
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe structural assembly switch of cell division protein FtsZ probed with fluorescent allosteric inhibitors†
Marta
Artola‡§
 a,
Laura B.
Ruíz-Avila‡¶
b,
Erney
Ramírez-Aportela
bc,
R. Fernando
Martínez
a,
Lidia
Araujo-Bazán
b,
Henar
Vázquez-Villa
a,
Mar
Martín-Fontecha
a,
María A.
Oliva
b,
A. Javier
Martín-Galiano||
b,
Pablo
Chacón
c,
María L.
López-Rodríguez
a,
José M.
Andreu
*b and
Sonia
Huecas
*b
a,
Laura B.
Ruíz-Avila‡¶
b,
Erney
Ramírez-Aportela
bc,
R. Fernando
Martínez
a,
Lidia
Araujo-Bazán
b,
Henar
Vázquez-Villa
a,
Mar
Martín-Fontecha
a,
María A.
Oliva
b,
A. Javier
Martín-Galiano||
b,
Pablo
Chacón
c,
María L.
López-Rodríguez
a,
José M.
Andreu
*b and
Sonia
Huecas
*b
aDept. Química Orgánica I, Facultad de Ciencias Químicas, UCM, 28040 Madrid, Spain
bCentro de Investigaciones Biológicas, CSIC, Ramiro de Maeztu 9, 28040 Madrid, Spain. E-mail: j.m.andreu@cib.csic.es; sonia@cib.csic.es
cInstituto de Química-Física Rocasolano, CSIC, Serrano 119, 20006 Madrid, Spain
First published on 21st October 2016
Abstract
FtsZ is a widely conserved tubulin-like GTPase that directs bacterial cell division and a new target for antibiotic discovery. This protein assembly machine cooperatively polymerizes forming single-stranded filaments, by means of self-switching between inactive and actively associating monomer conformations. The structural switch mechanism was proposed to involve a movement of the C-terminal and N-terminal FtsZ domains, opening a cleft between them, allosterically coupled to the formation of a tight association interface between consecutive subunits along the filament. The effective antibacterial benzamide PC190723 binds into the open interdomain cleft and stabilizes FtsZ filaments, thus impairing correct formation of the FtsZ ring for cell division. We have designed fluorescent analogs of PC190723 to probe the FtsZ structural assembly switch. Among them, nitrobenzoxadiazole probes specifically bind to assembled FtsZ rather than to monomers. Probes with several spacer lengths between the fluorophore and benzamide moieties suggest a binding site extension along the interdomain cleft. These probes label FtsZ rings of live Bacillus subtilis and Staphylococcus aureus, without apparently modifying normal cell morphology and growth, but at high concentrations they induce impaired bacterial division phenotypes typical of benzamide antibacterials. During the FtsZ assembly–disassembly process, the fluorescence anisotropy of the probes changes upon binding and dissociating from FtsZ, thus reporting open and closed FtsZ interdomain clefts. Our results demonstrate the structural mechanism of the FtsZ assembly switch, and suggest that the probes bind into the open clefts in cellular FtsZ polymers preferably to unassembled FtsZ in the bacterial cytosol.
Introduction
FtsZ is the organizer of bacterial cell division, a conserved self-assembling GTPase that forms the Z-ring marking the division site at mid cell; FtsZ is tethered to the inner face of the plasma membrane by FtsA and ZipA in Escherichia coli.1 The highly dynamic Z-ring recruits the divisomal machinery, including cell wall remodeling and chromosome segregation proteins, and it contracts during division.2–6 Electron cryo-tomography studies have shown a few7 or a small band of individual, laterally connected FtsZ filaments forming a ring parallel to the membrane.8 FtsZ filaments attached to different model membrane systems can self-organize, without motor proteins, into contractile ring structures8,9 and form dynamic chiral patterns driven by treadmilling of polar FtsZ polymers.10 The Z-ring is the subject of super resolution fluorescence microscopy and biophysical studies in different organisms, currently indicating that it is a patchy scaffold made of clusters of relatively disordered FtsZ protofilaments.11–17 The Z-ring is stabilized by a multi-layered protein network connecting the cell membrane to the chromosome in E. coli cells,18 where the septal cell wall synthesis provides constriction force.19Due to its key function and ubiquity FtsZ is an attractive target for discovering new antibiotics, which are urgently needed to fight the spread of pathogens resistant to current therapeutic options.20 The difluorobenzamide derivative PC190723 and its analogs are currently the best characterized FtsZ-targeting bacterial cell division inhibitors21–26 with effective anti-infective activity.27–30 Other small molecule25 FtsZ assembly modulators include nucleotide analogs31–33 and nucleotide replacing inhibitors,34–36 among many compounds reported to interact with FtsZ.37–39
FtsZ and its eukaryotic homolog tubulin are part of a superfamily of protein assembly machines that share homologous GTP-binding (N-terminal) and GTPase activating domains.40,41 Their subunits associate head to tail along protofilaments in which the GTPase active site is completed at the interface between consecutive monomers with a 4 nm spacing. FtsZ assembles into single protofilaments that laterally associate in different fashions, whereas tubulin forms microtubules. GTP hydrolysis is the chemical signal that triggers disassembly by weakening the association between subunits; this is coupled to FtsZ or tubulin subunit relaxation into different inactive conformations. In microtubules, subtle structural changes upon GTP hydrolysis lead to lattice strain generation,42 followed by disassembly and a large structural change of αβ-tubulin subunits that relax into their inactive curved conformation.43,44 Tubulin switching between curved and straight conformations substantially contributes to microtubule dynamic instability and the regulation by associated proteins.45
FtsZ has a distinct built-in assembly switch to polymerize into one-molecule wide protofilaments with a puzzlingly cooperative behavior, which was explained by FtsZ self-switching from an inactive monomer conformation into an actively associating filament conformation.46–49 Mapping FtsZ flexibility with computational approaches and point mutants suggested interdomain movements involving opening and closing the cleft located between the C-terminal domain and the nucleotide binding domain and modifying the axial association between FtsZ monomers.50 The quenching of an extrinsic fluorophore in one domain by a tryptophan residue in the other domain of a mutant FtsZ decreased upon assembly, supporting a movement apart of the two domains.51
Comparison of FtsZ crystal structures permits to identify distinct closed and open-cleft conformations, which suggests a possible structural mechanism for the FtsZ assembly switch. Structures of unassembled FtsZ from different organisms all showed very similar closed-cleft conformations.52 This is different in the more recent structure of FtsZ from Staphylococcus aureus (SaFtsZ), which has an open interdomain cleft and forms a protofilament in the crystal lattice.53,54 The C-terminal domain of SaFtsZ swings opening the cleft, rotating by ∼25° with respect to FtsZ from Bacillus subtilis (BsFtsZ), and main helix H7-loop T7 downshift by one helical turn.53 These changes would facilitate the formation of the tight interface between consecutive SaFtsZ monomers that buries the nucleotide and is thought to stabilize the filament. In addition, opening the cleft makes possible the binding of the small molecule cell division inhibitor PC190723,27 which stabilizes FtsZ polymers24 and reduces SaFtsZ assembly cooperativity possibly by an allosteric mechanism.54 However, additional structures, such as a closed-cleft structure of unassembled SaFtsZ, or an open-cleft structure of a filament of BsFtsZ, would be required to proof that the same protein can actually switch between both crystal conformations.
Large scale molecular dynamics simulations of SaFtsZ filaments have identified a coordinated Mg2+ ion as the key structural element in stabilizing GTP-filaments, and how the loss of contacts with the loop T7 from the next monomer in GDP-filaments leads to open interfaces that are prone to depolymerization. The interdomain cleft has been observed to relax into the closed conformation in isolated monomers and at filament minus-ends; and PC190723 bound into the cleft has been observed to allosterically induce tight intermonomer interfaces.55 These in silico observations support the crystal-based FtsZ switch mechanism, but a definitive proof that this mechanism operates in solution is still required.
