
Improving the performance of porous nickel foam for water oxidation using hydrothermally prepared Ni and Fe metal oxides†
Michelle P.
Browne
*ab,
Joana M.
Vasconcelos
ab,
João
Coelho
 ab,
Maria
O'Brien
ab,
Aurelie A.
Rovetta
ab,
Eoin K.
McCarthy
b,
Hugo
Nolan
ab,
Georg S.
Duesberg
ab,
Valeria
Nicolosi
ab,
Paula E.
Colavita
ab and
Michael E. G.
Lyons
ab
ab,
Maria
O'Brien
ab,
Aurelie A.
Rovetta
ab,
Eoin K.
McCarthy
b,
Hugo
Nolan
ab,
Georg S.
Duesberg
ab,
Valeria
Nicolosi
ab,
Paula E.
Colavita
ab and
Michael E. G.
Lyons
ab
aSchool of Chemistry, Trinity College Dublin, College Green, Dublin 2, Ireland. E-mail: brownem6@tcd.ie
bCentre for Research on Adaptive Nanostructures and Nanodevices (CRANN), Advanced Materials and BioEngineering Research (AMBER) Centre, Trinity College Dublin, Dublin 2, Ireland
First published on 9th December 2016
Abstract
Recent research trends have seen a rise in the interest in Transition Metal Oxides (TMO's) as catalysts for the Oxygen Evolution Reaction (OER). In particular, the use of inexpensive mixed TMO's such as Fe and Ni has gained much attention. While some techniques, such as electrodeposition, are widely investigated, reports into the fabrication of Fe, Ni/Fe and Ni oxides for the OER by alternative methods are sparse. To our knowledge, a study involving hydrothermal synthesis to produce all three of the aforementioned catalysts for the OER has yet to be undertaken. Additionally, the use of high surface area electrode supports to enhance the properties of the OER materials is very popular at present. Currently, industrial alkaline electrolysis uses Ni foam as an electrode due to its high porosity, three dimensional structure and low cost. Herein, the hydrothermal synthesis of nanoscale Fe, Ni/Fe and Ni oxide particles is reported. The resulting powders were subsequently used to modify Ni foam, an industrially compatible OER catalyst. The modified Ni foams exhibit improved OER capabilities when compared to the bare foam. Interestingly, the hydrothermal Ni oxide has the greatest enhancement in its OER activity compared to the mixed Ni/Fe and pure Fe oxides. Usually, electrodeposited NiFe oxides are superior OER catalysts compared to pure Ni and Fe oxides. Finally, the hydrothermal Ni oxide/Ni foam system offers a cheap and more active OER catalysts then the currently utilised anodic material for water electrolysis. This system could generate an increased amount of H2 gas for an economy searching for alternatives energy sources.
Introduction
Recently, materials fabricated from iron and nickel oxides via electrodeposition techniques have become of major interest for the use in the Oxygen Evolution Reaction (OER).1–5 The OER is the most kinetically demanding step in water electrolysis therefore if the overpotential associated with this reaction can be lowered, the overall efficiency of water electrolysis will increase. Hence, more hydrogen can be generated and utilised according to the hydrogen economy model.6 The power required to spit the water into O2 and H2 will be sourced from a renewable energy source such as solar cells, making the whole process environmentally friendly. Ni and Fe oxides are good candidates for the OER as they are cheap and widely available unlike the current industrially used materials, RuO2 and IrO2.7 Jaramillo et al. has published extensive work on benchmarking OER catalysts that includes various RuO2 and IrO2 materials.8–10 In one particular study, the OER activities of commercially available nano-particulate metal oxide powders drop casted onto carbon disk electrodes are examined.10 The overpotentials achieved for the commercial RuO2 and IrO2 materials at a current density of 10 mA cm−2 in 1 M NaOH was 0.38 ± 0.02 V and 0.38 ± 0.01 V, respectively.10Electrodeposited nickel oxides have been reported to exhibit OER overpotentials between 450 and 700 mV at 10 mA cm−2, depending on the Fe impurities in the electrolyte.2,11–14 Additionally, at the same current density (10 mA cm−2), iron oxides, also from electrodeposition processes, produce O2 at overpotentials of ∼700–800 mV.1,11 When these two metal oxides are combined in various ratios, synergetic effects can be observed in the OER performance of the daughter Ni/Fe oxides. The overpotential values can decrease dramatically to 350 mV and the TOF numbers take a sharp increase in value, from 0.006–0.06 s−1 for the parent oxides to 2 s−1 for the Ni/Fe oxide hybrid.11,15
Reports into the fabrication of Fe, Ni/Fe and Ni oxides for the OER by alternative methods are sparse compared with the popular electrodeposition techniques.15,16 To the author's knowledge, a study involving hydrothermal synthesis to produce all three of the aforementioned catalysts for the OER has yet to be undertaken. Hydrothermal synthesis involves the utilisation of supercritical temperature and pressure applied to an autoclave vessel containing aqueous solutions and metal salt precursors to fabricate metal or metal oxide nano-/mirco-particles.17–19
The OER catalyst produced by a hydrothermal synthetic route can be then deposited onto electrodes through various routes for OER studies.18–20 However, in literature, the hydrothermal synthesis of pure and mixed Ni and Fe oxides for OER have been sparse and no study exists comparing all three oxide for the OER fabricated by a hydrothermal route. Subsequently, a small set of studies are present in the literature that explore one or two of these aforementioned oxides. β-Ni(OH)2 was synthesised by Zhou et al. also through a hydrothermal process which exhibited an overpotential of 0.59 V at 10 mA cm−2 when deposited on an ITO substrate.21 Hydrothermal FeOx was produced by Yan et al., in order to investigate the OER performance of this material on a carbon fibre cloth support in 1 M KOH.22 The overpotentials exhibited at 10 mA cm−2 were 0.44 V, while the Tafel slope value was 93 mV per decade.
Chen et al. produced α-Ni(OH)2 and NiFe2O4 powder by a hydrothermal synthesis and investigated the OER activity of these materials on GC supports.16 Interestingly, the α-Ni(OH)2 displayed a superior OER performance when compared to the NiFe2O4. The overpotential at 10 mA cm−2 and Tafel slope for the α-Ni(OH)2 are 0.42 V and 47 mV per decade, respectively. While the NiFe2O4 does not reach a current density of 10 mA cm−2 and the Tafel slope has increased when compared to the α-Ni(OH)2 to 96 mV per decade. This result is the complete opposite than that observed for electrodeposited Ni(OH)2 and NiFe2O4 as the latter is always seen to be the optimum catalyst, regardless of the Fe impurities in the electrolyte.5,11,13,23 This variation in results for the same materials suggests that the fabrication technique plays a critical role in the design of OER catalysts. However, currently in the literature, there is no study which synthesises and investigates the OER performances of Ni, Ni/Fe and Fe oxide by the same hydrothermal route with only changing the relevant precursor salts. Another current hot topic in electrochemical energy applications is the use of high surface area supports to enhance the properties of the OER materials.24–27 Currently, industrial alkaline electrolysis uses Ni foam as an electrode due to its high porosity, three dimensional structure and its high abundancy.28
Herein, the synthesis of Fe, Ni/Fe and Ni oxides by a hydrothermal process is reported. The resulting materials were characterised by Transmission Electron Microscopy (TEM), X-ray diffraction (XRD) and X-ray Photoelectron Spectroscopy (XPS) to determine the morphology and nature of the metal oxides. The materials were sprayed onto high surface area Ni foams for investigation into the OER. This study investigates which hydrothermal powder (Ni, Ni/Fe or Fe oxide) is the most active towards the OER on Ni foams and secondly, whether the modification of the commercially used Ni foams with the hydrothermal particles can increase the OER activity.28 If so, this would increase the overall economic value of the powder/Ni foam system compared to just the stand alone catalysts. To ensure the materials are successfully deposited onto the foams, SEM-EDX analysis will be utilized before the commencement of the electrochemical measurements. XPS will be performed after OER to determine the change in oxidation states, if any, to gain insight into the active site of each catalyst.
Experimental
The materials and reagents used in these experiments were nickel metal foam (American Elements, PN: NI-M-01-FM), sulfuric acid (Sigma Aldrich, 95–97%, analytical grade), nickel sulfate (Sigma Aldrich, ≥99% metal basis, M 280.86 g mol−1), iron sulfate (Sigma Aldrich, >99% metal basis, M 278.01 g mol−1), mercury–mercuric oxide (Hg/HgO) reference electrode (CH instruments, cat no. 152), ethylenediaminetetraacetic acid (EDTA) (Sigma Aldrich), sodium hydroxide pellets (Sigma-Aldrich, ≥98%, reagent-grade), carbon tabs (Agar Scientific), copper tape (Agar Scientific), holey carbon/copper grids (S147, Agar Scientific), butanol (Sigma Aldrich, ≥99%, reagent-grade), Nafion (Sigma Aldrich), nickel wire (Sigma Aldrich) and Si wafers (SiMat, dopant P/boron, Si oxide layer 300 nm).The metal oxide powders were synthesised through a hydrothermal method. The method was adapted by a previously reported hydrothermal method for NiFe2O4.20 First, 0.2 g of EDTA was mixed with 20 ml of water. Then the relevant precursor metal salt was added into the EDTA/water and stirred until the salt was dissolved. The solution's pH was adjusted to pH 14 and poured into a Teflon lined autoclave. The autoclave was then sealed and placed inside an oven. The oven was ramped up to 160 °C and kept at this temperature for 16 hours. After this time, the oven was allowed to cool down to room temperature by natural convection. After synthesis, the resulting material was washed by centrifuging with water and then ethanol, three times. Then the material was allowed to dry in an oven for 1 hour at 100 °C.
