
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cationic pillar[6]arene/ATP host–guest recognition: selectivity, inhibition of ATP hydrolysis, and application in multidrug resistance treatment†
Guocan
Yu‡
a,
Jiong
Zhou‡
a,
Jie
Shen
b,
Guping
Tang
b and
Feihe
Huang
*a
aState Key Laboratory of Chemical Engineering, Center for Chemistry of High-Performance & Novel Materials, Department of Chemistry, Zhejiang University, Hangzhou 310027, P. R. China. E-mail: fhuang@zju.edu.cn; Fax: +86-571-8795-3189; Tel: +86-571-8795-3189
bDepartment of Chemistry, Institute of Chemical Biology and Pharmaceutical Chemistry, Zhejiang University, Hangzhou 310027, P. R. China
First published on 2nd March 2016
Abstract
Due to the differences in the cavity size of the hosts and the charge and length of the guests, a cationic water-soluble pillar[6]arene (WP6) selectively complexes with ATP to form a stable 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 inclusion complex WP6⊃ATP. This host–guest complexation was utilized to efficiently inhibit the hydrolysis of ATP, arising from the existence of the hydrophobic cavity of WP6. A folic acid functionalized diblock copolymer (FA-PEG-b-PAA) was employed to PEGylate WP6 to endow the polyion complex (PIC) micelles with specific targeting ability, preferentially delivering WP6 to folate receptor over-expressing KB cell. This host–guest complexation was further used to block the efflux pump to transport anticancer drugs out of cells by cutting off the energy source, which enhanced the efficacy of the cancer chemotherapy of DOX·HCl towards drug resistant MCF-7/ADR cell. This supramolecular method provides an extremely distinct strategy to potentially overcome multidrug resistance (MDR).
:1 inclusion complex WP6⊃ATP. This host–guest complexation was utilized to efficiently inhibit the hydrolysis of ATP, arising from the existence of the hydrophobic cavity of WP6. A folic acid functionalized diblock copolymer (FA-PEG-b-PAA) was employed to PEGylate WP6 to endow the polyion complex (PIC) micelles with specific targeting ability, preferentially delivering WP6 to folate receptor over-expressing KB cell. This host–guest complexation was further used to block the efflux pump to transport anticancer drugs out of cells by cutting off the energy source, which enhanced the efficacy of the cancer chemotherapy of DOX·HCl towards drug resistant MCF-7/ADR cell. This supramolecular method provides an extremely distinct strategy to potentially overcome multidrug resistance (MDR).
Introduction
The recognition and sensing of biologically important nucleosides and nucleotides using artificial receptors has attracted considerable attention over the past decades, as they make up the fundamental building blocks of all life forms.1 Among all nucleotides, the selective recognition of adenosine 5′-triphosphate (ATP) is of particular interest, because it is a universal energy source and is the intracellular mediator in many biological processes.2 For example, P-glycoprotein (P-gp) functions by increasing the efflux of drugs out of the cell against a concentration gradient and reducing the rate of influx of drugs by utilizing the energy from ATP hydrolysis, thereby decreasing intracellular levels of the drugs below their level of effectiveness, a phenomenon that leads to multidrug resistance (MDR).3 In cells, ATP is enzymatically hydrolyzed to PPi (pyrophosphate) and adenosine 5′-monophosphate (AMP) or Pi (orthophosphate) and adenosine 5′-diphosphate (ADP). This imposes certain conditions on a receptor to have excellent selectivity between these anions, especially for ATP. Most known artificial receptors for the detection of ATP use complementary hydrogen bonding for their recognition, which is greatly limited in the aqueous medium arising from the competitive hydrogen bonding of the solvent.4 Therefore, it is an urgent issue to search for a novel receptor to ATP with excellent selectivity.Analogous to crown ethers, cyclodextrins, calixarenes, cucurbiturils and other macrocycles,5 pillararenes,6 mainly including pillar[5]arenes and pillar[6]arenes, are a new kind of macrocycles composed of electron-donating 2,5-dialkoxybenzene units which are linked by methylene (–CH2–) bridges at their para-positions, forming a unique rigid pillar architecture. Due to their unique symmetrical structures and highly tunable functionalities, various interesting pillararene-based supramolecular systems, including liquid crystals,7 cyclic dimers,8 chemosensors,9 supramolecular polymers,10 drug delivery systems,11 metal–organic frameworks,12 transmembrane channels,6g cell glue13 and theranostic rotaxane6j have been constructed. Actually, highly tunable functionalities have endowed pillararenes with outstanding abilities to selectively bind different kinds of guests. It is known that the diameter of the internal cavity of pillar[5]arenes (∼4.7 Å) is smaller than that of pillar[6]arenes (∼6.7 Å).14 Therefore, pillar[6]arenes can complex with larger guest molecules to form inclusion complexes, such as azobenzene, 1,4-diazabicyclo[2.2.2]octane, tropylium tetrafluoroborate, adamantaneammonium, ATP and so on.15 For example, Diao et al. constructed an enzyme-responsive supramolecular system in water by using the recognition between an amphiphilic pillar[6]arene and ATP.15f
Herein, we investigate the host–guest complexation between cationic water soluble pillar[n]arenes (Fig. 1, n = 5 and 6, WP5 and WP6) and ribonucleotides (Fig. 1, AMP, ADP and ATP). Due to the differences in the cavity size of the hosts and the charge and length of the guests, WP6 bearing six trimethylammonium groups on both sizes selectively complex with ATP. This host–guest recognition motif is employed to efficiently inhibit the hydrolysis of ATP in the presence of alkaline phosphatase (CIAP). This can be potentially utilized to overcome MDR. PEGylation by a hydrophilic diblock copolymer poly(ethylene glycol)142-block-(polyacrylic acid)22 (Fig. 1, FA-PEG-b-PAA) endows the formed polyion complex (PIC) micelles with excellent targeting ability, preferentially delivering WP6 to folate receptor over-expressing cancer cells. The ATP-dependent drug efflux pump was abated efficiently due to the inhibition of ATP hydrolysis by the formation of a stable host–guest inclusion complex WP6⊃ATP. As a consequence, the efficacy of cancer chemotherapy of doxorubicin hydrochloride (DOX·HCl) was significantly improved towards drug resistant human breast cancer cells (MCF-7/ADR) in the presence of WP6.
