DOI:
10.1039/C5SC04751J
(Edge Article)
Chem. Sci., 2016,
7, 2604-2613
Chemistry informer libraries: a chemoinformatics enabled approach to evaluate and advance synthetic methods†
Received
9th December 2015
, Accepted 15th January 2016
First published on 26th January 2016
Abstract
Major new advances in synthetic chemistry methods are typically reported using simple, non-standardized reaction substrates, and reaction failures are rarely documented. This makes the evaluation and choice of a synthetic method difficult. We report a standardized complex molecule diagnostic approach using collections of relevant drug-like molecules which we call chemistry informer libraries. With this approach, all chemistry results, successes and failures, can be documented to compare and evolve synthetic methods. To aid in the visualization of chemistry results in drug-like physicochemical space we have used an informatics methodology termed principal component analysis. We have validated this method using palladium- and copper-catalyzed reactions, including Suzuki–Miyaura, cyanation and Buchwald–Hartwig amination.
Introduction
Organic synthesis continues to advance at a rapid pace, giving rise to new synthetic methods that enable transformations that would have been unthinkable only a few years ago. These new methods hold promise of applications across a diverse range of endeavours, from natural product synthesis to pharmaceuticals and materials, provided they work on the types of complex chemical structures commonly encountered within those disciplines.1 Most reported synthetic methods use simple commercial model substrates that perform in high yield to demonstrate scope, and there is little overlap between substrate sets from one publication to the next, making comparisons between methods difficult. For the chemist needing to choose among various reported methods to accomplish a given transformation in a complex synthesis, this incomplete and fragmented data set represents a significant barrier to utilization. To address this problem, we envisioned assembling collections of standard substrates representative of structures encountered in complex synthesis and using these to document the complete chemistry performance (both successes and failures) of different synthetic methods. In this work, we have created two substrate collections that are relevant to pharmaceutical research, which we have termed chemistry informer libraries, to evaluate this standardized diagnostic approach.2 Using these libraries, consisting of medium-complexity aryl pinacol boronates and high-complexity aryl halides, we have conducted a systematic study comparing the relative success rates of various reported catalytic methods in order to identify the best first-pass options for synthesis of pharmaceutically relevant compounds. We also demonstrate how these informer libraries can serve as diagnostic tools to improve the performance of chemistry methods in order to achieve broader utility and discuss how this approach may be combined with chemoinformatics tools to enable in depth explorations of structure–reactivity space.
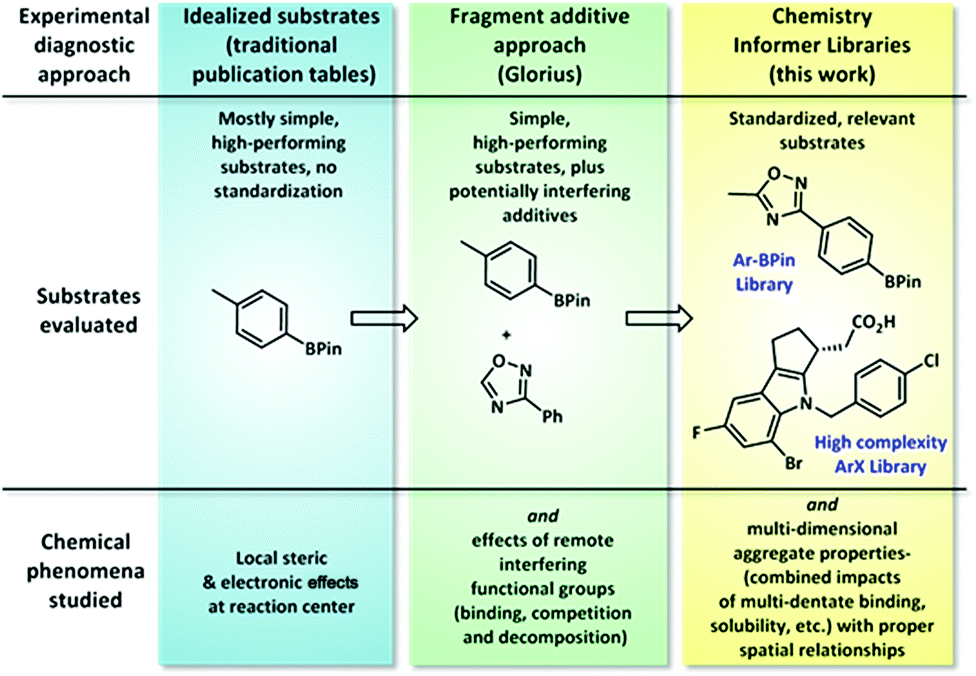
Typical synthetic publications employ simple model substrates to test the effects of local steric and electronic variations (Fig. 1). Recognizing the limitations of this approach, especially in regards to functional group compatibility, Glorius recently developed a diagnostic method in which molecular fragments are added to simple substrate combinations to determine which functionalities interfere with chemistry performance.3 This tool is operationally easy to execute and gives ready access to valuable reactivity data; however, we have found that approximating the chemical reactivity of a complex molecule as that of a collection of disconnected fragment structures often fails to account for important aggregate molecular properties, such as chelation effects and substrate solubility, that only arise in the context of whole molecules. In contrast, the chemistry informer library evaluates not only local steric and electronic effects and functional group compatibility, but also complex, whole molecule interactions.
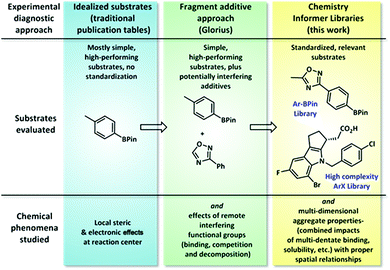 |
| | Fig. 1 Synthetic chemistry diagnostic approaches. Standard literature reports typically demonstrate simple, high-performing substrates; the Glorius “robustness” test probes the effects of potentially interfering molecular fragments on chemistry performance, and in this work we examine how standardized collections of full, complex molecules can be used to compare and improve literature synthetic methods. | |
Results and discussion
