
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancement of charge transport properties of small molecule semiconductors by controlling fluorine substitution and effects on photovoltaic properties of organic solar cells and perovskite solar cells†
Jae Hoon
Yun
ac,
Sungmin
Park
ad,
Jin Hyuck
Heo
e,
Hyo-Sang
Lee
af,
Seongwon
Yoon
b,
Jinback
Kang
g,
Sang Hyuk
Im
e,
Hyunjung
Kim
g,
Wonmok
Lee
h,
BongSoo
Kim
ai,
Min Jae
Ko
aj,
Dae Sung
Chung
*b and
Hae Jung
Son
*acf
aPhotoelectronic Hybrid Research Center, Korea Institute of Science and Technology, Seoul 02792, Republic of Korea. E-mail: hjson@kist.re.kr
bSchool of Chemical Engineering and Material Science, Chung-Ang University, Seoul 06974, Republic of Korea. E-mail: dchung@cau.ac.kr
cUniversity of Science and Technology (UST), Daejeon 34113, Republic of Korea
dDepartment of Chemistry, Korea University, Seoul 06974, Republic of Korea
eFunctional Crystallization Center (FCC), Department of Chemical Engineering, Kyung Hee University, Yongin-si 17104, Gyeonggi-do, Republic of Korea
fGreen School (School of Energy and Environment), Korea University, Seoul 02792, Republic of Korea
gDepartment of Physics, Sogang University, Seoul 04107, Republic of Korea
hDepartment of Chemistry, Sejong University, Seoul 05006, Republic of Korea
iDepartment of Science Education, Ewha Womans University, Seoul, 03760, Republic of Korea
jKU-KIST Graduate School of Converging Science and Technology, Korea University, Republic of Korea
First published on 27th July 2016
Abstract
We prepared a series of small molecules based on 7,7′-(4,4-bis(2-ethylhexyl)-4H-silolo[3,2-b:4,5-b′]dithiophene-2,6-diyl)bis(4-(5′-hexyl-[2,2′-bithiophene]-5-yl)benzo[c][1,2,5]thiadiazole) with different fluorine substitution patterns (0F–4F). Depending on symmetricity and numbers of fluorine atoms incorporated in the benzo[c][1,2,5]thiadiazole unit, they show very different optical and morphological properties in a film. 2F and 4F, which featured symmetric and even-numbered fluorine substitution patterns, display improved molecular packing structures and higher crystalline properties in a film compared with 1F and 3F and thus, 2F achieved the highest OTFT mobility, which is followed by 4F. In the bulk heterojunction solar cell fabricated with PC71BM, 2F achieves the highest photovoltaic performance with an 8.14% efficiency and 0F shows the lowest efficiency of 1.28%. Moreover, the planar-type perovskite solar cell (PSC) prepared with 2F as a dopant-free hole transport material shows a high power conversion efficiency of 14.5% due to its high charge transporting properties, which were significantly improved compared with the corresponding PSC device obtained from 0F (8.5%). From the studies, it is demonstrated that low variation in the local dipole moment and the narrow distribution of 2F conformers make intermolecular interactions favorable, which may effectively drive crystal formations in the solid state and thus, higher charge transport properties compared with 1F and 3F.
1. Introduction
Organic conjugated materials have been extensively investigated for applications in a variety of optoelectronic devices, such as light-emitting diodes (OLEDs), field-effect transistors (OFETs), photovoltaic cells (OPVs), sensors, optical amplifiers, and lasers.1–9 One key attraction of organic conjugated materials is the tunability of their physical and chemical properties through modifications of their molecular structures.10–13 Their use in place of conventional inorganic semiconductors offers opportunities to reduce manufacturing costs by implementing printing technologies involving solution-processable materials that are capable of covering large areas and compatible with flexible substrates.14–19Small molecule semiconductors have attracted significant research interest due to their advantages over polymeric semiconductors. Small molecule semiconductors provide well-defined structures, a high purity, a high degree of order, and reduced batch-to-batch variation.20 A representative application of small molecule semiconductors is their use as the electron-donating components in organic photovoltaic (OPV) devices. When blended with fullerene derivatives as electron acceptors, these materials can form a bulk heterojunction (BHJ) structure. The performances of solution-processed small-molecule-based BHJ OPVs (SM-OPVs) have steadily improved over the past several years, with power conversion efficiency (PCE) values in excess of 10% in single-layer BHJ solar cells.21 The notable performance enhancements are mostly attributed to the development of high-efficiency low-bandgap materials with a high charge carrier mobility.21–23 Small molecule semiconductors also show promise for their utility in organic field effect transistors (OFETs), which require well-ordered semiconductor channels. Unlike polymeric semiconductors, the low molecular weight of a small molecule semiconductor enables fine structural/morphological tuning, so that preferential molecular arrangements may be efficiently formed.24–26 The charge carrier mobilities of solution-processed small molecule semiconductors have improved continuously and now approach the mobilities of polymeric semiconductors.27–29
One effective synthetic strategy for achieving a low energy bandgap that simultaneously provides a good charge carrier mobility involves incorporating molecular electron donor (D) and acceptor (A) units in an alternating conjugated arrangement.30,31 The resulting push–pull effects along the molecular backbone facilitate intramolecular charge transfer, resulting in an electronically delocalized structure.32–34 This type of D–A small molecule material often exhibits a high hole mobility and good conductivity with potential utility as a dopant-free hole transport material (HTM) in efficient perovskite solar cells (PSCs), which have recently emerged as promising photovoltaic devices.35–38 Therefore, the rational molecular design of D–A structures has given rise to high-performance materials with physical properties that are appropriate for use in SM-OPVs as well as in PSCs. Thus far, a range of material designs have been presented and synthetic efforts have been applied toward controlling the optical, electrical and solid state assembly properties of a material by varying the D and A subunits to have different electron donating and accepting abilities. Thiophene-based fused aromatic building blocks, such as 4H-silolo[3,2-b:4,5-b′]dithiophene (DTS), benzo[1,2-b:4,5-b′]dithiophene (BDT), and oligothiophene derivatives, have emerged as attractive D units, and electron-deficient compounds of benzo[c][1,2,5]thiadiazole (BT) and 2-(1,1-dicyanomethylene)rhodanine have been used effectively as A units in high-performance SM-OPVs.39–42 A detailed understanding of the structure–property–function relationships within conjugated molecular materials has lagged behind the improvements in material design. It is particularly important to study the impacts of anchoring specific functional groups to a conjugated framework on the essential electrical properties, such as the charge carrier mobility. For example, the local dipole moment and electronic structures induced by functional groups can affect the intermolecular interactions and thus, the packing structures in a thin film.43–45
Herein, we prepared a series of conjugated molecules based on 7,7′-(4,4-bis(2-ethylhexyl)-4H-silolo[3,2-b:4,5-b′]dithiophene-2,6-diyl)bis(4-(5′-hexyl-[2,2′-bithiophene]-5-yl)benzo[c][1,2,5]thiadiazole) (DTS(BTTh2)2) that incorporate different numbers of fluorine atoms on the BT unit. Difluoro-substituted DTS(FBTTh2)2 compounds are used in high-performance solution-processed SM-OPVs.23,46,47 The small size of the fluorine atom generally renders it suitable for altering the energetics of molecular orbitals without introducing extraordinary steric demands.48–52 We examined how varying the fluorine group on an A unit influenced the properties of the solid-state ordering of the molecules and the charge carrier mobilities and photovoltaic performances in two different types of solar cells. The DTS(BTTh2)2-based compounds were first introduced as donor materials into the SM-OPVs, and subsequently, PSCs were prepared using the high mobility DTS(BTTh2)2 derivatives as a HTM without additives such as hygroscopic lithium bis(trifluoromethane sulfonyl) imide (Li-TFSi) and corrosive 4-tert-butylpyridine (t-BP). The properties of these solar cell devices were examined.