The material ink was prepared by mixing Nafion, ethanol and water in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :150:100 with 5 mg of each catalyst. The resulting ink was sonicated for 30 minutes and sprayed onto Ni foam electrodes. The support electrodes were prepared by cutting a 1 × 1 cm2 piece of nickel foam. A nickel wire was inserted into the foam to create a contact, which was sealed by Araldite. The electrodes were washed in a sonic bath in 3 M HCl for 5 min to remove any oxidised species present at the surface. A second wash was carried out in DI H2O for 5 min. Finally, the electrode was then dried under N2 flow. A volume of 2 ml of the relevant oxide material was deposited onto a Ni foam electrode using a spray gun. The weight of the Ni foam/Ni wire electrode was measured before and after the deposition of the relevant hydrothermal powder. The mass loading of the hydrothermal powders onto the electrodes were approximately 0.3–0.4 mg.
:150:100 with 5 mg of each catalyst. The resulting ink was sonicated for 30 minutes and sprayed onto Ni foam electrodes. The support electrodes were prepared by cutting a 1 × 1 cm2 piece of nickel foam. A nickel wire was inserted into the foam to create a contact, which was sealed by Araldite. The electrodes were washed in a sonic bath in 3 M HCl for 5 min to remove any oxidised species present at the surface. A second wash was carried out in DI H2O for 5 min. Finally, the electrode was then dried under N2 flow. A volume of 2 ml of the relevant oxide material was deposited onto a Ni foam electrode using a spray gun. The weight of the Ni foam/Ni wire electrode was measured before and after the deposition of the relevant hydrothermal powder. The mass loading of the hydrothermal powders onto the electrodes were approximately 0.3–0.4 mg.
Transmission Electron Microscopy (TEM) and Energy Dispersive X-ray (EDX) spectroscopy was performed on a FEI Titan 80–300 kV S/TEM using an EDAX EDX 30 mm2 detector using holey carbon grids. Scanning Electron Microscopy (SEM) was used to confirm the presence of the powders on the Ni foams using a Karl Zeiss Ultra Field Emission SEM at an accelerating voltage between 5 kV at a working distance between 1–5 mm. Elemental analysis on the Ni foams was also carried out using a 20 mm2 EDX detector from Oxford Instruments. The working distance for EDX was between 7.7–8.3 mm. Surface topography measurements were carried out via Atomic Force Microscopy (AFM, Asylum Research) and were performed in tapping mode with a frequency of 1 Hz and 512 scan lines using a silicon cantilever with Au conductive coating (NT-MDT) with a spring constant of 1.45–15.1 N m−1. Powder X-ray diffraction (XRD) was carried out using a Bruker Duo with a Cu Kα source (λ = 1.5418 Å). The reference patterns were matched to the experimental data in the Match! Software. XPS measurements were carried out using a VG Scientific ESCALab MKII system using an Al Kα X-ray source (1486.7 eV). The sample spot size was approximately 2 mm to allow for a large sample area. The analyser pass energy for the survey scans was 200 eV while a pass energy of 20 eV was used to obtain high resolution spectra of characteristic core levels, Ni 2p, Fe 2p, O 1s and C 1s. The binding energy scale was referenced to the C 1s peak of adventitious carbon at 284.8 eV. Casa XPS software was used to fit the high resolution core level peaks. All Raman spectroscopy measurements were performed using a Witec alpha 300R confocal Raman microscope using a 1800 line per mm grating and a 532 nm diode laser at an incident power of <10 mW and a spot size of 300 nm. BET isotherms were acquired in a Quantachrome Nova 4200e station using nitrogen at 77 K. The specific surface area was obtained by applying the BET equation to the linear portion of the isotherm curves. All samples were outgassed in nitrogen atmosphere at 150 °C for 16 hours before measurements.
All electrochemical experiments were undertaken in a standard three-electrode cell using a high performance digital potentiostat (CH model 1760 D Bi-potentiostat system monitored using CH1760D electrochemical workstation beta software). The working electrodes consisted of the hydrothermal powders on Ni foam. A graphite rod was employed as a counter-electrode and a mercury–mercuric oxide (Hg/HgO) reference electrode was used as a reference. Electrochemical measurements were taken at a constant temperature of 25 °C, using a thermal bath with the temperature maintained by a thermostat. All solutions were degassed with N2 for 15 minutes before commencing analysis to remove any dissolved oxygen present in the electrolyte. Linear sweep voltammetry (LSV) measurements were performed at a sweep rate of 5 mV s−1 in the forward oxidation direction between the potential limits of 0.0 V and 1.2 V vs. Hg/HgO. The stability tests were conducted by applying a current of 10 mA cm−2 to each of the catalysts for a period of time and measuring the corresponding potential. The turnover frequency (TOF) was calculated using the following equation:
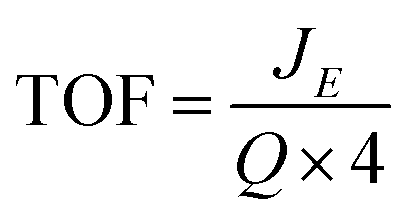
Results and discussion
The hydrothermal fabrication route yield Ni oxide, NiFe oxide and Fe oxide powders which were green, brown and dark brown in colour, respectively. After the synthesis of the Ni, Ni/Fe and Fe oxides by the EDTA-assisted hydrothermal route, various techniques were used to characterise the powders prior to any OER measurements. TEM-EDX was utilised to investigate the morphology and shape diameters of the hydrothermal materials fabricated, Fig. 1 and S1–S3.† AFM was utilised to obtain the height profile of the single hydrothermal particles, Fig. S4.† To help identify the structural and chemical composition of the metal oxides synthesised XRD, Fig. 2, Raman spectroscopy, Fig. S5,† and XPS, Fig. 3 and S6,† were undertaken. | ||
| Fig. 1 Transmission electron microscopy of (a) and (b) Fe, (c) and (d) Ni/Fe and (e) and (f) Ni oxide particles (black scale bar = 50 nm and pink scale bar = 20 nm). | ||
 | ||
| Fig. 2 X-ray diffraction patterns of (a) Fe oxide (b) Ni oxide (c) Ni/Fe oxide and (d) comparison XRD pattern of the three oxides. | ||
 | ||
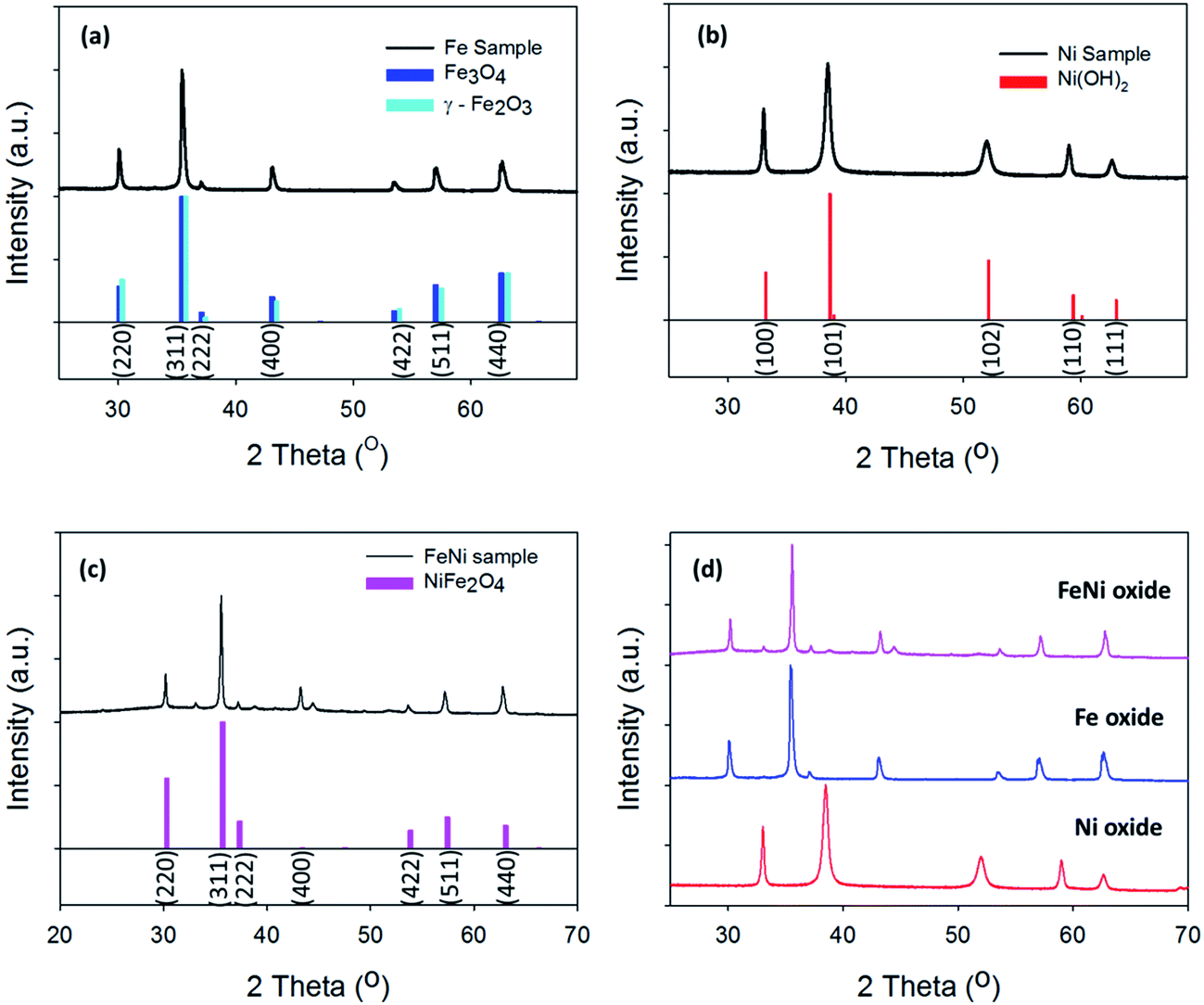