This work aimed to determine the structural switch of FtsZ as it assembles in solution or in bacterial cells. We have designed fluorescent benzamide analogs of PC190723 that specifically bind to FtsZ polymers and label the Z-ring in dividing cells. Changes in probe fluorescence anisotropy upon binding and dissociation monitor the opening and closing of the interdomain cleft during FtsZ assembly and disassembly, demonstrating the structural mechanism of the FtsZ activation switch.
Results and discussion
Design and selection of fluorescent probes for the PC190723 binding site of FtsZ
We constructed fluorescent PC190723 analogs targeting FtsZ based on two strategies (Fig. 1A), both keeping unmodified the 2,6-difluorobenzamide warhead,23,27 which we knew stabilizes FtsZ polymers rather than the thiazolopyridine tail.24 In one approach, the fluorescent tag was attached to the heterocyclic moiety through a rigid spacer or a flexible aliphatic chain, which replaces the chlorine atom of PC190723 analog 1 (previously known as 8j23,25). We employed dansyl (Ds), a coumarin derivative and nitrobenzoxadiazole (NBD) small fluorophores, giving compounds 2–5 (Fig. 1A and S1A†). Rigid compounds 2, 4, and 5 were synthesized starting from bromo derivative 18 by Sonogashira coupling with the corresponding terminal alkyne under microwave (MW) irradiation (Scheme 1). Thus, coupling of 18 with properly protected 2-propyn-1-amine, followed by trifluoroacetyl deprotection of derivative 19 and subsequent coupling of obtained amine 20 with Ds-chloride or 7-(diethylamino)coumarin-3-carboxylic acid yielded fluorescent compounds 2 or 4, respectively, whereas Sonogashira reaction of 18 with NBD alkyne 21 afforded final compound 5. The fluorescent derivative with a more flexible alkyl chain linker 3 was prepared by RANEY® Ni-hydrogenation of alkyne 19, followed by cleavage of trifluoroacetyl group and coupling of free amine with Ds-chloride. However, compounds 2–5 were inactive in fluorescence intensity and anisotropy tests of specific binding to FtsZ polymers (Fig. S1B†) and were not further explored. In another approach, the whole heterocyclic moiety of PC190723/1 was replaced by the fluorophore, which was attached to 2,6-difluorobenzamide in position 3 through an amino-ethoxy linker (Fig. 1A, compounds 6–13). In addition to the same small fluorophores, we employed pyridoxal, dapoxyl, bodipy-FL, tetramethylrhodamine (TAMRA), and the photoswitchable fluorophore ATTO565. Fluorescent derivatives 6–13 were synthesized by alkylation of 2,6-difluoro-3-hydroxybenzamide with N-Boc-2-bromo-1-ethylamine, followed by cleavage of the protecting group of 22 and subsequent coupling of the free amine 23 with the corresponding fluorophores as outlined in Scheme 1. The chemical structures and fluorescence emission spectra of compounds 2–13 are displayed in ESI Fig. S1A.†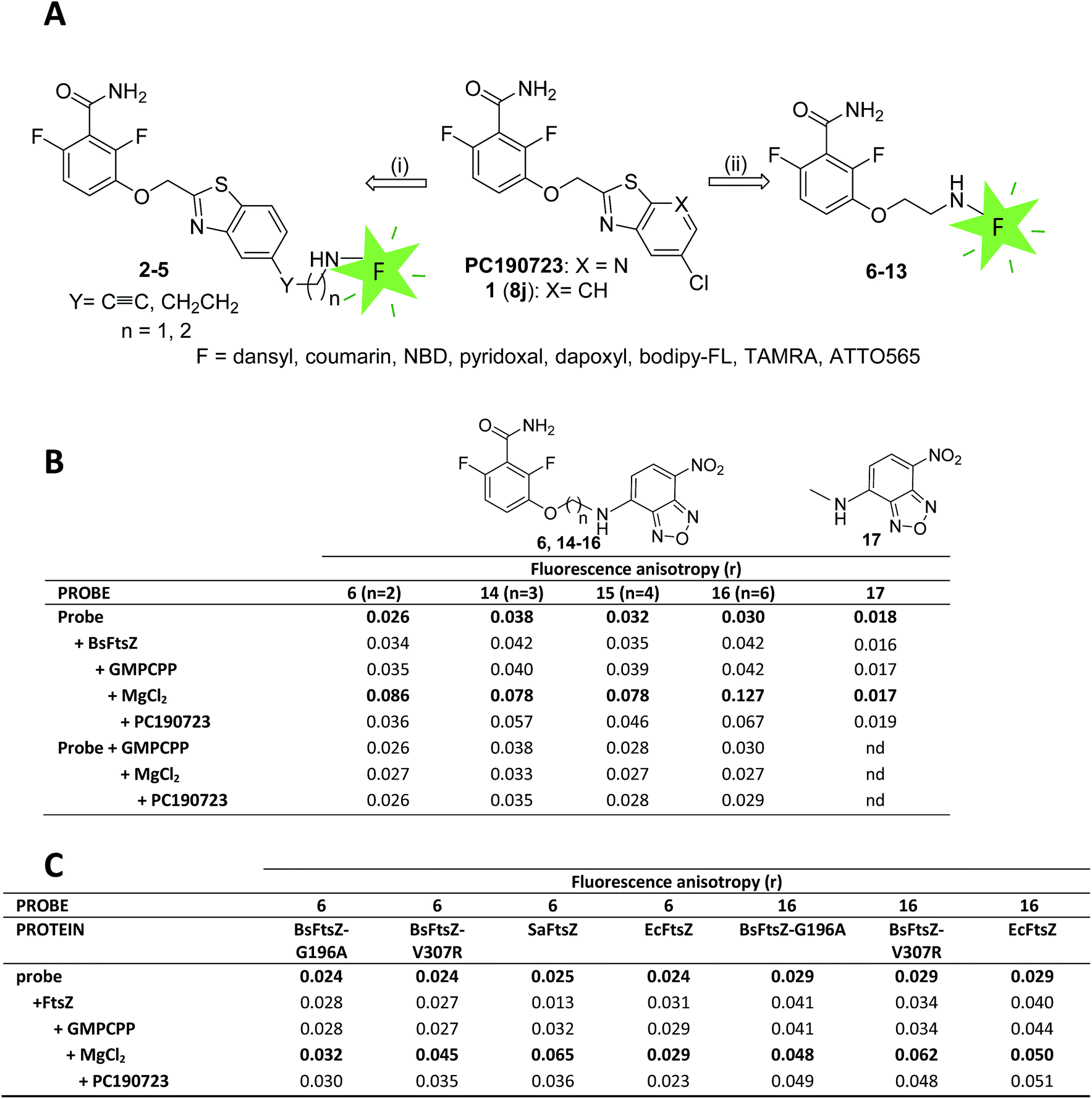 | ||
| Fig. 1 Fluorescent probe design, selection and validation. (A) Initial design for fluorescent analogs of benzamide antibiotics PC190723 and 8j. (B) Fluorescence anisotropy values of free and bound selected probes 6, 14–16 and inactive control methylamino-NBD fluorescent moiety 17. The values in the first row are from the free probe (10 μM). BsFtsZ (10 μM, unassembled) and GMPCPP (0.1 mM) were sequentially added, BsFtsZ polymerization was then induced by addition of MgCl2 (10 mM) and anisotropy values with steady state polymers recorded 5–10 minutes later. Non fluorescent PC190723 (10 μM) was subsequently added to displace the probe. The last three rows are controls made without protein to exclude probe interactions with GMPCPP, MgCl2 or PC190723. The r values are averages from ≥2 samples; standard deviation was typically ±0.002. (C) Anisotropy values of fluorescent probes 6 and 16 with PC190723-resistant mutant proteins BsFtsZ-G196A and BsFtsZ-V307R, PC190723-susceptible wild-type protein SaFtsZ and non-susceptible EcFtsZ. FtsZ polymers formation was confirmed by sedimentation, light scattering and electron microscopy tests in each case. | ||
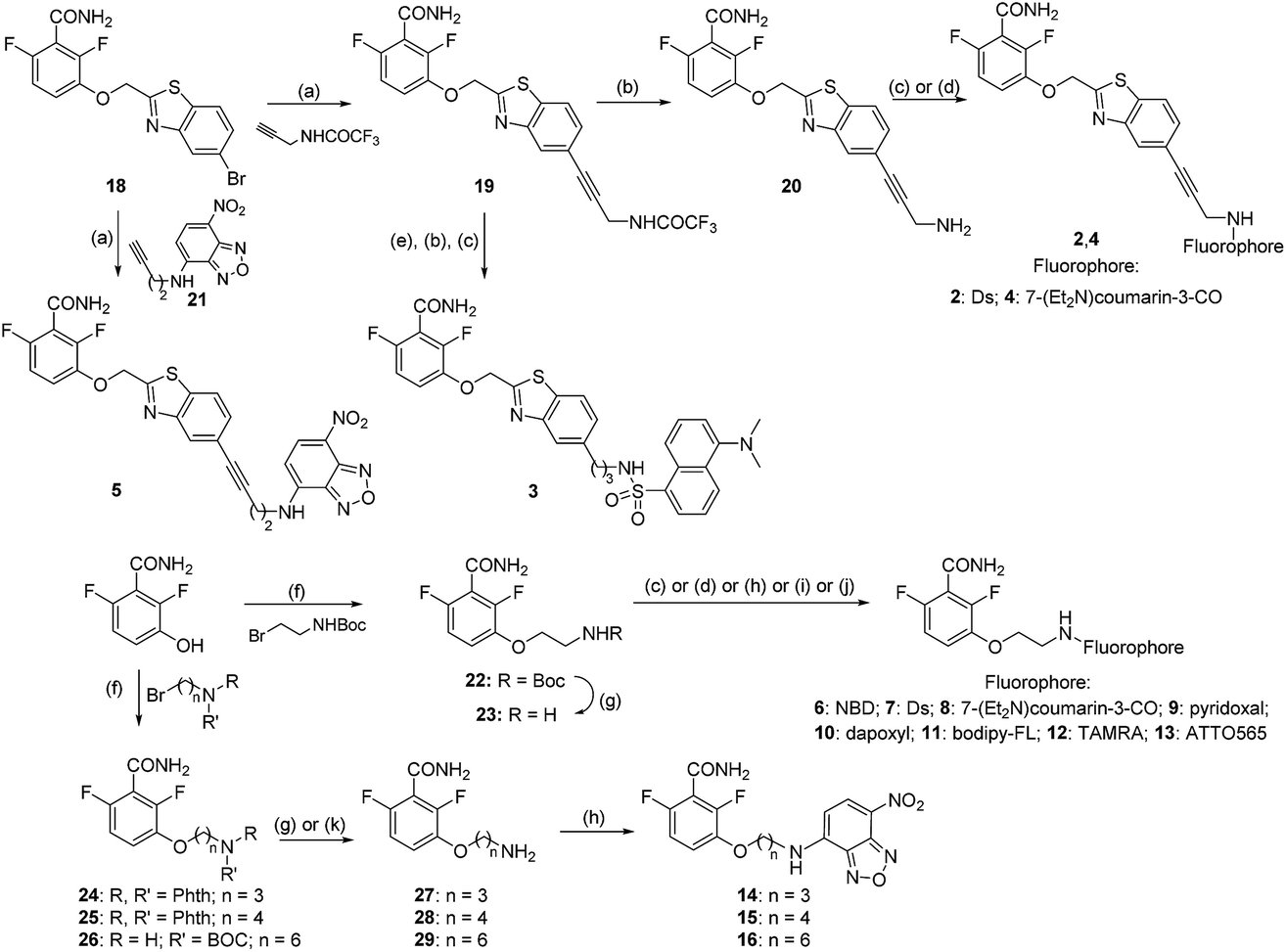 | ||
| Scheme 1 Synthesis of fluorescent compounds 2–16. Reagents and conditions: (a) Pd(PPh3)4, CuI, Et3N, DMF, MW, 100 °C, 45 min, 41–86%; (b) NH3 aq., MeOH, rt, 17 h, 99%; (c) Ds-Cl, Et3N, DCM, DMF, rt, 24 h, 26–28%; (d) 7-(Et2N)coumarin-3-COOH, PyBroP, DIPEA, DMF, rt, 4 h, 15–27%; (e) H2, RANEY®-Ni, THF, MeOH, rt, 3 h, 22%; (f) K2CO3, NaI, DMF, rt, 16 h, 57–87%; (g) TFA, DCM, rt, 1 h, 99%; (h) NBD-Cl, DIPEA, DMF, rt, 16 h, 23–30%; (i) (i) pyridoxal·HCl, NaHCO3, EtOH/H2O, rt, 2 h; (ii) NaBH3CN, rt, 3 h, 18%; (j) dapoxyl-NHS or bodipy-FL-NHS or TAMRA-NHS or ATTO-565-NHS, Et3N, DMF, rt, 4–16 h, 66–99%; (k) N2H4·H2O, EtOH, reflux, 2 h, 55–64%. | ||
