
Structural and kinetic changes to small-pore Cu-zeolites after hydrothermal aging treatments and selective catalytic reduction of NOx with ammonia†
Jonatan D.
Albarracin-Caballero‡
a,
Ishant
Khurana‡
a,
John R.
Di Iorio
a,
Arthur J.
Shih
a,
Joel E.
Schmidt
b,
Michiel
Dusselier
bc,
Mark E.
Davis
b,
Aleksey
Yezerets
d,
Jeffrey T.
Miller
a,
Fabio H.
Ribeiro
*a and
Rajamani
Gounder
 *a
*a
aCharles D. Davidson School of Chemical Engineering, Purdue University, 480 Stadium Mall Drive, West Lafayette, IN 47907, USA. E-mail: fabio@purdue.edu; rgounder@purdue.edu
bChemical Engineering, California Institute of Technology, 1200 E. California Boulevard, MC 210-41, Pasadena, CA 91125, USA
cCenter for Surface Chemistry and Catalysis, KU Leuven, Celestijnenlaan 200F, 3001 Heverlee, Belgium
dCummins Inc., 1900 McKinley Ave., MC 50183, Columbus, IN 47201, USA
First published on 7th December 2016
Abstract
Three small-pore, eight-membered ring (8-MR) zeolites of different cage-based topology (CHA, AEI, RTH), in their proton- and copper-exchanged forms, were first exposed to high temperature hydrothermal aging treatments (1073 K, 16 h, 10% (v/v) H2O) and then to reaction conditions for low temperature (473 K) standard selective catalytic reduction (SCR) of NOx with ammonia, in order to study the effect of zeolite topology on the structural and kinetic changes that occur to Cu-zeolites used in NOx abatement. UV-visible spectra were collected to monitor changes to Cu structure and showed that band intensities for isolated, hydrated Cu2+ cations (∼12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 cm−1) remain constant after hydrothermal aging, but decrease in intensity upon subsequent exposure to low temperature SCR reaction conditions. Standard SCR rates (per Cu, 473 K), activation energies, and reaction orders are similar between Cu-AEI and Cu-CHA zeolites before and after hydrothermal aging, although rates are lower after hydrothermal aging as expected from the decreases in intensity of UV-visible bands for Cu2+ active sites. For Cu-RTH, rates are lower (by 2–3×) and apparent activation energies are lower (by ∼2×) than for Cu-AEI or Cu-CHA. These findings suggest that the RTH framework imposes internal transport restrictions, effectively functioning as a one-dimensional framework during SCR catalysis. Hydrothermal aging of Cu-RTH results in complete deactivation and undetectable SCR rates, despite X-ray diffraction patterns and Ar micropore volumes (87 K) that remain unchanged after hydrothermal aging treatments and subsequent SCR exposure. These findings highlight some of the differences in low temperature SCR behavior among small-pore Cu-zeolites of different topology, and the beneficial properties conferred by double six-membered ring (D6R) composite building units. They demonstrate that deleterious structural changes to Cu sites occur after exposure to hydrothermal aging conditions and SCR reactants at low temperatures, likely reflecting the formation of inactive copper-aluminate domains. Therefore, the viability of Cu-zeolites for practical low temperature NOx SCR catalysis cannot be inferred solely from assessments of framework structural integrity after hydrothermal aging treatments, but also require Cu active site and kinetic characterization after hydrothermally aged zeolites are exposed to low temperature SCR reaction conditions.
500 cm−1) remain constant after hydrothermal aging, but decrease in intensity upon subsequent exposure to low temperature SCR reaction conditions. Standard SCR rates (per Cu, 473 K), activation energies, and reaction orders are similar between Cu-AEI and Cu-CHA zeolites before and after hydrothermal aging, although rates are lower after hydrothermal aging as expected from the decreases in intensity of UV-visible bands for Cu2+ active sites. For Cu-RTH, rates are lower (by 2–3×) and apparent activation energies are lower (by ∼2×) than for Cu-AEI or Cu-CHA. These findings suggest that the RTH framework imposes internal transport restrictions, effectively functioning as a one-dimensional framework during SCR catalysis. Hydrothermal aging of Cu-RTH results in complete deactivation and undetectable SCR rates, despite X-ray diffraction patterns and Ar micropore volumes (87 K) that remain unchanged after hydrothermal aging treatments and subsequent SCR exposure. These findings highlight some of the differences in low temperature SCR behavior among small-pore Cu-zeolites of different topology, and the beneficial properties conferred by double six-membered ring (D6R) composite building units. They demonstrate that deleterious structural changes to Cu sites occur after exposure to hydrothermal aging conditions and SCR reactants at low temperatures, likely reflecting the formation of inactive copper-aluminate domains. Therefore, the viability of Cu-zeolites for practical low temperature NOx SCR catalysis cannot be inferred solely from assessments of framework structural integrity after hydrothermal aging treatments, but also require Cu active site and kinetic characterization after hydrothermally aged zeolites are exposed to low temperature SCR reaction conditions.
1. Introduction
Leading emissions control strategies for the abatement of hazardous nitrogen oxide pollutants (NOx, x = 1, 2) in lean-burn and diesel engine exhaust involve their selective catalytic reduction (SCR) with ammonia, which is generated from the decomposition of urea stored in an on-board tank. Cu- and Fe-exchanged molecular sieves used to practice automotive SCR aftertreatment1–6 are required to retain sufficient SCR performance after excursions to high temperatures (>923 K) in the presence of steam (∼7% H2O (v/v)),7–10 conditions experienced during regeneration of particulate filters. The structural integrity of molecular sieve frameworks with medium pores (e.g., MFI,11–16 FER;11 ∼0.5 nm diam.) and large pores (e.g., BEA,13,14,17,18 FAU;14 >0.6 nm diam.) becomes compromised during hydrothermal aging;11–18 moreover, active sites within such frameworks can be poisoned chemically by residual hydrocarbons in exhaust streams. These deactivation issues are mitigated within small-pore, eight-membered ring (8-MR; <0.4 nm diam.) frameworks, which led to the advent of the aluminosilicate (SSZ-13) and silicoaluminophosphate (SAPO-34) compositions of the chabazite (CHA) topology8,19,20 as commercially used NOx SCR catalysts. Other small-pore molecular sieves with three-dimensional (e.g., AFX,13 AEI,21 KFI,13 SAV,22 SFW23,24) and two-dimensional (e.g., LEV,13 DDR,13 RTH25) pore connectivity have been considered as alternatives to CHA molecular sieves, based on observations that small-pore frameworks retain their structural integrity after exposure to hydrothermal aging conditions.Hydrothermal aging of zeolites leads to the removal of aluminum atoms from framework locations,13,26–28 which stabilize redox-active, extraframework Cu cations and ammonium species during SCR catalysis. Framework dealumination generally leads to sintering of extraframework alumina and mixed oxide domains with concomitant losses in microporous structure29–31 that may restrict molecular traffic to active sites. The effects of hydrothermal aging treatments on dealumination have been assessed by changes in Al coordination using solid-state 27Al magic angle spinning nuclear magnetic resonance (MAS NMR), and in long-range crystalline structure using X-ray diffraction (XRD) and micropore volume measurements.14,32–35 Structural changes upon dealumination are more severe in Cu-exchanged medium-pore and large-pore zeolites (e.g., Cu-MFI, Cu-BEA) than in small-pore zeolites (e.g., Cu-CHA, Cu-AEI),14,17,36 which are more recalcitrant to hydrothermal deactivation. Aluminum hydroxide species (Al(OH)3; ∼0.5 nm in diam.) formed upon dealumination at high temperatures are thought to be unable to diffuse through 8-MR windows in CHA13 and AEI36 (∼0.38 nm in diam.), which prevents the formation of larger extraframework alumina aggregates and allows for reincorporation of monomeric Al species within framework vacancy positions at low temperatures. Dealumination upon hydrothermal aging is also suppressed by the presence of extraframework cations (e.g., Cu, Na, Li, Mg),36–38 which remove Brønsted acid sites that are vulnerable locations for hydrolysis of framework bonds.39–41 Consequently, the ability of a zeolite framework to resist dealumination and retain its structural integrity upon hydrothermal aging has been used to identify promising candidates for practical NOx SCR catalysis.
Deactivation caused by hydrothermal aging of molecular sieves may also reflect changes to the structure and location of extraframework Cu cations, such as their aggregation into larger Cu oxide species (CuxOy), because the former isolated cations have been implicated as active sites for low temperature (473 K) SCR catalysis42–44 while the latter oxide clusters are unreactive.45 The disappearance of isolated Cu2+ cations upon hydrothermal aging of Cu-SSZ-13 has been inferred from the attenuation of absorption features characteristic of framework (T–O–T) vibrations (900 and 940 cm−1) perturbed by ion-exchanged Cu species in diffuse-reflectance infrared (DRIFTS) spectra, from decreases in the amount of NH3 desorbed from Lewis acidic Cu cations (∼553 K) in TPD experiments,32 and from decreases in electron paramagnetic resonance (EPR) signals for isolated Cu2+ cations.33 The aggregation of isolated Cu2+ cations into larger CuxOy domains upon hydrothermal aging has been detected by electron microscopy (TEM, SEM) and energy dispersive X-ray spectroscopy (EDX).32–34 Isolated Cu2+ cations have also been proposed to interact with extraframework Al species, formed via dealumination, to generate inactive copper-aluminate domains in hydrothermally aged Cu-SSZ-13, evident in extended X-ray absorption fine structure (EXAFS) spectra that show decreased Cu–Cu scattering distances and increased Cu–Al scattering distances, and in H2 temperature programmed reduction (TPR) profiles that show decreased intensities of lower temperature (500–670 K) reduction features for isolated Cu cations with the concomitant appearance of higher temperature (790–880 K) reduction features attributed to copper-aluminates.35 Additionally, 27Al MAS NMR spectra show decreased intensities for tetrahedral Al lines (δ ∼ 60 ppm) without concomitant increases in intensities for octahedral Al lines (δ ∼ 0 ppm), suggesting that interactions of Al with paramagnetic Cu render them invisible to NMR detection.14 These results provide evidence for one possible deactivation mechanism of Cu-SSZ-13 through loss of isolated Cu2+ active sites during hydrothermal aging, but do not account for structural changes to active sites that may result from subsequent exposure to standard SCR reactants. Thus, identifying new zeolite topologies that retain SCR reactivity after hydrothermal aging treatments requires knowledge of how such treatments, and subsequent exposure to SCR reaction conditions, affect the structures of both Cu active sites and the zeolite framework.
Here, we investigate the effects of hydrothermal aging and subsequent exposure to standard SCR reactants at low temperatures (473 K) on the structural and active site changes experienced by three different small-pore Cu-exchanged zeolites (Cu-CHA, Cu-AEI, Cu-RTH). Bulk characterization techniques, including XRD patterns and micropore volumes, reveal only subtle differences between Cu-zeolites before and after hydrothermal aging, and after subsequent exposure to low temperature SCR reaction conditions, and are unable to provide direct insight into the decreases in SCR reactivity measured on hydrothermally aged, small-pore Cu-zeolites. We provide evidence that exposure of hydrothermally aged catalysts to SCR reaction conditions at low temperatures causes further structural changes to active Cu sites that are detectable by UV-visible spectroscopy, consistent with the formation of mixed copper-aluminate domains via reaction with extraframework Al species formed upon dealumination during hydrothermal aging. These findings demonstrate that active site and structural characterization of hydrothermally aged Cu-zeolites after exposure to SCR reactants at low temperatures provide more accurate inferences about their catalytic behavior.