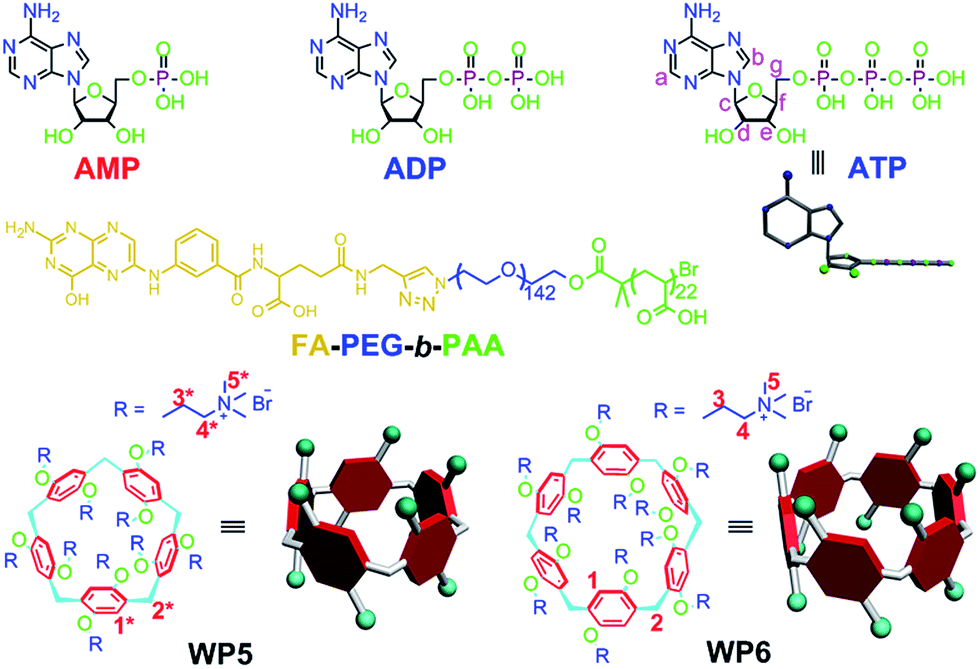 | ||
| Fig. 1 Chemical structures of AMP, ADP, ATP, WP5, WP6 and FA-PEG-b-PAA. | ||
Results and discussion
The host–guest complexations between the pillararene hosts (WP5 and WP6) and guests (AMP, ADP and ATP) were first studied by 1H NMR spectroscopy. Compared with the free guest (AMP, ADP or ATP), negligible chemical shift changes were observed for the protons on these ribonucleotide guests in the presence of WP5 (Fig. 2b, S10 and S12†). The reason was that the cavity of WP5 was too small to encapsulate the adenosine group. On the other hand, almost no chemical shifts were observed for the protons on AMP (or ADP) in the presence of WP6 (ESI, Fig. S14 and S16†) either, suggesting that the complexations between WP6 and AMP and between WP6 and ADP were not strong enough to include the adenosine group into the cavity of WP6. However, the resonance peaks related to protons Ha, Hb, Hd, He, Hf and Hg of ATP disappeared, caused by a broadening effect after complexation upon addition of 1.0 equiv. of WP6 (Fig. 2d). The reason was that these protons were located within the cavity of WP6 and were shielded by the electron-rich cyclic structure by forming an inclusion complex between WP6 and ATP.6e On the other hand, extensive broadening effect and upfield shifts were observed for the peak corresponding to proton Hc due to complexation dynamics.14 The results obtained from 1H NMR spectra demonstrated that WP6 could selectively complex with ATP, because the cavity size of WP6 was suitable for ATP and the binding affinity was strong enough to form an inclusion host–guest complex mainly through electrostatic interactions and π–π interactions between benzene rings and adenine.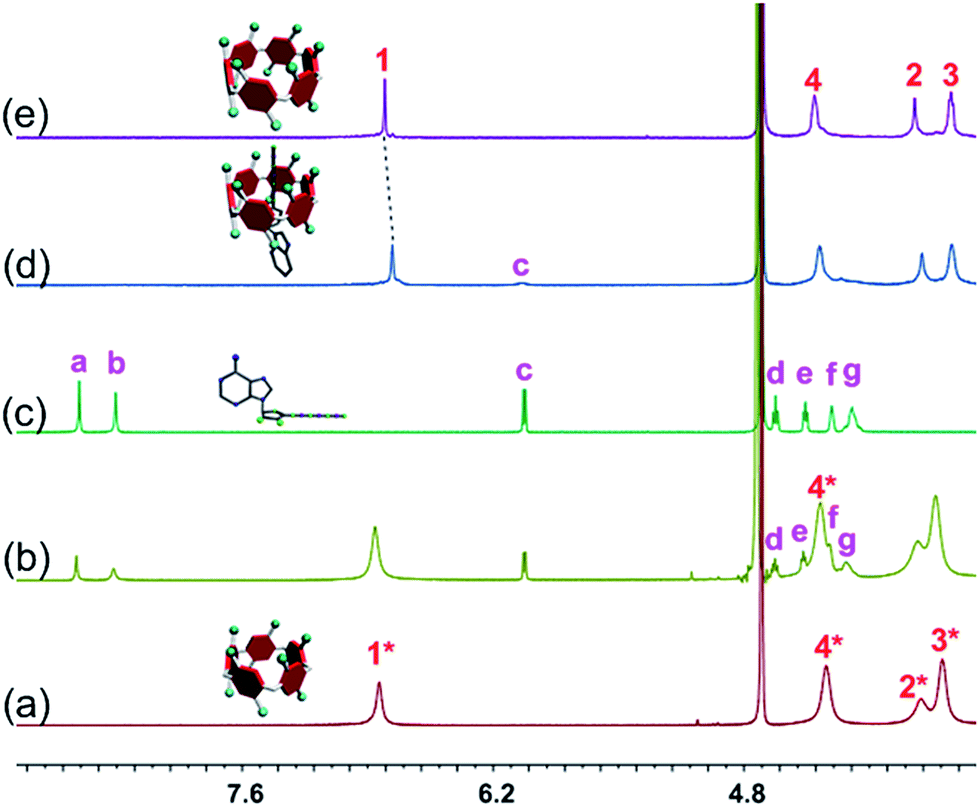 | ||
| Fig. 2 Partial 1H NMR spectra (400 MHz, D2O, 295 K): (a) WP5 (1.00 mM); (b) WP5 (1.00 mM) and ATP (1.00 mM); (c) ATP (1.00 mM); (d) WP6 (1.00 mM) and ATP (1.00 mM); (e) WP6 (1.00 mM). | ||
31P NMR spectroscopy was also conducted to further verify the size-selective host–guest complexation between WP6 and ATP (ESI, Fig. S11, S13, S15, S17 and S18†). No changes corresponding to the phosphorus signals of AMP (or ADP) were observed upon addition of WP5 or WP6 (3 equiv.). This was also true for the case of WP5 and ATP. These phenomena were in good agreement with the results obtained from 1H NMR studies (Fig. 2, S10, S12, S14 and S16†). On the contrary, the peaks related to the resonances of α-ATP, β-ATP and γ-ATP all became broad and showed chemical shift changes upon addition of WP6 (ESI, Fig. S18†), indicating successful complexation. A possible reason was that ATP was located in the cavity of WP6 upon formation of an inclusion complex and shielded by its electron-rich cyclic structure.