Principal component analysis
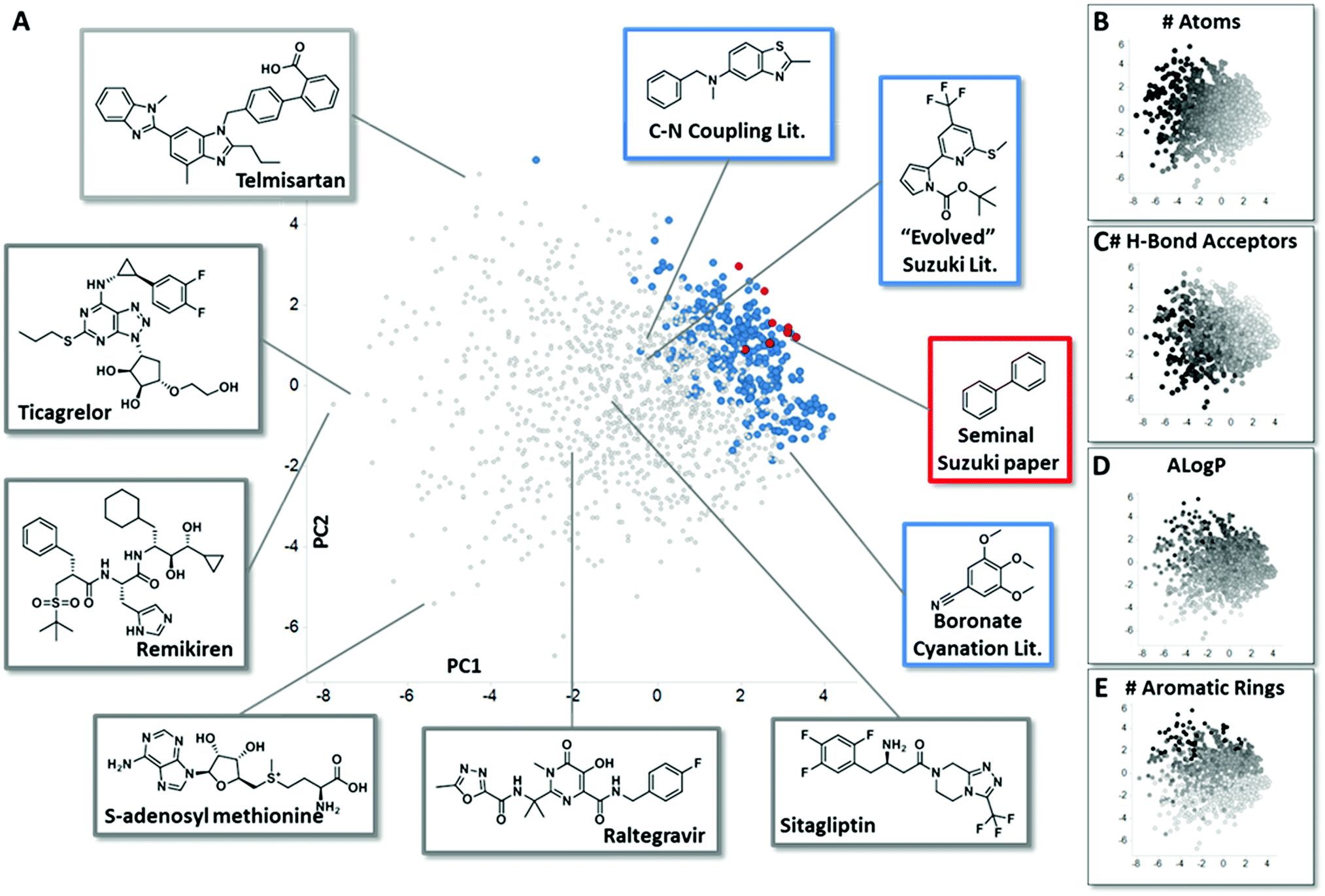
In order to assemble chemistry informer libraries representative of problems encountered in pharmaceutical research, we required a means to assess these molecules and their potential reaction products in the context of the chemical space occupied by actual small molecule drugs. To this end, we employed principal component analysis (PCA), an informatics method that has proven fruitful in reducing complex multidimensional chemical space to two or more dimensions that capture the variation in the higher dimensional space, allowing meaningful visualization.4 Performing a PCA on physicochemical properties has proven especially useful in evaluating relationships between sets of compounds.5 We trained our PCA with 14 physical chemical properties including molecular weight, number of rings, hydrogen bond donors, hydrogen bond acceptors, fraction sp3 character and Alog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P.6 We first used this PCA to compare the chemical space of FDA-approved small molecule drugs with products from the literature reports for the three different synthetic methodologies that are explored in this paper. We visualize the first two principal components (Fig. 2A, 58% of variance explained), and also depict the physical property trends that represent the greatest loadings for PC1 (Fig. 2B and C) and PC2 (Fig. 2D and E) for this space.7 This analysis reveals that literature compounds are generally clustered tightly in one area of chemical space due to their low molecular weight, and for their given size, high aromatic content, high lipophilicity, and low number of hydrogen bond donors/acceptors relative to marketed drugs. Furthermore, from analysing additional components (PC4 vs. PC3, ESI†), we see that some drugs have greater variation in properties, such as number of stereo atoms and rotatable bonds compared to the literature compounds. We highlight the seminal Suzuki–Miyaura coupling paper8 (Fig. 2A, red) to demonstrate that more recent Suzuki–Miyaura coupling methodology papers,9 as well as the leading Buchwald–Hartwig C–N coupling publications and the more recently reported aryl boronate cyanation reaction (all in blue), occupy a broader coverage of pharmaceutically relevant chemical space. Seen in this context, our goal in creating the chemistry informer libraries was to assemble compound test sets that could claim an even higher complexity and broader coverage of drug-likeness as demonstrated using PCA. Beyond this, we aspired to use chemoinformatics tools such as PCA to gain deeper insight into the reactivity patterns that would result from utilization of informer libraries with different synthetic methods, and use this knowledge to improve the applicability of modern synthetic methods to drug synthesis.
P.6 We first used this PCA to compare the chemical space of FDA-approved small molecule drugs with products from the literature reports for the three different synthetic methodologies that are explored in this paper. We visualize the first two principal components (Fig. 2A, 58% of variance explained), and also depict the physical property trends that represent the greatest loadings for PC1 (Fig. 2B and C) and PC2 (Fig. 2D and E) for this space.7 This analysis reveals that literature compounds are generally clustered tightly in one area of chemical space due to their low molecular weight, and for their given size, high aromatic content, high lipophilicity, and low number of hydrogen bond donors/acceptors relative to marketed drugs. Furthermore, from analysing additional components (PC4 vs. PC3, ESI†), we see that some drugs have greater variation in properties, such as number of stereo atoms and rotatable bonds compared to the literature compounds. We highlight the seminal Suzuki–Miyaura coupling paper8 (Fig. 2A, red) to demonstrate that more recent Suzuki–Miyaura coupling methodology papers,9 as well as the leading Buchwald–Hartwig C–N coupling publications and the more recently reported aryl boronate cyanation reaction (all in blue), occupy a broader coverage of pharmaceutically relevant chemical space. Seen in this context, our goal in creating the chemistry informer libraries was to assemble compound test sets that could claim an even higher complexity and broader coverage of drug-likeness as demonstrated using PCA. Beyond this, we aspired to use chemoinformatics tools such as PCA to gain deeper insight into the reactivity patterns that would result from utilization of informer libraries with different synthetic methods, and use this knowledge to improve the applicability of modern synthetic methods to drug synthesis.