| Fig. 3 X-ray photoelectron analysis before OER of (a) Fe 2p3/2 core level for the Fe oxide sample (cyan = Fe2O3, blue = Fe3O4) (b) Fe 2p3/2 core level for the Ni/Fe oxide sample (pink = NiFe2O4) (c) Ni 2p3/2 core level for the Ni oxide sample (green = Ni metal, red = Ni(OH)2). | ||
Physical characterisation
The morphology of the resulting Ni/Fe powders was investigated through TEM. Fig. 1 shows typical TEM images for the Fe, Ni/Fe and Ni oxide particles synthesised in this work on holey carbon grids. For the pure Fe, Fig. 1(a) and (b) and Ni oxides, Fig. 1(e) and (d), the particle shapes for these materials are round and hexagonal, respectively. The EDX analysis confirms the presence of only Fe and Ni in the relevant samples and no cross contamination is evident, Fig. S1 and S2.† Interestingly, for the mixed Ni/Fe oxide a plethora of particle morphologies are formed, including, hexagonal, square, rod and round, Fig. 1(c) and (d). The contribution of both Fe and Ni are detected by EDX in the mixed sample, Fig. S3.†Additionally, information about the size distribution of the particles was also extracted from the TEM images by using the ImageJ software. The particle sizes calculated were 41 ± 6 nm, 200 ± 108 nm and 105 ± 20 nm for the Fe, Ni/Fe and Ni oxides, respectively, Table S1.† The wide variety of particle shapes for the mixed Ni/Fe oxide may be attributed to the larger range in particle size, compared to the pure Fe and Ni oxide particles, which exhibit a tighter size distribution and appear greater homogeneity from the TEM images.
Fig. S4† shows the AFM topography image and respective 3D profile of individual Fe, Ni/Fe and Ni oxide particles. The average height profiles of the Fe, Ni/Fe and Ni particles after hydrothermal synthesis on Ti supports can be observed in Table S2.† The average height of the Fe, Ni/Fe and Ni oxides nano-materials are 3 ± 0.6, 21 ± 3 and 20.8 ± 5.3, respectively. The average height profile indicates that the introduction of Ni salt induces a bigger grain salt and consequently a higher height profile.
Fig. 2(a)–(c) shows the XRD graphs for the hydrothermal Fe, Ni and Ni/Fe oxide powders, respectively. Subsequently, a comparison graph of the XRD patterns for the three powders is shown in Fig. 2(d). Powder XRD patterns obtained for the pure and mixed Ni/Fe oxide powders are in good agreement with the reference patterns. The XRD for the pure Fe oxide material reveals that the particles may consist of both the Fe3O4 and the γ-Fe2O3 phases, Fig. 2(a).29 The XRD peaks positioned at the 2θ values of 30.01°, 35.41°, 37.09°, 43.05°, 53.40°, 57.20° and 62.61° can be indexed to both the Fe3O4 and γ-Fe2O3 (COD no: 96-901-3530 and 96-900-6317) planes of (220), (311), (222), (400), (422), (511) and (440), respectively. The hydrothermal pure Ni oxide sample matches the hexagonal Ni(OH)2 phase (COD no. 96-101-1135). The Miller indices of the hexagonal Ni(OH)2 phase, which are (100), (101), (102), (110) and (111) can be observed in the PXRD of the Ni oxide material at the 2θ values of 33.36°, 38.67°, 52.16°, 59.36° and 63.01°, Fig. 2(b).
The XRD pattern for the mixed hydrothermal prepared Ni/Fe oxide can be observed in Fig. 2(c). The Ni/Fe oxide can be indexed to the structure of NiFe2O4 phase. The diffraction peaks for the Ni/Fe material at the 2θ values of 30.01°, 35.41°, 37.09°, 43.05°, 53.40°, 57.20° and 62.61° correspond to the NiFe2O4 (COD no. 96-230-0296) lattice planes of (220), (311), (222), (400), (422), (511) and (440), respectively. The addition of the diffraction peak at the 2θ value of ∼44.30° can also be observed in previous XRD patterns reported in the literature for hydrothermal prepared NiFe2O4, which may only be visible in highly crystalline NiFe2O4.30 However, the small peaks at the 2θ values of ∼33° and ∼38° are not indexed to NiFe2O4 and may correspond to planes in Ni(OH)2, which would indicate that this material may have Ni(OH)2 residues. Of general note is the highly crystalline nature of the materials, as evidenced by the sharp and intense peaks in the XRD pattern. This result is in direct contrast with electrodeposited materials reported previously in the literature. Trotochaud et al. utilised XRD analysis for mixed Ni/Fe oxide electrodeposited samples and the XRD patterns exhibit greater noise and broader diffraction peaks, which indicates a more amorphous structure when compared to the Ni/Fe oxides in this study.13
Additional confirmation that the mixed Ni/Fe oxide material synthesised though the hydrothermal technique was indeed NiFe2O4, was achieved by Raman spectroscopy, Fig. S5.† Ni(OH)2 and NiFe2O4 have very distinct Raman modes, therefore if Ni(OH)2 was present it would be easily detected by this technique.11,14,31,32 The Ni(OH)2 species has one peak at approximately 465 cm−1 which corresponds to the Eg(R) mode from the Ni–OH bond stretching.33,34 The blue shift in this peak is due to impurities/disorder in the lattice causing a slight alteration in the Ni–O bonds. This shift is only by ∼65–70 cm−1, at most.1 Typical Raman peaks associated with NiFe2O4 are found at 207, 355, 483, 574 and 706 cm−1 which correspond to the Raman active vibrational modes of T2g, Eg, T2g, T2g and A1g, respectively.35,36
Fig. S5† shows the Raman spectrum for the hydrothermal Ni/Fe oxide material. From the Raman analysis, the hydrothermal Ni/Fe oxide exhibits characteristic peaks of NiFe2O4 with no peaks corresponding to normal or disordered Ni(OH)2. The detection of the Raman modes associated with NiFe2O4 and not Ni(OH)2 indicates that the mixed Ni/Fe oxide films is composed mainly of NiFe2O4.
To further identify the composition and oxidation states of the hydrothermal materials XPS was utilised. In Fig. S6,† the survey scans for all three hydrothermally prepared materials are presented. The Ni 2p core level can be observed in both the Ni and Ni/Fe oxide at a binding energy of ∼860 eV, while this core level is absent in the Fe oxide survey spectra.3,37 Subsequently, the Fe 2p core is present in the Fe and Ni/Fe oxide core levels at ∼710 eV, while this core level is not observed in the Ni oxide survey spectra.37 This is also an indication there has been no cross contamination between samples during synthesis.