Isothermal titration calorimetry (ITC) experiments were further performed to provide thermodynamic insight into the inclusion complexation between the pillar[n]arenes (n = 5, 6) and ribonucleotides. As shown in Table 1, the enthalpy and entropy changes were obtained (ΔH° > 0; TΔS° > 0; |ΔH°| < |TΔS°|), indicating that the complexations were driven by entropy changes. From comparison of the association constant (Ka) values of these host–guest systems, we know that WP6⊃ATP exhibited the highest binding affinity, which further confirmed that WP6 selectively complexed with ATP. Furthermore, NOE correlation signals were observed between proton Hc on the ribose unit of ATP and proton H1 on the benzene rings of WP6 from the 2D NOESY NMR spectrum (ESI, Fig. S26†), indicating that ATP penetrated into the cavity of WP6 to form a [2]pseudorotaxane-type inclusion complex. Fig. S27† shows the energy-minimized structure of WP6⊃ATP with the guest molecule tightly wrapped with WP6. Noticeably, the anionic segment of ATP locates on the upper side of WP6 to successfully achieve multivalent electrostatic interactions with the cationic trimethylammonium groups. On the other hand, the repeating units of WP6 rotate around the methylene bridges, changing its appearance from the pillar structure to a conical structure due to the asymmetric complexation.9b The ribose group threads into the cavity of WP6 and it is surrounded by the benzene rings. Furthermore, the largest part (adenine group) of the ATP guest is situated at the relatively larger side of the conic WP6. The information obtained from molecular modeling is in good agreement with the NMR results mentioned above. Moreover, fluorescence titration experiments were carried out to provide evidence for the interactions between WP6 and ATP. As shown in Fig. S25,† addition of ATP to a phosphate buffer solution of WP6 resulted in a substantial decrease in the intensity of the emission band at 325 nm caused by host–guest complexation between WP6 and ATP.
| Host | Guest | 10−3Ka/M−1 | ΔH/kcal mol−1 | ΔS/cal mol−1 K−1 |
|---|---|---|---|---|
| a Microcalorimetric titration experiments were conducted in PBS (pH = 7.4) at 298.15 K by titration of AMP (ADP or ATP) (2.00 mM, 10 μL per injection) into the solution of WP5 (or WP6) (0.100 mM). | ||||
| WP5 | AMP | 2.78 ± 0.35 | 85.9 ± 6.7 | 304 |
| WP5 | ADP | 3.92 ± 0.43 | 64.3 ± 5.1 | 232 |
| WP5 | ATP | 26.2 ± 3.30 | 8.99 ± 0.76 | 50.4 |
| WP6 | AMP | 2.86 ± 0.23 | 42.8 ± 2.2 | 159 |
| WP6 | ADP | 4.38 ± 0.43 | 16.9 ± 1.4 | 73.4 |
| WP6 | ATP | 152 ± 16 | 4.58 ± 0.42 | 39.1 |
From the above discussions, we can know that the adenosine group of the guest is included in the cavity of WP6 for WP6⊃ATP while this group is not in the cavity of WP6 for WP6⊃AMP and WP6⊃ADP. This should be ascribed to the differences in charge and length of these three guests. The length of the ATP is appropriate to maximize both multiple electrostatic interactions between the anionic segment of ATP and cationic ammonium units and π–π interactions between benzene rings and the adenine group, as shown by the energy-minimized structure of WP6⊃ATP (ESI, Fig. S27†).