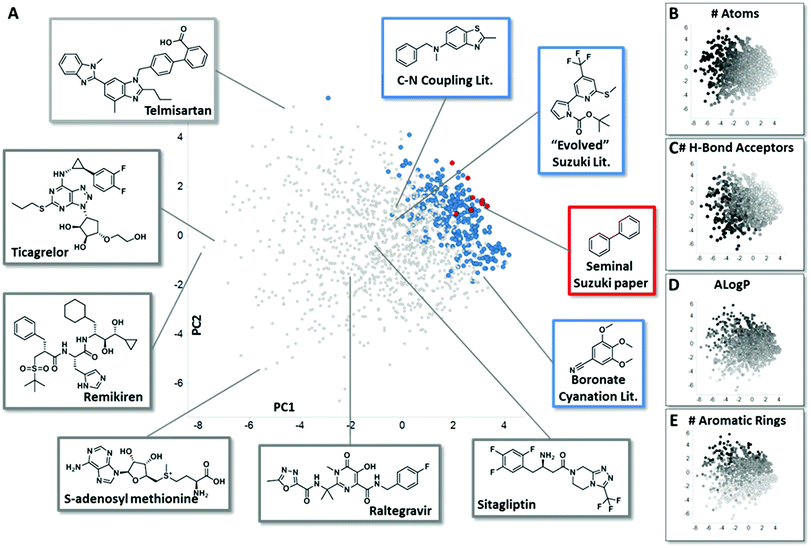 |
| | Fig. 2 Principal component analysis (PCA) comparing small molecule drugs with products described in synthetic literature reports. (A) shows a principal component analysis used to visualize the physicochemical space occupied by marketed small molecule drugs compounds (grey) and products described in literature reports for the several reaction types evaluated in this work: in red are compounds that were prepared in the seminal Suzuki–Miyaura paper, and in blue are the products formed in leading literature Suzuki–Miyaura reports, recent methods reported for the conversion of aryl pinacol boronates into aryl nitriles and recent Buchwald–Hartwig C–N coupling methods. Representative structures from both the small molecule drugs collection and literature reports are highlighted. Fig. 2B–E depict how properties with the greatest contribution to PC1 and PC2 are mapped by the PCA, with black representing the highest values for each parameter. See ESI† for full details. | |
Aryl and heteroaryl pinacol boronate informer library
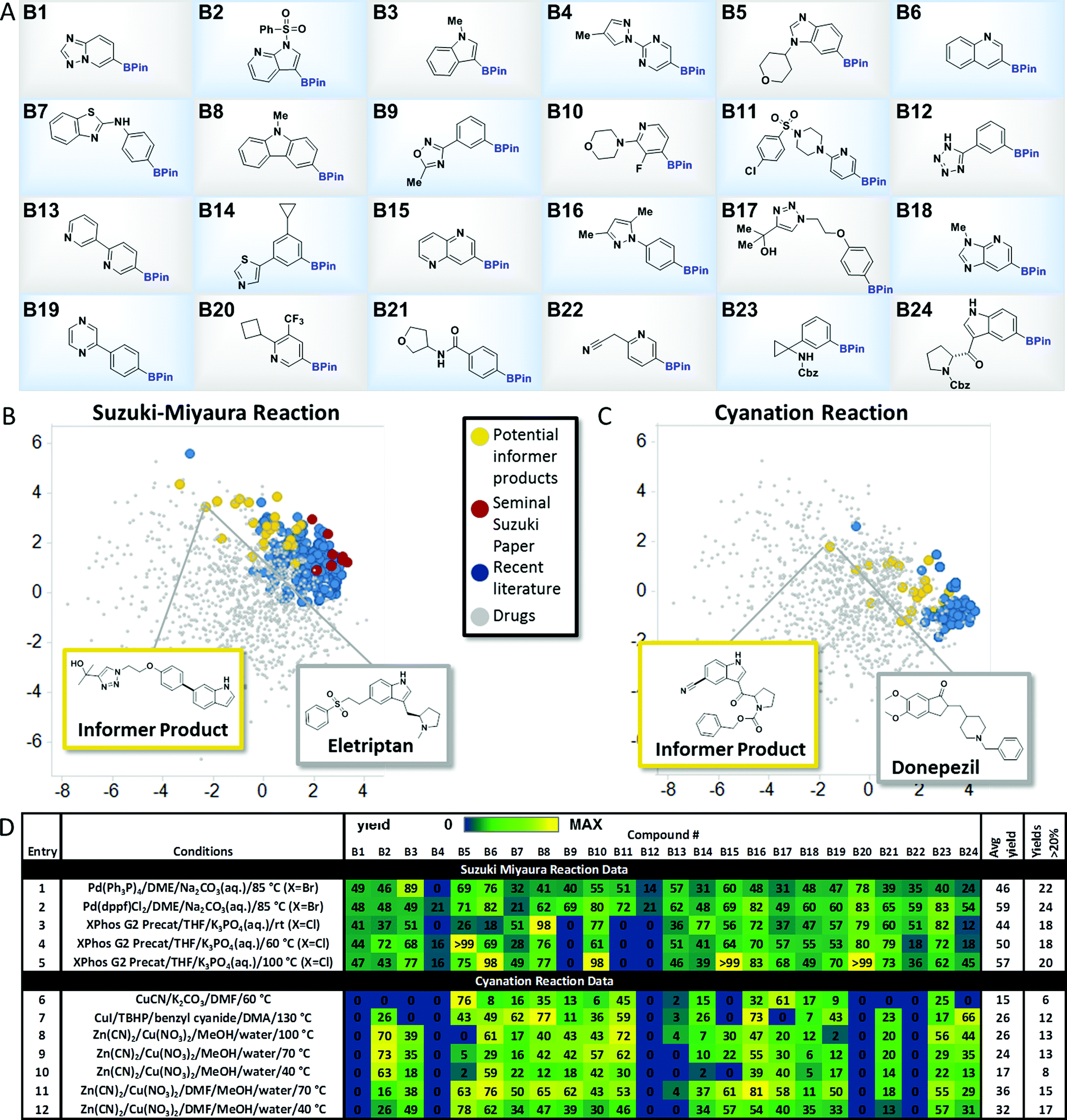
To explore the merits of the chemistry informer library concept, we curated a collection of heterocycle-containing pinacol boronates that contained the most commonly encountered rings in drugs10 as well as functional groups that are important in drug structures, such as amines, amides, sulfonamides, sulfones, alcohols, nitriles and aryl halides (Fig. 3A). Thousands of diverse pinacol boronates are available from commercial vendors, largely driven by their use in the venerable Suzuki–Miyaura reaction, popular in medicinal chemistry labs for decades.11 The Suzuki–Miyaura coupling is well established to work well on pharmaceutically relevant structures, so we chose this reaction to validate the performance of the boronate informer library and set a benchmark to which other methods could be compared. Since the discovery of the Suzuki–Miyaura reaction 35 years ago, significant efforts by many laboratories have led to new generations of Pd catalysts.12 We chose to examine whether the evolution of the Suzuki–Miyaura coupling methodology could be correlated, using our boronate informer library, with increased applicability in the pharmaceutical space. To this end, using the retrospective literature analysis as a comparative backdrop (Fig. 3B, red and blue dots), we created a standardized test set of potential Suzuki–Miyaura products with the boronate informer library, using 6-bromoindole as the coupling partner (Fig. 3B, yellow dots), for which the PCA indicates overlap of chemical space with the most evolved literature as well as increased coverage of relevant drug-like space that was not systematically explored in synthetic methods papers.13
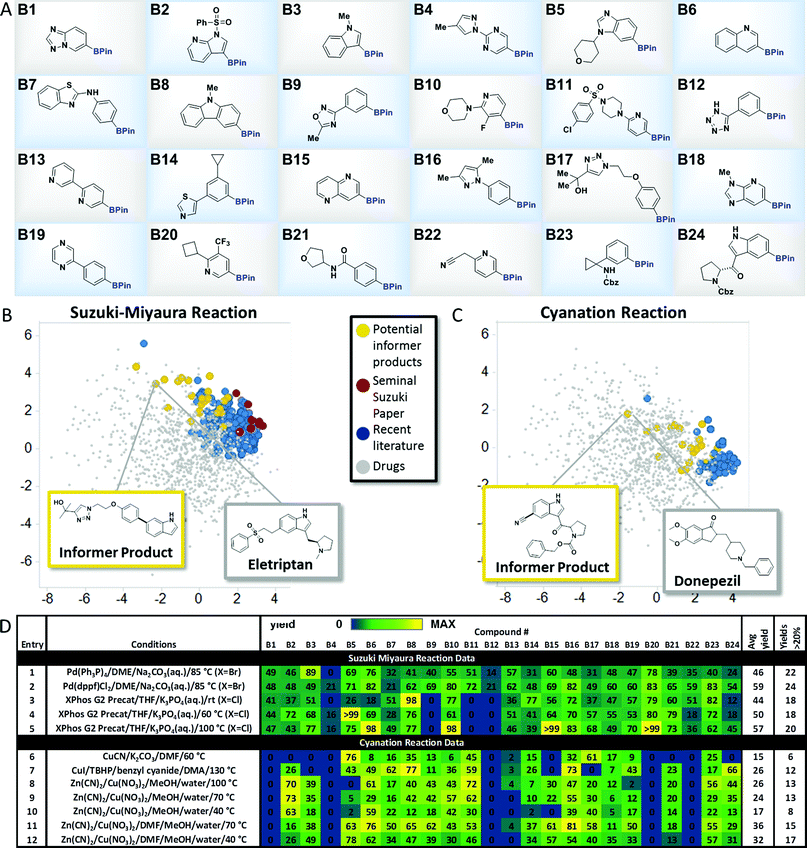 |
| | Fig. 3 Aryl and heteroaryl pinacol boronate informer library. (A) displays the 24-member boronate ester library plated in 1 mL vials in 10 μmol quantities, used for Suzuki–Miyaura and cyanation chemistry evaluation. (B) and (C) display the principal component analysis of potential products formed from Suzuki-Miyaura coupling and cyanation reactions, respectively, of the informer library (in yellow), literature products (in blue and red), and marketed drugs (in grey). (D) gives experimental results for Suzuki–Miyaura coupling reactions (entries 1–5) and cyanation reactions (entries 6–12) of the informer library members along with statistics for each method. For experimental details, see the ESI.† | |
Suzuki–Miyaura reaction
With this test set, we evaluated several landmark Suzuki–Miyaura reaction methods from the literature to systematically compare their performance (Fig. 3D). In order to facilitate these studies, experiments were conducted using parallel microscale reaction techniques developed previously in our laboratories.14 These experiments demonstrated that even the earliest published method, using tetrakis(triphenylphosphine) palladium (5 mol%) as the catalyst with inorganic base in an ethereal solvent at elevated temperature, gave synthetically useful15 amounts of products with all but two of the informer library members when 6-bromoindole was used as the coupling partner, with an average yield across the library of 46% (Fig. 3D, entry 1). Subsequently, we tested (1,1′-bis(diphenylphosphino)ferrocene)palladium(II)chloride [(dppf)PdCl2] under similar conditions and found that this bidentate phosphine palladium catalyst that has found extensive application in natural product and drug synthesis16 gave successful outcomes with all 24 members and a higher overall average yield (entry 2).17 Despite their strengths, both of these catalysts exhibit limited scope with aryl chloride substrates. Buchwald and co-workers recently reported a highly reactive system (XPhos G2 precatalyst, THF, aqueous K3PO4) that is capable of coupling sensitive boronic acids with aryl halides, including chlorides, at room temperature.9e Since it was not clear at the outset whether room temperature would be optimal for boronate ester coupling partners with such diverse structures, we ran the informer library, now with 6-Cl-indole as the halide coupling partner, at room temperature, 60 °C and 100 °C (Fig. 3D, entries 3, 4 and 5). At room temperature this highly active catalyst provided only 18 products in useful yield, although increasing the temperature to 100 °C gave success with 2 additional problematic substrates.18 Taken as a whole, the results of the informer library study of the Suzuki–Miyaura reaction validated the performance of the individual library members and confirmed the generality of the Suzuki–Miyaura methodology for pharmaceutically relevant substrates. These studies also showed that, despite the important extension to aryl chlorides enabled by ligands such as XPhos, certain substrates, including oxadiazole B9 and tetrazole B12, appeared to perform better with older generation catalysts in conjunction with aryl bromide coupling partners.