2. Results and discussion
2.1. Synthesis and physical properties
Scheme 1 illustrates the synthetic route to the five DTS (BTTh2)2 derivatives. Compounds 3 and 6 were reacted with a DTS ditin-compound in a Stille coupling condensation to provide the target molecules 0F and 4F, respectively. Compound 4 was prepared from the C–C coupling reaction of 4,7-dibromo-5-fluorobenzo[c][1,2,5]thiadiazole with (5′-hexyl-[2,2′-bithiophene]-5-yl)trimethylstannane. Compound 4 was coupled with a DTS mono-stannyl compound to afford compounds 9, which were then brominated using N-bromosuccinimide (NBS) and stannylated using hexamethylditin. Finally, 1F or 3F were prepared by C–C coupling reactions of compound 11 with 3 or 6, respectively. Compared with 2F, 0F and 1F showed better solubilities in chlorinated solvents, such as chloroform and chlorobenzene; however, 3F and 4F were poorly soluble in these solvents. The solubility of the compounds was related to the number of fluorine atom substitutions and the molecular symmetry generated by the presence of fluorine. A detailed description of the synthetic procedures and data are presented in the ESI.† All compounds were characterized by 1H/13C NMR spectroscopy, mass spectrometry, FT-IR spectrum, and elemental analysis (see the ESI†). The thermal properties of the compounds were evaluated using thermal gravimetric analysis (TGA) under a N2 atmosphere. The 5% weight loss (Td) of compounds occurred between 427 °C and 445 °C (Fig. S1, ESI†), indicating that these compounds provided excellent thermal stability.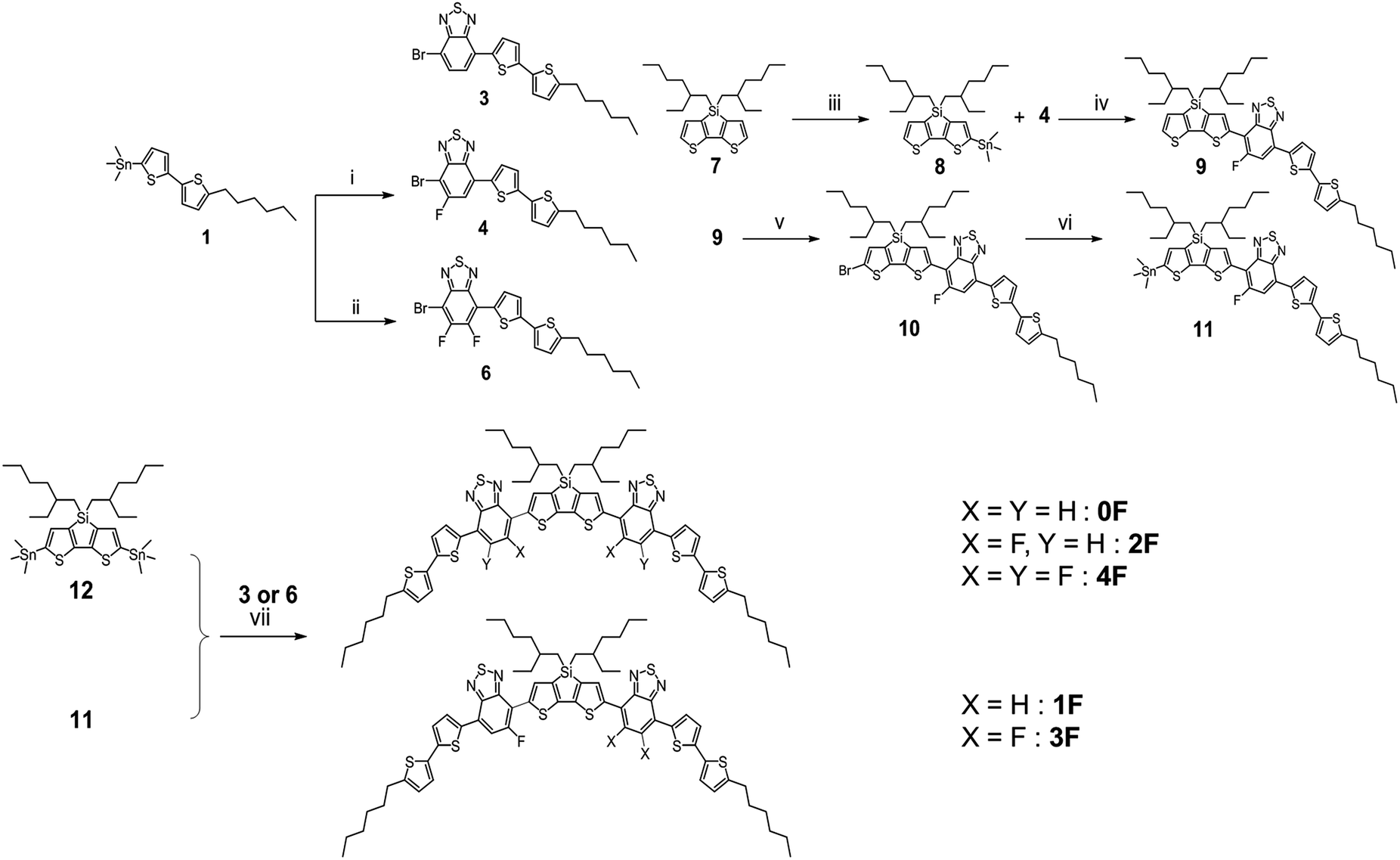 | ||
| Scheme 1 Synthetic procedure used to prepare 0F, 1F, 2F, 3F, and 4F: (i) Pd[PPh3]4, 4,7-dibromo-5-fluorobenzo[c][1,2,5]thiadiazole (2), toluene, 80 °C, 40 h. (ii) Pd[PPh3]4, 4,7-dibromo-5,6-difluorobenzo[c][1,2,5]thiadiazole (5), toluene, 160 °C, 1 h, microwave. (iii) n-BuLi, Me3SnCl, THF, R.T., 12 h. (iv) Pd[PPh3]4, toluene, 160 °C, 1 h, microwave. (v) NBS, CHCl3, R.T., 24 h. (vi) Pd[PPh3]4, hexamethylditin, toluene, 160 °C, 1 h, microwave. (vii) Pd[PPh3]4, toluene, 160 °C, 1 h, microwave. | ||
2.2. Photophysical and electrochemical properties
The photophysical and electrochemical data are summarized in Table 1. Cyclic voltammetry (CV) was performed to study the electrochemical properties of 0–4F in a dichloromethane solution (Fig. 1(a)). The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels were estimated from the onset oxidation and reduction potential. As the fluorine substitution number increased, the HOMO and LUMO energy levels gradually decreased. The HOMO levels of 0F, 1F, 2F, 3F, and 4F were −5.06, −5.06, −5.12, −5.15, and −5.18 eV, respectively. All molecules showed good oxidative stability under two reversible redox couples in solution, suggesting their potential utility as electron-donating and hole-transporting materials. The energy bandgap (Eelecg) obtained from the HOMO and LUMO energy levels increased from 0F (1.79 eV) to 4F (1.85 eV).| Solution | Film | E optg (eV) | Δλonseta (nm) | LUMO (eV) | HOMO (eV) | E elecg (eV) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| λ absonset (nm) | λ absmax (nm) | λ absonset (nm) | λ absmax (nm) | α(λmax) × 104 cm−1 | ||||||
| a Obtained from the λabsonset difference between the solution and film states. | ||||||||||
| 0F | 671 | 591 | 730 | 670 | 6.41 | 1.70 | 59 | −3.27 | −5.06 | 1.79 |
| 1F | 667 | 589 | 739 | 685 | 5.79 | 1.68 | 72 | −3.30 | −5.06 | 1.76 |
| 2F | 663 | 587 | 759 | 683 | 5.64 | 1.63 | 96 | −3.32 | −5.12 | 1.80 |
| 3F | 654 | 582 | 731 | 674 | 7.74 | 1.70 | 77 | −3.32 | −5.15 | 1.83 |
| 4F | 644 | 579 | 745 | 663 | 5.15 | 1.66 | 101 | −3.33 | −5.18 | 1.85 |
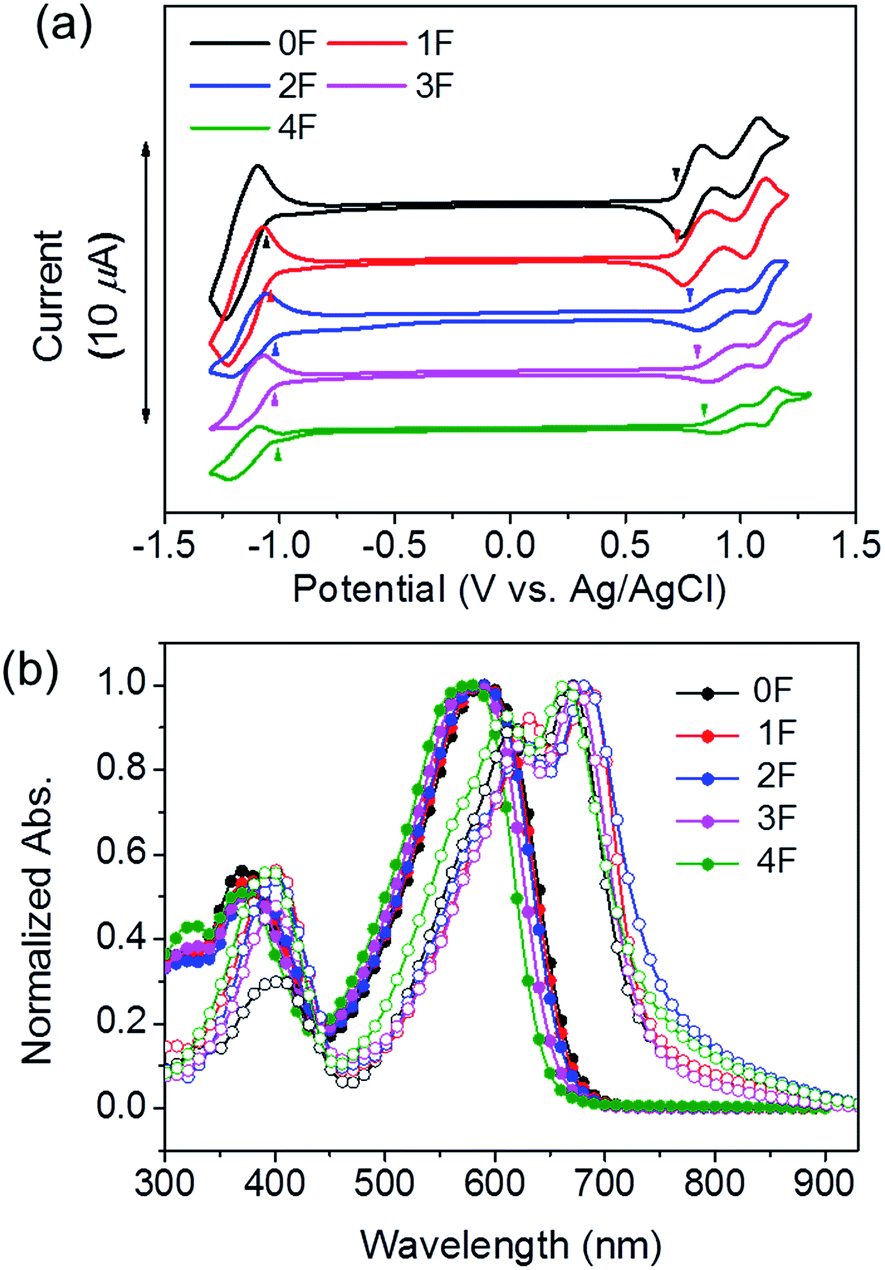 | ||
| Fig. 1 (a) Cyclic voltammograms of 0–4F dichloromethane solutions containing 0.1 mol L−1 Bu4NPF6 and a scan rate of 50 mV s−1. (b) UV-vis absorption spectra of 0–4F chloroform solutions (filled circles) and films (open circles). | ||
The absorption spectra of 0–4F in dilute chloroform solutions and in thin films are displayed in Fig. 1(b). The solution spectra revealed that increasing the number of fluorine substitutions resulted in a blue shift in both the absorption maximum (λabsmax) and onset points (λabsonset); therefore, the onset absorption was blue-shifted by 27 nm from 0F (λabsonset = 671 nm) to 4F (λabsonset = 644 nm). In the solid state, each compound exhibited a broad optical absorption band with distinct high- and low-energy bands attributed to localized π–π* transitions, and internal charge transfer respectively. The transition from the solution to the solid state in the thin films led to a red shift in the optical absorption spectra and the emergence of a fine structure within the low-energy band. In particular, 1F–4F displayed absorption bands that were more red-shifted than that of 0F. This red shift was attributed to the planarization of the conjugated backbone and the formation of efficient intermolecular interactions within the film.53–55 It should be noted that among the fluorinated molecules, the 2F and 4F films exhibited a greater extent of peak broadening and a bathochromic shift than did the 1F and 3F films. We surmised that the 2F and 4F films were characterized by a higher degree of ordered structures in the solid state. The distinctive molecular ordering in the films was reflected by the optical energy bandgaps (Eoptg) of the compound. The Eoptg values, estimated from the onset of the film absorption, were 1.70, 1.68, 1.63, 1.70, and 1.66 eV for 0F, 1F, 2F, 3F, and 4F. The compounds prepared with symmetric arrangements of an even number of fluorine atoms displayed lower Eoptg values than the compounds prepared with an asymmetrical arrangement of odd numbers of fluorine atoms. These results differed from the CV results, probably because the CV measurements performed in solution did not reflect intermolecular interactions in the solid state. As shown in Table 1, 3F had a higher absorption coefficient at the maximum absorption peak (7.74 × 104 cm−1) as compared with other compounds, and 4F showed the lowest value of 5.15 × 104 cm−1. Given the high photon flux of the solar spectrum within the range 500–800 nm, such different absorption coefficients are expected to affect the short-circuit currents in solar cell devices.