From the XRD analysis of the Fe oxide, Fig. 2(a), this material's composition is still not fully determined. The XRD indicates that the Fe oxide is either made up of Fe3O4, Fe2O3 or a mixture of the two. From the high resolution core level analysis of the Fe 2p3/2 peak, both the Fe3O4 and the γ-Fe2O3 phases can be detected on the surface of the Fe oxide sample, Fig. 3(a). The Fe 2p3/2 peak of the hydrothermal Fe oxide was fitted with contributions correlating to both Fe3+ (Fe2O3) and to Fe2+ (Fe3O4), Fig. 3(a).38 The Ni/Fe oxide hydrothermal material, Fig. 3(b), displays the fitting of the Fe 2p3/2 core level to a NiFe2O4 multiplet set with no evidence of contributions relating to any other Fe species. The Ni species, synthesised by this hydrothermal process, determined by the XRD is in agreement with the XPS analysis, Fig. 2(b). The Ni 2p3/2 core-level of the Ni oxide was fitted to a Ni2+ (Ni(OH)2) multiplet set, with only very minor contributions from Ni0 (Ni metal) and no evidence of higher oxidation states, Fig. 3(c).39 The exact percentages of each oxidation state found on the surface of the different catalysts can be seen in Table S3.† Brunauer–Emmett–Teller measurements of the powders were untaken to calculate the BET surface area of the powders, Fig. S7.† In Fig. S7(a)† the BET isotherms for Ni(OH)2 can be observed. This curves resembles a type IV isotherm, suggesting that the material under analysis is a mesoporous (2–50 nm according to IUPAC) adsorbent.40 A multipoint BET analysis resulted in a surface area of 91.08 m2 g−1 for Ni(OH)2. In comparison, the NiFe2O4 and Fe2O3/Fe3O4 samples exhibit much lower surface areas when compared to the Ni(OH)2 material of 34.35 and 20.45, respectively. Subsequently, the pure iron oxide (Fe2O3/Fe3O4) presents the typical shape of a type III isotherm, which is characteristic of non-porous materials where the interactions between adsorbate–adsorbent are very weak in comparison with the adsorbant–adsorbant interactions.41
The OER activity of the hydrothermal powders fabricated was investigated using Ni foam substrates. As previous mentioned, the powders were loaded on to the foams using a spray gun, Fig. 4(a) and S8.† To ensure the adhesion of the powders to the foams, SEM-EDX imaging was performed before any electrochemical analysis, Fig. S9.† In the SEM images, Fig. S9(a)–(c),† Ni(OH)2 powder can be observed distributed evenly on the Ni foam substrate. From Fig. S9(b),† little or no aggregation of the powders is seen to occur on the Ni foam. EDX mapping analysis of the Ni(OH)2 on the metal Ni foam was also undertaken, Fig. S10,† with the Ni Kα map showing a high intensity of elemental Ni. The position of the Ni(OH)2 particles can be observed in the O Kα map as the metallic Ni foam does not have a large elemental oxygen signal. Thus, the areas with a clear oxygen signal correspond to Ni(OH)2 particles.
The XPS, Fig. 3, and XRD, Fig. 2, analysis of Ni/Fe oxide indicate that NiFe2O4 is present and the SEM images of this material sprayed onto the Ni foam can be observed in Fig. S9(d)–(f).† SEM imaging of the Ni/Fe oxide particles on Ni foam reveals a range of sizes and shapes for this mixed material, Fig. S9(d)–(f).† This is consistent with the TEM images in Fig. 1(c) and (d) showing a wide size distribution. Additionally, it is clear from Fig. S9(d) and (e)† that Ni/Fe oxide powders aggregate on the Ni foam substrate. Again, EDX mapping was utilised to confirm the presence of the Ni/Fe oxide on the foam. Unlike the evenly spread Ni(OH)2, the mixed Ni/Fe sample seems to aggregate on the substrate. The Fe and O Kα maps, Fig. S11,† show the Ni/Fe oxide material is dispersed throughout the foam.
SEM analysis was also performed after the deposition of the hydrothermal Fe oxide onto the Ni foam in order to ensure the powder was successfully deposited. The SEM imaging of the Fe oxide on the Ni foam is presented in Fig. S9(g)–(i).† The SEM image, Fig. S9(h),† shows that the Fe oxide is homogenously positioned on the foam. In Fig. S12,† the EDX mapping of the hydrothermal Fe oxides can be observed. The Ni Kα map only highlights the Ni Foam but from the Fe Kα map the position of the Fe oxide particles on the foam can be seen. The Fe Kα map also shows the homogeneous nature of the Fe particles alongside the SEM analysis in Fig. S8.† No O Kα map was recorded to show the position of the Fe oxide as there is no Fe in the Ni foam substrate; directly mapping the Fe Kα peak intensity is sufficient to determine the location of the Fe oxide particles. SEM analysis was also untaken on the bare Ni foams to ensure no particles were present before deposition of the hydrothermal materials. Fig. S13† shows that no particles can be observed on the bare Ni foam and, consequently, all particles seen in the SEM Fig. S9,† and EDX imaging, Fig. S10–S12,† are indeed the hydrothermal oxide materials.
OER performance
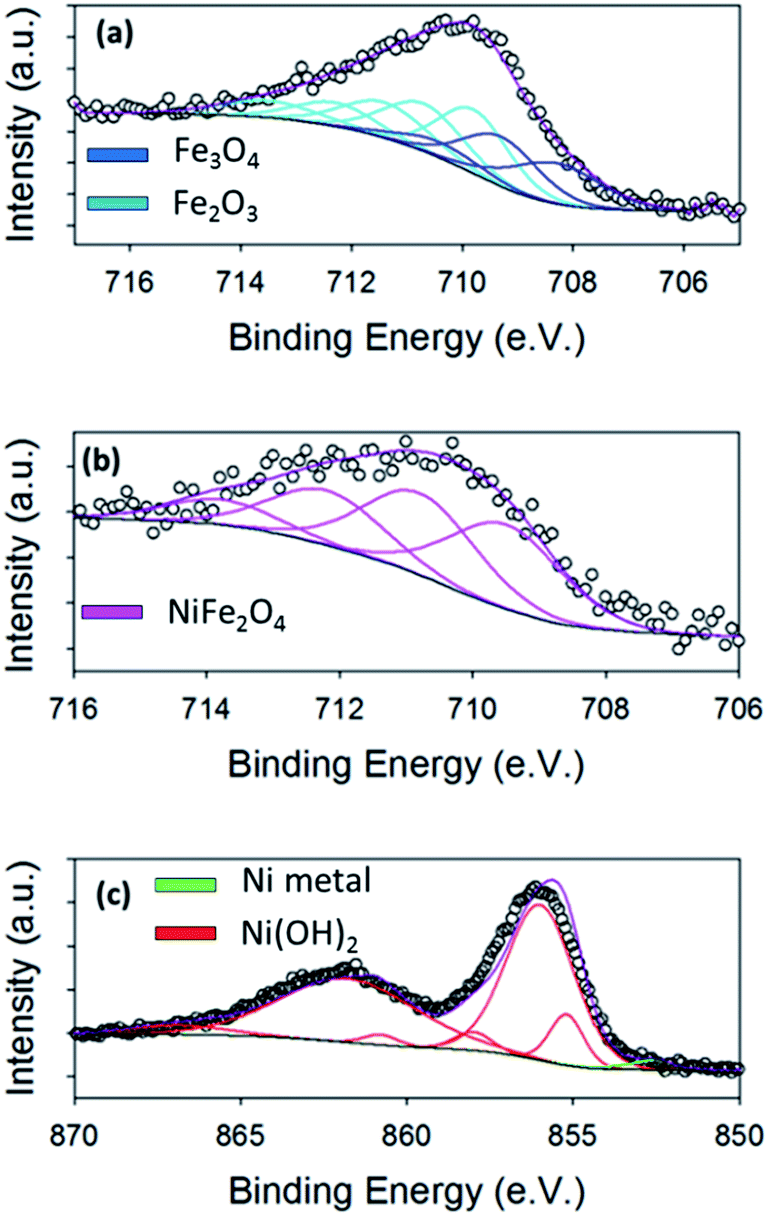
After SEM, the OER performances of the bare Ni foam and the foam modified with the hydrothermal nano-materials were examined. The Ni foam substrate itself is active towards the OER; as it reaches an overpotential value of 0.421 V at 10 mA cm−2, Fig. 4(b). Interestingly, this overpotential value is, in fact, lower than overpotentials achieved by electrodeposited Ni oxide on various planar substrates and by hydrothermally prepared NiFe2O4 on 3D glassy carbon supports.1,14,42 Hence, as already stated, the use of Ni foams are used in commercial electrolysers as catalysts in their own right.28 This observation raises the question; would doping or modification of the Ni foams be advantageous for the OER? Or, are the Ni foams a better OER catalysts with no modification?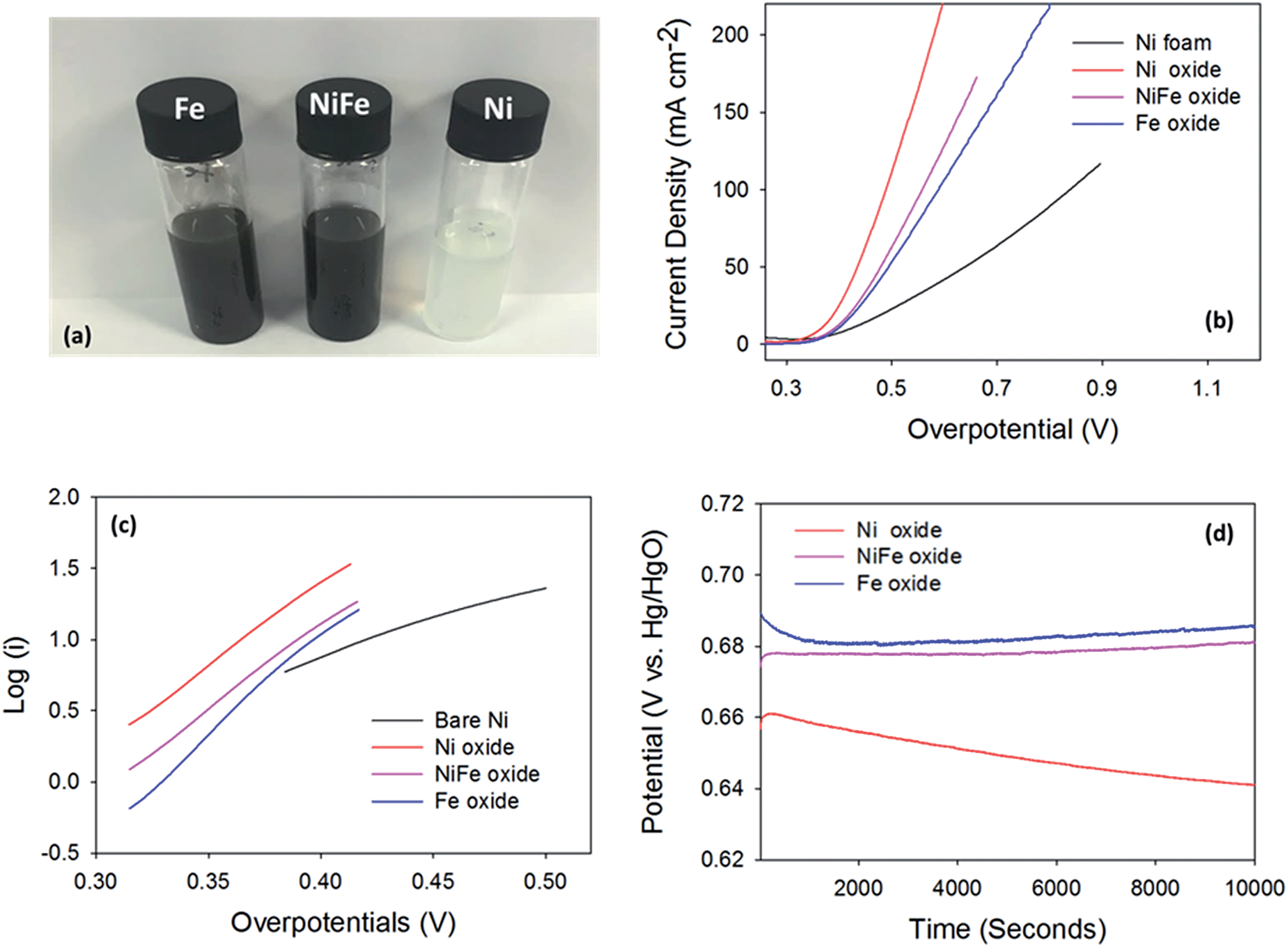 | ||