More interestingly, the hydrolysis of ATP was inhibited efficiently in the presence of WP6, in sharp comparison with the case of free ATP (ESI, Fig. S28–S30†). As indicated by 31P NMR spectra, the hydrolysis rate of ATP slowed down efficiently by forming a stable host–guest inclusion complex WP6⊃ATP in the presence of CIAP (6 U mL−1) (ESI, Fig. S30†). The reason was that ATP was located in the hydrophobic cavity of WP6, preventing ATP from being hydrolyzed.16 The development of MDR to a variety of chemotherapeutic agents is one of the major challenges for efficient cancer treatment. One of the major causes of MDR is the over-expression of the drug efflux pump, the ATP-binding cassette superfamily membrane proteins, which transport anticancer drugs out of the cells and result in drug resistance by utilizing the energy from ATP hydrolysis.17 Among various ATP-binding cassette superfamily membrane proteins, P-gp known as the ATP-dependent drug efflux pump which is upregulated in the plasma membrane of all MDR cancer cells, is probably the best characterized ABC transporter.18 If the hydrolysis of ATP can be inhibited, the source of the chemical energy is cut off, resulting in the blocking of the efflux pump. On the other hand, the anticancer drugs can hardly be transported out of the cells through other energy-dependent approaches due to the shortage of energy, thus the efficacy of the cancer chemotherapy can be improved significantly.
In order to specifically block the cancer cells rather than normal cells, a folic acid functionalized hydrophilic diblock copolymer FA-PEG-b-PAA was chosen to PEGylate the cationic WP6. Here WP6 worked as a supramolecular cross-linker to interact with the negatively charged carboxylate segments of the diblock copolymer through electrostatic interactions in the buffer, forming stable polyion complex (PIC) micelles where WP6 molecules and the anionic carboxylates are in the core of the PIC micelles, and the PEG blocks are on the surfaces of the micelles.19 FA groups decorated on the surfaces of the PIC micelles endow these nanovehicles with the ability to specifically deliver WP6 to folate receptor (FR) over-expressing cancer cells, because FR is a well-known tumor-associated receptor that is over-expressed in many tumors, including those of the breast, lung, kidney, and brain (e.g., HeLa and KB cell lines), relative to normal tissue. On the other hand, PEGylation could also be employed to reduce the cytotoxicity of the cationic WP6.
In order to monitor the in vitro cell accumulation of the ternary PIC micelles as well as to confirm targeting delivery of these nanocarriers to FR over-expressing cancer cells, DOX·HCl as a commonly used anticancer drug with strong fluorescence was loaded into the PIC micelles formed by FA-PEG-b-PAA and WP6. TEM was employed to reveal the morphology of the ternary self-assembly formed by FA-PEG-b-PAA, WP6 and DOX·HCl (charge ratio r = 1:1). As shown in Fig. S31a,† spherical aggregates were observed with diameters ranging from 100 to 170 nm, in good agreement with the results obtained from DLS (166 nm, ESI, Fig. S31b†). It should be emphasized that the DOX·HCl loaded ternary PIC micelles were quite stable in the buffer and showed no structural changes for several weeks.
Confocal laser scanning microscopic (CLSM) and flow cytometry investigations were conducted to verify whether the FA moieties decorated on the surface of the PIC micelles could guide these nanocarriers containing WP6 preferentially to FR over-expressing KB cells, rather than FR low-expressing A549 cells. As shown in Fig. 3a, KB cells exhibited strong intracellular DOX·HCl fluorescence after incubation with DOX·HCl (5.00 μg mL−1) loaded ternary PIC micelles for 1 h, and the fluorescence intensity became much stronger associated with extension of incubation time to 2 h. However, A549 cells showed weak fluorescence signal under the same experimental conditions (Fig. 3a). Moreover, red dots corresponding to the ternary PIC micelles affixed on the membrane of KB cells, indicating strong interactions between FA groups on the surfaces of the ternary PIC micelles and FRs on the cell membrane. Flow cytometry investigations demonstrated that KB cells always had faster uptake rate and higher intracellular accumulation than A549 cells (Fig. 3b and c). The mean fluorescence intensity was three times greater in KB cells than that in A549 cells (31.7 ± 0.93 vs. 10.6 ± 0.78) after incubation with the ternary PIC micelles for 4 h (ESI, Fig. S32†). These phenomena confirmed that the FA modified ternary PIC micelles could specifically enter the KB cells aided by the interaction of the FA moieties with FRs on the KB cells, possibly via receptor-mediated endocytosis.