2.3. Theoretical calculations
Density functional theory (DFT) calculations using Gaussian 09 at the B3LYP level with a 6-311(d,p) basis set were used to investigate the conformational and electronic properties of the compounds 0–4F. The optimized structures and HOMO and LUMO orbital diagrams of all compounds are shown in Fig. 2 and S2.† The lowest energy conformational isomer was nearly coplanar and adopted a bent banana-type structure. 0–4F commonly exhibited fully delocalized HOMOs and LUMOs across the molecular backbone. The calculated HOMO and LUMO values are summarized in Fig. S2.† As the number of fluorine atom substitutions increased, the HOMO and LUMO energy levels decreased, in agreement with the CV experimental results. 0F–2F showed similar energy bandgaps of ∼1.93 eV, and the energy bandgap increased slightly from 2F (1.93 eV) to 4F (1.98 eV). This trend differed from the results obtained from the absorption measurements, in which 2F and 4F exhibited lower Eoptg values than the other compounds 0F, 1F, and 3F. This disagreement most likely arose because the calculations assumed isolated molecules in the gas phase, whereas the optical properties were affected by intermolecular interactions in the solid state.56–592F is known to assume an inverted banana shape in the crystal structure rather than a banana shape. The inverse banana favors effective π–π interactions.60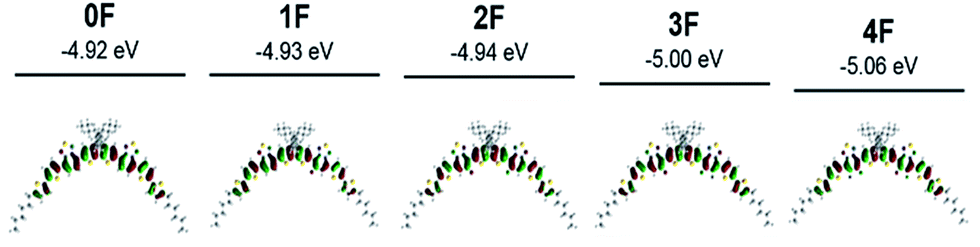 | ||
| Fig. 2 Optimized banana type structures and their HOMO orbital diagrams for compounds 0F–4F calculated by density functional theory (DFT). | ||
2.4. Charge transport
The charge transport characteristics of compounds 0–4F were explored by fabricating and analyzing bottom gate/top contact field effect transistors. Considering the given HOMO levels of all compounds, Au source/drain electrodes were selected to render near-ohmic junctions for hole injection/extraction. The transfer characteristics of the compounds presented in Fig. 3 suggested the operation of a typical p-type dominant charge transport mechanism. The charge carrier mobility values of all compounds measured in the saturation mode are summarized in Fig. 3, together with the other transistor parameters. 2F and 4F, which featured symmetric fluorine substitutions, displayed obviously higher charge carrier mobilities compared to the other derivatives, with maximum mobilities of 0.22 cm2 V−1 s−1 and 0.06 cm2 V−1 s−1, respectively. These mobility values were also higher than those of other D–A-type small molecule semiconductors, such as DTS-(PTTh2)2,61 suggesting that a more favorable crystalline structure was achieved in films of DTS-(BTTh2)2, especially by introducing symmetric and even-numbered fluorine substitutions. The subthreshold swing (S) of the FET was related to the trap density bywhere q is the electronic charge, k is Boltzmann's constant, and T is the temperature.62 Based on this equation, we estimated the interface trap densities of 0F, 1F, 2F, 3F, and 4F-based OFETs to be 8.90 × 1012, 6.52 × 1012, 1.60 × 1012, 5.62 × 1012, and 3.86 × 1012 cm−2, respectively. The estimated trap densities closely followed the trend in the charge carrier mobility, again confirming the presence of less energetic disorder in 2F and 4F.
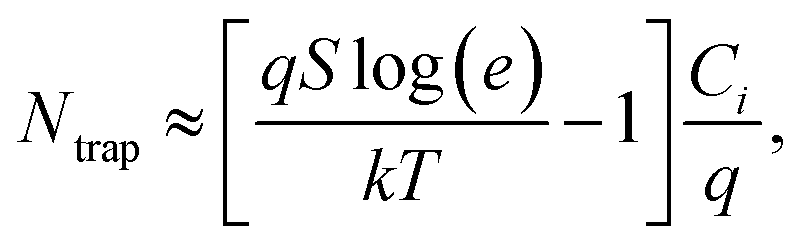
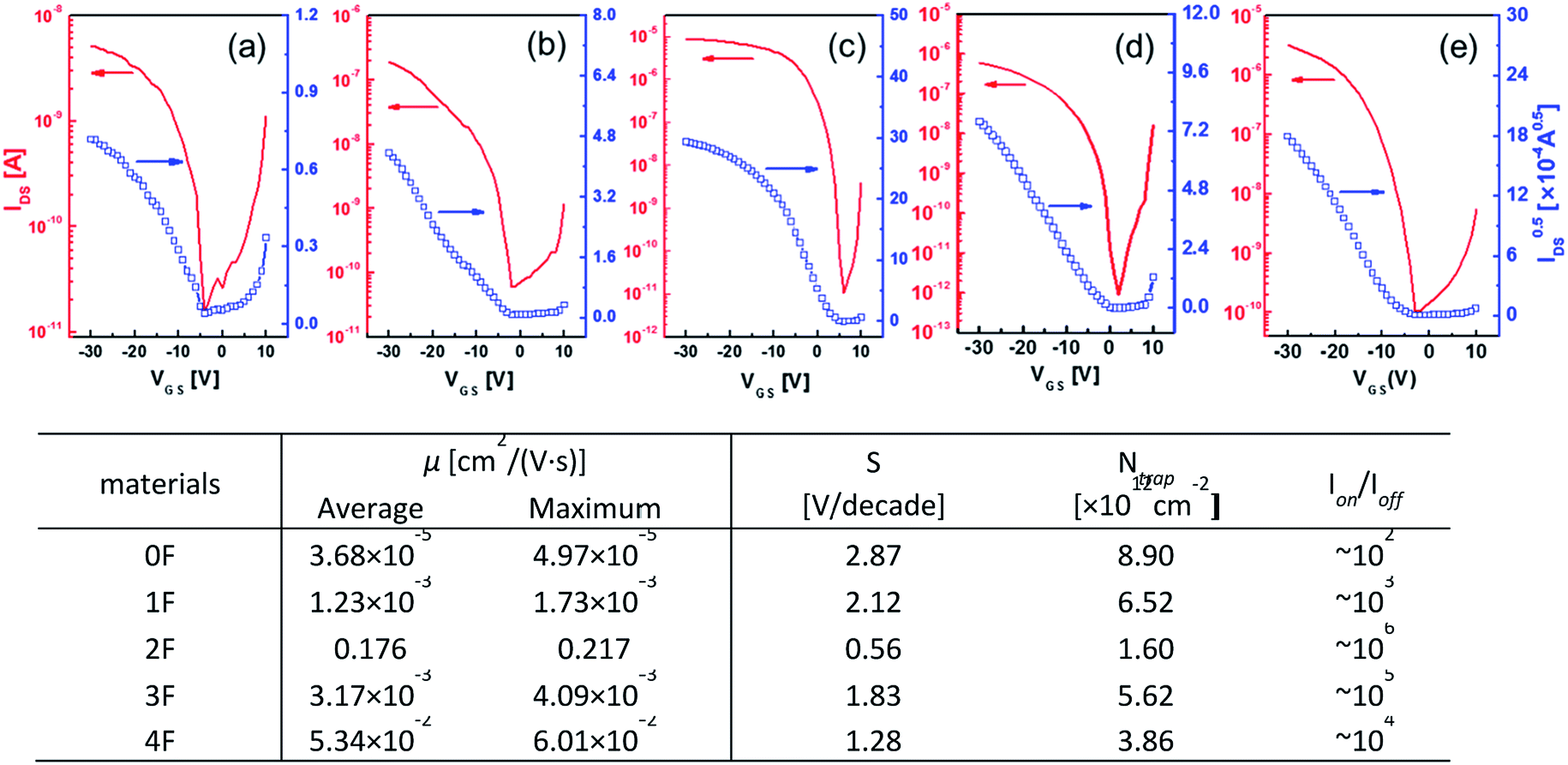 | ||
| Fig. 3 Transfer characteristics of the OFETs prepared with (a) 0F, (b) 1F, (c) 2F, (d) 3F, and (e) 4F films fabricated on OTS-modified substrates and the device performances of OFETs fabricated with 0F–4F. | ||
Grazing incidence X-ray diffraction (GIXD) studies were conducted to understand the overall superiority of 2F and 4F, in terms of charge carrier mobility, compared to other derivatives. Fig. 4 shows the 2D-GIXD patterns of all pristine compounds with their corresponding out-of-plane and in-plane profiles. The out-of-plane diffraction peaks contained a (001) diffraction peak, indicating that the DTS-(BTTh2)2 molecules stacked with their branched silyl alkyl groups on the substrate surface. The (001) peak intensity was highest for 2F and 4F. The 2D GIXD patterns obtained from 2F and 4F (as well as 3F, in part) displayed numerous reflection spots along the qz direction at a given qxy, suggesting that the DTS-(BTTh2)2 molecules adopted a three-dimensional multi-layered structure.63 Therefore, symmetric and even-numbered fluorine substitution patterns appeared to facilitate solid-state intermolecular interactions. A quantitative analysis of the crystal structures of all compounds was obtained by conducting pole figure analyses. Pole figures plot the distribution of diffraction peaks as a function of all possible crystallite orientations. The pole figures shown in Fig. 4(h) were normalized to the (200) scattering intensity of each sample in order to compare the orientations. The pole figures revealed that all the compound films adopted a preferential edge-on orientation, except for the case of 0F, in which no diffraction peaks were observed. Most of the crystallites in the film were preferentially oriented within a polar angle of 10°, indicating the preferential formation of an edge-on orientation in all compounds. Geometrical effects in each sample were corrected by multiplying the intensity profile of the pole figure by a factor of sin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ω, as shown in Fig. 4(i). Here, the relative population profile was normalized by the relative degree of crystallinity (rDoC) to facilitate a comparison among samples. The rDoC values of the compounds were calculated by integrating the relative population distributions, with consideration for the X-ray exposure time, beam footprint, sample thickness, and semiconductor volume fraction.64 The calculated rDoC values were 1.0, 4.58, 1.46, and 3.1 for the 1F, 2F, 3F, and 4F films, respectively. The large rDoC values of 2F and 4F may have been induced by the significant enhancement in the intermolecular interactions. As shown in Fig. 4(i), the 2F film exhibited a significant edge-on population, with a maximum at about ω = 6°. The 4F film showed pretty well-developed edge-on population profiles, with a maximum at about ω = 15°, although a broad mixed population was present as well (ω > 20°). The other derivatives displayed very broad random distributions that corresponded to a nearly constant population across the range ω = 20–70°. In addition to the molecular orientation profiles, we calculated Herman's orientation factors (S200) from the pole figures.65 These values ranged from −0.5 to 1, where a high value indicated high-quality edge-on orientations among the crystallites. The S200 values of the compounds are summarized in Fig. 4(j), revealing that S200 reached its highest values for 2F and 4F. Morphological analyses of the AFM topographies were further conducted in parallel with the crystalline analyses. Fig. 4(k)–(o) shows the AFM height images of all compounds. 2F and 4F appeared to form very large continuous terrace-like crystalline features (>500 nm) whereas the other derivatives formed small nodular crystallites (100 nm) with a much less featured morphology. Overall, the crystalline/morphological analyses revealed that 2F and 4F, which featured symmetric/even-numbered fluorine substitution patterns, could adopt a very well-developed preferential edge-on orientation, thereby forming well-connected crystalline morphologies with percolation pathways for charge carriers that resulted in high charge carrier mobilities.