Among these compounds, the NBD–benzamide derivative 6 showed a marked 3.3-fold fluorescence anisotropy increase, with nearly constant intensity, in the presence of polymers of BsFtsZ (Fig. 1B). Polymers were assembled from monomers by addition of GMPCPP (a GTP analog that more efficiently induces FtsZ assembly12 and is hydrolyzed about 10-fold slower) and magnesium. The anisotropy returned near the value of the free probe upon addition of the non-fluorescent PC190723 and did not change in the absence of the protein (Fig. 1B). These anisotropy measurements indicated a restriction of the rotational mobility of the fluorescent NBD moiety of the probe upon specific binding to BsFtsZ polymers, rather than binding to monomers. The isolated fluorescent part methylamino-NBD (17), employed as a negative control lacking the active moiety, did not change its anisotropy. In stark contrast with the NBD tagged 6, the compounds with other fluorophores (7–10, 12 and 13) either did not show a significant anisotropy increase (>1.5 fold) with BsFtsZ polymers, or the increase was non-specific, or had high anisotropy values in the absence of protein that indicated aggregation of the free probes. Only the bodipy-FL derivative 11 showed a 1.7-fold specific increase, but its absolute anisotropy values were very low (Fig. S1B†).
Biochemical validation of fluorescent probe 6
We further tested the specificity of probe 6 measuring its anisotropy changes, which required additional FtsZ proteins, two PC190723-resistant BsFtsZ mutants, as well as two other FtsZs from bacterial species known to be susceptible or resistant to PC190723 action (Fig. 1C). The FtsZ mutation G196A confers resistance to PC190723 in S. aureus and B. subtilis cells.21,26,27 Therefore we constructed BsFtsZ-G196A and observed a nearly complete inhibition of the fluorescence anisotropy change of 6 with the mutant FtsZ polymers compared to the wild type. On the other hand, PC190723 susceptible bacteria have V307 in their FtsZ, whereas intrinsically resistant organisms carry R307 or H307 residues.21 We thus constructed BsFtsZ-V307R and consequently observed a strong reduction of the anisotropy of the probe in the presence of these mutant polymers with respect to the wild type protein. We then confirmed that polymers of FtsZ from PC190723-susceptible S. aureus bind compound 6, whereas polymers of FtsZ from non-susceptible E. coli (EcFtsZ) do not significantly modify the probe fluorescence anisotropy (Fig. 1C). Thus our results with purified FtsZs and probe 6 nicely recapitulate the corresponding bacterial cells susceptibility to the antibiotic PC190723, which supports in vitro applications of the fluorescent benzamide probe.We also examined the effects of probe 6 on GTP-induced assembly and the GTPase activity of BsFtsZ polymers, finding that it can also behave as a weak FtsZ polymer stabilizer. Probe 6 enhanced BsFtsZ polymer formation similarly to the non-fluorescent moiety 2,6-difluoro-3-methoxibenzamide (DFMBA; Fig. S2†), reducing the critical concentration (Cr) values required for cooperative BsFtsZ polymerization in ligand excess. The GTPase rate of BsFtsZ also showed a cooperative activation above the Cr to a GTP hydrolysis rate of 2.17 ± 0.37 min−1 by polymerized BsFtsZ, which was reduced by 6 to 0.49 ± 0.08 min−1 (Fig. S2†) and by DFMBA to a similar value of 0.40 ± 0.06 min−1. The similar potency to the benzamide moiety (that requires ∼102-fold higher concentration than the complete ligand PC190723 (ref. 24)) suggests that the flexibly attached NBD fluorophore of 6 does not replace the PC190723 thiazolopyridine tail in the binding site of FtsZ polymers, or that its interactions balance into a negligible binding affinity increase. The first possibility is compatible with the observations that the fluorescence intensity of the probe does not change but the anisotropy increases upon binding to BsFtsZ polymers (Fig. 1B and S1†). As expected for probe binding to the BsFtsZ polymers rather than to monomers, we found a lack of probe 6 co-sedimentation with BsFtsZ monomers in analytical centrifugation experiments, made at similar protein and probe concentrations as the anisotropy experiments (Fig. S3†).