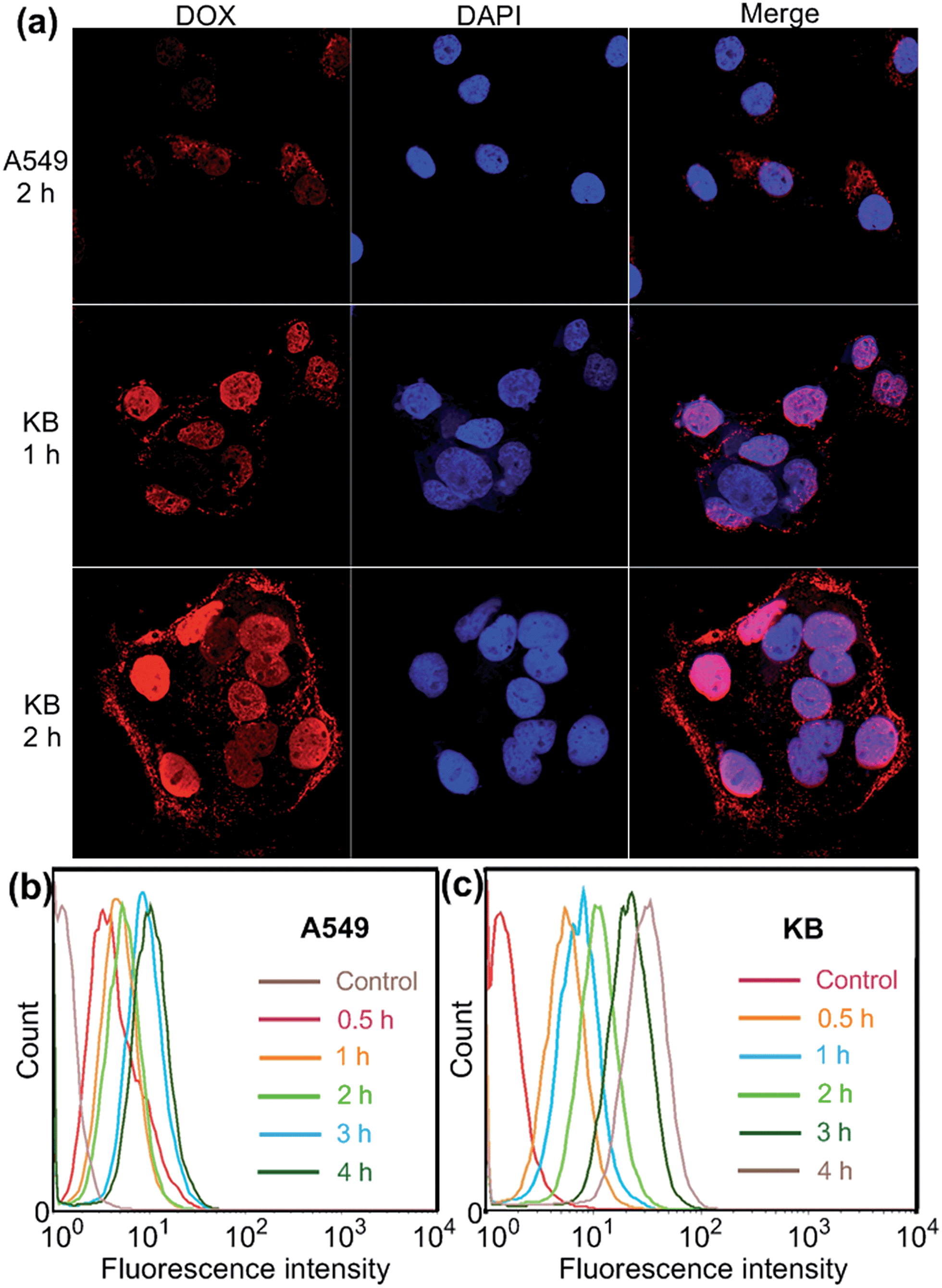 | ||
| Fig. 3 (a) Confocal laser scanning microscopy (CLSM) images of A549 and KB cells incubated with DOX·HCl (5.00 μg mL−1) loaded WP6/FA-PEG-b-PAA ternary PIC micelles. Flow cytometry showing increasing uptake of DOX·HCl (5.00 μg mL−1) loaded WP6/FA-PEG-b-PAA ternary PIC micelles over a 4 h period using A549 (b) and KB (c) cells. | ||
Induced by the hydrophobicity, calcein acetoxymethyl ester (calcein-AM) can diffuse across the cell membranes, and be hydrolyzed to hydrophilic fluorescent calcein by esterase in the cytoplasm, so it can be employed as an excellent probe to test the operation of the efflux pumps in cell membranes by monitoring the fluorescence changes of the cell culture.20 Fluorescence images provided visual colour changes of the cells stained with calcein in the presence and absence of WP6 by culturing the cell for 48 h. As shown in Fig. 4a, the colour of the cells in the absence of WP6 became weaker with the extension of culture time, indicating that calcein had a fast and profound release. In a parallel study by fluorescence microscopic observation, a similar phenomenon was observed for cells under the same incubation conditions in the presence of FA-PEG-b-PAA. While the fluorescence intensity of the cells changes much slower in the presence of WP6 owning to the inhibition of ATP hydrolysis by forming WP6⊃ATP. Fig. 4b shows the release profile of calcein from the drug resistant MCF-7/ADR cells. Regardless of the presence of WP6, the release rate and the total release of calcein from cells were much higher than those in the presence of WP6 by determining the fluorescence intensity of the cell culture. Accompanied with the enhancement of the concentration of WP6 from 100 to 250 μM, the release rate and the total release of calcein from cells slowed down efficiently caused by the inhibition of ATP hydrolysis (Fig. 4b). The ATP-dependent calcein release underlined the significant role of WP6 in the influence of the efflux pump by cutting off the energy source from ATP hydrolysis due to the formation of the host–guest inclusion complex.