Pinacol boronate cyanation reaction
We next sought to identify a relatively new transformation that has received much less “field testing” on molecules of significance to pharmaceutical synthesis. Recently, the discovery of direct C–H borylation methods19 has improved access to pinacol boronates, enabling their use as part of late stage functionalization strategies wherein the boronate functionality becomes a gateway to introduce a diverse array of groups, such as fluorine, alkyl, perfluoroalkyl, amines, ethers, etc., that can modulate physicochemical and pharmacological properties.20 In conjunction with this interest in C–H borylation, several groups have recently reported methods for the cyanation of aryl boronates to effect the late stage introduction of nitriles.21 Evaluating the chemical space explored by boronate cyanation reactions in the literature (Fig. 3C) revealed that the types of product structures used to exemplify the chemistry (shown in blue) were for the most part low molecular weight, simple arenes bearing little resemblance to drug-like molecules (shown in grey). In contrast, potential products derived from the cyanation of pinacol boronate informer library substrates (shown in yellow), although small in size relative to some drugs, sampled a broader cross-section of drug-like structural chemical space.
We chose three recently published cyanation protocols with diverse sets of reagents and conditions (Fig. 3D, entries 6, 7 and 8) to evaluate against the boronate informer library. The method published by Hartwig21a (using Zn(CN)2, Cu(NO3)2 and CsF base with a methanol/water solvent mixture at 100 °C) gave the best results, with 13 products obtained in useful yields and a 26% average yield across the library. The best literature performance still left much to be desired, however, especially when benchmarked against the previous Suzuki–Miyaura coupling results. We examined whether the informer library diagnostic might also be applied to optimize this method to increase its scope. Decreasing the reaction temperature to 70 °C (entry 9) gave very similar overall results, while performing the reactions at 40 °C resulted in lower conversions across the library (entry 10). Our observation that the MeOH/water solvent system was not ideal for dissolving the medium-complexity informer substrates led us to examine the use of solubilizing co-solvents. We found that running the reactions in the presence of dimethylformamide (DMF) co-solvent resulted in significant increases in reaction yield and hit rate across the library. Using DMF at 70 °C (entry 11) gave 15 out of 24 products and an improved overall average yield of 36% across the library; reducing the reaction temperature to 40 °C (entry 12) gave the highest hit rate (17 out of 24 substrates) and a similar overall average yield. These results support the use of informer libraries as a tool to evolve existing methods toward increased generality. In this particular case, the use of drug fragment-like boronates identified solubility as being a key factor in determining reaction outcome. Beyond this, the reactivity trends embedded within the informer library results revealed some curious observations that could prove useful to researchers looking to develop improved methods. For example, with the exception of tetrazole-containing boronate B12, which also gave poor results in the Suzuki–Miyaura coupling, all of the poorest performing members of the informer library (B1, B4, B13, B20 and B22) contained the Bpin moiety in a meta position to a nitrogen atom in a six-membered heterocyclic ring. Although the reasons for this are not clear, future work on cyanation methods should include examples of these types of structures given their prevalence in drugs.
High complexity aryl halide informer library
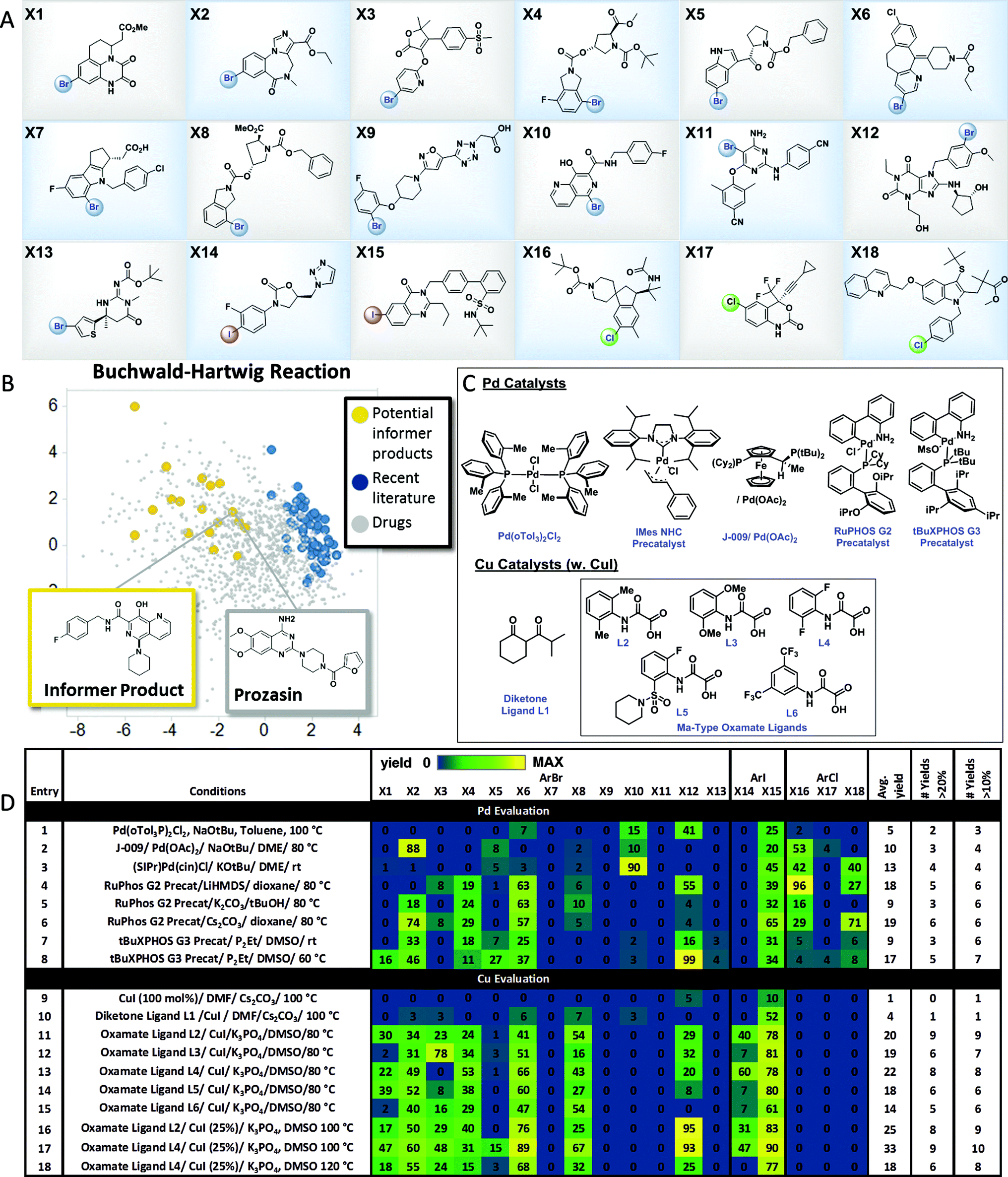
The results with the pinacol boronate informer library validated the concept of using collections of moderately complex drug fragments to systematically probe reaction utility. In order to explore the use of an informer library concept within significantly more complex drug-like space, we assembled a collection of 18 higher complexity aryl halides (3 chlorides, 13 bromides and 2 iodides) from the Merck & Co., Inc. compound collection (Fig. 4A). These compounds represent the penultimate intermediates from various drug discovery and development programs and thus ideally typify pharmaceutical substrates. In order to demonstrate the utility of this informer library, we chose to evaluate the C–N coupling reaction employing piperidine as the nucleophile. Fig. 4B plots the PCA of reaction products in six representative C–N coupling methodology literature reports22 (shown in blue) as compared to drug-like chemical space (grey). In contrast, the potential reaction products resulting from C–N coupling of the high complexity aryl halide informer library with piperidine (in yellow) are clearly more dispersed within pharmaceutically relevant space that has not been systematically explored in previous reports.