| Fig. 4 (a) Photograph of the Fe, Ni/Fe and Ni oxide dispersions used to spray onto the Ni foams. (b) OER polarisation curves, (c) Tafel curves and (d) stability tests of the Ni, Ni/Fe, Fe and oxide catalysts in 1 M NaOH electrolyte. | ||
Currently in literature, as previously mentioned, electrodeposited nickel ferrite (NiFe2O4) has an improved OER performance when compared to electrodeposited nickel or iron oxides, achieving typical OER overpotentials of 350 mV at 10 mA cm−2.11 Nickel hydroxide (Ni(OH)2) has been reported to exhibit overpotentials between 450–700 mV at a current density of 10 mA cm−2, while Fe2O3 evolves oxygen at an overpotential of ∼700 mV, at the same current density. In this study, the overpotential achieved by the bare Ni foam in 1 M NaOH (0.421 V at 10 mA cm−2) is, in fact, already superior to previously studied electrodeposited Ni and Fe oxides. In a study by Chen et al., hydrothermal Ni(OH)2 and NiFe2O4 particles were synthesised and deposited on to glassy carbon substrates.16 This work yielded an interesting observation that the hydrothermally prepared Ni(OH)2 exhibited an overpotential of 420 mV at a current density of 10 mA cm−2, which was superior to that of NiFe2O4. Consequently, the NiFe2O4 can be deemed a less active catalyst than the Ni(OH)2, when produced through hydrothermal methods.
The water oxidation capabilities of the nano-size hydrothermal oxides deposited on Ni foams, in this study, were investigated to determine if the hydrothermally prepared Ni/Fe oxide is a superior OER catalyst to the parent oxides, like electrodeposited films, and if the Ni foam is a good support for these materials i.e. if the overpotentials at the fixed current density of 10 mA cm−2 decreases to lower values towards the thermodynamic potential for OER. The OER performance of the hydrothermal materials on Ni foams are shown in Fig. 4(b)–(d).
It is immediately apparent that all three oxide particle types on the Ni foam show enhanced OER performance than the bare Ni foam, Fig. 4(b). Therefore the modification of the Ni foam, which is commercially used as an anodic material in electrolyzers, does enhance the OER activity of the Ni foam. Interestingly, for these hydrothermally prepared catalysts, the Ni oxide on the Ni foam had an improved OER activity when compared to the Ni/Fe and Fe oxide powders on the same Ni foam substrate. The Ni, Ni/Fe and Fe oxides on Ni foam exhibit overpotentials of 0.360 ± 0.004 V, 0.377 ± 0.01 V and 0.390 ± 0.002 V at 10 mA cm−2, respectively. The overpotentials at 10 mA cm−2 achieved for the Ni and NiFe oxide samples are lower when compared to the commercialised IrO2 and RuO2 catalysts investigated by Jaramillo et al.10 The enhancement of the pure Ni oxide (Ni(OH)2) over the mixed NiFe oxide (NiFe2O4) and the pure Fe oxide (Fe2O3/Fe3O4) could be related to the Ni(OH)2 exhibiting the largest BET surface area, Fig. S7.† In fact the exact trend for the OER observed by these three materials is mirrored in the BET results.
High current densities were achieved by these catalysts, reaching approximate values of 64, 34 and 29 mA cm−2 at an overpotential of just 0.45 V for the Ni, Ni/Fe and Fe oxides, respectively. The OER activity for the pure Ni(OH)2 is extremely encouraging when compared to other Ni oxide modified Ni foam electrodes reported in literature. For example, Han et al. prepared NiO on Ni foam by an electrochemical deposition method and achieved an overpotential of 0.44 V at 10 mA cm−2.43 When compared to other literature values for Fe oxides, this hydrothermal Fe oxide achieved one of the best overpotentials.1,44 For example, Louie et al., reported that for electrodeposited Fe oxide the exhibited overpotential was 550 mV at 10 mA cm−2.1 This may be due to the increased crystallinity of the Fe oxide, seen by XRD and TEM, compared with amorphous electrodeposited Fe oxide films.45 The Fe oxide in this study is also a superior OER catalysts when compared to the hydrothermal FeOx reported by Yan et al. The Fe oxide in this study evolves O2 at overpotentials values of ∼50 mV lower than that of the FeOx produced by Yan et al.22
Tafel analysis was also performed on the hydrothermal materials in this study, Fig. 4(c). From the Tafel slopes, mechanistic information about the rate determining step for each of the materials can be extracted. The Tafel slope is a measure of the sensitivity of the OER rate with respect to change in OER driving force (the applied potential), therefore the lower the slope value, the better the catalyst is at evolving O2. From Fig. 4(c), the Tafel slope calculated for the bare Ni foam is 240 mV per decade. Theoretically the Tafel slope for an electron transfer reaction proceeding at a porous electrode is exactly twice that at a planar electrode, which is 120 mV per decade. Hence a Tafel slope of 240 mV for a porous electrode corresponds to 120 at a planar electrode which corresponds to the first electron transfer step being rate determining for the bare unmodified Ni foam.46–49
The Tafel slopes calculated for the hydrothermal Ni, Ni/Fe and Fe oxide catalysts are 75, 80 and 65 mV per decade, respectively. The numerical value of the Tafel slope calculated for a material can reveal mechanistic information about the rate determining step for OER. A Tafel slope of ∼60 mV per decade indicates that a chemical reaction is the rate determining step leading to oxygen evolution for all of these hydrothermal materials. Even more significantly, the low Tafel slopes observed for the hydrothermal nano-materials on the porous Ni if divided by 1/2 will be even lower indicating that the OER activity is even more effective when compared to theoretical theory. Hence, the modifying of the Ni foams significantly improves the Tafel slope value.