Effects of increasing the spacer lengths in probe and model complexes suggest an extended binding site for inhibitors at FtsZ's interdomain cleft
As the X-ray structure of the SaFtsZ–PC190723 complex became available, showing a narrow pocket with apparently little room for chemical modification,27,53 we analyzed the effects of varying the length of the amino-ethoxy linker of probe 6, up to 6 methylene subunits in compounds 14–16 (n = 3, 4, 6; Fig. 1B, Scheme 1). Increasing the chain length to three and four carbons in 14 and 15, respectively, practically did not modify the probe anisotropy in the presence of BsFtsZ polymers, suggesting a similar recognition mode as for the probe with the two carbon linker (Fig. 1B); but it decreased the reversibility of binding upon addition of PC190723 from 98% (in 6) to 55% and 82% (for 14 and 15, respectively), suggesting a higher level of non-specific binding as the probes became less polar. Compounds 14 and 15 enhanced GTP-induced BsFtsZ assembly similarly to 6 above. In contrast, increasing the chain length to six carbons in 16 gave a larger anisotropy change with BsFtsZ polymers, which was 70% reversible (Fig. 1B), and also higher anisotropy values with BsFtsZ mutants and EcFtsZ (Fig. 1C). However, an enhanced BsFtsZ assembly was not observed with 16, suggesting a different mode of binding with the longer linker.We determined a binding stoichiometry of one molecule of probe 6 per protein subunit in BsFtsZ polymers (assembled with GTP or GMPCPP) using polymer sedimentation and spectrophotometric measurements. We employed fluorescence anisotropy titrations (Fig. S4†) to measure the apparent binding affinity of 6, Kd = 29 ± 3 μM (with GTP) and Kd = 26 ± 3 μM (with GMPCPP). The Kd values of the other probes were: 14, 13 ± 3 μM; 15, 11 ± 1 μM; 16, 12 ± 2 μM (with GMPCPP). The maximal anisotropy values were 0.22 to 0.27 in all cases, which is compatible with partially restricted wobbling of the fluorophore in the bound probes.56
In order to gain structural insight into the binding mode of these NBD–difluorobenzamide probes in comparison with PC190723, we performed docking experiments to obtain a first guess of the complexes into the binding site localized along the cleft formed between the C-terminal domain and helix H7 of SaFtsZ. Molecular dynamics (MD) simulations were employed to assess the reliability of the highest scoring docking complexes (see ESI† Methods for details). In all the cases, the complexes remain stable during the course of the 500 ns simulation after an initial relaxing period. In the first part of the simulation (up to 130 ns) the ligands usually accommodate the NBD moiety to the final equilibrium position inside the cleft (see RMSD trajectories in Fig. S5† to monitor the relative flexibility and stability of the complexes). The final MD SaFtsZ complex structures with 6, 15 and 16 (n = 2, 4 and 6, respectively) are shown in Fig. 2 and the entire trajectories can be directly visualized in the accompanying ESI videos (see ESI Movies 1 to 4†). Probes 6 and 15 share interactions with residues from loop T7 and beta strands S8–S9 (Fig. 2) already observed in the atomic structure of the SaFtsZ–PC190723 complex27,53,54 and confirmed with MD simulations.55 These contacts are critical to suppress disassembly by keeping the interdomain cleft open and to allosterically induce tight intermonomer interfaces, thus stabilizing the filament.54,55 However, we observed different poses of the new fluorescent moiety, which did not replace the PC190723 heterocyclic tail in the binding site. NBD partially exposed the nitro group to the solvent in 6 and less in 15. Strikingly, we found two alternative binding modes for 16 with the six carbon spacer, both different from PC190723 binding (Fig. 2). In this case the interactions with loop T7 and beta strands S8–S9 are missing, as the ligand has displaced upwards along the cleft (see Fig. S6† for superposition with the PC190723 complex). The two alternative modes switched the positions between benzamine and NBD in the binding site. These models disclose a distinct binding mode to the other probes studied, which could explain the different interactions of 16 with FtsZ that we had experimentally observed. The results and the models thus suggest an extension beyond the canonical PC190723 binding site that could be available for binding of inhibitors along the FtsZ interdomain cleft.