ω, as shown in Fig. 4(i). Here, the relative population profile was normalized by the relative degree of crystallinity (rDoC) to facilitate a comparison among samples. The rDoC values of the compounds were calculated by integrating the relative population distributions, with consideration for the X-ray exposure time, beam footprint, sample thickness, and semiconductor volume fraction.64 The calculated rDoC values were 1.0, 4.58, 1.46, and 3.1 for the 1F, 2F, 3F, and 4F films, respectively. The large rDoC values of 2F and 4F may have been induced by the significant enhancement in the intermolecular interactions. As shown in Fig. 4(i), the 2F film exhibited a significant edge-on population, with a maximum at about ω = 6°. The 4F film showed pretty well-developed edge-on population profiles, with a maximum at about ω = 15°, although a broad mixed population was present as well (ω > 20°). The other derivatives displayed very broad random distributions that corresponded to a nearly constant population across the range ω = 20–70°. In addition to the molecular orientation profiles, we calculated Herman's orientation factors (S200) from the pole figures.65 These values ranged from −0.5 to 1, where a high value indicated high-quality edge-on orientations among the crystallites. The S200 values of the compounds are summarized in Fig. 4(j), revealing that S200 reached its highest values for 2F and 4F. Morphological analyses of the AFM topographies were further conducted in parallel with the crystalline analyses. Fig. 4(k)–(o) shows the AFM height images of all compounds. 2F and 4F appeared to form very large continuous terrace-like crystalline features (>500 nm) whereas the other derivatives formed small nodular crystallites (100 nm) with a much less featured morphology. Overall, the crystalline/morphological analyses revealed that 2F and 4F, which featured symmetric/even-numbered fluorine substitution patterns, could adopt a very well-developed preferential edge-on orientation, thereby forming well-connected crystalline morphologies with percolation pathways for charge carriers that resulted in high charge carrier mobilities.
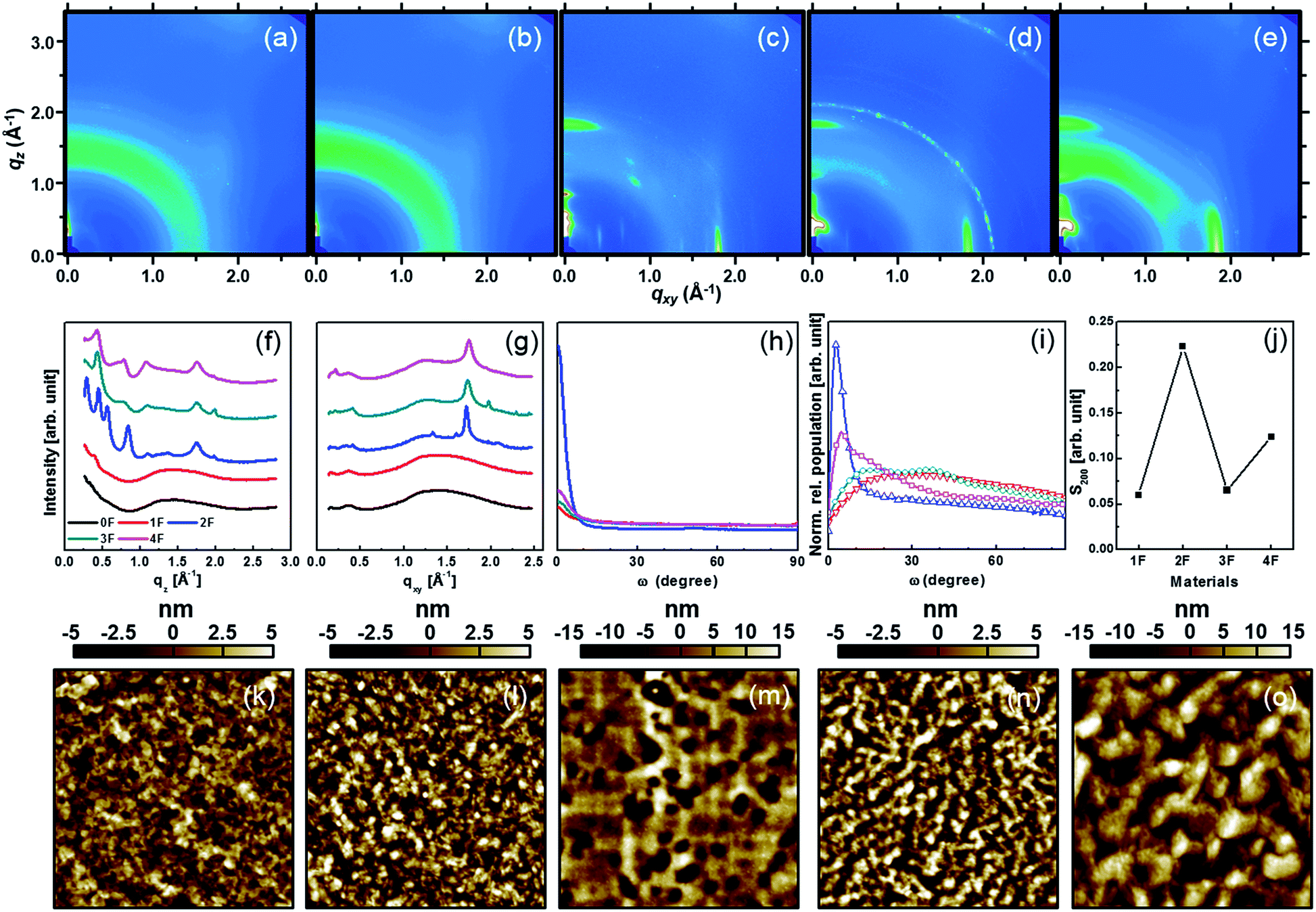 | ||
| Fig. 4 2D-GIXD patterns of (a) 0F, (b) 1F, (c) 2F, (d) 3F, and (e) 4F films, GIXD patterns in (f) out-of-plane and (g) in-plane directions, (h) the pole figures, (i) the pole figures multiplied by a factor of sinω, and (j) the Herman's orientation factors as a function of the number of fluorine substitutions. AFM topography images of films consisting of (k) 0F, (l) 1F, (m) 2F, (n) 3F, and (o) 4F. | ||
2.5. Solar cells
The solar cell properties were examined by fabricating bulk heterojunction solar cells with a device structure of ITO/PEIE/SM:PC71BM/MoO3/Ag. The photoactive layer was fabricated by spin-casting a solution of the compound:PC71BM mixture dissolved in a chlorobenzene (CB):1,8-diiodooctane (DIO) (99.6:0.4 vol%) cosolvent. The optimized compound:PC71BM ratio that gave the best solar cell performance was 3:2 for 0F, 1F, and 4F and 6:5 for 3F (see the ESI† for details about the device fabrication process). The photovoltaic performances of the prepared solar cell devices were investigated under simulated AM 1.5 G 1 Sun illumination (100 mW cm−2). The current density–voltage (J–V) curves obtained from the solar cell devices are shown in Fig. 5(a), and the characteristic solar cell data are summarized in Table 2. 2F provided the best solar cell performance, with the highest measured PCE value of 8.14%, a short circuit current (Jsc) of 15.37 mA cm−2, a fill factor (FF) of 0.664, and an open-circuit voltage (Voc) of 0.80 V. These values were 6 times the corresponding values obtained from the nonfluorinated compound 0F. 0F provided the lowest efficiency, 1.28%, with a Jsc of 4.21 mA cm−2, a FF of 0.407, and a Voc of 0.75 V. The single fluorine addition in going from 2F to 3F lowered the performance with respect to 2F, affording a PCE of 6.84% for 3F. 4F, which had one more fluorine substitution than 3F, exhibited a slightly higher efficiency of 7.06% compared to 3F, mainly due to an increase in the Voc (0.86 V) and FF (0.698) values. Therefore, the compounds that incorporated an even number of fluorine atoms, 2F and 4F, generally exhibited enhanced solar cell performances compared with the analogous compound bearing an odd number of fluorine atoms, 1F and 3F, primarily due to the improved Voc and greater FF values. The higher Voc values of the 2F and 4F-based solar cells agreed well with their deeper HOMO energy levels estimated from the electrochemical measurements (Table 1), and we concluded that symmetrical even-numbered fluorine substitutions increased Voc more effectively. Overall, fluorinated compounds achieved improved FF values compared to the nonfluorinated 0F. Among the fluorinated compounds, 2F and 4F showed higher FF values (2F: 0.664, 4F: 0.698) compared with 1F and 3F (1F: 0.520, 3F: 0.628). Fig. 5(b) shows the external quantum efficiency (EQE) spectra of the SMs-OPV devices. The EQE spectra of the devices reflected the absorption properties of each compound. 2F exhibited larger EQE values than the other compounds. Its values were as high as 70% over the wavelength range 450–750 nm, which corresponded to the absorption spectrum of 2F. These results were consistent with the observation that the 2F-based device displayed the highest Jsc value among all solar cell devices. The Jsc,EQE values obtained by integrating the EQE spectra were close to the Jsc values determined from the J–V curves (Table 2), indicating that the J–V measurements were highly reliable.