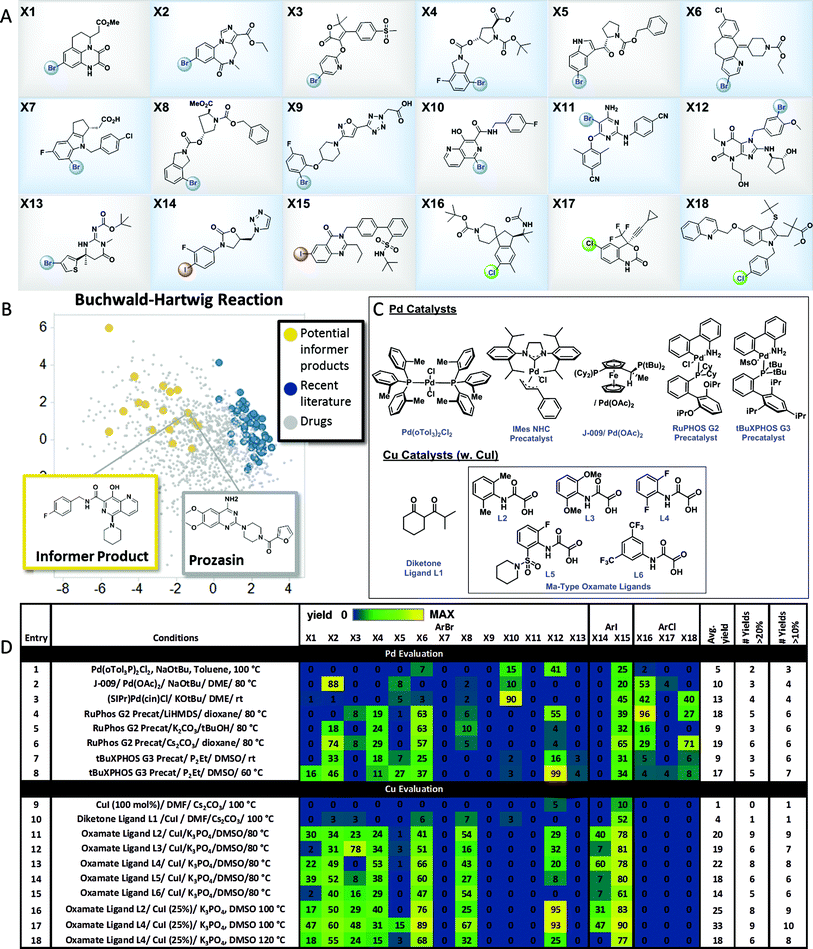 |
| | Fig. 4 High-complexity aryl halide informer library. (A) displays the 18-member aryl halide array, plated in 2.5 μmol quantities in 250 μL “microvials”, used for Pd and Cu C–N coupling chemistry evaluation. (B) displays the principal component analysis of potential products formed from C–N coupling reactions of the informer library (in yellow), literature products (in blue and red), and marketed drugs (in grey). (C) shows structures for the catalysts employed in the Pd and Cu C–N reactivity study depicted in (D). For experimental details, see the ESI.† | |
Pd C–N coupling
Over the past two decades C–N coupling of aryl (pseudo)halides has become one of the most widely used classes of reactions in pharmaceutical synthesis.11b,23 Generations of new catalyst development have resulted in highly evolved systems that are capable of coupling a wide array of aryl halides and pseudohalides with diverse nitrogen-based nucleophiles such as amines, amides, and N-heterocycles. Despite this progress, in our laboratories the C–N coupling reaction has exhibited a relatively low success rate (∼45%) when applied in the final steps of drug synthesis. Using the high-complexity aryl halides informer library, we surveyed the landmark Pd C–N coupling literature, starting with the first conditions reported by both Buchwald and Hartwig employing ((o-Tol)3P)2PdCl2, NaOtBu and base with toluene as the solvent at 100 °C.22a,b These conditions gave a very low success rate against the informer library, resulting in formation of only 2 out of 18 products in greater than 20% yield (Fig. 4D, entry 1). Two subsequently reported Pd catalysts bearing electron-rich ligands capable of activating aryl chlorides, one a Josiphos bis(phosphine) ligand (entry 2)24 and the other a saturated carbene ligand (entry 3),22d showed only marginal improvements under similarly harsh reaction conditions. The electron-rich ligand RuPhos has been recommended as an optimal ligand for 2° amine coupling,25 and advances in Pd precatalyst design have allowed for the use of milder bases and reaction temperatures with this ligand.26 The combination of RuPHOS G2 precatalyst and LiHMDS/dioxane at 80 °C resulted in 5 compounds being prepared (entry 4). Varying solvent/base combinations (entries 5 and 6) with this catalyst gave only marginal improvements. We recently reported that the use of tBuXPhos G3 precatalyst in conjunction with the strong soluble phosphazene base P2Et in the polar solvent DMSO at ambient temperature gave broad scope for C–X coupling reactions on complex substrates, including some of the members of this informer library.27 This system exhibited low reactivity at room temperature (entry 7) but gave somewhat better results at 60 °C (entry 8), essentially equivalent to the best RuPhos results. The results of this study demonstrated how advances in Pd-catalyzed Buchwald–Hartwig coupling protocols over time have resulted in incremental improvements in applicability to drug-like intermediates. Since no method was able to give better than a 33% success rate, and even a hypothetical parallel evaluation of all 8 sets of Pd-catalyzed C–N coupling conditions would have resulted in only 50% of the compounds being prepared, this reaction is still far from a solved problem in high-complexity drug synthesis.