2. Experimental methods
2.1. Catalyst synthesis and treatment
A sample of CHA (SSZ-13) zeolite with a Si/Al ratio of 15 was made as reported elsewhere.46 Briefly, 28.4 g of N,N,N-trimethyl-1-adamantylammonium hydroxide (TMAdaOH, Sachem, 25 wt%) were mixed with 71.4 g of deionized water (18.2 MΩ), 0.87 g of aluminum hydroxide (SPI Pharma, 99.9 wt%, 0325 grade), and 34.6 g of 0.1 M sodium hydroxide (NaOH; Alfa Aesar), then stirred for 15 minutes at ambient conditions. 10 g of fumed silica (Cab-o-Sil M-5) were added to the mixture and stirred for 2 hours at ambient conditions. The final molar composition of the synthesis solution was 1SiO2/0.033Al2O3/0.20TMAdaOH/0.02NaOH/23.8H2O. All reagents were used without further purification. The resulting solution was transferred to eight Teflon-lined Parr autoclaves (45 mL each) and held at 433 K for 10 days under rotation.A sample of AEI (SSZ-39) zeolite with a Si/Al ratio of 9.5 was synthesized in a rotating oven at 413 K for 4 days as reported elsewhere,47 using cis-2,6-dimethylpiperidinium hydroxide as the organic structure directing agent (OSDA). The molar composition of the synthesis mixture was 1SiO2/0.017Al2O3/0.07OSDA/0.65OH−/0.58Na+/12.3H2O, obtained by mixing (aqueous) OSDA, NaOH (1 M, RT Baker), double distilled water, sodium silicate (N® type, PQ Corporation) and CBV500 (NH4-USY, Si/Al = 2.6, Zeolyst). A sample of RTH (SSZ-50) zeolite with a Si/Al ratio of 15 was made using the CBV720 synthesis protocol reported elsewhere.48–50
As-synthesized zeolites were washed alternately with deionized water and acetone, recovered via centrifugation, and dried at 323 K for 24 hours. The dried samples were then treated to 873 K (0.0083 K s−1) in air (Commercial grade, Indiana Oxygen) for 6 hours before ion-exchanging in an aqueous 0.1 M NH4NO3 solution (Sigma Aldrich; 1000 mL per g zeolite) at 353 K for 10 hours. NH4-exchanged zeolites were washed with deionized H2O, recovered via centrifugation, dried at 323 K for 24 hours, then treated at 823 K (0.0083 K s−1) in air for 6 hours to obtain H-form zeolites. Cu-exchanged CHA, AEI, and RTH zeolites were prepared via liquid phase ion-exchange of H-form zeolites using an aqueous 0.2 M Cu(NO3)2 solution (99.999% trace metals basis, Sigma-Aldrich; 150 mL per g zeolite) at ambient temperature for 4 hours. The pH during the exchange was not controlled and the final pH of the solution was ∼3.6.
Hydrothermal aging experiments were performed on Cu-zeolites in a three-zone horizontal tube furnace (Applied Test Systems Series 3210), in which each zone was equipped with independent temperature control (Watlow EZ-Zone PM Express). Once the furnace temperature reached 373 K, water was introduced via syringe pump (KD Scientific Legato 100) into a stream of flowing air (100 mL min−1, 99.999%, Indiana Oxygen), which was transferred to the furnace through stainless steel lines held at >373 K. Approximately 1 gram of catalyst was loaded into quartz boats held within the tube furnace and treated to 1073 K (0.033 K s−1) for 16 hours in flowing air (100 mL min−1, 99.999%, Indiana Oxygen) containing 10% (v/v) water. After treatment for 16 hours at 1073 K, water was removed from the flowing air stream while the sample was cooled to ambient.
2.2. Catalyst structural characterization
Powder diffraction patterns were collected using a Rigaku SmartLab diffractometer with a Cu Kα radiation source (1.76 kW), from 4 to 40° with a scan rate of 0.05° s−1 and a step size of 0.01°. Diffraction patterns are normalized so that the maximum peak intensity in each pattern is unity. The diffraction patterns were compared to reference patterns to confirm the RTH, CHA and AEI topologies.51Ar adsorption isotherms were used to determine micropore volumes on zeolite samples (87 K) using a Micromeritics ASAP 2020 Surface Area and Porosity Analyzer. Micropore volumes were obtained by converting adsorbed gas volumes (cm3 gcat−1 at STP) to liquid volumes assuming the liquid density of Ar at 87 K. Samples were pelleted and sieved to retain particles between 125–250 μm in diameter. Samples (0.03–0.05 g) were degassed by heating to 393 K (0.167 K s−1) under high vacuum (∼5 μm Hg) for 2 h, and then heating to 623 K (0.167 K s−1) under high vacuum (∼5 μm Hg) and holding for 9 h. Micropore volumes (cm3 gcat−1 at STP) were estimated from extrapolation of the linear volumetric uptake during the beginning of mesopore filling (∼0.08–0.30 P/P0) to zero relative pressure, which agreed with micropore volumes estimated from analyzing the semi-log derivative plot of the adsorption isotherm (∂(Vads)/∂(ln(P/P0)) vs. ln(P/P0)).
In order to quantify the fractions of framework and extraframework Al, 27Al magic angle spinning nuclear magnetic resonance (MAS NMR) spectra were recorded on H-form and Cu-form CHA, AEI and RTH zeolite samples. NMR spectra were collected using a Chemagnetics CMX-Infinity 400 spectrometer in a wide-bore 9.4 Tesla magnet (Purdue Interdepartmental NMR Facility) and were acquired at ambient conditions using a 2.3 μs pulse (equivalent to ca. 30 degrees), an acquisition time of 12.8 ms and a relaxation delay of 1 s, and were measured at 104.24 MHz and a MAS rate of 5 kHz. 1H decoupling was used during acquisition, employing two-pulse phase modulation (TPPM) scheme. Prior to packing in a 4 mm ZrO2 rotor, zeolite samples were hydrated by holding for >48 h in a desiccator containing a saturated potassium chloride (KCl) solution. All 27Al MAS NMR spectra are referenced to a static sample of AlCl3 dissolved in D2O (0 ppm 27Al line).
Diffuse reflectance UV-visible spectra were recorded under ambient conditions using a Varian UV-vis-NIR spectrophotometer (Cary 5000) with a diffuse reflectance accessory consisting of two ellipsoidal mirrors (Harrick Scientific Praying Mantis). Barium sulfate (BaSO4, 99.9%, Sigma-Aldrich) was used as the 100% reflectance standard. An ex situ sample holder was loaded with 0.1 g of sample, which was pelleted and sieved to retain particles between 125–250 μm in diameter. Spectra were collected from 7000 to 50000 cm−1 with a scan speed of 2000 cm−1 min−1, and spectra of the H-form zeolite was subtracted from those for corresponding Cu-zeolites to correct for contributions of absorption from the framework.
2.3. Brønsted acid site quantification using NH3 titration methods