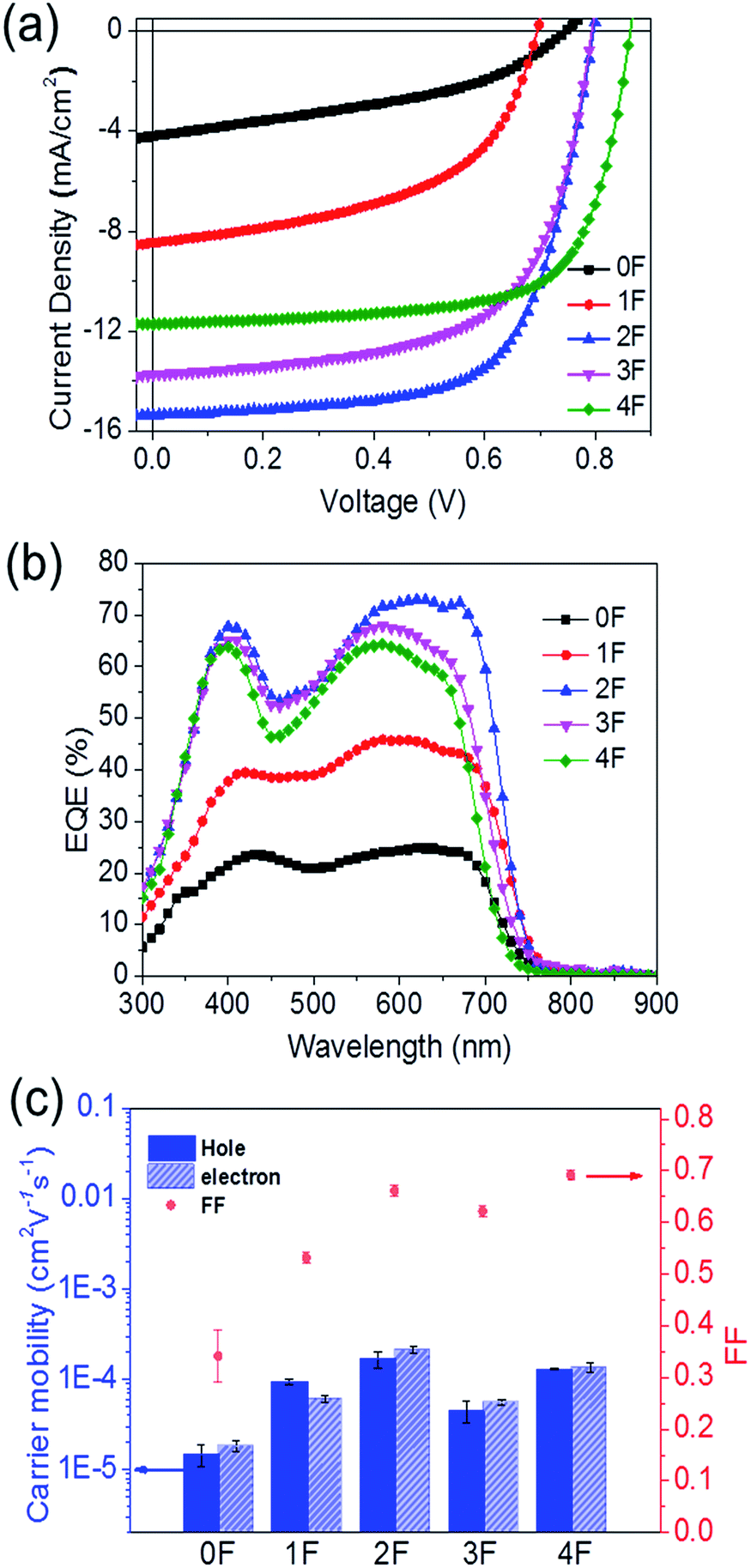 | ||
| Fig. 5 (a) The current density–voltage (J–V) characteristics and (b) external quantum efficiency (EQE) curves based on the 0F–4F/PC71BM devices. (c) Comparisons of SCLC hole and electron mobilities of the 0F–4F:PC71BM films, and the fill factor values of 0F–4F. | ||
| Active layer | V oc [V] | J sc [mA cm−2] | J sc [mA cm−2] | FFa | PCEa [%] | PCEmax [%] |
|---|---|---|---|---|---|---|
| a Average data with standard deviations obtained from nine devices. b The value extracted from the EQE spectra. | ||||||
| 0F | 0.76 ± 0.02 | 4.55 ± 0.41 | 5.01 | 0.34 ± 0.05 | 1.16 ± 0.09 | 1.28 |
| 1F | 0.70 ± 0.01 | 8.23 ± 0.24 | 9.30 | 0.53 ± 0.01 | 3.02 ± 0.03 | 3.07 |
| 2F | 0.81 ± 0.01 | 14.96 ± 0.39 | 14.37 | 0.66 ± 0.01 | 7.98 ± 0.09 | 8.14 |
| 3F | 0.78 ± 0.01 | 13.42 ± 0.22 | 12.84 | 0.62 ± 0.01 | 6.59 ± 0.14 | 6.84 |
| 4F | 0.86 ± 0.02 | 11.56 ± 0.15 | 11.51 | 0.69 ± 0.01 | 6.86 ± 0.14 | 7.06 |
GIXD studies were conducted using the compound:PC71BM blend films to understand the origin of the enhanced photovoltaic performances as a result of symmetric/even-numbered fluorination. Fig. S3† presents the 2D-GIXD patterns obtained from all blend films with their corresponding out-of-plane and in-plane profiles as well as the results of the pole analyses. All blend films showed typical (l00) lamellar-type stacking and distinct (010) π–π stacking along the out-of-plane and in-plane directions, respectively, suggesting close-to-edge-on orientations among all compounds. The crystalline orientations of the compounds were analyzed qualitatively by comparing the relative population profiles normalized by the rDoC values of the pole figures, as summarized in Fig. S3(g).† Unlike the pristine compound samples discussed in the previous section, a (010) π–π stacking peak was obtained in the pole figure analysis because the 3F-blend film did not give clear (l00) diffraction peaks. As shown in Fig. S3(g),† the 4F-blend film exhibited a relatively large edge-on population with a pronounced increase in the population at ω > 60°. All other blend films showed distinct edge-on populations only for ω > 80°. These results suggested that overall, all compounds adopted nearly randomly-oriented crystalline structures, with a slightly larger fraction of the edge-on population. The large number of fluorine moieties in 4F appeared to facilitate the formation of edge-on orientations among the compounds, even in the blend films. This effect will be further explored by our group in the near future. The values of rDoC are summarized in Fig. S3(h)† and display a clear decreasing trend in the order of 2F > 4F > 3F > 1F. The relatively large rDoC values of 2F and 4F suggested that the nanocrystalline domains of the compounds with symmetric/even-numbered fluorine substitutions were packed more densely in a defect-free manner, which could enhance percolated charge transport, as estimated from FF values. We argue that rather than merely indicating slightly different orientations among the compounds, the rDoC values have a dramatic effect on solar cell performances.
The influence of fluorination on the BHJ solar cell properties was investigated by collecting transmission electron microscopy (TEM) and atomic force microscopy (AFM) images of the compound:PC71BM BHJ film nanostructures in the solar cell devices. Fig. S4† shows the TEM and AFM surface topographic images of the BHJ blend films. The 0F and 1F blend films revealed large-scale phase separation, hundred nanometer-scale PC71BM-rich domains (Fig. S4(a) and (b),† darker phase), and a relatively high surface root-mean-square (RMS) roughness over the range 1.5–3.9 nm, as shown in Fig. S4(f) and (g).† By contrast, the 2–4F blend films displayed well-defined ten nanometer-scale phase separation and interpenetrating networks. The 2F and 4F films formed fine fibril structures, although their sizes and shapes differed, suggesting improved intermolecular packing and long-range ordered structures in the 2F and 4F blend films.
The charge carrier mobilities of the compound:PC71BM blend films were measured using the space charge limited current (SCLC) method. The 2F blend film exhibited the highest hole mobility of 1.70 × 10−4 cm2 V−1 s−1, followed by 4F (1.30 × 10−4 cm2 V−1 s−1). 1F and 3F exhibited lower hole mobility values of 9.38 × 10−5 and 4.47 × 10−5 cm2 V−1 s−1, respectively. 0F showed the lowest hole mobility of 1.50 × 10−5 cm2 V−1 s−1. These results agreed well with the OTFT mobility obtained from the pristine compound, suggesting that the intermolecular packing structure of the compound was retained in the BHJ film. The electron mobilities in the blend film followed a trend similar to the hole mobility trend. The electron mobility was highest in the 2F blend film, with a value of 2.11 × 10−4 cm2 V−1 s−1, and 4F revealed a slightly higher electron mobility of 1.35 × 10−4 cm2 V−1 s−1 compared to 1F and 3F. 1F and 3F exhibited similar electron mobility values of 6.06 × 10−5 cm2 V−1 s−1 and 5.60 × 10−5 cm2 V−1 s−1, respectively. 0F also showed the lowest electron mobility of 1.39 × 10−5 cm2 V−1 s−1. The poor charge carrier transporting properties of 0F were correlated with the amorphous packing structure in the film, as indicated in the GIXD results. All compounds displayed similar hole-to-electron mobility ratios within the range 1–1.5, indicating good charge balance between the holes and electrons in all solar cell devices. The different charge mobilities measured among the compounds could affect the FFs in the solar cells. Fig. 5(c) indicated a change in the SCLC hole and electron mobilities as a function of the fluorine substitution. Both mobilities were influenced by the symmetricity and numbers of the fluorine atom substitutions. Notable correlations between the FF values of the solar cells and the charge carrier mobilities were observed. Among the fluorinated compounds, 2F and 4F showed better transport properties and, thus, FFs compared with 1F and 3F. A comparison of the absorption coefficients of the compounds revealed that the Jsc values of the compounds depended on the charge mobility. The effects of the molecular arrangements and mobilities on the photocurrent properties of solar cells were estimated by comparing the Jsc values and the absorption properties of the compounds. Fig. S7† shows pristine film absorption profiles for 0F–4F (determined by the absorption coefficient and the film thickness, thickness of each film was adjusted to 70 ± 3 nm). The ratios of Jsc to integrated absorption were found to decrease in the order of 2F > 4F > 3F > 1F > 0F, in good agreement with the SCLC mobility trend observed across 0–4F. We expected that the remarkably different solar cell performances among the molecules were correlated with the improved charge transport properties, which may be influenced by the fluorine substitutions on the accepting BT units. As a result, the overall solar cell efficiency depended significantly on the symmetry of the fluorine incorporation pattern, and number (even or odd) of fluorine atom substitutions. The compounds 2F and 4F achieved higher solar cell performances compared with 1F and 3F.
Differences in the nanoscale morphology and charge transport properties of 0–4F pointed to the acceptor BT and its fluorine substitution pattern, which played a critical role in the intermolecular interactions and packing structure of the thin film.