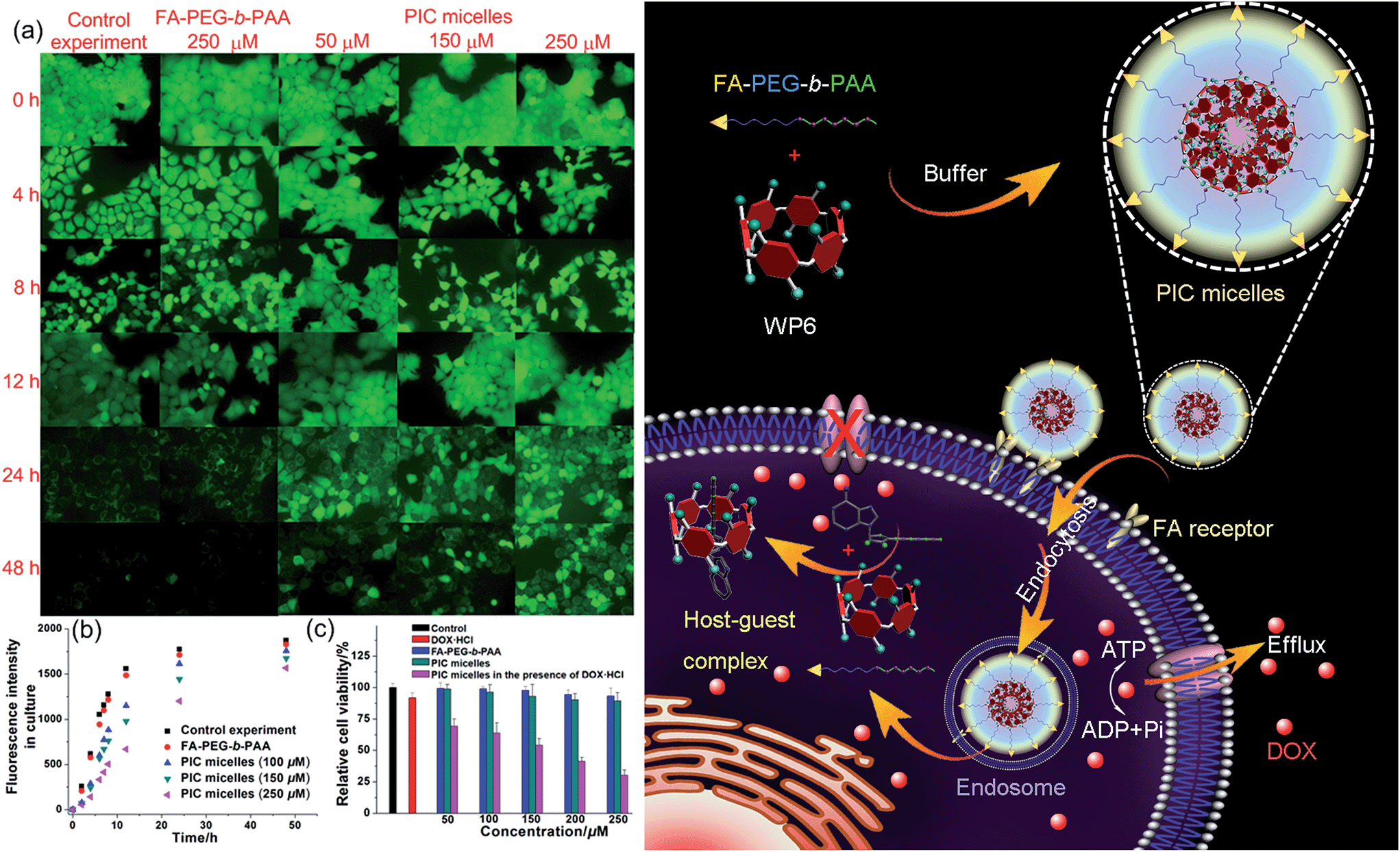 | ||
| Fig. 4 (a) Fluorescence images of the MCF-7/ADR cells stained with calcein-AM incubated without/with FA-PEG-b-PAA (250 μM), PIC micelles containing different amount of WP6. (b) Fluorescence intensity changes of the culture in the presence of FA-PEG-b-PAA or PIC micelles containing different amounts of WP6. (c) Cytotoxicity of DOX·HCl (25 μM), FA-PEG-b-PAA, PIC micelles, and DOX·HCl (25 μM) loaded PIC micelles with different concentrations of WP6 against MCF-7/ADR cells. Schematic illustration of the preparation of PIC micelles and possible mechanism to inhibit the efflux pump by forming a host–guest complex WP6⊃ATP in the cell. | ||
The efficacy of the cancer chemotherapy of DOX·HCl in the presence and absence of WP6 against MCF-7/ADR cell lines was carried out using 3-(4′,5′-dimethylthiazol-2′-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. As control experiments, the viabilities of MCF-7/ADR cells cultured with the diblock polymer FA-PEG-b-PAA or the PIC micelles were investigated, which showed minor cytotoxicity with concentrations ranging from 50 to 250 μM (Fig. 4c). On the other hand, the relative cell viability was 91.8% by culturing the cells with DOX·HCl (25 μM) (Fig. 4c). In stark contrast, the viability of MCF-7/ADR cells decreased from 69.2% to 30.3% accompanied with the increase of WP6 from 50 to 250 μM in the presence of DOX·HCl at the same concentration (25 μM). The reason was that the source of the chemical energy was cut off by forming the stable inclusion complex WP6⊃ATP, resulting in the blocking of the efflux pump and other energy-dependent approaches to transport DOX·HCl out of cells. Therefore, the intracellular concentration of DOX·HCl was higher in the presence of WP6 than those in the absence of WP6, resulting in the reduction of the corresponding relative cell viability, which indicated that the efficacy of the cancer chemotherapy DOX·HCl was improved significantly.
Conclusions
In summary, a cationic water-soluble pillar[6]arene (WP6) selectively complexed with ATP to form a stable 1:1 inclusion complex WP6⊃ATP mainly driven by entropy change. As a result, the hydrolysis of ATP was efficiently inhibited in the presence of alkaline phosphatase due to the existence of a hydrophobic cavity of WP6. A folic acid ended diblock polymer FA-PEG-b-PAA was employed to PEGylate the cationic pillar[6]arene WP6 to obtain PIC micelles in buffer, endowing them with specific targeting ability to deliver WP6 to folate receptor over-expressing cancer cells. The ATP-dependent efflux pump was blocked by cutting off the energy source from ATP hydrolysis due to the formation of the host–guest inclusion complex. Furthermore, MTT assay demonstrated that the efficacy of the anticancer drug DOX·HCl was improved effectively in the presence of PIC micelles. The present results pave a way to develop novel therapeutic agents, implying that supramolecular chemistry may be engineered into promising vehicles to overcome multidrug resistance in cancer therapy. More detailed biologic investigations need to be carried out to figure out the deeper-level mechanism of the MDR treatment.