The total number of Brønsted acid sites (H+) on H-form and Cu-exchanged zeolites was quantified by temperature programmed desorption (TPD) of NH3 on a gas-phase plug flow reactor, as described by Bates et al.,52 using a procedure described elsewhere.53 For H-form zeolites, NH3 saturation was performed via aqueous-phase exchange with NH4+ cations, as reported elsewhere.52 For Cu-exchanged zeolites, samples were saturated with 500 ppm NH3 diluted with He (99.999%, UHP, Indiana Oxygen) at 433 K for 2 h with a total flow rate of 350 mL min−1. Following this NH3 saturation step, the sample was flushed with 2.5–3.0% water in UHP He (wet purge) at 433 K for 8 h while maintaining the same total flow rate to desorb NH3 bound to non-protonic sites. Following the wet purge step, samples were heated to 820 K (0.167 K s−1) under flowing He (UHP, 350 mL min−1). The total moles of NH3 desorbed during the TPD experiment was measured using on-board calibrations in an MKS Multigas 2030 gas-phase FT-IR spectrometer.522.4. Kinetic measurements of standard SCR turnover rates
Standard selective catalytic reduction (SCR) kinetics were measured on a bench-top tubular glass reactor described elsewhere.52 All samples were sieved to a nominal size of 125–250 μm and diluted with silica gel to obtain a bed height of ∼2.5 cm. Steady-state kinetic data were collected at NO conversions below 20% (differential); thus, the entire catalyst bed was exposed to approximately the same gas concentrations. Under standard SCR conditions, the reactant gas mixture comprised 300 ppm NO (3.6% NO/Ar, Praxair), 300 ppm NH3 (3.0% NH3/Ar, Praxair), 7% CO2 (liquid, Indiana Oxygen), 10% O2 (99.5%, Indiana Oxygen), 2.5% H2O (deionized, 18.2 MΩ, introduced through saturator), and balance N2 (99.999% UHP, Indiana Oxygen). For all kinetic measurements, the total gas stream was maintained at a flow rate of 1.5 L min−1 and at ambient pressure (∼101 kPa). Apparent reaction orders were measured by independently varying partial pressures of NH3 (0.02–0.05 kPa), NO (0.02–0.05 kPa) or O2 (5–15 kPa) in the reactant gas stream, and adjusting the balance N2 to maintain a constant total gas flow rate and pressure. Apparent activation energies were measured under standard SCR conditions by varying the temperature between 444–476 K. Outlet gas concentrations were analyzed using on-board gas calibrations on an MKS MultigasTM 2030 gas-phase Fourier transform infrared (FTIR) spectrometer and NO, NO2, NH3, CO2, and H2O concentration data was recorded every 0.95 s. Kinetic measurements were recorded after waiting for outlet gas concentrations to reach steady-state, which typically occurred after 2–4 hours. Reactant pressures and temperatures were then varied over the course of 18 hours, and finally returned to initial conditions to verify that catalytic rates returned to their initial steady-state values and that the catalyst had not undergone any deactivation.3. Results and discussion
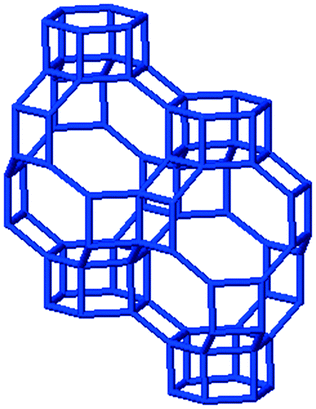
3.1. Structural features of CHA, AEI and RTH topologies
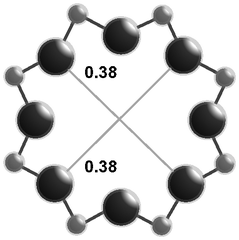
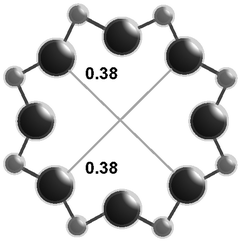
The salient structural features of the three molecular sieve framework topologies studied here are summarized in Table 1. The CHA framework46 has three-dimensional micropore interconnectivity and is formed by the repetitive stacking of a hexagonal array of planar 6-membered rings (6-MR) connected in an AABBCC-type stacking scheme that form hexagonal prisms (double 6-MR). These double 6-MR (D6R) units are ordered to form large chabazite cages that are ∼0.73 nm in diameter, which are limited by symmetric 8-MR windows that are ∼0.38 nm in diameter. The CHA framework contains only one crystallographically unique T-site and its unit cell contains 36 tetrahedrally-coordinated atoms (T-atoms) connected by 4-MR, 6-MR, and 8-MR units that are shared between adjacent cages.| Framework | CHA | AEI | RTH |
|---|---|---|---|
| a Structural information from the International Zeolite Association structural database.51 b Window diameter taken as the maximum diameter of a sphere that can diffuse through the framework.68 c Cage size taken as the maximum diameter of a sphere that be occluded within the framework.68 | |||
| Crystal topology |
|
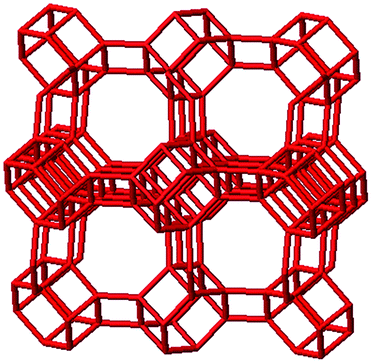
|
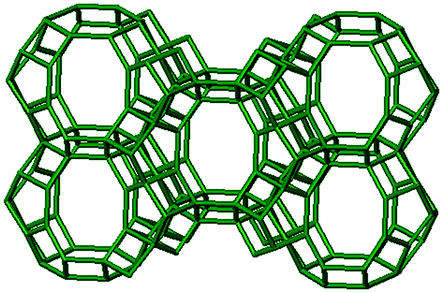
|
|
|
|
|
| Zeolite trade name | SSZ-13 (ref. 46) | SSZ-39 (ref. 54) | SSZ-50 (ref. 55) |
| Space group |
R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m m |
Cmcm | C2/m |
| Ring sizes (X-MR)a | 8, 6, 4 | 8, 6, 4 | 8, 6, 5, 4 |
| Number of unique T-sitesa | 1 | 3 | 4 |
| Connectivitya | 3-D | 3-D | 2-D |
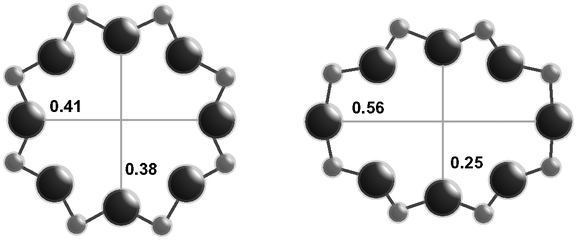
| Window diameterb (nm) | 0.38 × 0.38 | 0.38 × 0.38 | 0.54 × 0.25 |
| 0.41 × 0.38 | |||
| Cage diameterc (nm) | 0.73 | 0.73 | 0.81 |
The AEI framework54 also has three-dimensional micropore interconnectivity and is constructed from a hexagonal array of 6-MR units similar to CHA, but neighboring D6R units are rotated 180° with respect to each other (Table 1). The D6R units are ordered to form AEI cavities that are ∼0.73 nm in diameter and are contained within 4-MR, 6-MR, and 8-MR units, with access into AEI cavities limited by symmetric 8-MR windows that are ∼0.38 nm in diameter, as in the case of CHA. In contrast to the CHA unit cell, the AEI unit cell contains 48 T-atoms and three crystallographically-distinct T-sites.
The RTH framework55 is unique among the three small-pore zeolites studied here because it does not contain D6R building units, but instead is formed by two sets of three 4-MR that are connected via 5-MR linkages. These chained 4-MR and 5-MR periodic building units are repeated with simple translations to form RTH cavities that are 0.81 nm in diameter, and are contained within 4-MR, 5-MR, 6-MR, and 8-MR units. Consequently, the RTH unit cell (32 T-atoms) contains both symmetric (0.38 nm × 0.41 nm) and asymmetric (0.25 nm × 0.56 nm) 8-MR windows that result in only two-dimensional pore interconnectivity. RTH contains four crystallographically-distinct T-sites, three of which occupy positions accessible through either of the two 8-MR windows, and one that resides within the interconnected 4-MR chain and is inaccessible from the RTH cavity.
3.2. Synthesis and characterization of H-form and Cu-form zeolites before hydrothermal aging