The solid state packing structure depends on the molecular structure, relative energy, and electrostatics of the conformations adopted by the compound 0–4F. The equilibrium geometries and dipole moments of 0–4F in the gas phase were calculated using DFT methods. Fig. 6 shows the optimized structures of the possible molecular conformations, tabulated energies, and dipole moments of 0–4F. All compounds assumed a banana-shaped bent conformation as the energetically most stable structure. These results differed from the results obtained from the single crystal X-ray diffraction studies of 2F, in which the conjugated backbone formed an inverted banana shape with a high planarity and packed in a slip-stack manner. The inverted banana shape conformation induced high intermolecular overlap among the conjugated backbones, which facilitated π–π overlapping.58 Although a large fraction of 2F is present in a banana shape in the gas state, the conformation switches to an inverted banana shape in the solid state, thereby favoring efficient intermolecular interactions. The other compounds, 0F, 1F, 3F, and 4F, may behave similarly and undergo similar packing processes in the film state, although their fluorine substitution patterns influenced the intermolecular interactions. We compared the energy differences among the different conformers. A large energy increase from the banana shape to another conformation would suggest that few of the compounds would make this transition, and the banana shape would dominate. As shown in Fig. 6, 0F showed the highest energy increase from the banana shape to the inverted banana shape (6.53 kJ mol−1), followed by 1F (4.3 kJ mol−1). Notably, as more fluorine atoms were incorporated from 0F to 4F, the compounds displayed a smaller energy difference between the banana and inverted banana shapes, indicating that a greater fraction of the fluorinated compounds was present in an inverted banana shape, which favored efficient π–π interactions in the thin film. The predicted film conformations agreed with the experimental GIXD results, which indicated that 0F formed amorphous structures and provided the lowest charge carrier mobility in the OTFT studies. The energy difference between the banana shape and the zigzag conformation decreased from 0F to 4F. Therefore, the relative energy difference between the zigzag and the inverted banana shape was smaller for the fluorinated compounds. 4F had the smallest value of 0.36 kJ mol−1, whereas 0F had the largest value of 3.39 kJ mol−1. The energy differences of 1F and 3F depended on the molecular shape. The zigzag-shaped conformation formed by rotating the more fluorinated BT units was lower in energy than the conformation of the corresponding shape formed by rotating the non- or less fluorinated BT. Fig. 6 presents the magnitude and direction of the dipole moment. The compounds with a symmetrical conformation (0F, 2F, and 4F) showed a dipole direction perpendicular to the linear molecule, whereas the asymmetrically shaped compounds 1F and 3F exhibited distinct dipole directions that depended on the conformation. In the zigzag conformation, 1F and 3F assumed two distinct isomers due to their asymmetric chemical structures, and the isomers displayed different dipole moments relative to one another. The asymmetric 1F and 3F molecules displayed more diverse dipole moments and directions, as well as a greater number of conformations, which could be related to their poor crystalline properties in the thin film state, as shown in the GIXD patterns (Fig. 4). The 1F and 3F diffraction patterns did not reveal any notable peaks corresponding to the long-range ordering, such as the lamellar-like packing structures. By contrast, peaks due to the alkyl stacking interactions as well as the well-defined π–π stacking interactions were observed in the 2F and 4F films. The molecules tended to organize themselves in such a way as to minimize the net dipole moment. Molecules with a symmetric linear conformation favorably assembled in an antiparallel manner and efficiently formed a lamellar-like packing structure. Although all compounds with the inverted banana conformation could efficiently dimerize and form a lamellar-like structure, molecules with different conformations were expected to coexist in a certain distribution. Because the zigzag shapes were relatively stable compared with the inverted banana shape, they were present in some fraction in the thin film. The zigzag shape was able to form more favorable π–π stacking structures compared with the banana shape because it was somewhat linear. The zigzag structures in 1F and 3F may not form an effective alkyl stacking due to their diverse dipole moments and orientations present in these molecular distributions. For example, a combination of inverted banana and zigzag configurations (1F zigzag-2) in 1F can form a lamellar-like packing structure that aligns the dipole moments of the molecules in a single direction, thereby increasing the energy of the system. The broad distribution of conformers and the relatively large dipole moments of 1F and 3F increase the probability of assuming this type of molecular structure, consistent with the observed low crystallinities of 1F and 3F in the thin film state. By contrast, 2F and 4F were present in a narrow distribution of conformations, and the dipole moments were lower than those of 1F and 3F. Therefore, the probability of forming alkyl stacking interactions among the different conformations in 2F (or 4F) was relatively low. Even dimerization between the different conformations yielded an overall dipole and energy enhancements that were not as high as the corresponding values for 1F and 3F. The narrower distribution of conformers in 2F and 4F and their more favorable dipole–dipole interactions suggested that these compounds should display better assembly properties in the thin film states.
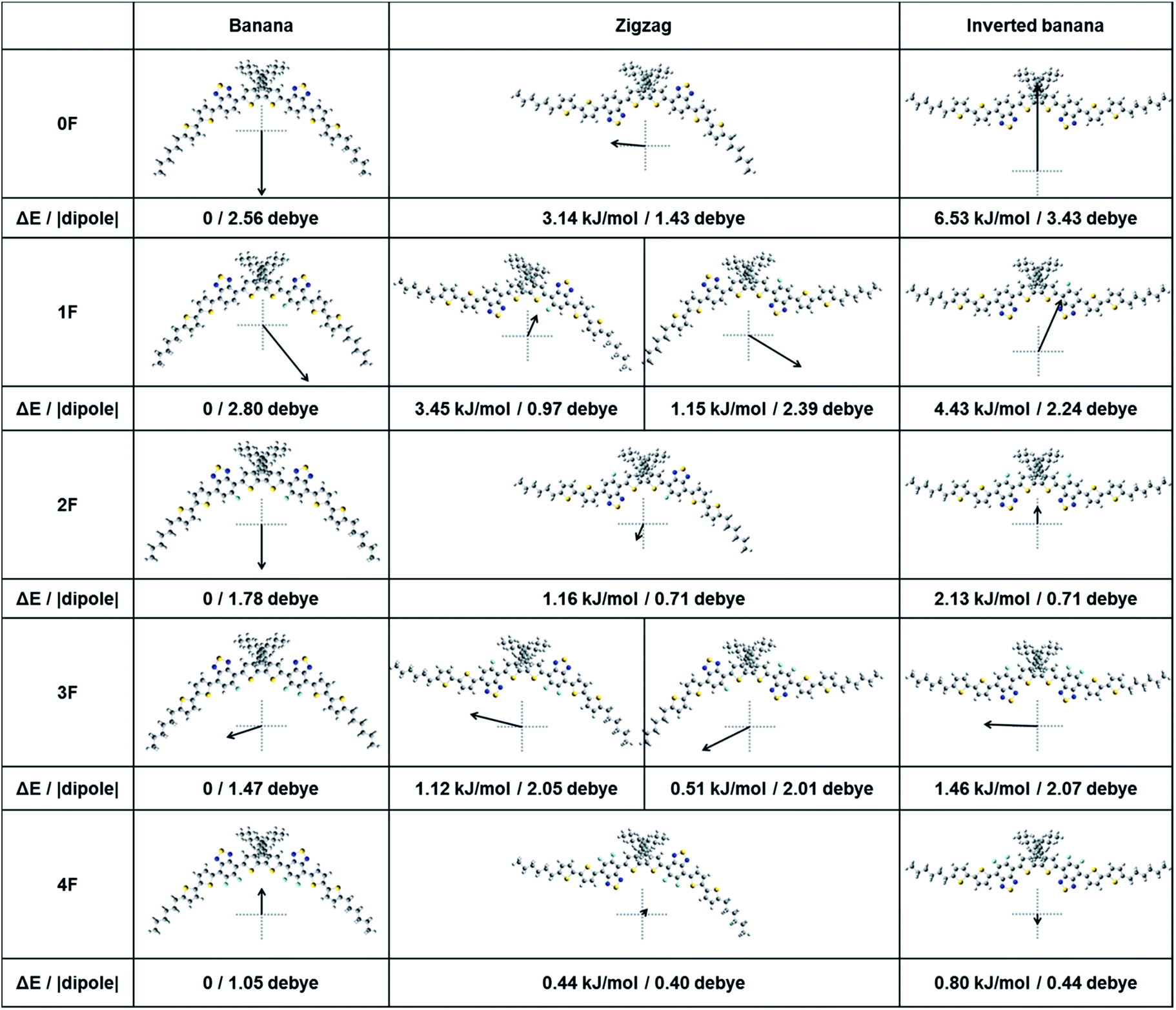 | ||
| Fig. 6 Optimized molecular conformations, energy difference from the banana shape conformer of each compound and their dipole moments for compounds 0F–4F. | ||
The advantages of the 2F molecular conformations, packing structures, and mobility were exploited by preparing planar heterojunction perovskite solar cells (PHJ PSCs) using 2F as a dopant-free hole transporting material (HTM). The fabricated cell has the architecture of FTO/TiO2/MAPbI3/HTM/Au. The device preparation steps are described in detail in the ESI.† For comparison purposes, a PSC was fabricated by replacing 2F with 0F in the same device structure prepared under otherwise identical processing conditions. Fig. 7(a) shows a representative SEM cross-sectional image of the 2F-based PHJ PSC. Fig. 7(b) displays the corresponding energy band diagrams of each component, revealing that the holes generated in MAPbI3 could be effectively transported to the 0F and 2F HTMs. The photovoltaic performances of these two solar cell devices under a simulated AM 1.5 G 1 Sun illumination (100 mW cm−2) are represented by the current density–voltage (J–V) curves plotted in Fig. 7(c) and (d). The photovoltaic properties of the solar cells are summarized in Table 3. The PHJ PSC prepared with 2F provided a PCE (η) of 14.2% under forward scan conditions (scan rate = 50 mV s−1), with an open circuit voltage (Voc) of 0.99 V, a short circuit current density (Jsc) of 20.0 mA cm−2, and a fill factor (FF) of 0.72. An efficiency of 14.7% was obtained under reverse scan conditions, with a Voc of 0.99 V, a Jsc of 20.1 mA cm−2, and a FF of 0.74, thereby yielding an average PCE (ηavg) of 14.45%. By contrast, the parameters obtained from the 0F-based device were all notably lower. The PSC was characterized by a Voc of 0.93 V, a Jsc of 17.0 mA cm−2, and a FF of 0.54, yielding an efficiency of 8.5% under the forward scan conditions and a Voc of 0.94 V, a Jsc of 17.4 mA cm−2, a FF of 0.57, and an efficiency of 9.4% under reverse scan conditions; the resulting ηavg was 8.95%. It is notable that the 2F based PSC device shows much smaller J–V hysteresis compared with the PSC with 0F. Fig. 7(e) shows the current density variation of 0F and F2 based devices with a light soaking time under optimal bias voltage (Vopt) (Vopt = 0.64 V for the 0F based device and Vopt = 0.80 V for the 2F based device) of maximum power (Pmax) point under 1 Sun (100 mW cm−2) illumination of continuous light. Both devices exhibit the same tendency that the current density (J) is quickly saturated with a light soaking time and maintains stability under the current experimental time window. Fig. S5† shows histograms of the efficiencies and the PCEs averaged from 40 devices are 5.14 ± 2.08% and 11.18 ± 1.89% for the 0F and 2F PSCs, respectively. The superior performance of the 2F-based device was attributed to the enhancement in the FF value (averaged from forward and reverse scans) from 55.6% to 72.6% and the Jsc value from 17 to 20 mA cm−2. The higher Voc of the 2F-based device, which was 50 mV higher than the value measured from 0F, is attributed to the lower HOMO energy level of 2F compared with 0F.