Finally, the stability of the hydrothermal materials was investigated using chronoamperometry over a period of 10000 seconds, Fig. 4(d). The Ni/Fe and the Fe oxide electrodes show great stability over this time range as no increase in potential occurred, which indicates the active material was not physically breaking off the electrode or corroding. The Ni(OH)2 material on the foam exhibits a decrease in potential over time. This results suggests that the Ni(OH)2 material is becoming more active for the OER with time. The increase in activity may be due to the Fe impurities in the NaOH electrolyte modifying the catalyst surface with increased time under OER conditions or the β-Ni(OH)2 is being generated over long polarisation.1,11,50
Turnover frequency (TOF) numbers are regularly used to compare OER catalysts in literature.5,47–49 Typical TOF values previously reported for electrodeposited pure nickel and iron oxides are between 0.065–0.006 s−1 and 0.0015–0.0009 s−1, respectively.11,44 In Fig. 5, the TOF values for the hydrothermal material in this study can be observed. The TOF values obtained in this study for the Ni and Fe oxides are 0.283 ± 0.008 and 0.10 ± 0.04 s−1, respectively. The TOF values for the hydrothermally prepared Ni(OH)2 and Fe3O4/Fe2O3 are higher than those for the same materials produced by electrochemical techniques.11 The individual hydrothermal particles on the Ni foam could provide a larger surface area for the OER to occur than a continuous film produced by electrodeposition.
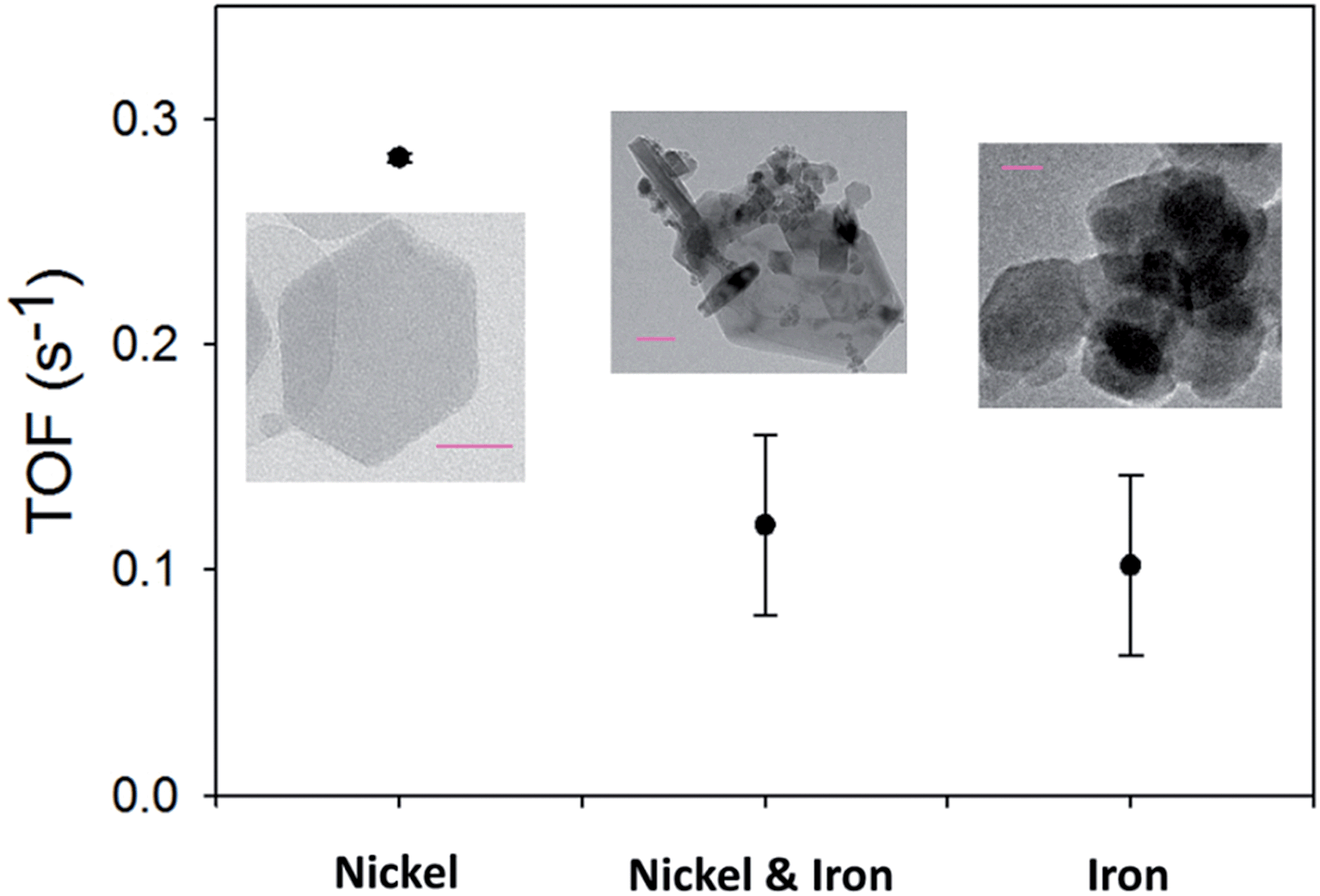 | ||
| Fig. 5 TOF values for the hydrothermal materials. | ||
Typical TOF values reported for mixed Ni/Fe oxides are higher than the TOF values for the parent oxides (nickel or iron oxide) and can achieve values up to 2–3 s−1. TOF values for hydrothermally prepared Ni/Fe oxide materials have yet to be reported. The TOF value for the Ni/Fe oxide, in this study, is 0.120 ± 0.04 s−1, which is lower than the Ni(OH)2 and similar to the Fe3O4/Fe2O3. This is not a surprising result, as the overpotentials, in this study, also reveal that the Ni(OH)2 on the Ni foam to be a superior catalyst to the Ni/Fe and Fe oxides.
The modification of the Ni foam, which is already used as a commercial catalyst, improved the OER performance compared to the bare Ni foam. All three hydrothermal materials synthesised in this study greatly enhanced the Ni foam's OER capability. The Ni(OH)2/Ni foam system exhibited the lowest overpotentials at 10 mA cm−2 and the stability test shows that this value improved with time.
Additionally, pre-treatments of these materials i.e. ultrasonic irradiation or liquid phase exfoliation for layered metal oxides, could further enhance the OER performance of these materials.51 These aforementioned techniques are being used to create 2D materials from their bulk counterparts, which have recently been shown to have superior electrochemical properties than their bulk counterparts.15,52,53
Another important aspect of OER catalysts is to determine the active site for oxygen evolution or a change in species on the surface. To investigate the species/oxidation state of our hydrothermal materials after OER, high resolution XPS was carried out on planar Ti substrates so-as to eliminate any signal from the Ni foam which would interfere with the signal from the hydrothermal materials. The resulting XPS graphs can be observed in Fig. 6.