Powder XRD patterns of H-form AEI, CHA, and RTH zeolites (Fig. S.1, ESI†) were consistent with reported diffraction patterns for these topologies51 and did not show diffraction peaks for phase impurities. Ar adsorption isotherms (87 K) on H-form zeolites (Fig. S.2, ESI†) gave micropore volumes (Table 2) consistent with the AEI,54 CHA,46 and RTH55 topologies. 27Al MAS NMR spectra of H-form zeolites (Fig. S.3.1–S.3.3, ESI†) show predominantly Al incorporated into tetrahedral framework positions (Alf, δ ∼ 60 ppm), with Alf/Altot values quantified to be 0.85 for H-CHA, 0.85 for H-AEI and 0.94 for H-RTH (Table 2). The number of protons per framework Al atom (H+/Alf, Table 2) measured by NH3 TPD (Fig. 2) on H-form zeolites was 0.95 and 0.85 for H-CHA and H-AEI, respectively, indicating that nearly every framework Al generated a proton. In contrast, the H+/Alf value was much lower on H-RTH (0.60, Table 2), suggesting either that some framework Al atoms generate H+ sites that are inaccessible to NH3, or that not all Al atoms are associated with a corresponding proton site. In the RTH framework, it is plausible that some H+ sites are inaccessible to NH3, which has a kinetic diameter (∼0.26 nm)56 that is larger than one of the dimensions of the distorted RTH window (0.56 nm × 0.25 nm), and because one of the four T-sites in RTH is in a location that is inaccessible from the RTH cage. Infrared spectra collected after H-RTH was exposed to NH3 (433 K), however, showed complete disappearance of Brønsted acidic OH stretches (Fig. S.4, ESI†) indicating that all H+ sites are accessible to NH3. Therefore, the H+/Alf value of 0.60 on H-RTH reflects the presence of distorted Al structures that do not generate H+ sites, but are otherwise detected as Alf species in NMR spectra, as noted previously.45,57,58| Sample | Si/Al ratioa | Cua wt% | Cu/Al ratioa | V ads,micro (cm3 g−1) | V ads,meso (cm3 g−1) | H+/Al ratioc | Alf/Altotd | H+/Alf |
|---|---|---|---|---|---|---|---|---|
| a Elemental composition determined by atomic absorption spectroscopy (AAS). b Micropore and mesopore volumes determined from Ar adsorption isotherms (87 K) (Fig. S.2, ESI). c Number of H+ sites quantified by selective NH3 titration and temperature-programmed desorption. d Fraction of tetrahedrally coordinated Al determined from 27Al MAS NMR (Fig. S.3.1–S.3.3, ESI). | ||||||||
| H-CHA | 15 | — | — | 0.18 | 0.04 | 0.95 | 0.85 | 1.10 |
| Cu-CHA | 15 | 0.7 | 0.12 | 0.17 | 0.05 | 0.72 | 0.90 | — |
| H-AEI | 9.5 | — | — | 0.20 | 0.01 | 0.85 | 0.85 | 1.00 |
| Cu-AEI | 9.5 | 1.7 | 0.17 | 0.19 | 0.01 | 0.54 | 0.91 | — |
| H-RTH | 15 | — | — | 0.20 | 0.05 | 0.60 | 0.94 | 0.61 |
| Cu-RTH | 15 | 0.7 | 0.11 | 0.17 | 0.04 | 0.38 | 0.98 | — |
Powder XRD patterns of AEI, CHA, and RTH zeolites after Cu exchange do not show significant changes in structure compared to their respective H-form zeolites or the presence of bulk CuxOy (Fig. S.1, ESI†). The micropore volume of each Cu-exchanged zeolite decreased slightly (Table 2; Fig. S.2, ESI†) due to the presence of extraframework Cu cations, which occupy a small, but detectable, fraction of the void volume. Gaseous NH3 titration53,59 of residual H+ sites on Cu-CHA (Cu/Al = 0.12) shows that H+ sites are replaced with an exchange stoichiometry of two protons per Cu, reflecting the presence of only divalent Cu2+ cations (Table 2, Fig. 2). This result is consistent (within experimental error) with the sequential exchange of isolated Cu2+ at paired Al sites until saturation followed by subsequent exchange of monovalent [CuOH]+ at isolated Al sites.42,60,61 Cu-RTH (Cu/Al = 0.11) shows an H+/Cu exchange stoichiometry of two that suggests only Cu2+ sites are present, while Cu-AEI (Cu/Al = 0.17) shows an H+/Cu exchange stoichiometry between 1 and 2 that suggests a mixture of Cu2+ and [CuOH]+ sites are present. UV-visible spectra of hydrated Cu-AEI, Cu-CHA, and Cu-RTH zeolites (Fig. 3) show absorption bands characteristic of d–d transitions for hydrated Cu2+ complexes (∼12500 cm−1) and broad bands for metal–ligand charge transfer (35000–47000 cm−1), which are convoluted by zeolitic framework metal–oxygen charge transfer (36750 and 43500 cm−1) and Cu–O charge transfer (∼42000 cm−1).62–64 An additional feature is present at ∼25000 cm−1 in the UV-vis spectrum of Cu-RTH, but not in spectra of either Cu-AEI or Cu-CHA, and appears in a region attributed to Cu–O charge transfer in small Cu oxide clusters.63
3.3. Standard SCR kinetics of Cu-zeolites before hydrothermal aging
Rates of NO consumption (473 K, per Cu) during standard SCR (equimolar NO and NH3, with O2 as the oxidant) are shown in Table 3 and plotted in Fig. 4. The measured NO consumption rate (per Cu) was similar on Cu-AEI and Cu-CHA (within 1.3×), although direct quantitative comparison of these turnover rates is not rigorously justified since they appear to be measured in different kinetic regimes, reflected in the different apparent NH3 reaction orders of −0.5 and −0.1 on Cu-CHA and Cu-AEI (Table 3), respectively. The measured NO consumption rate was lower (by 2.2–2.9×) on Cu-RTH than on either Cu-AEI or Cu-CHA (Table 3). At first glance, the similar turnover rates (per Cu) on Cu-CHA, Cu-AEI and Cu-RTH (within 3×, 473 K) seem reminiscent of standard SCR turnover rates (473 K) that have been reported to be insensitive to the zeolite topology (CHA, BEA, MFI), as a consequence of the solvation of Cu cations by NH3 during low temperature SCR conditions.42| Sample | Exposure to SCR gases | V ads,micro (cm3 g−1) | V ads,meso (cm3 g−1) | H+/Altot ratiob | Alf/Altot | Standard SCR rate (per total Cu, 473 K)c | E app (kJ mol−1) | NO ordere | O2 ordere | NH3 ordere |
|---|---|---|---|---|---|---|---|---|---|---|
| a Micropore and mesopore volumes determined from Ar adsorption isotherms (87 K) (Fig. 6). b Number of H+ sites quantified by selective NH3 titration and temperature-programmed desorption. c Units of 10−3 mol NO (mol Cu)−1 s−1. d Errors ± 7 kJ mol−1. e Errors are ± 0.1. *n.d., not detectable (<0.3 × 10−3 mol NO (mol Cu)−1 s−1). | ||||||||||
| CHA | ||||||||||
| H-Form | 0.18 | 0.04 | 0.95 | 0.85 | ||||||
| Cu-Form | Before | 0.17 | 0.05 | 0.72 | 0.90 | |||||
| After | 0.17 | 0.03 | 0.70 | 3.1 | 56 ± 5 | 0.4 | 0.6 | −0.5 | ||
| Cu-Form aged | Before | 0.15 | 0.01 | 0.16 | 0.84 | |||||
| After | 0.15 | 0.07 | 0.14 | 2.2 | 51 ± 5 | 0.5 | 0.4 | −0.1 | ||
| AEI | ||||||||||
| H-Form | 0.20 | 0.01 | 0.85 | 0.85 | ||||||