Cu C–N coupling
Having explored Pd-catalyzed C–N coupling methods, we next turned our attention to conditions employing Cu catalysts. Although traditionally Cu-based methods have been employed more commonly in the coupling of amide and N-heterocyclic substrates, some literature precedent points to their utility in amine couplings.23 Reported Cu-based systems possess several attractive features when viewed from a pharmaceutical perspective, including the common use of polar aprotic solvents and mild inorganic bases. We first evaluated a variation of the original Ullmann conditions28 which are stoichiometric in Cu (Fig. 4D, entry 9): these conditions gave no hits across the library. Moving on to one of the earliest reported examples from Buchwald and coworkers22c of a Cu-catalyzed C–N coupling, using CuI in conjunction with the diketone ligand L1 as the catalyst in DMF solvent with Cs2CO3 base, gave two hits (entry 10). The Ma group has reported oxamate-based ligands that give greatly increased reactivity in Cu-catalyzed amine couplings.22f Using the reported conditions employing L2 as the ligand (entry 11) gave the best overall performance observed thus far, with 9 total products generated in usable yields. Hoping to improve on these results, we prepared a series of analogs of the original oxamate ligand L2 and used the informer library as a diagnostic to determine whether any of these exhibited increased reactivity and scope (entries 12–18). Ligand L4 gave similar results to L2, with 8 compounds prepared in usable yields (entry 13). Noting the extremely clean profiles of these reactions, we evaluated both the L2 and L4 systems again at 100 °C, and found that L4 gave 9 compounds at 33% average yield, the best reaction profile for any Pd or Cu conditions we evaluated (entry 17). Further increases in temperature were not beneficial (entry 18). The successful coupling of 9 out of 15 aryl bromides and iodides in the informer library with one set of conditions clearly substantiates the notion that Cu is an excellent choice for C–N coupling in highly complex synthesis, although significant room for improvement, including the extension to aryl chlorides, remains.
Comparison of the whole molecule informer library and the fragment-based robustness test
The informer library approach to reaction appraisal carries high experimental demands, requiring access to complex molecules for experimentation as well as the need for scale-up, isolation and characterization of all new products in order to enable quantitative LC analysis.29 By comparison, the simpler fragment-based “robustness” test involves adding potentially interfering fragments to simple model reactions and then measuring the reaction performance relative to a control that does not have the fragments present. This method measures both the decrease in product formation, indicating reaction inhibition, and the decrease in the amount of the fragment itself remaining at the end of reaction, indicating decomposition pathways. In order to compare the two diagnostic methods, we chose 3 boronates (B9, B11 and B12) that failed in the Suzuki–Miyaura reaction with 6-chloroindole using the XPhos G2 precatalyst system at 100 °C (see ESI† for details on all fragment-based experiments). In this study, we found that fragments containing oxadiazole and 4-chlorophenylsulfonamide functionalities significantly diminished the reaction yield of a model Suzuki–Miyaura coupling reaction, mirroring the performance observed in the informer library. In contrast, a tetrazole-containing additive did not inhibit the test Suzuki–Miyaura coupling reaction, nor was it consumed in the reaction. Similarly, we studied the failure of the C–N coupling of halide X7 with piperidine, first using the simplest fragment approach, and found that none of the potentially interfering fragments that could be derived from the deconstruction of complex molecule X7 diminished the reactivity of a simple C–N coupling. Instead, we considered it more likely that the local steric and electronic demands of the core of X7 were responsible for the low reactivity and confirmed that 1-benzyl-7-bromoindole indeed showed very low (<1% LC area percent) product formation. These studies show that the simplest, most experimentally accessible fragment-based method may significantly under-predict reaction failures in complex systems. It is likely that in such cases the aggregate of steric and electronic properties of the reacting core, the presence of various functional groups and the overall physical properties of the entire molecule will prove deterministic of reaction success.
The future of chemistry informer libraries
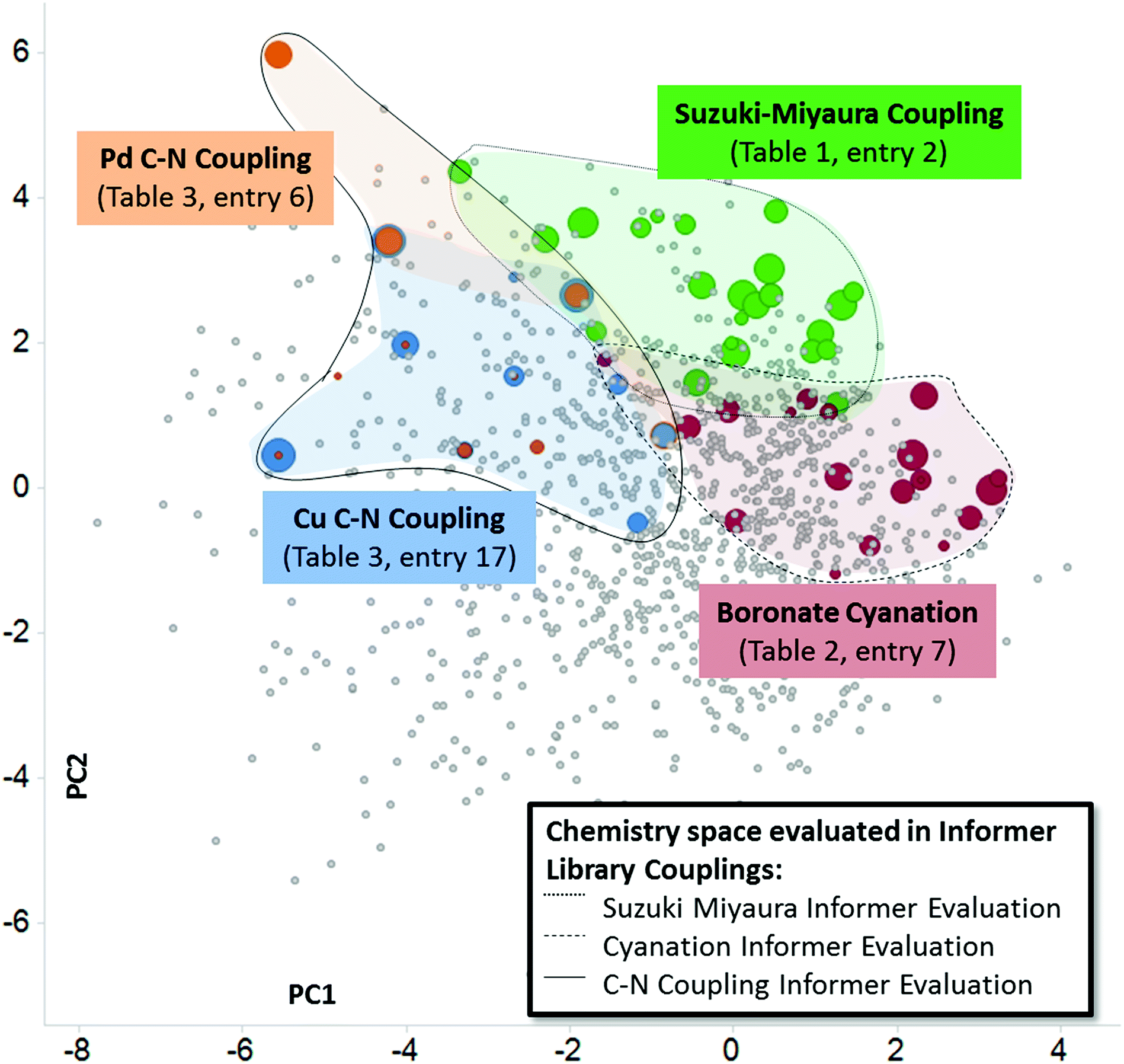
In this work we have used heat maps, shown in Fig. 3 and 4, to provide a visualization of individual reaction performance as well as reactivity patterns and trends for different synthetic methods. As useful as such depictions may be, they ignore the richness of structural information embedded in the individual informer library members and products. Fig. 5 depicts one way in which both reactivity and structural information may be visualized together. By mapping the experimental yields (magnitude represented by size of circles) for particular informer library products onto the two-dimensional PCA of marketed drugs, different chemistry methods and even entirely different reaction types can be represented in a single graph to reveal chemical reactivity trends and relevance within a given region of structural space where the products share similar physicochemical characteristics. Fig. 5 illustrates the possibilities of systematically exploring structure–reactivity relationships within complex chemical space with the aid of modern chemoinformatics techniques. Obviously, the data sets acquired in this work are much too limited to enable the construction of quantitative, predictive structure–reactivity models. Currently, despite significant advances made in the high-throughput screening of reactions on a small scale30 the number of chemistry informer library compounds that can be evaluated is limited by practical considerations arising from the need to isolate and characterize products in order to quantify reaction yields. However, this limitation may not remain for long.31 In this case, one could then assemble and test the reactivity of larger informer sets that would provide a statistical representation of diverse structural motifs in a complex molecule setting. Mining the rich data sets that would result from such an exercise would provide a holistic picture of structure–reactivity relationships that would encompass the various levels of detail found in Fig. 1, including: (1) information on local steric and electronic effects at reaction centers, (2) identification of interfering structural elements, and (3) the effects of aggregate molecular properties, such as potential chelation motifs or bulk physicochemical characteristics.