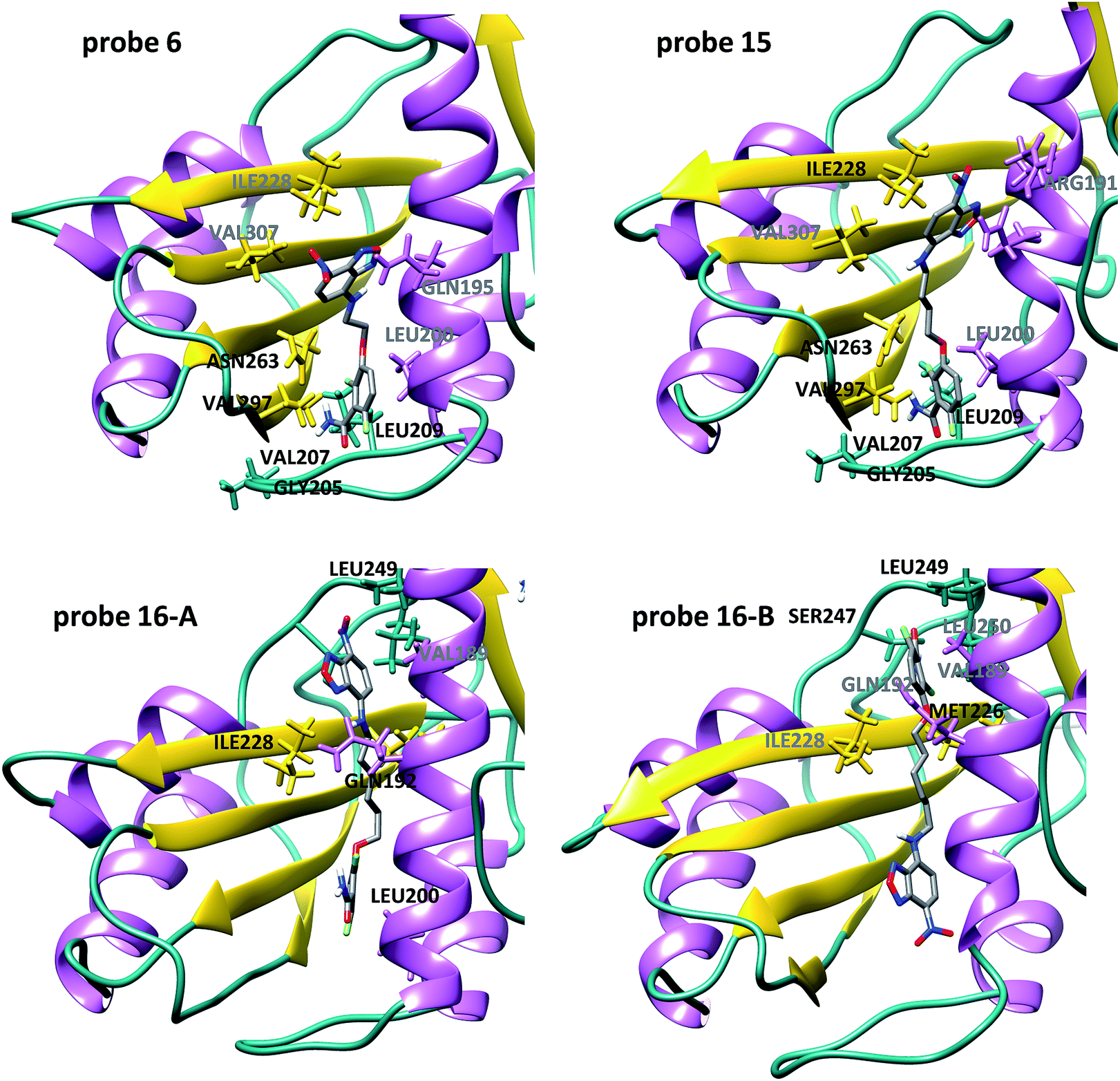 | ||
| Fig. 2 Binding modes of difluorobenzamide–NBD probes into the FtsZ interdomain cleft. Molecular dynamics snapshots of the final equilibrated SaFtsZ model complexes with 6, 15 and 16. Residues labelled in black and grey correspond to stable and weak probe contacts respectively observed during the last 100 ns of the MD simulations. For 6 and 15, stable H-bonds between the amino group and V207 (from loop T7) and N263 (beta strand S8), as well as H-bonds between the carbonyl group and G205 and L209 (loop T7) are observed during the simulations. Hydrophobic interactions with V297 (beta strand S9) also contribute to the benzamide binding. Interactions with I228, and to a lesser extent with V307 and R191, help to keep the wobbling fluorophore in the cleft. However, in the case of 16 there are two potential poses; main contributors to the binding are I228 and L249 for NBD (mode A) and M226, S247 and L249 for the benzamide (mode B). | ||
Fluorescent benzamide derivatives label the Z-ring in dividing bacterial cells
We tested whether our probes would enter live bacterial cells and stain FtsZ subcellular structures using fluorescence microscopy. Interestingly, probe 6 (≤50 μM) labelled a mid cell band in dividing cells (Fig. 3A) but did not appreciably modify B. subtilis cell morphology or growth (Fig. S7;† the MIC, minimal inhibitory concentration for growth, was 800 μM). The probe labelling pattern is similar to that of the FtsZ-GFP fusion in B. subtilis SU570 cells observed with conventional wide-field fluorescence microscopy.12,35,36 Addition of non-fluorescent PC190723 abolished any specific labelling and induced the characteristic filamentous cell morphology expected; and the methylamino-NBD negative control 17 gave no intracellular labelling. Higher concentrations of 6 (≥200 μM) induced helical division events and filamentation, which are characteristic of benzamide antibiotics targeting FtsZ.25 Similar results were obtained with probes 14–16 (Fig. S7;† MIC > 250 μM, above compounds solubility in culture medium). Treatment of the cells with a cell division inhibitor targeting another binding site in FtsZ (GTP replacing compound 28 (ref. 36)), resulted in FtsZ delocalization into punctate foci and irregularly spaced rings that were visualized with 6 or with anti-FtsZ antibody (Fig. S8†). In the case of S. aureus, which is also susceptible to PC190723 action, we observed in the spherical cells a mid cell band at low 6 concentrations, whereas higher concentrations induced abnormal divisions and larger bacterial size (Fig. 3B) that are characteristic of division arrest.25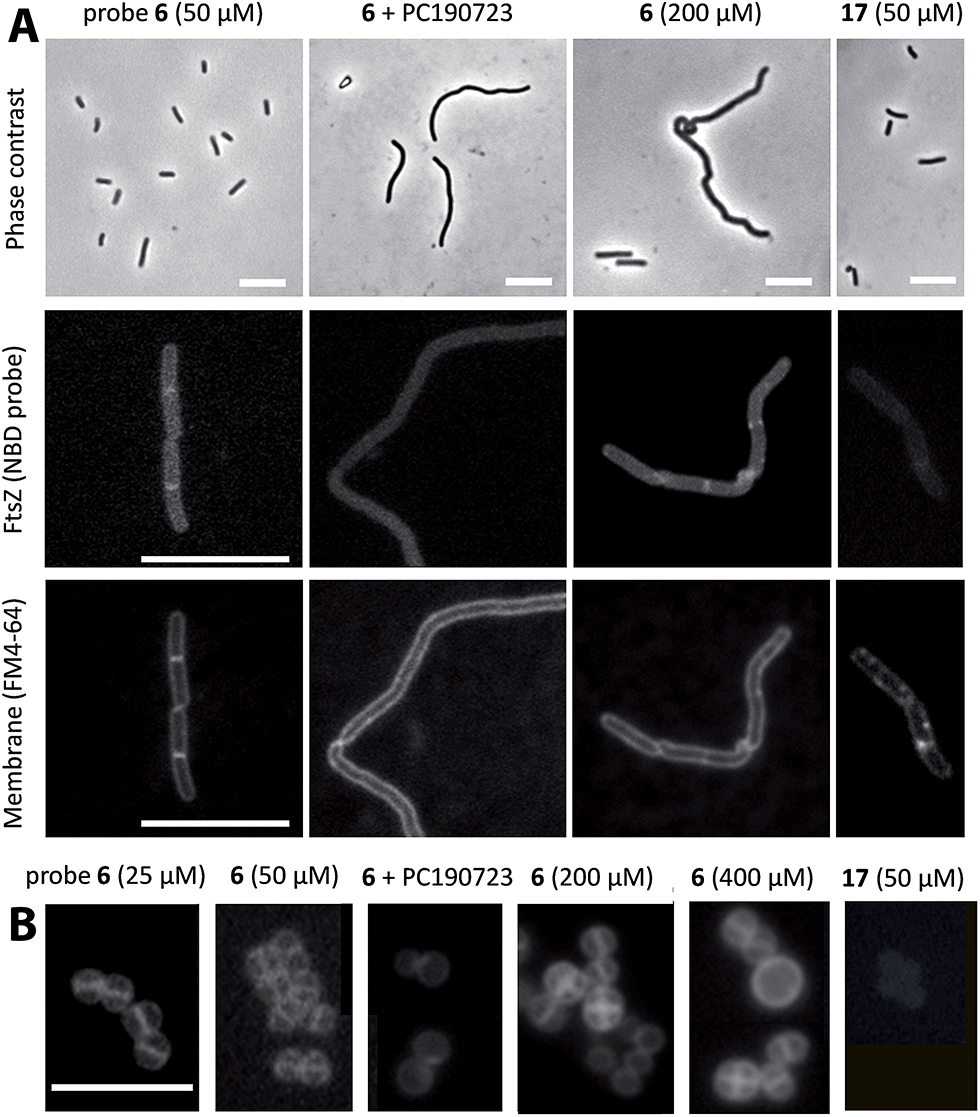 | ||
| Fig. 3 Imaging FtsZ rings in bacterial cells with fluorescent benzamides. (A) B. subtilis 168 cells were treated with 50 μM 6 (1 h; untreated controls gave similar results), with 50 μM 6 plus 25 μM PC190723 (3 h), with 200 μM 6 (3 h) or with 50 μM 17 control fluorophore (1 h) and observed by phase contrast microscopy (top row). FtsZ was directly visualized with 6 (middle row) and membrane with FM4-64 (bottom row) by fluorescence microscopy. Bars, 10 μm. See Fig. S7† for growth curves and similar experiments with probes 14–16. (B) A similar experiment with S. aureus Mu50 cells treated with different concentrations of 6 and controls, observed with fluorescence microscopy. Bar, 5 μm. | ||
We interpret that assembled FtsZ in the divisomal Z-ring is being imaged in growing cells with the NBD–benzamide probes at low concentration, whereas higher concentrations impair FtsZ function and cell division as expected from benzamide inhibitors. Small molecules able to label FtsZ assemblies in living cells may have distinct potential advantages for cytological and biophysical studies of bacterial division, including the ease of use with wild-type cells and the smaller size of the chemical probes compared with FtsZ-fluorescent protein fusions.57
Mechanism of the FtsZ activation switch: opened interdomain cleft during assembly detected by fluorescent probe 6