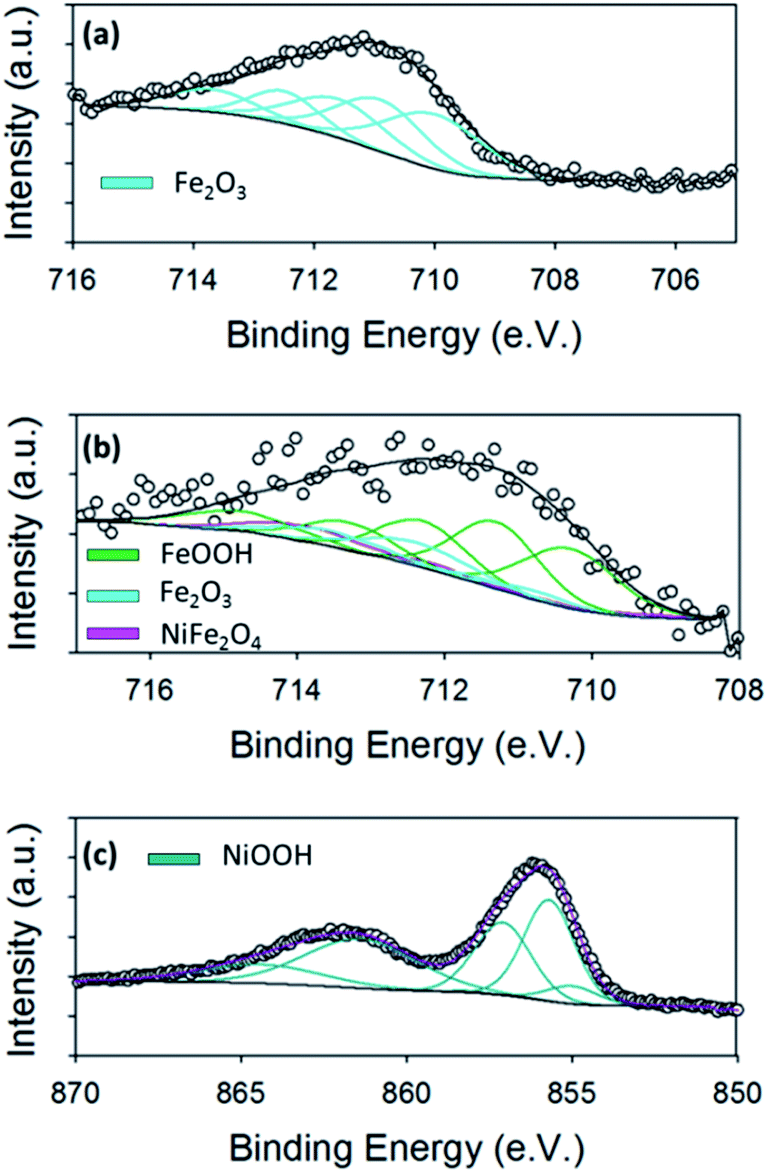 | ||
| Fig. 6 XPS analysis of (a) Fe 2p3/2 core level for the Fe oxide sample (cyan = Fe2O3) (b) Fe 2p3/2 core level for the Ni/Fe oxide sample (pink = NiFe2O4, cyan = Fe2O3, green = FeOOH) (c) Ni 2p3/2 core level for the Ni oxide sample (dark cyan = NiOOH). All spectra were taken after OER measurements had been carried out. | ||
Fig. 6(a) shows the high resolution Fe 2p3/2 core level for the hydrothermal Fe oxide. It is evident from the Fe 2p3/2 core level of the Fe material before, Fig. 3(a), and after the OER, Fig. 6(a), that the oxidation of the Fe oxide has occurred from increase in the binding energy. To determine the exact nature of the Fe oxide after OER, known parameters from literature were fitted to the spectrum.38 The Fe 2p3/2 peak for the Fe oxide was fitted with contributions correlating to only Fe2O3 (Fe3+). These observations may suggest that the Fe2O3 may be the active site for the OER in the hydrothermal Fe oxide, in this study, which is consistent with literature findings.54
The ex situ XPS was also used to determine the surface chemistry of the Ni/Fe oxide after OER and if the oxidation states differed from that previously reported in the literature for electrodeposited Ni/Fe oxide.11 The Fe 2p3/2 core level for the Ni/Fe oxide after OER can be observed in Fig. 6(b). The fitting parameters reveal that this sample, after OER, consists of contributions from NiFe2O4, Fe2O3 and FeOOH.38 From a previous paper by the group on electrodeposited Ni/Fe oxide under the exact same conditions, the best electrodeposited Ni/Fe oxide catalyst reveals that after OER the surface composition of NiFe2O4 is 90.5%, while the value for this species for the hydrothermal Ni/Fe oxide is 9.5%, Table S4.†11 This major decrease in NiFe2O4 for the hydrothermal Ni/Fe oxide may account for the slight decrease in OER performance when compared to its electrochemical fabricated counterpart. The NiFe2O4 may corrode or break down into Fe2O3 and FeOOH at higher potentials, which are detected by XPS, Fig. 6(b). This iron oxide species are known to be not as active as NiFe2O4 for oxygen evolution.1,23
XPS was performed for the hydrothermal Ni oxide in order to determine if oxidation of the Ni occurred. Fig. 6(c) shows the for Ni 2p3/2 core level for the Ni oxide sample. The fitting of this core level is known to be rather complex. The fitting of the Ni 2p3/2 core level was accomplished by using parameters established by Grosvenor et al.39,55 These curve fitting parameters were determined from the combination of a multitude of different resources; including using reference samples to acquire quality spectra, theoretical modelling and using other known literature fitting procedures.37–39 The fitting of the Ni oxide material after OER revealed that the predominate Ni species is NiOOH. From literature, NiOOH is reported to be the active site for OER for Ni oxides.56,57 The enhanced OER activity observed by NiOOH is thought to be generated from the increased crystallinity of the lattice when is it oxidised from Ni2+ to Ni3+.56 The crystallinity may either be induced by the dehydration of the Ni(OH)2 when polarised in base due to or by the insertion of Fe impurities from the base into the Ni oxide lattice, however the exact mechanism for the generation of the NiOOH active site is not known.11,58 Of course, if the NiOOH is produced from an exchange of Fe ion impurities in the lattice then the formula of the active site would be Ni−xFe+xO(H).
Regardless of the mechanism undertaken to generate O2 at the active site, the results show that the hydrothermal Ni oxide on Ni foam is a superior dopant in comparison to the hydrothermal Ni/Fe or Fe oxide also produced in this study. Subsequently, the Ni oxide significantly improves the OER capabilities of the already commercially used Ni foam at a cheap cost.
Conclusion
In summary, various metal oxide powders were synthesised through a hydrothermal method. The powders were characterised by TEM, XRD, Raman spectroscopy and XPS. The composition of the three powders were determined to be Ni(OH)2, NiFe2O4 and Fe3O4/Fe2O3 for pure Ni, mixed Ni/Fe and pure Fe oxides, respectively. TEM imaging of these materials revealed the Ni(OH)2 and Fe3O4/Fe2O3 powders to be hexagonal and round in shape, respectively; while the mixed NiFe2O4 material had a varied morphology. Furthermore, the Ni(OH)2 and Fe3O4/Fe2O3 nanoparticles had a much tighter size distribution than the NiFe2O4.The OER performances of the hydrothermal powders on Ni foams were evaluated by examining the overpotential required to generate a current of 10 mA cm−2, Tafel slopes, stability and TOF numbers. Ni foams were used as the underlying support as this material is currently used as a water oxidation catalyst in commercial electrolysis. The modification of the Ni foam with our hydrothermally prepared powders allowed us to investigate whether such a simple technique is viable to enhance the OER performance of a commercial catalyst.
The Ni(OH)2 modified Ni foam exhibited the best values for overpotential at 10 mA cm−2, TOF values and Tafel slopes when compared to the NiFe2O4 and Fe3O4/Fe2O3 modified Ni foam and the bare Ni foam. More interestingly, from the stability tests, it can be seen that the overpotential at the fixed current density of 10 mA cm−2 improves with time. The BET results show that the material with the highest specific area is the pure Ni oxide followed by the mixed NiFe oxide and finally the Fe oxide. This trend is consistent with the OER results which indicates that the Ni oxide has more active sites.
The enhancement in the Ni(OH)2 was also deemed to be due to the formation of NiOOH during OER, as revealed by XPS. NiOOH has been reported in literature to be the active site for OER in Ni oxides. Mixed Ni/Fe oxides produced via electrochemical means typically outperform Ni(OH)2 as OER catalysts. However, the hydrothermal NiFe2O4 fabricated in this study was found to exhibit inferior OER performance than the Ni(OH)2. This was explained due to the breakdown of the NiFe2O4 into FeOOH and Fe2O3 during OER, as shown by XPS, which are less active catalysts for the OER. Finally, although the hydrothermal Fe oxide did not exhibit the best OER activity within this study, this material achieved one of the best overpotentials and TOF numbers when compared to other literature values for hydrothermal Fe oxides. This may be due to the increased crystallinity and uniformity of the Fe oxide nanoparticles, as evidenced by XRD and TEM, compared to the more varied Fe oxides formed via electrodeposition.
In conclusion, it was demonstrated that a commercial OER catalyst such as Ni foam can be readily modified with metal oxide nanoparticles to enhance the performance of the electrode. The active sites for each metal oxide system in this study were identified and related to the observed trend in OER activity.
Author contributions
The manuscript was written by M. P. B. All authors have given approval to the final version of the manuscript. M. P. B. did the synthesis, electrochemical analysis, XPS, SEM-EDX, and XRD. J. M. V. did the AFM analysis. E. M. C. did the TEM/EDX. M. O. B. did the Raman analysis. A. A. S. sprayed the powders onto the Ni foams. M. P. B. also did all the data analysis/treatment for the electrochemical analysis, XPS, XRD, Raman and the EDX. J. M. V. did the data analysis for the AFM. J. C. did the BET experimental and data analysis.Conflicts of interest
The authors declare no competing financial interests.Funding sources
This publication has emanated in part from research conducted with the financial support of Science Foundation Ireland (SFI) under Grant Number SFI/10/IN.1/I2969, SFI/12/IP/1273, and PI10/IN.1/I3030.Acknowledgements
We would like to thank the staff in the AML, CRANN for help with the SEM analysis. We would also like to thank Dr Brendan Twamley for help with the XRD analysis.References
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS PubMed.
- M. K. Bates, Q. Jia, H. Doan, W. Liang and S. Mukerjee, ACS Catal., 2016, 6, 155–161 CrossRef CAS.
- H. Ali-Löytty, M. W. Louie, M. R. Singh, L. Li, H. G. Sanchez Casalongue, H. Ogasawara, E. J. Crumlin, Z. Liu, A. T. Bell, A. Nilsson and D. Friebel, J. Phys. Chem. C, 2016, 120, 2247–2253 Search PubMed.