Acknowledgements
This work was supported by National Basic Research Program (2013CB834502), the National Natural Science Foundation of China (21434005, 91527301), the Fundamental Research Funds for the Central Universities, the Key Science Technology Innovation Team of Zhejiang Province (2013TD02), and Open Project of State Key Laboratory of Supramolecular Structure and Materials (sklssm201611).Notes and references
- (a) M. I. Simon, M. P. Strathmann and N. Gautam, Science, 1991, 252, 802 CrossRef CAS PubMed; (b) A. Y. Ting, K. H. Kain, R. L. Klemke and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 15003 CrossRef CAS PubMed.
- (a) S. M. Butterfield and M. L. Waters, J. Am. Chem. Soc., 2003, 125, 9580 CrossRef CAS PubMed; (b) D. H. Lee, S. Y. Kim and J.-I. Hong, Angew. Chem., Int. Ed., 2004, 43, 4777 CrossRef CAS PubMed; (c) M. B. Yaffe, Nat. Rev. Mol. Cell Biol., 2002, 3, 177 CrossRef CAS PubMed; (d) S. K. Kim, D. H. Lee, J. I. Hong and J. Yoon, Acc. Chem. Res., 2009, 42, 23 CrossRef CAS PubMed.
- (a) A. L. Davidson, J. Bacteriol., 2002, 184, 1225–1233 CrossRef CAS PubMed; (b) C. F. Higgins and K. J. Linton, Nat. Struct. Mol. Biol., 2004, 11, 918–926 CrossRef CAS PubMed; (c) M. F. Fromm, Trends Pharmacol. Sci., 2004, 25, 423–429 CrossRef CAS PubMed.
- (a) A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515 CrossRef CAS PubMed; (b) J. L. Sessler, V. Kral, T. V. Shishkanova and P. A. Gale, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4848 CrossRef CAS PubMed; (c) X. Zuo, S. Song, J. Zhang, D. Pan, L. Wang and C. Fan, J. Am. Chem. Soc., 2007, 129, 1042 CrossRef CAS PubMed.
- (a) K. Kim, N. Selvapalam, Y. H. Ko, K. M. Park, D. Kim and J. Kim, Chem. Soc. Rev., 2007, 36, 267 RSC; (b) C. Schmuck, T. Rehm, K. Klein and F. Gröhn, Angew. Chem., Int. Ed., 2007, 46, 1693 CrossRef CAS PubMed; (c) Z. Niu and H. W. Gibson, Chem. Rev., 2009, 109, 6024 CrossRef CAS PubMed; (d) W. Jiang, A. Schäfer, P. C. Mohr and C. A. Schalley, J. Am. Chem. Soc., 2010, 132, 2309 CrossRef CAS PubMed; (e) D.-S. Guo, K. Wang, Y.-X. Wang and Y. Liu, J. Am. Chem. Soc., 2012, 134, 10244 CrossRef CAS PubMed; (f) K. Zhu, V. N. Vukotic and S. J. Loeb, Angew. Chem., Int. Ed., 2012, 51, 2168 CrossRef CAS PubMed; (g) A. G. Campaña, D. A. Leigh and U. Lewandowska, J. Am. Chem. Soc., 2013, 135, 8639 CrossRef PubMed; (h) J.-F. Ayme, J. E. Beves, C. J. Campbell and D. A. Leigh, Chem. Soc. Rev., 2013, 42, 1700 RSC; (i) R. Barat, T. Legigan, I. Tranoy-Opalinski, B. Renoux, E. Péraudeau, J. Clarhaut, P. Poinot, A. E. Fernandes, V. Aucagne, D. A. Leigh and S. Papot, Chem. Sci., 2015, 6, 2608–2613 RSC.
- (a) T. Ogoshi, S. Kanai, S. Fujinami, T. A. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022 CrossRef CAS PubMed; (b) D. Cao, Y. Kou, J. Liang, Z. Chen, L. Wang and H. Meier, Angew. Chem., Int. Ed., 2009, 48, 9721 CrossRef CAS PubMed; (c) M. Xue, Y. Yang, X. Chi, Z. Zhang and F. Huang, Acc. Chem. Res., 2012, 45, 1294 CrossRef CAS PubMed; (d) H. Li, D.-X. Chen, Y.-L. Sun, Y. Zheng, L.-L. Tan, P. S. Weiss and Y.-W. Yang, J. Am. Chem. Soc., 2013, 135, 1570 CrossRef CAS PubMed; (e) J. Fan, H. Deng, J. Li, X. Jia and C. Li, Chem. Commun., 2013, 49, 6343 RSC; (f) W. Si, Z.-T. Li and J.-L. Hou, Angew. Chem., Int. Ed., 2014, 53, 4578 CrossRef CAS PubMed; (g) W.-B. Hu, H.-M. Yang, W.-J. Hu, M.-L. Ma, X.-L. Zhao, X.-Q. Mi, Y. A. Liu, J.-S. Li, B. Jiang and K. Wen, Chem. Commun., 2014, 50, 10460 RSC; (h) Z.-Y. Li, Y. Zhang, C. W. Zhang, L.-J. Chen, C. Wang, H. Tan, Y. Yu, X. Li and H.-B. Yang, J. Am. Chem. Soc., 2014, 136, 8577 CrossRef CAS PubMed; (i) W.-B. Hu, W.-J. Hu, Y. A. Liu, J.-S. Li, B. Jiang and K. Wen, Org. Lett., 2015, 17, 2940 CrossRef CAS PubMed; (j) X. Xiao, G. Nie, X. Zhang, D. Tian and H. Li, Chem.–Eur. J., 2016, 22, 941 CrossRef CAS PubMed; (k) B. Yuan, J.-F. Xu, C.-L. Sun, H. Nicolas, M. Schonhoff, Q.-Z. Yang and X. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 3679 CrossRef CAS PubMed; (l) G. Yu, D. Wu, Y. Li, Z. Zhang, L. Shao, J. Zhou, Q. Hu, G. Tang and F. Huang, Chem. Sci., 2016 10.1039/C6SC00036C.