| Cu-Form | Before | 0.19 | 0.01 | 0.54 | 0.91 | |||||
| After | 0.18 | 0.00 | 0.50 | 4.1 | 46 ± 5 | 0.5 | 0.4 | −0.1 | ||
| Cu-Form aged | Before | 0.17 | 0.06 | 0.16 | 0.78 | |||||
| After | 0.16 | 0.02 | 0.15 | 1.9 | 49 ± 5 | 0.5 | 0.4 | 0.0 | ||
| RTH | ||||||||||
| H-Form | 0.20 | 0.05 | 0.60 | 0.94 | ||||||
| Cu-Form | Before | 0.17 | 0.04 | 0.38 | 0.98 | |||||
| After | 0.17 | 0.03 | 0.39 | 1.4 | 28 ± 5 | 0.4 | 0.4 | −0.1 | ||
| Cu-form aged | Before | 0.17 | 0.10 | 0.07 | 0.72 | |||||
| After | 0.17 | 0.08 | 0.00 | n.d.* | — | — | — | — | ||
Apparent activation energies (Table 3) estimated from rate data collected between 444–476 K (Fig. 4) were similar on Cu-AEI (46 ± 5 kJ mol−1) and Cu-CHA (56 ± 5 kJ mol−1), and in a range previously reported for standard SCR activation energies on Cu-CHA (Si/Al = 35, Cu/Al = 0–0.31).44,45 Apparent activation energies were much lower on Cu-RTH (28 ± 5 kJ mol−1), however, and approximately half of the value measured on Cu-CHA, characteristic of severe intrazeolite mass transfer limitations. Both the CHA and AEI frameworks contain three-dimensional pore systems interconnected by symmetric 8-MR windows (0.38 nm diameter), but the RTH framework is a two-dimensional pore system with a limiting asymmetric 8-MR ring of size (0.25 nm) similar to the kinetic diameter of the SCR reactants (∼0.3 nm). In effect, the RTH framework appears to behave as a one-dimensional pore system for this reaction, in which reactants preferentially diffuse through the symmetric 8-MR window. Internal diffusion limitations have been proposed to account for the lower NOx conversions (423–573 K) in two-dimensional, small-pore LEV and DDR zeolites, when compared to three-dimensional small-pore CHA zeolites.13 Thus, while small-pore zeolites show improved hydrothermal stability over medium and large-pore zeolites,14,17 considerations of pore connectivity and limiting aperture sizes are also critical in determining the reactivity of Cu sites located within them.
The number of residual H+ sites (per Altot) on Cu-CHA before and after exposure to SCR gases was 0.72 and 0.70 ± 0.05, respectively. Similarly, the residual H+/Alf value on Cu-AEI and Cu-RTH changed only from 0.54 to 0.50 ± 0.05 and from 0.38 to 0.39 ± 0.05, respectively (Table 3). Therefore, exposure to SCR gases did not significantly change the number of residual H+ sites on Cu-AEI, Cu-CHA and Cu-RTH, indicating that framework Al remained largely intact on Cu-zeolites after NOx SCR catalysis. Ar micropore and mesopore volume measurements (87 K) on Cu-AEI, Cu-CHA and Cu-RTH before and after exposure to SCR gases were unchanged (Table 3, Fig. 6), with micropore volumes for both Cu-CHA and Cu-RTH of 0.17 cm3 g−1 and for Cu-AEI of 0.18–0.19 cm3 g−1 before and after exposure to SCR gases, respectively. Similarly, the mesopore volume for Cu-AEI, Cu-CHA and Cu-RTH zeolites before and after exposure to SCR gases were the same, within experimental error, between 0.03–0.05 cm3 g−1 (Table 3). Taken together, these characterization data of Cu-zeolites that have not been exposed to hydrothermal aging treatments indicate that minimal changes to H+ or Cu sites, or the zeolite framework, occur after exposure to low temperature standard SCR reaction conditions (473 K).
3.4. Characterization of Cu-form zeolites before and after hydrothermal aging
Severe hydrothermal aging treatments of each Cu-exchanged zeolite, performed to reproduce the effects experienced during a 135000 mile lifetime (1073 K, 10% (v/v) H2O, 16 h),34 did not result in detectable loss of long-range structure as inferred from powder XRD patterns (Fig. 5). After hydrothermal aging treatments, the Ar micropore volumes decreased by only ∼10% on Cu-AEI and Cu-CHA, but remained constant on Cu-RTH (Table 3, Fig. 6). 27Al MAS NMR spectra of Cu-zeolites after hydrothermal aging (Fig. 1) show decreased intensities for tetrahedrally-coordinated Al lines (Alf, ∼60 ppm; Table 3) and increased intensities in octahedrally-coordinated Al lines (Alex, ∼0 ppm; Fig. 1), indicating the formation of extraframework Al species from framework dealumination. Framework dealumination occurred to greater extents on Cu-RTH (∼25% loss in Alf) than on either Cu-AEI or Cu-CHA (∼7% loss in Alf), although we note that Al quantification from NMR spectra of Cu-zeolites will be affected by species that are not detected because of interactions with paramagnetic Cu. Moreover, hydrothermal aging of Cu-RTH results in the appearance of a broad shoulder at ∼40–50 ppm reflecting penta-coordinated or distorted tetrahedral Al,65 which did not occur in either Cu-AEI or Cu-CHA. After hydrothermal aging treatments, XRD lines shifted to higher angles on Cu-CHA and Cu-AEI (Fig. 5) reflecting lattice contraction upon extraction of a small amount of framework Al (∼7% by 27Al NMR), while XRD lines shifted to lower angles on Cu-RTH (Fig. 5) reflecting lattice expansion that appears to arise from the more extensive dealumination that formed persistent partial-extraframework aluminum species in distorted coordination environments (∼40–50 ppm in 27Al NMR). After hydrothermal aging, each Cu-zeolite sample showed a ∼70–80% decrease in the number of H+ sites measured by NH3 TPD (Table 3; Fig. S.5, ESI†), in spite of the only minor decreases in Alf intensity observed in the 27Al MAS NMR spectra, which may reflect structural changes to extraframework Al species caused by the hydration treatments used prior to recording NMR spectra. These findings demonstrate that characterization of the bulk structure (e.g. XRD patterns, micropore volumes) or Al atoms (27Al MAS NMR) are insufficient to describe the local site and structural changes caused by hydrothermal aging treatments,36 and serve as a reminder for the need to use techniques that probe and quantify active sites directly (e.g. base titration of proton sites) to accurately detect such structural changes.59,66,67
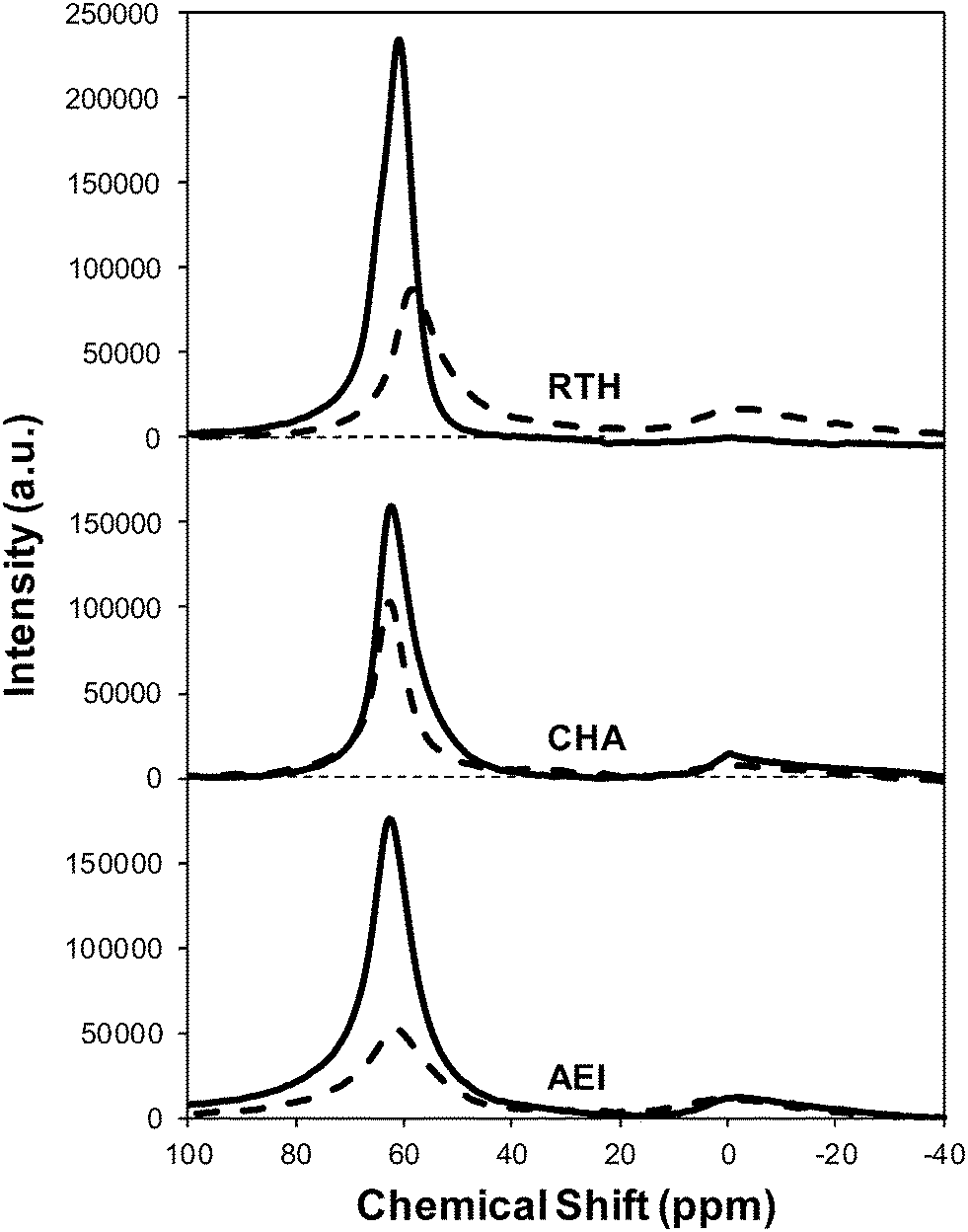 | ||
| Fig. 1 27Al MAS NMR spectra of hydrated fresh (solid) and aged (dashed) Cu-form of RTH, CHA and AEI zeolites. | ||
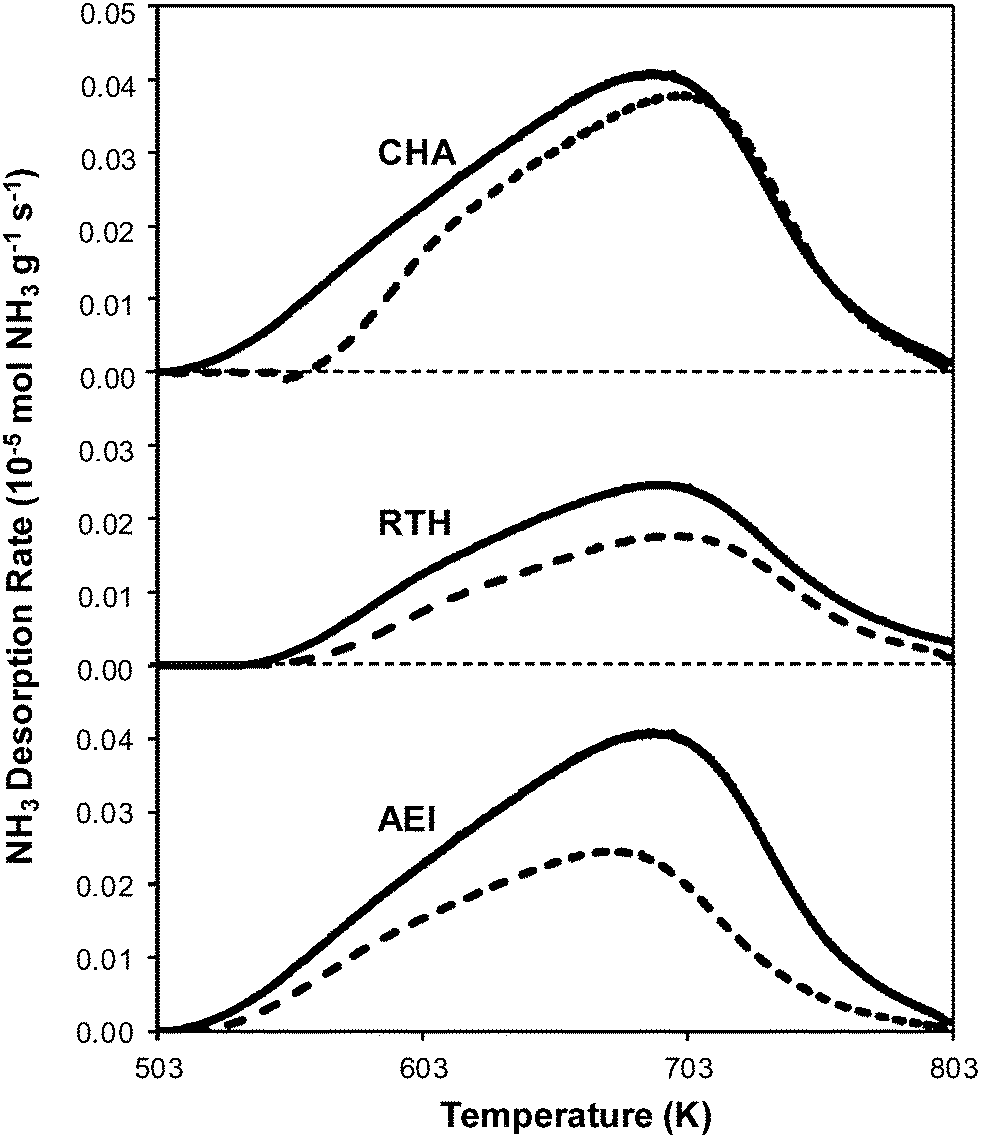 | ||
| Fig. 2 NH3 desorption rates as a function of temperature on H-form (solid) and fresh Cu-form (dashed) on AEI, CHA, and RTH zeolites. | ||