In order to test the structural mechanism for the FtsZ activation switch we measured the fluorescence anisotropy changes of probe 6 during GMPCPP-induced assembly and subsequent disassembly upon nucleotide consumption (Fig. 4). Crystal structures of BsFtsZ monomers show a closed cleft, as the majority of FtsZs in the Protein Data Bank (PDB) for which filament structures have not been obtained. Fittingly, GMPCPP addition to unassembled BsFtsZ triggered a gradual anisotropy increase (Fig. 4A, red line) as light-scattering polymers assembled (in an identically prepared sample, Fig. 4B, red line); the probe anisotropy reached a plateau and later went down near the anisotropy level of the free probe as the polymers disassembled upon nucleotide exhaustion. Anisotropy time courses with 6 at 2 μM and 10 μM were coincident, suggesting that the rate limiting process for the anisotropy increase is FtsZ assembly rather than a slow binding of the probe. Addition of equimolar PC190723 at the plateau reduced the anisotropy of 6, indicating a relatively weak binding affinity of the probe, and then enhanced polymer scattering as expected (Fig. 4A, blue lines). Thus, probe 6 at low concentration did not perturb GMPCPP-induced BsFtsZ assembly, as monitored by light scattering (Fig. 4B, red and black traces), although higher compound concentrations enhance GTP-induced assembly (Fig. S2†). Electron micrographs showed similar filamentous toroidal condensates in the absence and presence of the probe, which were different from the filament bundles observed with PC190723, indicating that probe 6 did not modify the BsFtsZ polymer morphology under these conditions (Fig. S9†). As technical controls for the anisotropy time courses we employed non-susceptible EcFtsZ, and the assembly of the eukaryotic homolog tubulin into microtubules, both of which gave negligible probe anisotropy changes (Fig. S10†).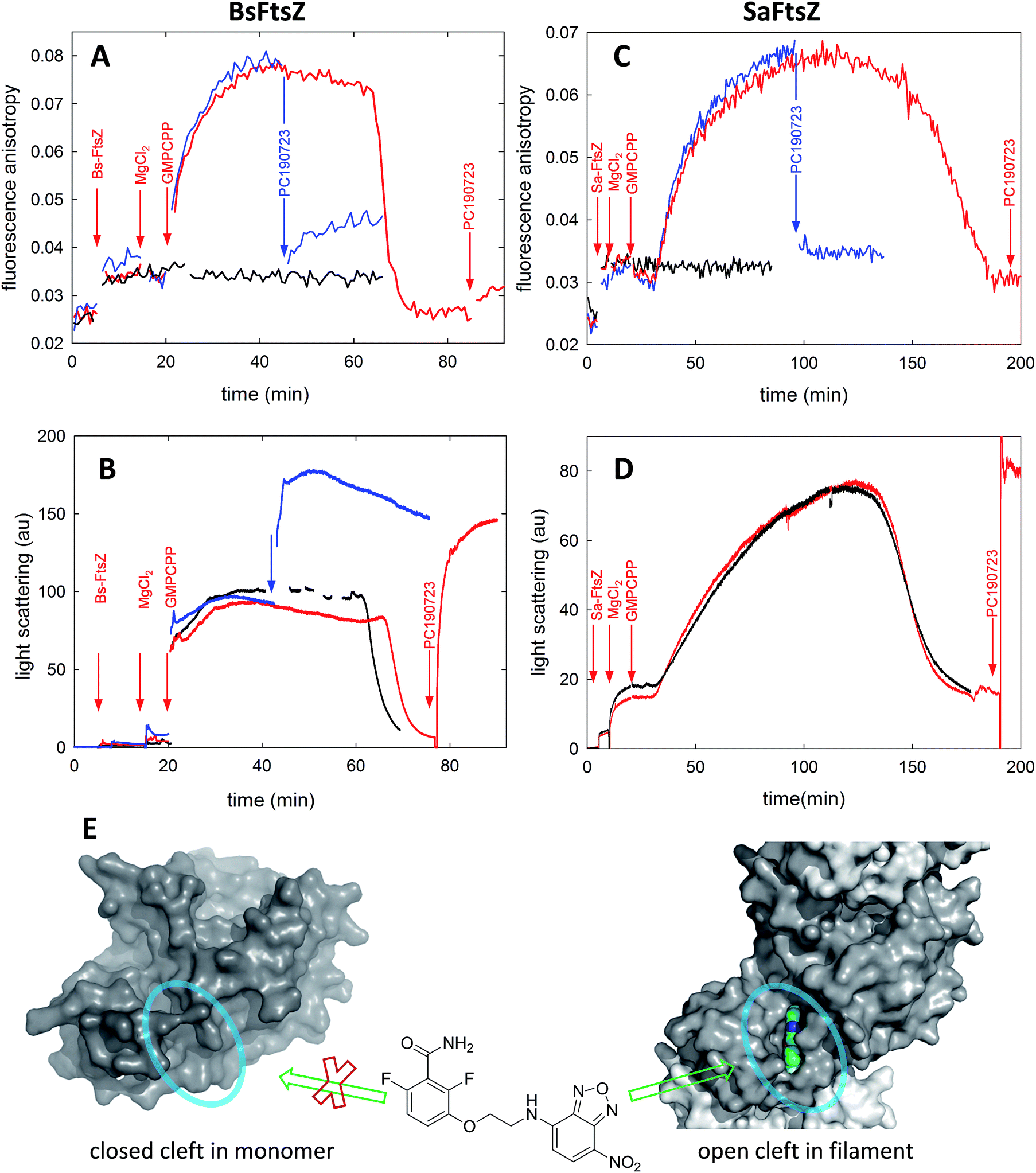 | ||
| Fig. 4 FtsZ assembly monitored with fluorescent probe 6. (A) Anisotropy time courses of probe 6 during BsFtsZ assembly. Assembly was initiated by adding GMPCPP. BsFtsZ (10 μM), MgCl2 (10 mM), GMPCPP (0.1 mM) and PC190723 (10 μM) were successively added to 6 (10 μM), as indicated by the arrows. The red sample was allowed to assemble and disassemble upon nucleotide consumption, whereas PC190723 was added to the blue sample at the plateau anisotropy, and the black sample lacked GMPCPP. (B) Replicate experiments in which assembly was monitored by the right angle light scattering of BsFtsZ polymers, except the black sample that is a control made without probe. (C) Probe 6 fluorescence anisotropy time courses during SaFtsZ assembly, made similarly to panel A. (D) Light scattering time courses of SaFtsZ with (red) and without probe (black). Notice that the probe minimally perturbs FtsZ assembly. Due to technical limitations for simultaneously measuring scattering and anisotropy with our spectrofluorometers it is difficult to ascertain whether the anisotropy changes are simultaneous with or lag after the scattering changes. (E) Scheme interpreting the results. The benzamide probe binds into the interdomain cleft that opens upon FtsZ assembly, thus it binds to FtsZ polymers (Fig. S2†) rather than to FtsZ monomers (Fig. S3†). This is exemplified by the closed cleft crystal structures of BsFtsZ monomers on the left side (represented here by PDB 2vxy), which cannot fit the parent compound PC190723 and the open cleft (encircled) X-ray structure of SaFtsZ forming crystal filaments that bind PC190723, which is shown on the right side by the grey protein surface and colored ligand (PDB entries 3vob, 4dxd). | ||
A similar experiment performed on SaFtsZ gave comparable results (Fig. 4C and D). Notice, however, that a monomeric structure has not been reported for this protein, but the structures forming crystal filaments that are currently available show a characteristically open cleft (for example, PDB 3voa) where PC190723 binds (PDB 3vob, 4dxd). Thus the BsFtsZ and SaFtsZ results with probe 6 complement each other. The straightforward interpretation of our results is that the binding and dissociation events of fluorescent probe 6 monitor the opening and closing of the interdomain cleft upon FtsZ assembly and disassembly respectively. Therefore both BsFtsZ and SaFtsZ monomers have predominantly closed interdomain clefts that open in polymers (Fig. 4E).