- J. A. Carrasco, J. Romero, M. Varela, F. Hauke, G. Abellan, A. Hirsch and E. Coronado, Inorg. Chem. Front., 2016, 3, 478–487 RSC.
- M. Gong and H. Dai, Nano Res., 2015, 8, 23–39 CrossRef CAS.
- M. P. Browne, H. Nolan, G. S. Duesberg, P. E. Colavita and M. E. G. Lyons, ACS Catal., 2016, 6, 2408–2415 CrossRef CAS.
- M. Browne, R. J. Cullen, R. L. Doyle, P. E. Colavita and M. E. G. Lyons, ECS Trans., 2013, 53, 59–77 CrossRef.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- S. Jung, C. C. L. McCrory, I. M. Ferrer, J. C. Peters and T. F. Jaramillo, J. Mater. Chem. A, 2016, 4, 3068–3076 CAS.
- M. P. Browne, S. Stafford, M. O'Brien, H. Nolan, N. C. Berner, G. S. Duesberg, P. E. Colavita and M. E. G. Lyons, J. Mater. Chem. A, 2016, 4, 11397–11407 CAS.
- D. A. Corrigan, J. Electrochem. Soc., 1987, 134, 377–384 CrossRef CAS.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- S. Klaus, Y. Cai, M. W. Louie, L. Trotochaud and A. T. Bell, J. Phys. Chem. C, 2015, 119, 7243–7254 CAS.
- F. Song and X. Hu, Nat. Commun., 2014, 5, 4477–4485 CAS.
- H. Chen, J. Yan, H. Wu, Y. Zhang and S. Liu, J. Power Sources, 2016, 324, 499–508 CrossRef CAS.
- T. Yang, K. Li, L. Pu, Z. Liu, B. Ge, Y. Pan and Y. Liu, Biosens. Bioelectron., 2016, 86, 129–134 CrossRef CAS PubMed.
- T. Mousavand, S. Takami, M. Umetsu, S. Ohara and T. Adschiri, J. Mater. Sci., 2006, 41, 1445–1448 CrossRef CAS.
- T. Adschiri, Y. Hakuta and K. Arai, Ind. Eng. Chem. Res., 2000, 39, 4901–4907 CrossRef CAS.
- N. Kasapoğlu, A. Baykal, M. S. Toprak, Y. Köseoğlu and H. Bayrakdar, Turk. J. Chem., 2007, 31, 659–666 Search PubMed.
- X. Zhou, Z. Xia, Z. Zhang, Y. Ma and Y. Qu, J. Mater. Chem. A, 2014, 2, 11799–11806 CAS.
- F. Yan, C. Zhu, S. Wang, Y. Zhao, X. Zhang, C. Li and Y. Chen, J. Mater. Chem. A, 2016, 4, 6048–6055 CAS.
- S. Klaus, M. W. Louie, L. Trotochaud and A. T. Bell, J. Phys. Chem. C, 2015, 119, 18303–18316 CAS.
- D. Cai, D. Wang, B. Liu, L. Wang, Y. Liu, H. Li, Y. Wang, Q. Li and T. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 5050–5055 CAS.
- J. Yang, E. Zhang, X. Li, Y. Yu, J. Qu and Z.-Z. Yu, ACS Appl. Mater. Interfaces, 2016, 8, 2297–2305 CAS.
- B. You, N. Jiang, M. Sheng, M. W. Bhushan and Y. Sun, ACS Catal., 2016, 6, 714–721 CrossRef CAS.
- Q.-C. Zhu, F.-H. Du, S.-M. Xu, Z.-K. Wang, K.-X. Wang and J.-S. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 3868–3873 CAS.
- J. Luo, J.-H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N.-G. Park, S. D. Tilley, H. J. Fan and M. Grätzel, Science, 2014, 345, 1593–1596 CrossRef CAS PubMed.
- S. Komaba, T. Mikumo, N. Yabuuchi, A. Ogata, H. Yoshida and Y. Yamada, J. Electrochem. Soc., 2010, 157, A60–A65 CrossRef CAS.
- M. Fu, Q. Jiao and Y. Zhao, J. Mater. Chem. A, 2013, 1, 5577–5586 CAS.
- J. L. Bantignies, S. Deabate, A. Righi, S. Rols, P. Hermet, J. L. Sauvajol and F. Henn, J. Phys. Chem. C, 2008, 112, 2193–2201 CAS.
- J. Desilvestro, D. A. Corrigan and M. J. Weaver, J. Phys. Chem., 1986, 90, 6408–6411 CrossRef CAS.
- D. S. Hall, D. J. Lockwood, S. Poirier, C. Bock and B. R. MacDougall, J. Phys. Chem. A, 2012, 116, 6771–6784 CrossRef CAS PubMed.
- Y. L. Lo and B. J. Hwang, Langmuir, 1998, 14, 944–950 CrossRef CAS.
- J. Landon, E. Demeter, N. İnoğlu, C. Keturakis, I. E. Wachs, R. Vasić, A. I. Frenkel and J. R. Kitchin, ACS Catal., 2012, 2, 1793–1801 CrossRef CAS.
- P. Stelmachowski, A. Kopacz, P. Legutko, P. Indyka, M. Wojtasik, L. Ziemiański, G. Żak, Z. Sojka and A. Kotarba, Catal. Today, 2015, 257(1), 111–116 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- A. P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. McIntyre, Surf. Interface Anal., 2004, 36, 1564–1574 CrossRef CAS.
- A. P. Grosvenor, M. C. Biesinger, R. S. C. Smart and N. S. McIntyre, Surf. Sci., 2006, 600, 1771–1779 CrossRef CAS.
- K. S. Sing, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- S. Lowell, Characterization of Porous Solids and Powders: Surface Area, Pore Size and Density, Springer, 2004 Search PubMed.
- Y. Li, M. Zhao, Y. Zhao, L. Song and Z. Zhang, Part. Part. Syst. Charact., 2016, 33, 158–166 CrossRef CAS.
- G.-Q. Han, Y.-R. Liu, W.-H. Hu, B. Dong, X. Li, X. Shang, Y.-M. Chai, Y.-Q. Liu and C.-G. Liu, Appl. Surf. Sci., 2015, 359, 172–176 CrossRef CAS.
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, J. Am. Chem. Soc., 2012, 134, 17253–17261 CrossRef CAS PubMed.
- P.-S. Li and H. Teng, J. Chin. Inst. Chem. Eng., 2007, 38, 267–273 CrossRef CAS.
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 13801 CrossRef PubMed.
- M. E. G. Lyons, R. L. Doyle, D. Fernandez, I. J. Godwin, M. P. Browne and A. Rovetta, Electrochem. Commun., 2014, 45, 56–59 CrossRef CAS.
- M. E. G. Lyons, R. L. Doyle, D. Fernandez, I. J. Godwin, M. P. Browne and A. Rovetta, Electrochem. Commun., 2014, 45, 60–62 CrossRef CAS.
- R. L. Doyle, I. J. Godwin, M. P. Brandon and M. E. G. Lyons, Phys. Chem. Chem. Phys., 2013, 15, 13737–13783 RSC.
- S. Klaus, Y. Cai, M. W. Louie, L. Trotochaud and A. T. Bell, J. Phys. Chem. C, 2015, 119, 7243–7254 CAS.
- J. Coelho, B. Mendoza-Sánchez, H. Pettersson, A. Pokle, E. K. McGuire, E. Long, L. McKeon, A. P. Bell and V. Nicolosi, 2D Materials, 2015, 2, 025005 CrossRef.
- N. Han, F. Zhao and Y. Li, J. Mater. Chem. A, 2015, 3, 16348–16353 CAS.
- H. Liang, F. Meng, M. Cabán-Acevedo, L. Li, A. Forticaux, L. Xiu, Z. Wang and S. Jin, Nano Lett., 2015, 15, 1421–1427 CrossRef CAS PubMed.
- J. Y. C. Chen, L. Dang, H. Liang, W. Bi, J. B. Gerken, S. Jin, E. E. Alp and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 15090–15093 CrossRef CAS PubMed.
- M. C. Biesinger, B. P. Payne, L. W. M. Lau, A. Gerson and R. S. C. Smart, Surf. Interface Anal., 2009, 41, 324–332 CrossRef CAS.
- M. E. G. Lyons and M. P. Brandon, Int. J. Electrochem. Sci., 2008, 3, 1386–1424 CAS.
- O. Diaz-Morales, D. Ferrus-Suspedra and M. T. M. Koper, Chem. Sci., 2016, 7, 2639–2645 RSC.
- A. Delahaye-Vidal and M. Figlarz, J. Appl. Electrochem., 1987, 17, 589–599 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Further electrochemical measurements and the characterization data including the XPS, AFM, BET, SEM-EDX, and Raman can be found. See DOI: 10.1039/c6se00032k |
| This journal is © The Royal Society of Chemistry 2017 |