In contrast to the dramatic changes observed for H+ sites on each Cu-zeolite after hydrothermal aging, the identity and coordination of Cu species appear to remain unchanged as inferred from UV-vis spectra (Fig. 3). UV-vis absorption bands for Cu2+ d–d transitions (∼12500 cm−1) appear identical for hydrated Cu-zeolites before and after hydrothermal aging, without any new features observed in the region for Cu oxide clusters (∼25000 cm−1). Slight changes in the intensities of absorbance bands characteristic of metal–ligand charge transfer (35000–47000 cm−1) are observed for each Cu-zeolite after hydrothermal aging, which may reflect changes in the zeolite structure caused by removal of framework aluminum atoms. Taken together, these results indicate that hydrothermal aging treatments of Cu-exchanged CHA, AEI and RTH zeolites cause framework dealumination and a decrease in the numbers of corresponding H+ sites, but do not result in detectable changes to the exchanged Cu cations or to the long-range structural order in the zeolite framework.
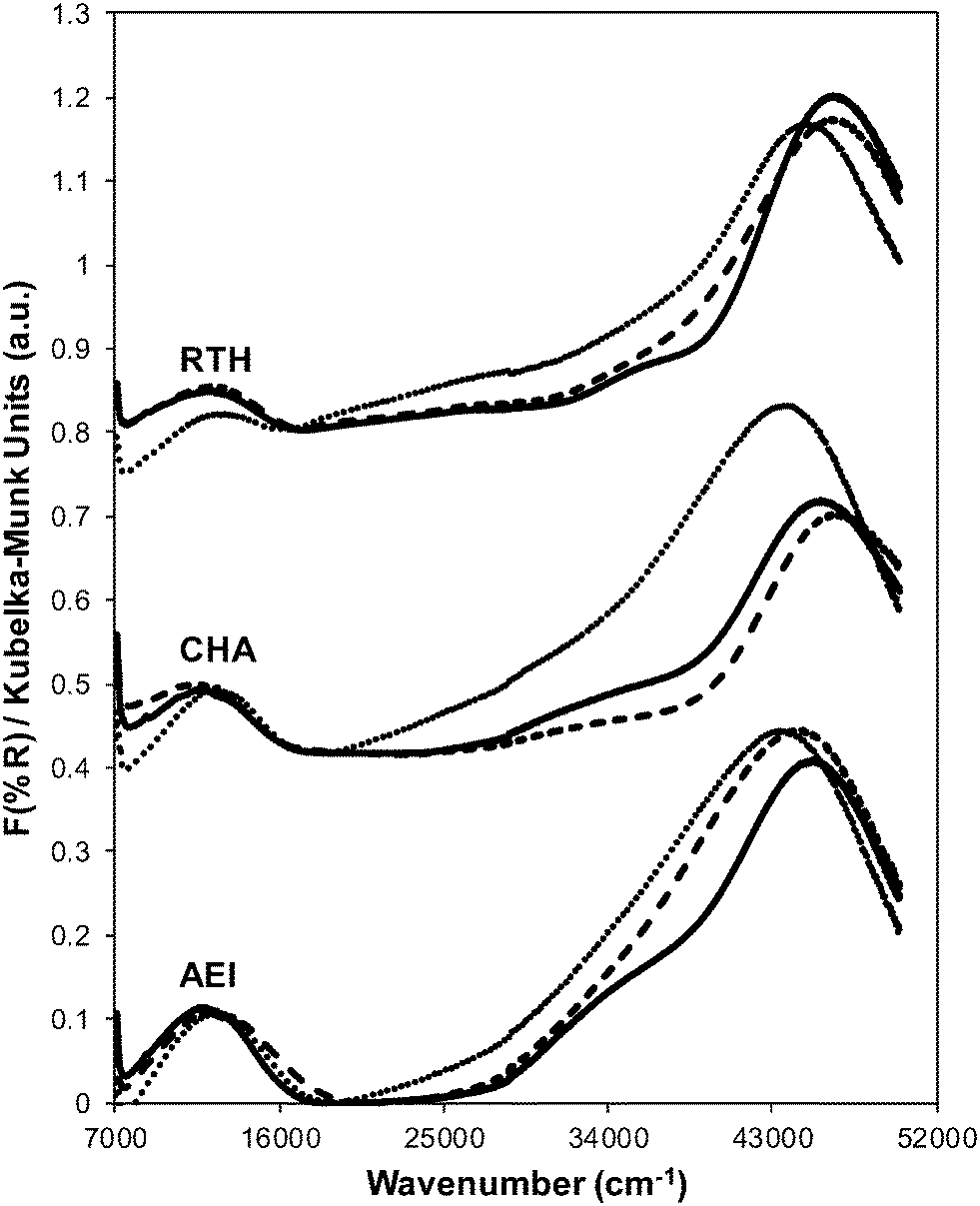 | ||
| Fig. 3 UV-vis spectra on hydrated fresh Cu-form before SCR (solid), aged Cu-form before SCR (dashed), and aged after SCR (dotted) on RTH, CHA and AEI zeolites. Spectra are offset for clarity (CHA: by 0.4 a.u., RTH: by 0.8 a.u.). | ||
3.5. Standard SCR kinetics of Cu-form zeolites before and after hydrothermal aging
The standard SCR rate (per Cu, 473 K) measured on hydrothermally aged Cu-AEI decreased by ∼50% compared to the rate measured on Cu-AEI prior to aging (Table 3, Fig. 4). The standard SCR rate measured on hydrothermally aged Cu-CHA decreased by ∼25% compared to the rate measured on Cu-CHA prior to aging (Table 3, Fig. 4). The standard SCR rate on Cu-RTH, however, was not measureable (<0.3 × 10−3 mol NO (mol Cu)−1 s−1) after hydrothermal aging despite the presence of isolated, hydrated Cu2+ species detected in its UV-vis spectrum (Fig. 3). As a result, apparent activation energies and reaction orders could not be measured on Cu-RTH subjected to hydrothermal aging treatments. Although hydrothermally aged Cu-RTH shows undetectable SCR rates, XRD patterns and Ar micropore volumes indicate virtually no changes to Cu-RTH before and after aging. Thus, assessments of long-range structural features by XRD and micropore volume after Cu-zeolites have been hydrothermally aged cannot be used as accurate predictors of SCR catalytic behavior.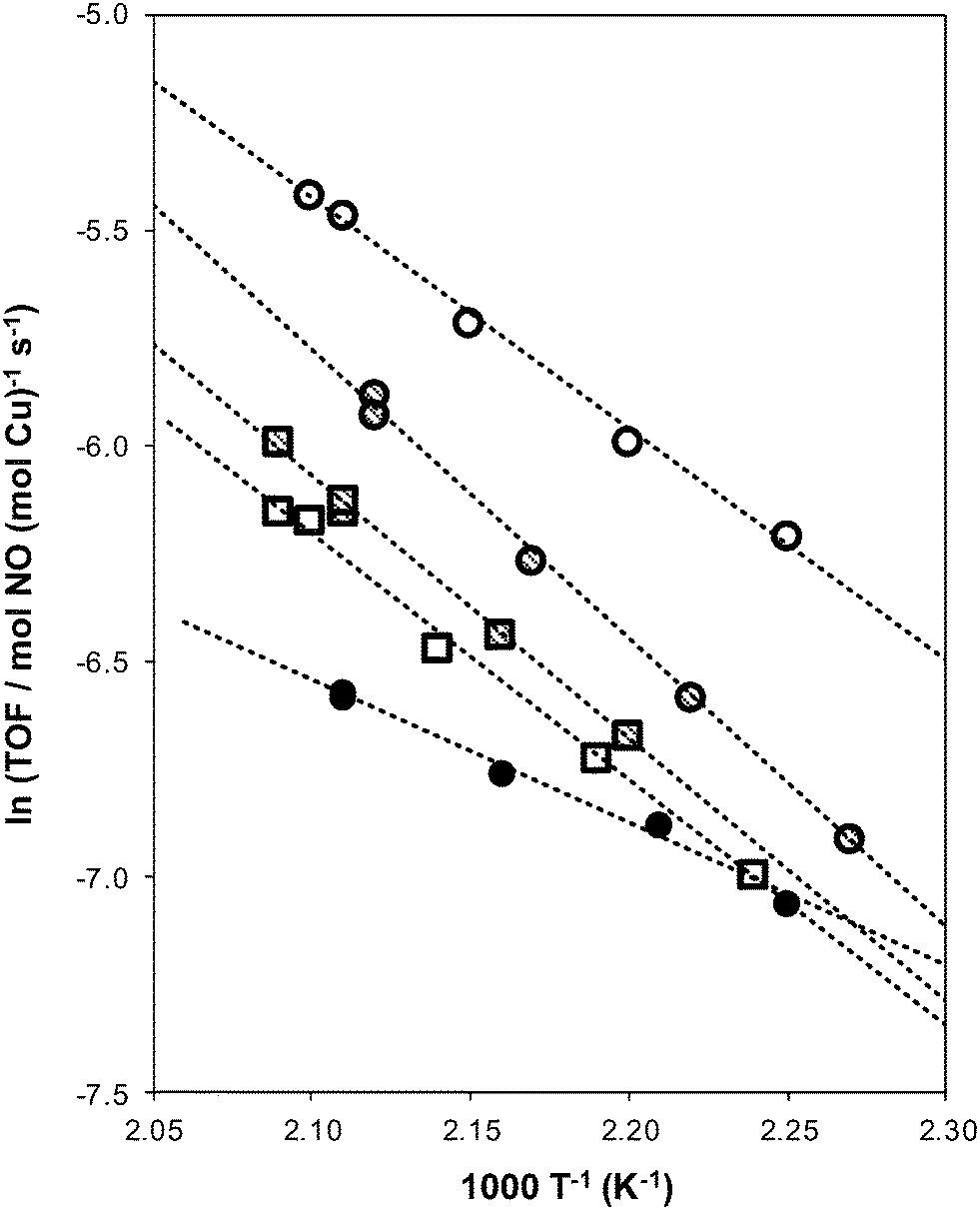 | ||
| Fig. 4 Dependence of standard SCR turnover rates (per Cu) on temperature for fresh (circles) and aged (squares) Cu-form AEI (hollow), CHA (cross hatched), and RTH (filled circles) zeolites. | ||
Hydrothermal aging treatments did not affect the apparent activation energies on either Cu-AEI (46–49 kJ mol−1) or Cu-CHA (51–56 kJ mol−1), nor the apparent NO (0.5), O2 (0.4) and NH3 (∼0) orders on Cu-AEI and the apparent NO (0.4–0.5) and O2 (0.4–0.6) orders on Cu-CHA (Table 3). The apparent NH3 order measured on Cu-CHA (−0.5) became less negative after hydrothermal aging (−0.1, Table 3); we surmise that structural changes caused by hydrothermal aging and exposure to SCR gases led to a change in operation to a new kinetic regime characterized by weaker NH3 inhibition. Turnover rates were similar between the hydrothermally-aged Cu-CHA and Cu-AEI samples (1.9–2.2 × 10−3 mol NO (mol Cu)−1 s−1) and the apparent reaction orders and activation energies were identical for both samples (Table 3), providing evidence that these rate data were measured in equivalent kinetic regimes. These data indicate that the Cu species that remain active on both CHA and AEI after hydrothermal aging behave catalytically similar, which may be linked to the nature of the Cu2+ exchange sites at the 6-MR windows of D6R composite building units that are found in both CHA and AEI.
3.6. Characterization of Cu-form zeolites before and after hydrothermal aging, and after exposure to NOx SCR
Ar adsorption isotherms (87 K) and micropore volumes (Fig. 6, Table 3) of Cu-zeolites after hydrothermal aging were indistinguishable before and after exposure to low temperature standard SCR reaction conditions (473 K). XRD patterns were also similar for hydrothermally-aged Cu-zeolites before and after exposure to standard SCR gases (Fig. 5). These characterization data indicate that further structural changes to the zeolite framework did not occur when aged Cu-zeolites were exposed to standard SCR gas mixtures. The number of H+ sites on hydrothermally-aged Cu-AEI and Cu-CHA zeolites were also similar before and after exposure to standard SCR gas mixtures (Table 3), but H+ sites were no longer detectable (<0.03 H+/Al, Table 3) on hydrothermally-aged Cu-RTH exposed to SCR gases. UV-vis spectra of hydrothermally-aged Cu-CHA and Cu-RTH zeolites after exposure to standard SCR reactants (Fig. 3) showed a reduction in Cu2+ d–d transition intensity (∼12500 cm−1) and concomitant increases in intensity for broad absorption bands between 20000–40000 cm−1. The spectra of Cu-AEI, however, retained similar d–d transition intensity after aging and exposure to SCR reactants, with an increase in intensity in the metal–ligand charge transfer region (35000–47000 cm−1) that is also observed for Cu-CHA, but not for Cu-RTH.
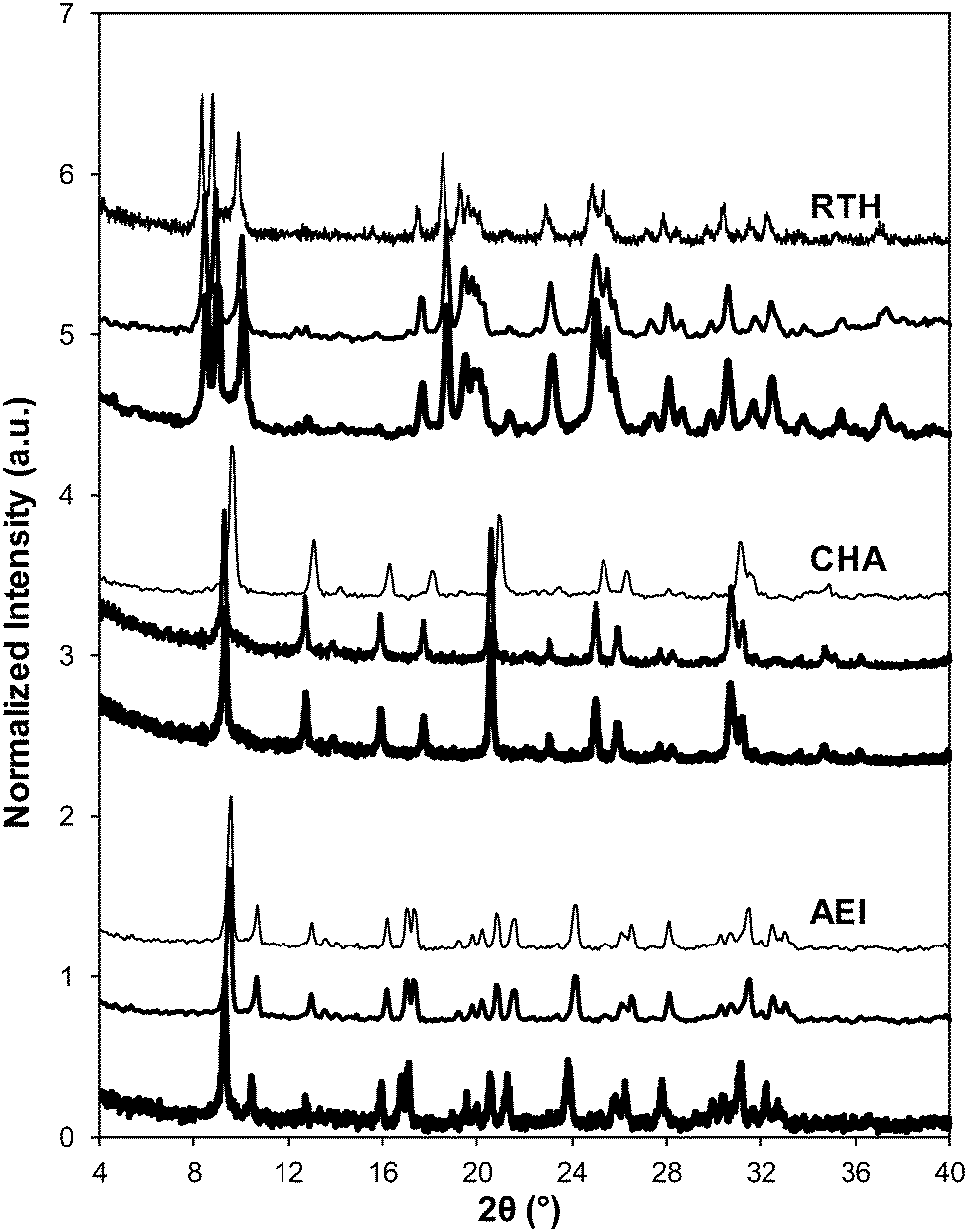 | ||
| Fig. 5 Powder XRD patterns of fresh Cu-form before SCR (dark), aged Cu-form before SCR (medium), and aged Cu-form after SCR (light) on AEI, CHA, and RTH zeolites. Diffraction patterns are normalized so that the maximum peak intensity in each pattern is unity, and offset for clarity. | ||
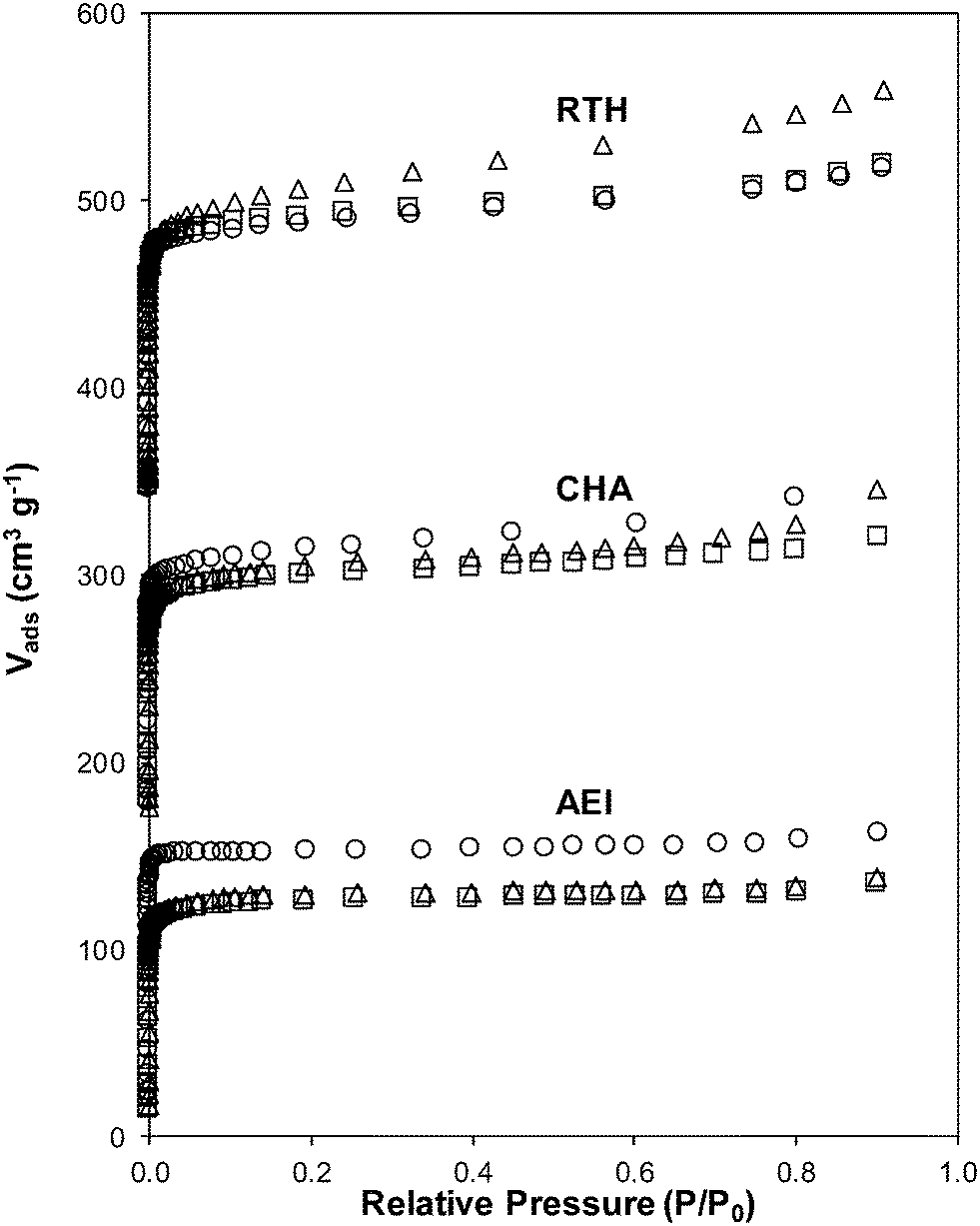 | ||
| Fig. 6 Ar adsorption isotherms (87 K) on fresh Cu-form before SCR (circles), aged Cu-form before SCR (triangles), aged Cu-form after SCR (squares) on AEI, CHA, and RTH zeolites. Isotherms are vertically offset for clarity (CHA: 160 cm3 g−1, RTH: by 320 cm3 g−1). | ||