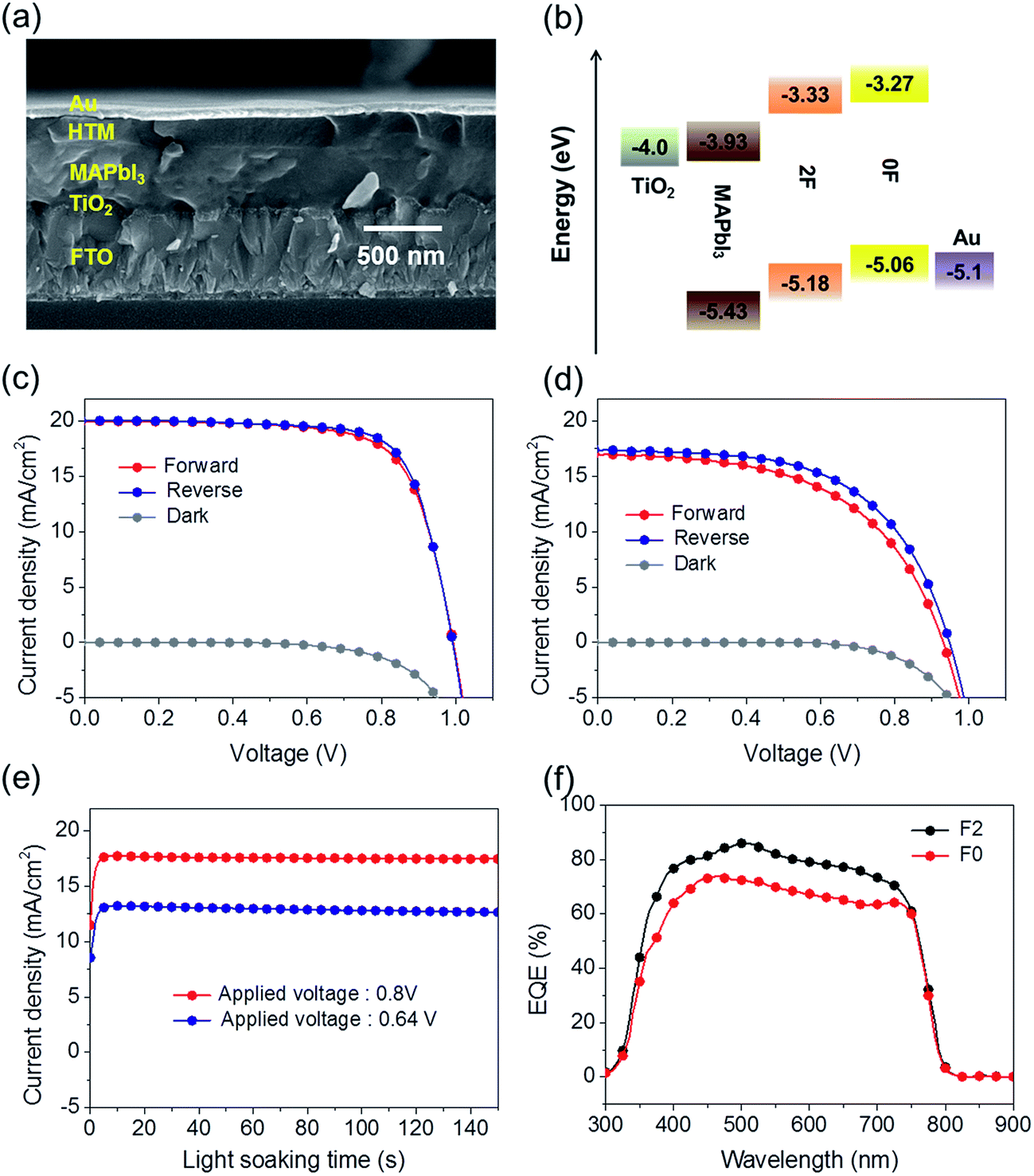 | ||
| Fig. 7 (a) The SEM cross-section image and (b) energy diagram of the perovskite solar cell device. The current density–voltage (J–V) characteristics based on the solar cells with (c) 2F and (d) 0F. (e) current density (J) variation of the 0F and 2F based PSCs with a light soaking (1 Sun) time under the applied bias voltage (Vopt) at maximum power point (Pmax) and (f) EQE spectra of the 0F and 2F based PSCs. | ||
| Scan direction | V oc [V] | J sc [mA cm−2] | FF | PCE [%] | PCE (ηavg) | |
|---|---|---|---|---|---|---|
| 0F | Forward | 0.93 | 17.0 | 0.54 | 8.50 | 8.95 |
| Reverse | 0.94 | 17.4 | 0.57 | 9.40 | ||
| 2F | Forward | 0.99 | 20.0 | 0.72 | 14.20 | 14.45 |
| Reverse | 0.99 | 20.1 | 0.74 | 14.70 |
The improved photocurrent properties in the 2F device most likely arose from the higher hole transporting ability of 2F. The EQE spectra shown in Fig. 7(f) revealed that the PHJ PSC prepared with 2F offered a better EQE value in the range of 350–800 nm compared with the 0F device. The Jsc of the 2F device was calculated by integrating the EQE data across the AM1.5G solar spectrum (100 mW cm−2) and was found to be 19.3 mA cm−2 higher than the value of 16.6 mA cm−2 obtained from the 0F-based device, a difference that was comparable to the difference in Jsc obtained from the respective J–V curves.
3. Conclusions
Four DTS(BTTh2)2 small molecules with different fluorine substitution patterns were prepared, and their charge transport and solar cell properties were examined. The optical, electronic, and morphological properties of these compounds were compared to understand the significance and effect of the symmetricity and number of fluorine atoms incorporated. 2F and 4F displayed better edge-on orientations and higher crystalline properties in the film state compared with 1F and 3F; thus, 2F achieved the highest OTFT mobility of 0.217 cm2 V−1 s−1 among the compounds. When blended with PC71BM, 0F–4F exhibited similar random orientations; however, 2F and 4F, which featured symmetric and even-numbered fluorine substitution patterns, displayed greater order among the nanocrystalline domains than 1F or 3F, resulting in higher charge transport properties in the blend films. As a result, 2F achieved the highest photovoltaic performances with an 8.14% efficiency due to its improved FF and photocurrent properties. Furthermore, the PSC fabricated using 2F as a dopant-free HTM displayed a high PCE of 14.45% by virtue of its high charge transport properties, which were significantly better than those obtained from the corresponding 0F-based PSC device (8.95%). The low variation in the local dipole moment and the narrow distribution of 2F conformers facilitated intermolecular interactions, which could enhance/drive crystal formation in the solid state compared with 1F and 3F. The results suggested that the molecular conformations favored upon introducing substitution groups, such as fluorine, are important factors in the design of high-performance small-molecule materials: in addition to the electron-donating or -accepting properties of a molecular building blocks, factors that affect the intermolecular interactions in the solid state must be considered during the design of push–pull-type charge transporting materials.Acknowledgements
This work was supported by New and Renewable Energy Program of the Korea Institute of Energy Technology Evaluation and Planning (KETEP) grant funded by the Ministry of Knowledge Economy (MKE) (20163030013620), the Global Frontier R&D Program on Center for Multiscale Energy System funded by the National Research Foundation under the Ministry of Science, and the National Research Foundation of Korea Grant, funded by the Korean Government (MSIP) (2016, University-Institute corporation program).References
- K. H. Cheon, J. Cho, Y. H. Kim and D. S. Chung, ACS Appl. Mater. Interfaces, 2015, 7, 14004–14010 CAS.
- V. Grenier, A. S. Walker and E. W. Miller, J. Am. Chem. Soc., 2015, 137, 10894–10897 CrossRef CAS PubMed.
- M. Gross, D. C. Muller, H.-G. Nothofer, U. Scherf, D. Neher, C. Brauchle and K. Meerholz, Nature, 2000, 405, 661–665 CrossRef CAS PubMed.
- V. G. Kozlov, V. Bulovic, P. E. Burrows and S. R. Forrest, Nature, 1997, 389, 362–364 CrossRef CAS.
- J. R. Lawrence, G. A. Turnbull and I. D. W. Samuel, Appl. Phys. Lett., 2002, 80, 3036 CrossRef CAS.
- G. Li, V. Shrotriya, J. Huang, Y. Yao, T. Moriarty, K. Emery and Y. Yang, Nat. Mater., 2005, 4, 864–868 CrossRef CAS.
- G. Long, X. Wan, B. Kan, Y. Liu, G. He, Z. Li, Y. Zhang, Y. Zhang, Q. Zhang, M. Zhang and Y. Chen, Adv. Energy Mater., 2013, 3, 639–646 CrossRef CAS.
- J. Yao, C. Yu, Z. Liu, H. Luo, Y. Yang, G. Zhang and D. Zhang, J. Am. Chem. Soc., 2016, 138, 173–185 CrossRef CAS PubMed.
- X. Zhang, H. Bronstein, A. J. Kronemeijer, J. Smith, Y. Kim, R. J. Kline, L. J. Richter, T. D. Anthopoulos, H. Sirringhaus, K. Song, M. Heeney, W. Zhang, I. McCulloch and D. M. DeLongchamp, Nat. Commun., 2013, 4, 2238 Search PubMed.
- Y. Liu, Z. A. Page, T. P. Russell and T. Emrick, Angew. Chem., Int. Ed., 2015, 54, 11485–11489 CrossRef CAS PubMed.
- J. Mei, H. Kim do, A. L. Ayzner, M. F. Toney and Z. Bao, J. Am. Chem. Soc., 2011, 133, 20130–20133 CrossRef CAS PubMed.
- J. W. Sun, J. Y. Baek, K.-H. Kim, C.-K. Moon, J.-H. Lee, S.-K. Kwon, Y.-H. Kim and J.-J. Kim, Chem. Mater., 2015, 27, 6675–6681 CrossRef CAS.
- A. T. Yiu, P. M. Beaujuge, O. P. Lee, C. H. Woo, M. F. Toney and J. M. Frechet, J. Am. Chem. Soc., 2012, 134, 2180–2185 CrossRef CAS PubMed.
- Y. Diao, B. C. Tee, G. Giri, J. Xu, H. Kim do, H. A. Becerril, R. M. Stoltenberg, T. H. Lee, G. Xue, S. C. Mannsfeld and Z. Bao, Nat. Mater., 2013, 12, 665–671 CrossRef CAS PubMed.
- S. Hong, H. Kang, G. Kim, S. Lee, S. Kim, J.-H. Lee, J. Lee, M. Yi, J. Kim, H. Back, J.-R. Kim and K. Lee, Nat. Commun., 2016, 7, 10279 CrossRef CAS PubMed.
- J. Huang, C.-Z. Li, C.-C. Chueh, S.-Q. Liu, J.-S. Yu and A. K. Y. Jen, Adv. Energy Mater., 2015, 5, 1500406 CrossRef.
- S. Mandal, G. Dell’Erba, A. Luzio, S. G. Bucella, A. Perinot, A. Calloni, G. Berti, G. Bussetti, L. Duò, A. Facchetti, Y.-Y. Noh and M. Caironi, Org. Electron., 2015, 20, 132–141 CrossRef CAS.
- T. Sekitani, U. Zschieschang, H. Klauk and T. Someya, Nat. Mater., 2010, 9, 1015–1022 CrossRef CAS PubMed.