- I. Nierengarten, S. Guerra, M. Holler, J.-F. Nierengarten and R. Deschenaux, Chem. Commun., 2012, 48, 8072 RSC.
- Z. Zhang, G. Yu, C. Han, J. Liu, X. Ding, Y. Yu and F. Huang, Org. Lett., 2011, 13, 4818 CrossRef CAS PubMed.
- (a) N. L. Strutt, R. S. Forgan, J. M. Spruell, Y. Y. Botros and J. F. Stoddart, J. Am. Chem. Soc., 2011, 133, 5668 CrossRef CAS PubMed; (b) G. Yu, Z. Zhang, C. Han, M. Xue, Q. Zhou and F. Huang, Chem. Commun., 2012, 48, 2958 RSC.
- (a) Z. Zhang, Y. Luo, J. Chen, S. Dong, Y. Yu, Z. Ma and F. Huang, Angew. Chem., Int. Ed., 2011, 50, 1397 CrossRef CAS PubMed; (b) C. Li, Chem. Commun., 2014, 50, 12420 RSC; (c) S. Wang, Y. Wang, Z. Chen, Y. Lin, L. Weng, K. Han, J. Li, X. Jia and C. Li, Chem. Commun., 2015, 51, 3434 RSC.
- (a) H. Zhang, X. Ma, K. T. Nguyen and Y. Zhao, ACS Nano, 2013, 7, 7853 CrossRef CAS PubMed; (b) Q. Duan, Y. Cao, Y. Li, X.-Y. Hu, T. Xiao, C. Lin, Y. Pan and L. Wang, J. Am. Chem. Soc., 2013, 135, 10542 CrossRef CAS PubMed; (c) Y. Cao, X.-Y. Hu, Y. Li, X. Zou, S. Xiong, C. Lin, Y.-Z. Shen and L. Wang, J. Am. Chem. Soc., 2014, 136, 10762 CrossRef CAS PubMed; (d) G. Yu, W. Yu, Z. Mao, C. Gao and F. Huang, Small, 2015, 11, 919 CrossRef CAS PubMed.
- N. L. Strutt, D. Fairen-Jimenez, J. Iehl, M. B. Lalonde, R. Q. Snurr, O. K. Farha, J. T. Hupp and J. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 17436 CrossRef CAS PubMed.
- (a) G. Yu, Y. Ma, C. Han, Y. Yao, G. Tang, Z. Mao, C. Gao and F. Huang, J. Am. Chem. Soc., 2013, 135, 10310 CrossRef CAS PubMed; (b) H. Chen, J. Fan, X. Hu, J. Ma, S. Wang, J. Li, Y. Yu, X. Jia and C. Li, Chem. Sci., 2015, 6, 197 RSC.
- G. Yu, C. Han, Z. Zhang, J. Chen, X. Yan, B. Zheng, S. Liu and F. Huang, J. Am. Chem. Soc., 2012, 134, 8711 CrossRef CAS PubMed.
- (a) T. Ogoshi, H. Kayama, D. Yamafuji, T. Aoki and T.-a. Yamagishi, Chem. Sci., 2012, 3, 3221 RSC; (b) T. Ogoshi and T.-a. Yamagishi, Eur. J. Org. Chem., 2013, 2961 CrossRef CAS; (c) W. Chen, Y. Zhang, J. Li, X. Lou, Y. Yu, X. Jia and C. Li, Chem. Commun., 2013, 49, 7956 RSC; (d) N. Song and Y.-W. Yang, Sci. China: Chem., 2014, 57, 1185 CrossRef CAS; (e) D. Xia, G. Yu, J. Li and F. Huang, Chem. Commun., 2014, 50, 3606 RSC; (f) J. Zhou, M. Chen and G. Diao, Chem. Commun., 2014, 50, 11954 RSC.
- (a) G. Yu, J. Yang, D. Xia and Y. Yao, RSC Adv., 2014, 4, 18763 RSC; (b) B. Hua, J. Zhou and G. Yu, Tetrahedron Lett., 2015, 56, 986 CrossRef CAS.
- (a) M. M. Gottesman, T. Fojo and S. E. Bates, Nat. Rev. Cancer, 2002, 2, 48 CrossRef CAS PubMed; (b) G. Taubes, Science, 2008, 321, 356 CrossRef CAS PubMed; (c) N. Z. Kuhn and L. Nagahara, Mol. Pharm., 2011, 8, 1994 CrossRef CAS PubMed; (d) I. Brigger, C. Dubernet and P. Couvreur, Adv. Drug Delivery Rev., 2012, 64, 24 CrossRef.
- (a) M. Dean and T. Annilo, Annu. Rev. Genomics Hum. Genet., 2005, 6, 123 CrossRef CAS PubMed; (b) P. D. Eckford and F. J. Sharom, Chem. Rev., 2009, 109, 2989 CrossRef CAS PubMed.
- (a) S. Fukushima, K. Miyata, N. Nishiyama, N. Kanayama, Y. Yamasaki and K. Kataoka, J. Am. Chem. Soc., 2005, 127, 2810 CrossRef CAS PubMed; (b) C. Li, T. Wu, C. Hong, G. Zhang and S. Liu, Angew. Chem., Int. Ed., 2012, 51, 455 CrossRef CAS PubMed.
- Z. Mao, R. Cartier, A. Hohl, M. Farinacci, A. Dorhoi, T.-L. Nguyen, P. Mulvaney, J. Ralston, S. H. E. Kaufmann, H. Möhwald and D. Wang, Nano Lett., 2011, 11, 2153 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, NMR spectra, and other materials. See DOI: 10.1039/c6sc00531d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |