G. Liang, M. Ma, Y. Gao and B. Xu, J. Mater. Chem., 2007, 17, 850–854 RSC.
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856–1862 RSC.
- E. R. Draper, L. L. E. Mears, A. M. Castilla, S. M. King, T. O. McDonald, R. Akhtar and D. J. Adams, RSC Adv., 2015, 5, 95369–95378 RSC.
- L. Chen, G. Pont, K. Morris, G. Lotze, A. Squires, L. C. Serpell and D. J. Adams, Chem. Commun., 2011, 47, 12071–12073 RSC.
- L. Chen, J. Raeburn, S. Sutton, D. G. Spiller, J. Williams, J. S. Sharp, P. C. Griffiths, R. K. Heenan, S. M. King, A. Paul, S. Furzeland, D. Atkins and D. J. Adams, Soft Matter, 2011, 7, 9721–9727 RSC.
- J. Raeburn, C. Mendoza-Cuenca, B. N. Cattoz, M. A. Little, A. E. Terry, A. Zamith Cardoso, P. C. Griffiths and D. J. Adams, Soft Matter, 2015, 11, 927–935 RSC.
- A. Mahler, M. Reches, M. Rechter, S. Cohen and E. Gazit, Adv. Mater., 2006, 18, 1365–1370 CrossRef CAS.
- T. Liebmann, S. Rydholm, V. Akpe and H. Brismar, BMC Biotechnol., 2007, 7, 88 CrossRef PubMed.
- A. Delville, J. Grandjean and P. Laszlo, J. Phys. Chem., 1991, 95, 1383–1392 CrossRef CAS.
- M. Wallace, A. Z. Cardoso, W. J. Frith, J. A. Iggo and D. J. Adams, Chem. – Eur. J., 2014, 20, 16484–16487 CrossRef CAS PubMed.
- I. O. Shklyarevskiy, P. Jonkheijm, P. C. M. Christianen, A. P. H. J. Schenning, A. Del Guerzo, J. P. Desvergne, E. W. Meijer and J. C. Maan, Langmuir, 2005, 21, 2108–2112 CrossRef CAS PubMed.
- O. Cala, F. Guillière and I. Krimm, Anal. Bioanal. Chem., 2014, 406, 943–956 CrossRef CAS PubMed.
- V. Lozano, R. Hernandez, A. Arda, J. Jimenez-Barbero, C. Mijangos and M.-J. Perez-Perez, J. Mater. Chem., 2011, 21, 8862–8870 RSC.
- C. Cruz, E. J. Cabrita and J. A. Queiroz, J. Chromatogr. A, 2011, 1218, 3559–3564 CrossRef CAS PubMed.
- D. E. Woessner, Concepts Magn. Reson., 2001, 13, 294–325 CrossRef CAS.
- H. Maki, K. Yoshida, H. Nariai and M. Mizuhata, Colloids Surf., A, 2015, 471, 1–10 CrossRef CAS.
- E. R. Draper, J. R. Lee, M. Wallace, F. Jäckel, A. J. Cowan and D. J. Adams, Chem. Sci., 2016, 7, 6499–6505 RSC.
- M. Raue, A. Bernet, M. Kuppers, S. Stapf, H. W. Schmidt, B. Blumich and T. Mang, in Intelligent Hydrogels, ed. G. Sadowski and W. Richtering, Springer International Publishing, 2013, vol. 140, pp. 45–51 Search PubMed.
- P. G. Daniele, C. Foti, A. Gianguzza, E. Prenesti and S. Sammartano, Coord. Chem. Rev., 2008, 252, 1093–1107 CrossRef CAS.
- H. A. Tajmir-Riahi and J. T. Agbebavi, Carbohydr. Res., 1993, 241, 25–35 CrossRef CAS.
- M. Khabiri, B. Minofar, J. Brezovský, J. Damborský and R. Ettrich, J. Mol. Model., 2013, 19, 4701–4711 CrossRef CAS PubMed.
- Z. W. Dai, L. S. Wan, X. J. Huang, J. Ling and Z. K. Xu, J. Phys. Chem. C, 2011, 115, 22415–22421 CAS.
- J. P. Jacobsen and K. Schaumburg, J. Magn. Reson., 1977, 28, 191–201 CAS.
- L. Matějka, Polym. Bull., 1991, 26, 109–116 CrossRef.
- M. Harsch, F. Herzog and J. Karger-Kocsis, J. Compos. Mater., 2008, 42, 2299–2309 CrossRef CAS.
- A. M. Castilla, M. Wallace, L. L. E. Mears, E. R. Draper, J. Doutch, S. Rogers and D. J. Adams, Soft Matter, 2016, 12, 7848–7854 RSC.
- J. Kříž and J. Dybal, J. Phys. Chem. B, 2004, 108, 9306–9314 CrossRef.
- J. A. Sáez, B. Escuder and J. F. Miravet, Chem. Commun., 2010, 46, 7996–7998 RSC.
- M. Goto, H. Hayashi, I. Miyahara, K. Hirotsu, M. Yoshida and T. Oikawa, J. Biol. Chem., 2006, 281, 34365–34373 CrossRef CAS PubMed.
- B. Escuder, J. F. Miravet and J. A. Sáez, Org. Biomol. Chem., 2008, 6, 4378–4383 CAS.
- L. Chen, T. O. McDonald and D. J. Adams, RSC Adv., 2013, 3, 8714–8720 RSC.
- L. Wang, J. Mei, X. Zhang, M. Zhu, J. Wang and L. Wang, RSC Adv., 2014, 4, 1193–1196 RSC.
- M. A. Greenfield, J. R. Hoffman, M. O. De La Cruz and S. I. Stupp, Langmuir, 2010, 26, 3641–3647 CrossRef CAS PubMed.
- J. Shi, Y. Gao, Y. Zhang, Y. Pan and B. Xu, Langmuir, 2011, 27, 14425–14431 CrossRef CAS PubMed.
- S. Roy, N. Javid, P. W. J. M. Frederix, D. A. Lamprou, A. J. Urquhart, N. T. Hunt, P. J. Halling and R. V. Ulijn, Chem. – Eur. J., 2012, 18, 11723–11731 CrossRef CAS PubMed.
- V. J. Nebot, J. J. Ojeda-Flores, J. Smets, S. Fernández-Prieto, B. Escuder and J. F. Miravet, Chem. – Eur. J., 2014, 20, 14465–14472 CrossRef CAS PubMed.
- M. Jaspers, A. E. Rowan and P. H. J. Kouwer, Adv. Funct. Mater., 2015, 25, 6503–6510 CrossRef CAS.
- Y. Feng, M. Taraban and Y. B. Yu, Soft Matter, 2012, 8, 11723–11731 RSC.
- T. Otsuka, T. Maeda and A. Hotta, J. Phys. Chem. B, 2014, 118, 11537–11545 CrossRef CAS PubMed.
- B. Ozbas, J. Kretsinger, K. Rajagopal, J. P. Schneider and D. J. Pochan, Macromolecules, 2004, 37, 7331–7337 CrossRef CAS.
- P. Trigo-Mouriño, C. Merle, M. R. M. Koos, B. Luy and R. R. Gil, Chem. – Eur. J., 2013, 19, 7013–7019 CrossRef PubMed.
- Å. Östlund, D. Bernin, L. Nordstierna and M. Nydén, J. Colloid Interface Sci., 2010, 344, 238–240 CrossRef PubMed.
- Y. Mitrev, S. Simova and D. Jeannerat, Chem. Commun., 2016, 52, 5418–5420 RSC.
- G. H. Nancollas, J. Chem. Soc., 1956, 735–743 Search PubMed.
- M. DeFronzo and R. J. Gillies, J. Biol. Chem., 1987, 262, 11032–11037 CAS.
- S. K. Mishra and N. Suryaprakash, Nucl. Magn. Reson., 2015, 44, 547–591 CAS.
- E. Bouhoutsos-Brown, D. Murk Rose and R. G. Bryant, J. Inorg. Nucl. Chem., 1981, 43, 2247–2248 CrossRef CAS.
- J. M. Aramini, T. Drakenberg, T. Hiraoki, Y. Ke, K. Nitta and H. J. Vogel, Biochemistry, 1992, 31, 6761–6768 CrossRef CAS PubMed.
- N. R. Krishna and V. Jayalakshmi, Prog. Nucl. Magn. Reson. Spectrosc., 2006, 49, 1–25 CrossRef CAS.
- Y. Koga, H. Katayanagi, J. V. Davies, H. Kato, K. Nishikawa and P. Westh, Bull. Chem. Soc. Jpn., 2006, 79, 1347–1354 CrossRef CAS.
- J. Raeburn, G. Pont, L. Chen, Y. Cesbron, R. Lévy and D. J. Adams, Soft Matter, 2012, 8, 1168–1174 RSC.
- R. Orbach, L. Adler-Abramovich, S. Zigerson, I. Mironi-Harpaz, D. Seliktar and E. Gazit, Biomacromolecules, 2009, 10, 2646–2651 CrossRef CAS PubMed.
- P. Giraudeau, V. Silvestre and S. Akoka, Metabolomics, 2015, 11, 1041–1055 CrossRef CAS.
- K. L. Morris, L. Chen, J. Raeburn, O. R. Sellick, P. Cotanda, A. Paul, P. C. Griffiths, S. M. King, R. K. O'Reilly, L. C. Serpell and D. J. Adams, Nat. Commun., 2013, 4, 1480 CrossRef PubMed.
- M. Liu, X. A. Mao, C. Ye, H. Huang, J. K. Nicholson and J. C. Lindon, J. Magn. Reson., 1998, 132, 125–129 CrossRef CAS.
- A. M. Brown, Comput. Methods Programs Biomed., 2001, 65, 191–200 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sm02404a |
| ‡ Current address: School of Chemistry, College of Science and Engineering, University of Glasgow, Glasgow, G12 8QQ, UK. |
| This journal is © The Royal Society of Chemistry 2017 |