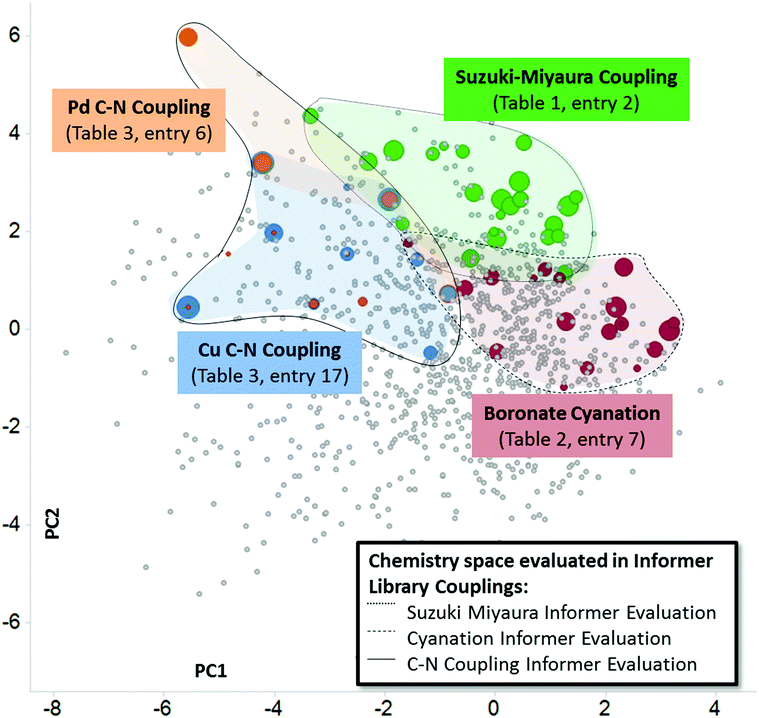 |
| | Fig. 5 Informer library reactivity data mapped onto relevant physicochemical PCA to produce a representation of structure–reactivity relationships. Each colored ball represents one molecule successfully prepared, and the size of the ball is proportional to the yield of the reaction. In this way, different reaction types can be directly compared for performance and relevance within a region of structural space. | |
Conclusions
We have demonstrated how a standardized chemistry informer library can allow direct head-to-head comparisons of different synthetic methods in complex structural space. In contrast to current methodology studies that tend to explore the reactivity of simple substrates, or total synthesis papers that tend to report only the positive results for the reactivity of specific complex substrates, our method comprehensively explores the relationship between reaction types and diverse complex substrates. The general application of this approach would enable synthetic practitioners to make better decisions about which synthetic methods to prioritize in their problem solving and give them a diagnostic that could reveal if a new method was significantly better than existing methods. Chemistry informer libraries could also provide insight to academic researchers, as well as funding agencies, into which synthetic methods are mature versus those, such as the C–N coupling reaction, that apparently still require significant innovative development to become broadly relevant to complex synthesis. The same analyses can point out specific gaps in substrate scope that need to be addressed. Finally, the chemistry informer library concept points toward a new way to study chemical reactivity, in which chemoinformatics tools are applied to large data sets generated from complex substrates to enable a holistic understanding of structure–reactivity relationships, from local steric and electronic effects at reaction centers to aggregate physicochemical properties arising from the entire substrate structure. Efforts in our labs towards further validating this chemoinformatics-enabled approach to reaction evaluation and optimization are underway. We eagerly anticipate that other laboratories will begin to adopt and expand upon these methods, and in doing so help drive the field to develop synthetic transformations with ever more powerful applications in complex molecule synthesis.32
Acknowledgements
We thank Meir Glick and Eric Fischer for helpful discussions, Mikhail Reibarkh for assistance with NMR experiments and Natalya Pissarnitski for assistance in compound purification.
Notes and references
- A. Nadin, C. Hattotuwagama and I. Churcher, Angew. Chem., Int. Ed., 2012, 51, 1114 CrossRef CAS PubMed.
- For related approaches, see:
(a) K. Zhang, S. El Damaty and R. Fasan, J. Am. Chem. Soc., 2011, 133, 3242 CrossRef CAS PubMed;
(b) E. N. Bess, A. J. Bischoff and M. S. Sigman, PNAS, 2014, 111, 14698 CrossRef CAS PubMed.
-
(a) K. D. Collins and F. Glorius, Nat. Chem., 2013, 5, 597 CrossRef CAS PubMed;
(b) K. D. Collins and F. Glorius, Acc. Chem. Res., 2015, 48, 619 CrossRef CAS PubMed;
(c) K. D. Collins and F. Glorius, Tetrahedron, 2013, 69, 7817 CrossRef CAS;
(d) K. D. Collins, A. Ruhling, F. Lied and F. Glorius, Chem. Eur. J., 2014, 20, 3800 CrossRef CAS PubMed;
(e) K. D. Collins and F. Glorius, Nat. Protoc., 2014, 9, 1348 CrossRef CAS PubMed.
- J. L. Medina-Franco, K. Martinez-Mayorga, M. A. Giulianotti, R. A. Houghten and C. Pinilla, Curr. Comput.–Aided Drug Des., 2008, 4, 322 CrossRef CAS.
- A. A. Shelat and R. K. Guy, Curr. Opin. Chem. Biol., 2007, 11, 244 CrossRef CAS PubMed.
- In order to generate a PCA visualization that was consistent across each analysis, we performed a PCA on all compounds, although in many of our figures only a subset of compounds is depicted.
- We visualize PC3 and PC4 in Fig. S1 in the ESI† (78% cumulative variance explained).
- N. Miyaura, T. Yanagi and A. Suzuki, Synth. Commun., 1981, 11, 513 CrossRef CAS.
-
(a) A. Zapf, R. Jackstell, F. Rataboul, T. Riermeier, A. Monsees, C. Fuhrmann, N. Shaikh, U. Dingerdissen and M. Beller, Chem. Commun., 2004, 38 RSC;
(b) N. Kudo, M. Perseghini and G. C. Fu, Angew. Chem., Int. Ed., 2006, 45, 1282 CrossRef CAS PubMed;
(c) C. J. O'Brien, E. A. B. Kantchev, C. Valente, C. N. Hadei, G. A. Chass, A. Lough, A. C. Hopkinson and M. G. Organ, Chem.–Eur. J., 2006, 12, 4743–4748 CrossRef PubMed;
(d) K. Billingsley and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 3358 CrossRef CAS PubMed;
(e) T. Kinzel, Y. Zhang and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 14073 CrossRef CAS PubMed;
(f) D. W. Robbins and J. F. Hartwig, Org. Lett., 2012, 14, 4266 CrossRef CAS PubMed;
(g) M. A. Düfert, K. L. Billingsley and S. L. Buchwald, J. Am. Chem. Soc., 2013, 135, 12877 CrossRef PubMed.
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845 CrossRef CAS PubMed.