Our solution studies thus demonstrate the structural mechanism of the FtsZ assembly switch, and suggest that the fluorescent probe detects cellular FtsZ filaments rather than the unassembled protein in the bacterial cytosol.
Conclusions
This work shows the FtsZ assembly switch in action as the cleft between the N- and C-terminal domains of cell division protein FtsZ is open for assembled subunits and closed for unassembled monomers. To this purpose we have developed new fluorescent analogs of the allosteric modulator PC190723, whose fluorescence anisotropy changes upon specific binding to FtsZ polymers. We have also found that these NDB-benzamide probes directly label FtsZ division rings in live bacterial cells. We envisage that competitive probe binding assays combined with cytological profiling methods58 may be employed to screen for allosteric inhibitors of FtsZ assembly in search of new antibiotics.Materials and methods
Detailed synthetic procedures, docking and molecular dynamics methods, FtsZs purification and assembly, FtsZ mutants, probe fluorescence anisotropy, biochemical, microbiological and microscopy methods are described in the ESI.† Experiments with BsFtsZ and EcFtsZ were made in 50 mM HEPES, 50 mM KCl, 1 mM EDTA, pH 6.8, plus 10 mM MgCl2 and 1 mM GTP or 0.1 mM GMPCPP at 25 °C. Experiments with SaFtsZ were made in 50 mM MES, 50 mM KCl, 1 mM EDTA, pH 6.5, complemented as for BsFtsZ. We gratefully acknowledge the Leibniz Supercomputing Centre under Partnership for Advanced Computing in Europe (PRACE) and the Red Española de Supercomputacion for providing high-performance computing resources.Acknowledgements
We thank David Juan for proteins purification. This work was supported by grants BFU2014-51823-R (JMA), BFU2013-44308-P (PC), SAF2013-48271-C2 (MLLR), CMS 2010/BMD-2353 (MLLR, PC, JMA), contracts Ramon y Cajal 2011-07900 (MAO), Miguel Servet (AJMG), postdoctoral MINECO contract FPDI-2013-16470 (RFM), doctoral fellowships FPI (LBRA), FPU (MA) and CSIC-JAE (ERA).References
- J. Lutkenhaus, S. Pichoff and S. S. Du, Cytoskeleton, 2012, 69, 778–790 CrossRef CAS PubMed.
- H. P. Erickson, D. E. Anderson and M. Osawa, Microbiol. Mol. Biol. Rev., 2010, 74, 504–528 CrossRef CAS PubMed.
- E. Xiao and E. D. Goley, Curr. Opin. Microbiol., 2016, 34, 90–96 CrossRef PubMed.
- A. J. F. Egan and W. Vollmer, Ann. N. Y. Acad. Sci., 2013, 1277, 8–28 CrossRef CAS PubMed.
- I. V. Hadjuk, C. D. A. Rodrigues and E. J. Harry, Semin. Cell Dev. Biol., 2016, 53, 2–9 CrossRef PubMed.
- D. P. Haeusser and W. Margolin, Nat. Rev. Microbiol., 2016, 14, 305–319 CrossRef CAS PubMed.
- Z. Li, M. J. Trimble, Y. V. Brun and G. J. Jensen, EMBO J., 2007, 26, 4694–4708 CrossRef CAS PubMed.
- P. Szwedziak, Q. Wang, T. A. M. Bharat, M. Tsim and J. Lowe, eLife, 2014, 3, 52 Search PubMed.
- M. Osawa and H. P. Erickson, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11000–11004 CrossRef CAS PubMed.
- M. Loose and T. J. Mitchison, Nat. Cell Biol., 2014, 16, 38–46 CrossRef CAS PubMed.
- G. Fu, T. Huang, J. Buss, C. Coltharp, Z. Hensel and X. A. Jie, PLoS One, 2010, 5, e12680 Search PubMed.
- M. P. Strauss, A. T. F. Liew, L. Turnbull, C. B. Whitchurch, L. G. Monahan and E. J. Harry, PLoS Biol., 2012, 10, e1001389 CAS.
- F. W. Si, K. Busiek, W. Margolin and S. X. Sun, Biophys. J., 2013, 105, 1976–1986 CrossRef CAS PubMed.
- J. S. Biteen, E. D. Goley, L. Shapiro and W. E. Moerner, ChemPhysChem, 2012, 13, 1007–1012 CrossRef CAS PubMed.
- V. W. Rowlett and W. Margolin, Biophys. J., 2014, 107, L17–L20 CrossRef CAS PubMed.
- S. J. Holden, T. Pengo, K. L. Meibom, C. F. Fernandez, J. Collier and S. Manley, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 4566–4571 CrossRef CAS PubMed.
- M. Jacq, V. Adam, D. Bourgeois, C. Moriscot, A. M. Di Guilmi, T. Vernet and C. Morlot, mBio, 2015, 6, e001108–e001115 CrossRef PubMed.
- J. Buss, C. Coltharp, G. Shtengel, X. X. Yang, H. Hess and J. Xiao, PLoS Genet., 2015, 11, 24 Search PubMed.
- C. Coltharp, J. Buss, T. M. Plumer and J. Xiao, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E1044–E1053 CrossRef CAS PubMed.
- K. Lewis, Nature, 2012, 485, 439–440 CrossRef CAS PubMed.
- D. J. Haydon, N. R. Stokes, R. Ure, G. Galbraith, J. M. Bennett, D. R. Brown, P. J. Baker, V. V. Barynin, D. W. Rice, S. E. Sedelnikova, J. R. Heal, J. M. Sheridan, S. T. Aiwale, P. K. Chauhan, A. Srivastava, A. Taneja, I. Collins, J. Errington and L. G. Czaplewski, Science, 2008, 321, 1673–1675 CrossRef CAS PubMed.
- L. G. Czaplewski, I. Collins, E. A. Boyd, D. Brown, S. P. East, M. Gardiner, R. Fletcher, D. J. Haydon, V. Henstock, P. Ingram, C. Jones, C. Noula, L. Kennison, C. Rockley, V. Rose, H. B. Thomaides-Brears, R. Ure, M. Whittaker and N. R. Stokes, Bioorg. Med. Chem. Lett., 2009, 19, 524–527 CrossRef CAS PubMed.
- D. J. Haydon, J. M. Bennett, D. Brown, I. Collins, G. Galbraith, P. Lancett, R. Macdonald, N. R. Stokes, P. K. Chauhan, J. K. Sutariya, N. Nayal, A. Srivastava, J. Beanland, R. Hall, V. Henstock, C. Noula, C. Rockley and L. Czaplewski, J. Med. Chem., 2010, 53, 3927–3936 CrossRef CAS PubMed.
- J. M. Andreu, C. Schaffner-Barbero, S. Huecas, D. Alonso, M. L. Lopez-Rodriguez, L. B. Ruiz-Avila, R. Nunez-Ramirez, O. Llorca and A. J. Martin-Galiano, J. Biol. Chem., 2010, 285, 14239–14246 CrossRef CAS PubMed.
- D. W. Adams, L. J. Wu, L. G. Czaplewski and J. Errington, Mol. Microbiol., 2011, 80, 68–84 CrossRef CAS PubMed.
- D. W. Adams, L. J. Wu and J. Errington, Mol. Microbiol., 2016, 99, 1028–1042 CrossRef CAS PubMed.
- C. M. Tan, A. G. Therien, J. Lu, S. H. Lee, A. Caron, C. J. Gill, C. Lebeau-Jacob, L. Benton-Perdomo, J. M. Monteiro, P. M. Pereira, N. L. Elsen, J. Wu, K. Deschamps, M. Petcu, S. Wong, E. Daigneault, S. Kramer, L. Z. Liang, E. Maxwell, D. Claveau, J. Vaillancourt, K. Skorey, J. Tam, H. Wang, T. C. Meredith, S. Sillaots, L. Wang-Jarantow, Y. Ramtohul, E. Langlois, F. Landry, J. C. Reid, G. Parthasarathy, S. Sharma, A. Baryshnikova, K. J. Lumb, M. G. Pinho, S. M. Soisson and T. Roemer, Sci. Transl. Med., 2012, 4, 126ra35 Search PubMed.
- M. Kaul, L. Mark, Y. Z. Zhang, A. K. Parhi, Y. L. Lyu, J. Pawlak, S. Saravolatz, L. D. Saravolatz, M. P. Weinstein, E. J. LaVoie and D. S. Pilch, Antimicrob. Agents Chemother., 2015, 59, 4845–4855 CrossRef CAS PubMed.
- A. J. Lepak, A. Parhi, M. Madison, K. Marchillo, J. VanHecker and D. R. Andes, Antimicrob. Agents Chemother., 2015, 59, 6568–6574 CrossRef CAS PubMed.
- M. Kaul, L. Mark, A. K. Parhi, E. J. LaVoie and D. S. Pilch, Antimicrob. Agents Chemother., 2016, 60, 4290–4296 CrossRef PubMed.
- T. Läppchen, V. A. Pinas, A. F. Hartog, G. J. Koomen, C. Schaffner-Barbero, J. M. Andreu, D. Trambaiolo, J. Lowe, A. Juhem, A. V. Popov and T. den Blaauwen, Chem. Biol., 2008, 15, 189–199 CrossRef PubMed.
- F. Marcelo, S. Huecas, L. B. Ruiz-Avila, F. J. Canada, A. Perona, A. Poveda, S. Martin-Santamaria, A. Morreale, J. Jimenez-Barbero and J. M. Andreu, J. Am. Chem. Soc., 2013, 135, 16418–16428 CrossRef CAS PubMed.
- S. Huecas, F. Marcelo, A. Perona, L. B. Ruiz-Avila, A. Morreale, F. J. Canada, J. Jimenez-Barbero and J. M. Andreu, ACS Chem. Biol., 2015, 10, 2382–2392 CrossRef CAS PubMed.
- A. Plaza, J. L. Keffer, G. Bifulco, J. R. Lloyd and C. A. Bewley, J. Am. Chem. Soc., 2010, 132, 9069–9077 CrossRef CAS PubMed.
- L. B. Ruiz-Avila, S. Huecas, M. Artola, A. Vergonos, E. Ramirez-Aportela, E. Cercenado, I. Barasoain, H. Vazquez-Villa, M. Martin-Fontecha, P. Chacon, M. L. Lopez-Rodriguez and J. M. Andreu, ACS Chem. Biol., 2013, 8, 2072–2083 CrossRef CAS PubMed.
- M. Artola, L. B. Ruiz-Avila, A. Vergoñós, S. Huecas, L. Araujo-Bazán, M. Martín-Fontecha, H. Vázquez-Villa, C. Turrado, E. Ramírez-Aportela, A. Hoegl, A. Nodwell, I. Barasoain, P. Chacón, S. A. Sieber, J. M. Andreu and M. L. Lopez-Rodriguez, ACS Chem. Biol., 2015, 10, 834–843 CrossRef CAS PubMed.
- C. Schaffner-Barbero, M. Martin-Fontecha, P. Chacon and J. M. Andreu, ACS Chem. Biol., 2012, 7, 268–276 CrossRef PubMed.
- P. Sass and H. Brotz-Oesterhelt, Curr. Opin. Microbiol., 2013, 16, 522–530 CrossRef CAS PubMed.
- T. den Blaauwen, J. M. Andreu and O. Monasterio, Bioorg. Chem., 2014, 55, 27–38 CrossRef CAS PubMed.
- E. Nogales, K. H. Downing, L. A. Amos and J. Lowe, Nat. Struct. Mol. Biol., 1998, 5, 451–458 CAS.
- M. A. Oliva and J. M. Andreu, eLS, 2014, DOI:10.1002/9780470015902.a0025586.
- R. Zhang, G. M. Alushin, A. Brown and E. Nogales, Cell, 2015, 162, 849–859 CrossRef CAS PubMed.
- R. M. Buey, J. F. Diaz and J. M. Andreu, Biochemistry, 2006, 45, 5933–5938 CrossRef CAS PubMed.
- L. M. Rice, E. A. Montabana and D. A. Agard, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5378–5383 CrossRef CAS PubMed.
- G. J. Brouhard and L. M. Rice, J. Cell Biol., 2014, 207, 323–334 CrossRef CAS PubMed.
- S. Huecas, O. Llorca, J. Boskovic, J. Martin-Benito, J. M. Valpuesta and J. M. Andreu, Biophys. J., 2008, 94, 1796–1806 CrossRef CAS PubMed.
- A. Dajkovic, A. Mukherjee and J. Lutkenhaus, J. Bacteriol., 2008, 190, 2513–2526 CrossRef CAS PubMed.
- E. R. Miraldi, P. J. Thomas and L. Romberg, Biophys. J., 2008, 95, 2470–2486 CrossRef CAS PubMed.
- G. Lan, A. Dajkovic, D. Wirtz and S. X. Sun, Biophys. J., 2008, 95, 4045–4056 CrossRef CAS PubMed.
- A. J. Martín-Galiano, R. M. Buey, M. Cabezas and J. M. Andreu, J. Biol. Chem., 2010, 285, 22554–22565 CrossRef PubMed.
- Y. Chen and H. P. Erickson, Biochemistry, 2011, 50, 4675–4684 CrossRef CAS PubMed.
- M. A. Oliva, D. Trambaiolo and J. Lowe, J. Mol. Biol., 2007, 373, 1229–1242 CrossRef CAS PubMed.
- T. Matsui, J. Yamane, N. Mogi, H. Yamaguchi, H. Takemoto, M. Yao and I. Tanaka, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2012, 68, 1175–1188 CAS.
- N. L. Elsen, J. Lu, G. Parthasarathy, J. C. Reid, S. Sharma, S. M. Soisson and K. J. Lumb, J. Am. Chem. Soc., 2012, 134, 12342–12345 CrossRef CAS PubMed.
- E. Ramirez-Aportela, J. R. Lopez-Blanco, J. M. Andreu and P. Chacon, Biophys. J., 2014, 107, 2164–2176 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, 3rd edn, 2006 Search PubMed.
- O. Kocaoglu and E. E. Carlson, Nat. Chem. Biol., 2016, 12, 472–478 CrossRef CAS PubMed.
- L. Araujo-Bazán, L. B. Ruiz-Avila, D. Andreu, S. Huecas and J. M. Andreu, Front. Microbiol., 2016, 7, 1558, DOI:10.3389/fmicb.2016.01558.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary Fig. S1 to S10, materials and methods, molecular dynamics Movies M1–M4. See DOI: 10.1039/c6sc03792e |
| ‡ These authors share first authorship of this work. |
| § Present address: Leiden Institute of Chemistry, Leiden University, Leiden, The Netherlands. |
| ¶ Present address: Max Planck Institute of Biochemistry, Martinsried, Germany. |
| || Present address: Centro Nacional de Microbiología-Instituto de Salud Carlos III, Madrid, Spain. |
| This journal is © The Royal Society of Chemistry 2017 |