- H. Youn, T. Lee and L. J. Guo, Energy Environ. Sci., 2014, 7, 2764–2770 CAS.
- Y. Lin, Y. Li and X. Zhan, Chem. Soc. Rev., 2012, 41, 4245–4272 RSC.
- B. Kan, M. Li, Q. Zhang, F. Liu, X. Wan, Y. Wang, W. Ni, G. Long, X. Yang, H. Feng, Y. Zuo, M. Zhang, F. Huang, Y. Cao, T. P. Russell and Y. Chen, J. Am. Chem. Soc., 2015, 137, 3886–3893 CrossRef CAS PubMed.
- B. Kan, Q. Zhang, M. Li, X. Wan, W. Ni, G. Long, Y. Wang, X. Yang, H. Feng and Y. Chen, J. Am. Chem. Soc., 2014, 136, 15529–15532 CrossRef CAS PubMed.
- A. K. K. Kyaw, D. H. Wang, V. Gupta, W. L. Leong, L. Ke, G. C. Bazan and A. J. Heeger, ACS Nano, 2013, 7, 4569–4577 CrossRef CAS PubMed.
- K. H. Jung, S. Y. Bae, K. H. Kim, M. J. Cho, K. Lee, Z. H. Kim, D. H. Choi, D. H. Lee, D. S. Chung and C. E. Park, Chem. Commun., 2009, 5290–5292 RSC.
- J. I. Park, J. W. Chung, J. Y. Kim, J. Lee, J. Y. Jung, B. Koo, B. L. Lee, S. W. Lee, Y. W. Jin and S. Y. Lee, J. Am. Chem. Soc., 2015, 137, 12175–12178 CrossRef CAS PubMed.
- T. Yamamoto and K. Takimiya, J. Am. Chem. Soc., 2007, 129, 2224–2225 CrossRef CAS PubMed.
- M. J. Kang, I. Doi, H. Mori, E. Miyazaki, K. Takimiya, M. Ikeda and H. Kuwabara, Adv. Mater., 2011, 23, 1222–1225 CrossRef CAS PubMed.
- H. Minemawari, T. Yamada, H. Matsui, J. Tsutsumi, S. Haas, R. Chiba, R. Kumai and T. Hasegawa, Nature, 2011, 475, 364–367 CrossRef CAS PubMed.
- Y. Yuan, G. Giri, A. L. Ayzner, A. P. Zoombelt, S. C. Mannsfeld, J. Chen, D. Nordlund, M. F. Toney, J. Huang and Z. Bao, Nat. Commun., 2014, 5, 3005 Search PubMed.
- T. Umeyama and H. Imahori, J. Mater. Chem. A, 2014, 2, 11545–11560 CAS.
- Z.-G. Zhang and J. Wang, J. Mater. Chem., 2012, 22, 4178 RSC.
- A. Ajayaghosh, Chem. Soc. Rev., 2003, 32, 181–191 RSC.
- B. Albinsson, M. P. Eng, K. Pettersson and M. U. Winters, Phys. Chem. Chem. Phys., 2007, 9, 5847–5864 RSC.
- H. Zhou, L. Yang and W. You, Macromolecules, 2012, 45, 607–632 CrossRef CAS.
- Y. Hua, B. Xu, P. Liu, H. Chen, H. Tian, M. Cheng, L. Kloo and L. Sun, Chem. Sci., 2016, 7, 2633–2638 RSC.
- Y. Liu, Z. Hong, Q. Chen, H. Chen, W. H. Chang, Y. M. Yang, T. B. Song and Y. Yang, Adv. Mater., 2016, 28, 440–446 CrossRef CAS PubMed.
- P. Qin, H. Kast, M. K. Nazeeruddin, S. M. Zakeeruddin, A. Mishra, P. Bäuerle and M. Grätzel, Energy Environ. Sci., 2014, 7, 2981–2985 CAS.
- C. Steck, M. Franckevičius, S. M. Zakeeruddin, A. Mishra, P. Bäuerle and M. Grätzel, J. Mater. Chem. A, 2015, 3, 17738–17746 CAS.
- N. Cho, K. Song, J. K. Lee and J. Ko, Chem.–Eur. J., 2012, 18, 11433–11439 CrossRef CAS PubMed.
- K. Sun, Z. Xiao, S. Lu, W. Zajaczkowski, W. Pisula, E. Hanssen, J. M. White, R. M. Williamson, J. Subbiah, J. Ouyang, A. B. Holmes, W. W. Wong and D. J. Jones, Nat. Commun., 2015, 6, 6013 CrossRef CAS PubMed.
- T. S. van der Poll, J. A. Love, T. Q. Nguyen and G. C. Bazan, Adv. Mater., 2012, 24, 3646–3649 CrossRef CAS PubMed.
- Q. Zhang, B. Kan, F. Liu, G. Long, X. Wan, X. Chen, Y. Zuo, W. Ni, H. Zhang, M. Li, Z. Hu, F. Huang, Y. Cao, Z. Liang, M. Zhang, T. P. Russell and Y. Chen, Nat. Photonics, 2015, 9, 35–41 CrossRef CAS.
- J. J. Intemann, K. Yao, F. Ding, Y. Xu, X. Xin, X. Li and A. K. Y. Jen, Adv. Funct. Mater., 2015, 25, 4889–4897 CrossRef CAS.
- A. Ojala, A. Petersen, A. Fuchs, R. Lovrincic, C. Pölking, J. Trollmann, J. Hwang, C. Lennartz, H. Reichelt, H. W. Höffken, A. Pucci, P. Erk, T. Kirchartz and F. Würthner, Adv. Funct. Mater., 2012, 22, 86–96 CrossRef CAS.
- C. Sutton, C. Risko and J.-L. Brédas, Chem. Mater., 2016, 28, 3–16 CrossRef CAS.
- V. Gupta, A. K. Kyaw, D. H. Wang, S. Chand, G. C. Bazan and A. J. Heeger, Sci. Rep., 2013, 3, 1965 Search PubMed.
- Y. Sun, J. Seifter, L. Huo, Y. Yang, B. B. Y. Hsu, H. Zhou, X. Sun, S. Xiao, L. Jiang and A. J. Heeger, Adv. Energy Mater., 2015, 5, 1400987 CrossRef.
- X. Liu, Y. Sun, B. B. Hsu, A. Lorbach, L. Qi, A. J. Heeger and G. C. Bazan, J. Am. Chem. Soc., 2014, 136, 5697–5708 CrossRef CAS PubMed.
- T. L. Nguyen, H. Choi, S. J. Ko, M. A. Uddin, B. Walker, S. Yum, J. E. Jeong, M. H. Yun, T. J. Shin, S. Hwang, J. Y. Kim and H. Y. Woo, Energy Environ. Sci., 2014, 7, 3040–3051 CAS.
- A. C. Stuart, J. R. Tumbleston, H. Zhou, W. Li, S. Liu, H. Ade and W. You, J. Am. Chem. Soc., 2013, 135, 1806–1815 CrossRef CAS PubMed.
- M. Zhang, X. Guo, S. Zhang and J. Hou, Adv. Mater., 2014, 26, 1118–1123 CrossRef CAS PubMed.
- H. Zhou, L. Yang, A. C. Stuart, S. C. Price, S. Liu and W. You, Angew. Chem., Int. Ed., 2011, 50, 2995–2998 CrossRef CAS PubMed.
- J. S. Kim, Z. Fei, S. Wood, D. T. James, M. Sim, K. Cho, M. J. Heeney and J.-S. Kim, Adv. Energy Mater., 2014, 4, 1400527 CrossRef.
- P. M. Oberhumer, Y.-S. Huang, S. Massip, D. T. James, G. Tu, S. Albert-Seifried, D. Beljonne, J. Cornil, J.-S. Kim, W. T. S. Huck, N. C. Greenham, J. M. Hodgkiss and R. H. Friend, J. Chem. Phys., 2011, 134, 114901 CrossRef PubMed.
- R. Steyrleuthner, M. Schubert, I. Howard, B. Klaumünzer, K. Schilling, Z. Chen, P. Saalfrank, F. Laquai, A. Facchetti and D. Neher, J. Am. Chem. Soc., 2012, 134, 18303–18317 CrossRef CAS PubMed.
- J. Clark, J.-F. Chang, F. C. Spano, R. H. Friend and C. Silva, Appl. Phys. Lett., 2009, 94, 163306 CrossRef.
- A. T. Haedler, H. Misslitz, C. Buehlmeyer, R. Q. Albuquerque, A. Kohler and H. W. Schmidt, ChemPhysChem, 2013, 14, 1818–1829 CrossRef CAS PubMed.
- R. Noriega, J. Rivnay, K. Vandewal, F. P. V. Koch, N. Stingelin, P. Smith, M. F. Toney and A. Salleo, Nat. Mater., 2013, 12, 1038–1044 CrossRef CAS PubMed.
- M. Schubert, D. Dolfen, J. Frisch, S. Roland, R. Steyrleuthner, B. Stiller, Z. Chen, U. Scherf, N. Koch, A. Facchetti and D. Neher, Adv. Energy Mater., 2012, 2, 369–380 CrossRef CAS.
- J. A. Love, C. M. Proctor, J. Liu, C. J. Takacs, A. Sharenko, T. S. van der Poll, A. J. Heeger, G. C. Bazan and T.-Q. Nguyen, Adv. Funct. Mater., 2013, 23, 5019–5026 CrossRef CAS.
- X. Liu, Y. Sun, L. A. Perez, W. Wen, M. F. Toney, A. J. Heeger and G. C. Bazan, J. Am. Chem. Soc., 2012, 134, 20609–20612 CrossRef CAS PubMed.
- S. Nam, D. S. Chung, J. Jang, S. H. Kim, C. Yang, S.-K. Kwon and C. E. Park, J. Electrochem. Soc., 2010, 157, H90–H93 CrossRef CAS.
- W. H. Lee, J. A. Lim, D. H. Kim, J. H. Cho, Y. Jang, Y. H. Kim, J. I. Han and K. Cho, Adv. Funct. Mater., 2008, 18, 560–565 CrossRef.
- M. R. Hammond, R. J. Kline, A. A. Herzing, L. J. Richter, D. S. Germack, H.-W. Ro, C. L. Soles, D. A. Fischer, T. Xu, L. Yu, M. F. Toney and D. M. DeLongchamp, ACS Nano, 2011, 5, 8248–8257 CrossRef CAS PubMed.
- R. Kroon, A. Diaz de Zerio Mendaza, S. Himmelberger, J. Bergqvist, O. Backe, G. C. Faria, F. Gao, A. Obaid, W. Zhuang, D. Gedefaw, E. Olsson, O. Inganas, A. Salleo, C. Muller and M. R. Andersson, J. Am. Chem. Soc., 2014, 136, 11578–11581 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details including synthesis and experimental method. See DOI: 10.1039/c6sc02448c |
| This journal is © The Royal Society of Chemistry 2016 |