-
(a)
M. L. Crawley and B. M. Trost, Applications of transition metal catalysis in drug discovery and development: an industrial perspective, Wiley, Hoboken, 2012 Search PubMed;
(b) D. G. Brown and J. Boström, J. Med. Chem., 2015, 58 DOI:10.1021/acs.jmedchem.5b01409 CAS.
- For reviews on the Suzuki–Miyaura coupling, see:
(a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS;
(b) A. Suzuki, J. Organomet. Chem., 1999, 576, 147 CrossRef CAS;
(c) F. Bellina, A. Carpita and R. Rossi, Synthesis, 2004, 2419 CAS;
(d)
C. Valente and M. G. Organ, in Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, ed. D. G. Hall, Wiley, Chichester, 2nd edn, 2011 Search PubMed.
- While many Suzuki–Miyaura reactions have been reported in the literature for complex drug-like structures or natural products, there is no standardized
reactivity data for these compounds, and in many cases no attempt is made to determine the actual yields of reactions.
-
(a) C. S. Shultz and S. W. Krska, Acc. Chem. Res., 2007, 40, 1320 CrossRef CAS PubMed;
(b) S. D. Dreher, P. G. Dormer, D. L. Sandrock and G. A. Molander, J. Am. Chem. Soc., 2008, 130, 9257 CrossRef CAS PubMed.
- For a typical synthesis of an investigational drug compound in a medicinal chemistry project, a solution yield of >20% can reliably provide a usable amount of material for initial biochemical testing after purification.
- R. Rossi, F. Bellina, M. Lessi, C. Manzini, G. Marianetti and L. A. Perego, Curr. Org. Chem., 2015, 19, 1302 CrossRef CAS.
- Selected Suzuki–Miyaura coupling conditions were repeated on a larger scale to confirm product structures and to obtain UV response factors for solution yield determination. See ESI† for more details.
- The XPhos G2 precatalyst has been reported to give good results in Suzuki–Miyaura couplings of arylboronic acids with nitrogen-rich, unprotected heterocycles at elevated temperature: see ref. 9g.
-
(a) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS PubMed;
(b) J. F. Hartwig, Chem. Soc. Rev., 2011, 40, 1992 RSC;
(c) J. F. Hartwig, Acc. Chem. Res., 2012, 45, 864 CrossRef CAS PubMed;
(d) S. M. Preshlock, B. Ghaffari, P. E. Maligres, S. W. Krska, R. E. Maleczka and M. R. Smith, J. Am. Chem. Soc., 2013, 135, 7572 CrossRef CAS PubMed.
-
(a) M. A. Larsen and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 4287 CrossRef CAS PubMed;
(b) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev. 10.1039/c5cs00628g Search PubMed.
-
(a) C. W. Liskey, X. Liao and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 11389 CrossRef CAS PubMed;
(b) G. Zhang, L. Zhang, M. Hu and J. Cheng, Adv. Synth. Catal., 2011, 353, 291 CrossRef CAS;
(c) P. Anbarasan, J. Neumann and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 519 CrossRef CAS PubMed;
(d) J. Kim, J. Choi, K. Shin and S. Chang, J. Am. Chem. Soc., 2012, 134, 2528 CrossRef CAS PubMed;
(e) Y. Luo, Q. Wen, Z. Wu, J. Jin, P. Lu and Y. Wang, Tetrahedron, 2013, 69, 8400 CrossRef CAS. For a related protocol which is reported only for boronic acid substrates, see: Z. H. Zhang and L. S. Liebeskind, Org. Lett., 2006, 8, 4331 CrossRef CAS PubMed.
-
(a) A. S. Guram, R. A. Rennels and S. L. Buchwald, Angew. Chem., Int. Ed., 1995, 34, 1348 CrossRef CAS;
(b) J. Louie and J. F. Hartwig, Tetrahedron Lett., 1995, 36, 3609 CrossRef CAS;
(c) A. Shafir and S. L. Buchwald, J. Am. Chem. Soc., 2006, 128, 8742 CrossRef CAS PubMed;
(d) N. Marion, O. Navarro, J. Mei, E. D. Stevens, N. M. Scott and S. P. Nolan, J. Am. Chem. Soc., 2006, 128, 4101 CrossRef CAS PubMed;
(e) C. T. Yang, Y. Fu, Y. B. Huang, J. Yi, Q. X. Guo and L. Liu, Angew. Chem., Int. Ed., 2009, 48, 7398 CrossRef CAS PubMed;
(f) Y. Zhang, X. Yang, Q. Yao and D. Ma, Org. Lett., 2012, 14, 3056 CrossRef CAS PubMed.
- I. P. Beletskaya and A. V. Cheprakov, Organometallics, 2012, 31, 7753 CrossRef CAS.
- Q. Shen, T. Ogata and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 6586 CrossRef CAS PubMed.
- D. Maiti, B. P. Fors, J. L. Henderson, Y. Nakamura and S. L. Buchwald, Chem. Sci., 2011, 2, 57 RSC.
- N. C. Bruno, M. T. Tudge and S. L. Buchwald, Chem. Sci., 2013, 4, 916 RSC.
- A. B. Santanilla, M. Christensen, L.-C. Campeau, I. W. Davies and S. D. Dreher, Org. Lett., 2015, 17, 3370 CrossRef PubMed.
- J. Lindley, Tetrahedron, 1984, 40, 1433 CrossRef CAS.
- Using the pre-plated libraries of informer compounds (18–24 compounds per library), the experimental evaluation of a given reaction method, including reaction set-up, quantitative LC analysis and data workup, requires 3–4 hours. However, in order to enable quantitative LC analysis, the generation of all reaction products at preparative scale, isolation and full characterization requires on the order of one to two weeks of experimental effort per library.
-
(a) J. R. Schmink, A. Bellomo and S. Berritt, Aldrichimica Acta, 2013, 46, 71 Search PubMed;
(b) K. D. Collins, T. Gensch and F. Glorius, Nat. Chem., 2014, 6, 859 CrossRef CAS PubMed;
(c)
A. Gordillo, S. Titlbach, C. Futter, M. L. Lejkowski, E. Prasetyo, L. T. A. Rupflin, T. Emmert and S. A. Schunk, Ullmann's Encyclopedia of Industrial Chemistry, 2014, p. 1 Search PubMed;
(d) A. B. Santanilla, E. R. Regalado, T. Pereira, M. Shevlin, K. Bateman, L. C. Campeau, J. Schneeweis, S. Berritt, Z.-C. Shi, P. Nantermet, Y. Liu, R. Helmy, C. J. Welch, P. Vachal, I. W. Davies, T. Cernak and S. D. Dreher, Science, 2015, 347, 49 CrossRef PubMed.
-
(a) Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai, Y. Hitora, K. Takada, S. Matsunaga, K. Rissanen and M. Fujita, Nature, 2013, 495, 461 CrossRef CAS PubMed;
(b) N. Zigon, M. Hoshino, S. Yoshioka, Y. Inokuma and M. Fujita, Angew. Chem., Int. Ed., 2014, 54, 9033 CrossRef PubMed;
(c) T. R. Ramadhar, S.-L. Zheng, Y.-S. Chen and J. Clardy, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 46 CAS;
(d) T. R. Ramadhar, S.-L. Zheng, Y.-S. Chen and J. Clardy, Chem. Commun., 2015, 51, 11252 RSC.
- A. Milo, A. J. Neel, F. D. Toste and M. S. Sigman, Science, 2015, 347, 737 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc04751j |
|
| This journal is © The Royal Society of Chemistry 2016 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
James F.
Dropinski
b,
Kevin D.
Dykstra
c,
Bing
Li
c,
Daniel A.
DiRocco
b,
Eric C.
Streckfuss
c,
Louis-Charles
Campeau
b,
Tim
Cernak
a,
James F.
Dropinski
b,
Kevin D.
Dykstra
c,
Bing
Li
c,
Daniel A.
DiRocco
b,
Eric C.
Streckfuss
c,
Louis-Charles
Campeau
b,
Tim
Cernak