The Cu structure in Cu-CHA, Cu-AEI and Cu-RTH, characterized by UV-vis spectra, showed hardly any changes after hydrothermal aging treatments, but showed noticeable decreases in Cu2+ intensity upon subsequent exposure to low temperature SCR reaction conditions. These findings provide evidence that hydrothermal aging causes removal of Al from framework to extraframework positions, and that further structural changes continue to occur in the presence of SCR reactants at low temperatures (473 K) because NH3 facilitates the solvation and mobility of Cu cations.42 We speculate that an inactive copper aluminate phase (CuAlxOy) forms as a result of interactions of active Cu sites with extraframework Al(OH)3 species, as proposed previously.14,17,35 UV-vis spectra of hydrothermally-aged Cu-RTH reveal decreased intensities for hydrated Cu2+ d–d transitions along with increases in new charge transfer bands between 20000–40000 cm−1 that may reflect CuAlxOy species and account for decreases in SCR rate. Interestingly, any remaining H+ sites in Cu-RTH upon hydrothermal aging and subsequent exposure to standard SCR reactants become inaccessible to NH3, suggesting that Cu active sites in RTH, which appear to catalyze SCR in a diffusion-limited regime before hydrothermal aging, also become inaccessible to SCR reactants after hydrothermal aging.
4. Conclusions
CHA and AEI zeolites are similar in structure, with three-dimensional micropore systems connected by symmetric 8-MR windows (0.38 nm diameter), while the RTH framework is a two-dimensional pore system with constrained, asymmetric 8-MR windows (0.56 nm × 0.25 nm) that limit access in one dimension and effectively causes the RTH framework to behave as a one-dimensional pore system for NOx SCR with NH3. As a result, standard SCR turnover rates (per Cu, 473 K) and apparent activation energies are similar between Cu-CHA and Cu-AEI, but turnover rates are lower (by ∼2–3×) and apparent activation energies are lower (by ∼2×) on Cu-RTH. Hydrothermal aging causes dealumination of Cu-CHA, Cu-AEI and Cu-RTH, evident in a decrease in the fraction of Alf determined from 27Al MAS NMR spectra and corresponding decreases in the number of H+ sites quantified by NH3 TPD, but does not cause noticeable changes in the bulk framework structure assessed by XRD and micropore volume or in the Cu structure by UV-vis spectroscopy. The number of active Cu sites, however, decreased after hydrothermally aged samples were subsequently exposed to low temperature standard SCR reactants, evident in changes to UV-vis spectra and concomitant decreases in standard SCR turnover rates. Hydrothermal aging causes removal of Al from framework to extraframework positions, and further structural changes continue to occur in the presence of ammonia at low temperatures (473 K), which solvate and mobilize extraframework cations to facilitate the formation of inactive copper-aluminate phases (CuAlxOy). These structural changes appear to occur more readily in Cu-RTH than either Cu-AEI or Cu-CHA, providing further evidence linking the presence of double six-membered rings (D6R) in small-pore molecular sieve frameworks to increased resistance to active site and framework structural changes upon hydrothermal aging.Bulk structural characterization of small-pore zeolites after hydrothermal aging treatments cannot be used to accurately infer catalytic behavior for low temperature NOx SCR with NH3. This is evident in the case of hydrothermally-aged Cu-RTH, which deactivates completely upon exposure to standard SCR reactants but is characterized by similar bulk properties (XRD, micropore volume) before and after hydrothermal aging. Probes of Al structure (e.g., 27Al MAS NMR) reveal that octahedrally-coordinated Al species are formed after hydrothermal aging of Cu-zeolites, but in amounts that are unable to account for the much larger disappearance in Brønsted acid sites titrated by NH3, providing another reminder that methods to directly probe active sites are needed to assess their structural changes. We conclude that more accurate assessments of molecular sieve framework topologies that are viable for practical NOx SCR catalysis require quantification and characterization of Al and Cu site structures after hydrothermally aged samples are exposed to low temperature SCR reaction conditions. We expect that holistic approaches to active site characterization, especially of Al and Cu sites, in Cu-zeolites after hydrothermal aging and subsequent exposure to low temperature SCR reaction conditions will be able to provide more accurate guidance about molecular sieve topologies that are viable candidates for practical SCR technologies.
Acknowledgements
We acknowledge the financial support provided by the National Science Foundation GOALI program under award number 1258715-CBET. RG acknowledges the financial support from a Ralph E. Powe Junior Faculty Enhancement Award from the Oak Ridge Associated Universities (ORAU). MD acknowledges Research Foundation Flanders (FWO) for postdoctoral funding. We thank Dr. John Harwood (Purdue Interdepartmental NMR Facility) for assistance collecting the NMR spectra, and Dr. Atish A. Parekh for helpful technical discussions. We also thank Daniel Gonzalez (Universidad Nacional de Colombia), through the Undergraduate Research Experience Purdue-Colombia (UREP-C) program, for experimental assistance constructing the apparatus to perform hydrothermal aging treatments and performing some aging experiments on CHA zeolites. We also thank Sachem, Inc. for providing the organic structure-directing agent used to synthesize SSZ-13.References
- S. Brandenberger, O. Kröcher, A. Tissler and R. Althoff, Catal. Rev.: Sci. Eng., 2008, 50, 492–531 CAS.
- T. Komatsu, M. Nunokawa, I. S. Moon, T. Takahara, S. Namba and T. Yashima, J. Catal., 1994, 148, 427–437 CrossRef CAS.
- G. Delahay, S. Kieger, N. Tanchoux, P. Trens and B. Coq, Appl. Catal., B, 2004, 52, 251–257 CrossRef CAS.
- G. Delahay, E. A. Villagomez, J.-M. Ducere, D. Berthomieu, A. Goursot and B. Coq, ChemPhysChem, 2002, 3, 686–692 CrossRef CAS PubMed.
- J. H. Baik, S. D. Yim, I.-S. Nam, Y. S. Mok, J.-H. Lee, B. K. Cho and S. H. Oh, Top. Catal., 2004, 30, 37–41 CrossRef.
- J.-H. Park, H. J. Park, J. H. Baik, I.-S. Nam, C.-H. Shin, J.-H. Lee, B. K. Cho and S. H. Oh, J. Catal., 2006, 240, 47–57 CrossRef CAS.
- K. Kamasamudram, N. W. Currier and X. Chen, Catal. Today, 2010, 151, 212–222 CrossRef CAS.
- J. H. Kwak, R. G. Tonkyn, D. H. Kim, J. Szanyi and C. H. F. Peden, J. Catal., 2010, 275, 187–190 CrossRef CAS.
- S. T. Korhonen, D. W. Fickel, R. F. Lobo, B. M. Weckhuysen and A. M. Beale, Chem. Commun., 2011, 47, 800–802 RSC.
- A. M. Beale, F. Gao, I. Lezcano-Gonzalez, C. H. F. Peden and J. Szanyi, Chem. Soc. Rev., 2015, 44, 7371–7405 RSC.
- K. Rahkamaa-Tolonen, T. Maunula, M. Lomma, M. Huuhtanen and R. L. Keiski, Catal. Today, 2005, 100, 217–222 CrossRef CAS.
- Q. Ye, L. Wang and R. T. Yang, Appl. Catal., A, 2012, 427, 24–34 CrossRef.
- D. W. Fickel, E. D'Addio, J. A. Lauterbach and R. F. Lobo, Appl. Catal., B, 2011, 102, 441–448 CrossRef CAS.
- J. H. Kwak, D. Tran, S. D. Burton, J. Szanyi, J. H. Lee and C. H. F. Peden, J. Catal., 2012, 287, 203–209 CrossRef CAS.
- K. C. C. Kharas, H. J. Robota and D. J. Liu, Appl. Catal., B, 1993, 2, 225–237 CrossRef CAS.
- S. A. Gómez, A. Campero, A. Martínez-Hernández and G. A. Fuentes, Appl. Catal., A, 2000, 197, 157–164 CrossRef.
- P. G. Blakeman, E. M. Burkholder, H. Y. Chen, J. E. Collier, J. M. Fedeyko, H. Jobson and R. R. Rajaram, Catal. Today, 2014, 231, 56–63 CrossRef CAS.
- S. Shwan, R. Nedyalkova, J. Jansson, J. Korsgren, L. Olsson and M. Skoglundh, Ind. Eng. Chem. Res., 2012, 51, 12762–12772 CrossRef CAS.
- P. J. Andersen, J. E. Bailie, J. L. Casci, H. Y. Chen, J. M. Fedeyko, R. K. Shin Foo and R. R. Rajaram, US Pat., 0290963, 2010 Search PubMed.
- I. Bull, R. S. Boorse, W. M. Jaglowski, G. S. Koermer, A. Moini, J. A. Patchett, W. Xue, P. Burk, J. C. Dettling and M. T. Caudle, US Pat., 0226545, 2008 Search PubMed.
- M. Moliner, C. Franch, E. Palomares, M. Grill and A. Corma, Chem. Commun., 2012, 48, 8264–8266 RSC.
- A. Lorena Picone, S. J. Warrender, A. M. Z. Slawin, D. M. Dawson, S. E. Ashbrook, P. A. Wright, S. P. Thompson, L. Gaberova, P. L. Llewellyn, B. Moulin, A. Vimont, M. Daturi, M. B. Park, S. K. Sung, I.-S. Nam and S. B. Hong, Microporous Mesoporous Mater., 2011, 146, 36–47 CrossRef CAS.
- D. Xie, L. B. McCusker, C. Baerlocher, S. I. Zones, W. Wan and X. Zou, J. Am. Chem. Soc., 2013, 135, 10519–10524 CrossRef CAS PubMed.
- T. M. Davis, A. T. Liu, C. M. Lew, D. Xie, A. I. Benin, S. Elomari, S. I. Zones and M. W. Deem, Chem. Mater., 2016, 28, 708–711 CrossRef CAS.
- D. Jo, J. Bin Lim, T. Ryu, I.-S. Nam, M. A. Camblor and S. B. Hong, J. Mater. Chem. A, 2015, 3, 19322–19329 CAS.
- S. Altwasser, J. Jiao, S. Steuernagel, J. Weitkamp and M. Hunger, Stud. Surf. Sci. Catal., 2004, 154B, 1212–1213 CrossRef CAS.
- M. Nielsen, R. Y. Brogaard, H. Falsig, P. Beato, O. Swang and S. Svelle, ACS Catal., 2015, 5, 7131–7139 CrossRef CAS.
- K. Ehrhardt, M. Suckow and W. Lutz, Stud. Surf. Sci. Catal., 1995, 94, 179–186 CrossRef CAS.
- L. Ma, Y. Cheng, G. Cavataio, R. W. McCabe, L. Fu and J. Li, Appl. Catal., B, 2014, 156–157, 428–437 CrossRef CAS.
- M. V. L. Pereira, A. Nicolle and D. Berthout, Catal. Today, 2015, 258, 424–431 CrossRef.
- L. R. Aramburo, L. Karwacki, P. Cubillas, S. Asahina, D. A. M. de Winter, M. R. Drury, I. L. C. Buurmans, E. Stavitski, D. Mores, M. Daturi, P. Bazin, P. Dumas, F. Thibault-Starzyk, J. A. Post, M. W. Anderson, O. Terasaki and B. M. Weckhuysen, Chem. – Eur. J., 2011, 17, 13773–13781 CrossRef CAS PubMed.
- D. Wang, Y. Jangjou, Y. Liu, M. K. Sharma, J. Luo, J. Li, K. Kamasamudram and W. S. Epling, Appl. Catal., B, 2015, 165, 438–445 CrossRef CAS.
- J. Wang, Z. Peng, H. Qiao, L. Han, W. Bao, L. Chang, G. Feng and W. Liu, RSC Adv., 2014, 4, 42403–42411 RSC.
- S. J. Schmieg, S. H. Oh, C. H. Kim, D. B. Brown, J. H. Lee, C. H. F. Peden and D. H. Kim, Catal. Today, 2012, 184, 252–261 CrossRef CAS.
- W. Su, Z. Li, Y. Peng and J. Li, Phys. Chem. Chem. Phys., 2015, 17, 29142–29149 RSC.
- M. Dusselier, M. A. Deimund, J. E. Schmidt and M. E. Davis, ACS Catal., 2015, 5, 6078–6085 CrossRef CAS.
- F. Gao, Y. Wang, N. M. Washton, M. Kollar, J. Szanyi and C. H. F. Peden, ACS Catal., 2015, 5, 6780–6791 CrossRef CAS.
- A. V. Kucherov and A. A. Slinkin, Zeolites, 1986, 6, 175–180 CrossRef CAS.
- G. T. Kerr, J. Phys. Chem., 1967, 71, 4155–4156 CrossRef CAS.
- G. T. Kerr, J. Catal., 1969, 15, 200–204 CrossRef CAS.
- M. C. Silaghi, C. Chizallet, E. Petracovschi, T. Kerber, J. Sauer and P. Raybaud, ACS Catal., 2015, 5, 11–15 CrossRef CAS.
- C. Paolucci, A. A. Parekh, I. Khurana, J. R. Di Iorio, H. Li, J. D. Albarracin Caballero, A. Shih, T. Anggara, W. N. Delgass, J. T. Miller, F. H. Ribeiro, R. Gounder and W. F. Schneider, J. Am. Chem. Soc., 2016, 138, 6028–6048 CrossRef CAS PubMed.
- T. V. W. Janssens, H. Falsig, L. F. Lundegaard, P. N. R. Vennestrøm, S. Rasmussen, P. G. Moses, F. Giordanino, E. Borfecchia, K. A. Lomachenko, C. Lamberti, S. Bordiga, A. Godiksen, S. Mossin and P. Beato, ACS Catal., 2015, 5, 2832–2845 CrossRef CAS.
- F. Gao, N. M. Washton, Y. Wang, M. Kollár, J. Szanyi and C. H. F. Peden, J. Catal., 2015, 331, 25–38 CrossRef CAS.
- S. A. Bates, A. A. Verma, C. Paolucci, A. A. Parekh, T. Anggara, A. Yezerets, W. F. Schneider, J. T. Miller, W. N. Delgass and F. H. Ribeiro, J. Catal., 2014, 312, 87–97 CrossRef CAS.
- S. I. Zones, US Pat., 4544538A, 1985 Search PubMed.
- M. Dusselier, J. E. Schmidt, R. Moulton, B. Haymore, M. Hellums and M. E. Davis, Chem. Mater., 2015, 27, 2695–2702 CrossRef CAS.
- J. E. Schmidt, M. A. Deimund, D. Xie and M. E. Davis, Chem. Mater., 2015, 27, 3756–3762 CrossRef CAS.
- J. E. Schmidt, M. W. Deem and M. E. Davis, Angew. Chem., Int. Ed., 2014, 53, 8372–8374 CrossRef CAS PubMed.
- J. E. Schmidt, M. A. Deimund and M. E. Davis, Chem. Mater., 2014, 34, 7774–7779 Search PubMed.
- C. Baerlocher, W. H. Baur, J. M. Bennett, H. Gies, J. B. Higgins, R. Kirchner and D. H. Olson, Zeolites, 1996, 17, 1–230 CrossRef.
- S. A. Bates, A. A. Verma, C. Paolucci, A. A. Parekh, T. Anggara, A. Yezerets, W. F. Schneider, J. T. Miller, W. N. Delgass and F. H. Ribeiro, J. Catal., 2014, 312, 87–97 CrossRef CAS.
- J. R. Di Iorio, S. A. Bates, A. A. Verma, W. N. Delgass, F. H. Ribeiro, J. T. Miller and R. Gounder, Top. Catal., 2015, 58, 424–434 CrossRef CAS.
- S. I. Zones, Y. Nakagawa, S. T. Evans and G. S. Lee, US Pat, 5958370A, 1999 Search PubMed.
- G. S. Lee and S. I. Zones, US Pat, 6605267B1, 2003 Search PubMed.
- G. Centi and S. Perathoner, Appl. Catal., A, 1995, 132, 179–259 CrossRef CAS.
- R. Gounder, A. J. Jones, R. T. Carr and E. Iglesia, J. Catal., 2012, 286, 214–223 CrossRef CAS.
- A. I. Biaglow, D. J. Parrillo, G. T. Kokotailo and R. J. Gorte, J. Catal., 1994, 148, 213–223 CrossRef CAS.
- S. A. Bates, W. N. Delgass, F. H. Ribeiro, J. T. Miller and R. Gounder, J. Catal., 2014, 312, 26–36 CrossRef CAS.
- J. R. Di Iorio and R. Gounder, Chem. Mater., 2016, 28, 2236–2247 CrossRef CAS.
- C. Paolucci, J. R. Di Iorio, F. H. Ribeiro, R. Gounder and W. F. Schneider, Adv. Catal., 2016, 59, 1–107 Search PubMed.
- J. Texter, D. H. Strome, R. G. Herman and K. Klier, J. Phys. Chem., 1977, 81, 333–338 CrossRef CAS.
- F. Giordanino, P. N. R. Vennestrøm, L. F. Lundegaard, F. N. Stappen, S. Mossin, P. Beato, S. Bordiga and C. Lamberti, Dalton Trans., 2013, 42, 12741–12761 RSC.
- K. L. Fujdala, I. J. Drake, A. T. Bell and T. D. Tilley, J. Am. Chem. Soc., 2004, 126, 10864–10866 CrossRef CAS PubMed.
- J. A. van Bokhoven, A. L. Roest, D. C. Koningsberger, J. T. Miller, G. H. Nachtegaal and A. P. M. Kentgens, J. Phys. Chem. B, 2000, 104, 6743–6754 CrossRef CAS.
- A. I. Biaglow, D. J. Parrillo, G. T. Kokotailo and R. J. Gorte, J. Catal., 1994, 148, 213–223 CrossRef CAS.
- R. Gounder and E. Iglesia, J. Catal., 2011, 277, 36–45 CrossRef CAS.
- M. D. Foster, I. Rivin, M. M. J. Treacy and O. D. Friedrichs, Microporous Mesoporous Mater., 2006, 90, 32–38 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6re00198j |
| ‡ J. D. A.-C. and I. K. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |