DOI:
10.1039/C5SC04312C
(Edge Article)
Chem. Sci., 2016,
7, 2856-2863
One-step selective hydroxylation of benzene to phenol with hydrogen peroxide catalysed by copper complexes incorporated into mesoporous silica–alumina†
Received
12th November 2015
, Accepted 5th January 2016
First published on 5th January 2016
Abstract
Benzene was hydroxylated with hydrogen peroxide (H2O2) in the presence of catalytic amounts of copper complexes in acetone to yield phenol at 298 K. At higher temperatures, phenol was further hydroxylated with H2O2 by catalysis of copper complexes to yield p-benzoquinone. The kinetic study revealed that the rate was proportional to concentrations of benzene and H2O2, but to the square root of the concentration of a copper(II) complex ([Cu(tmpa)]2+: tmpa = tris(2-pyridylmethyl)amine). The addition of a spin trapping reagent resulted in formation of a spin adduct of hydroperoxyl radical (HO2˙), as observed by EPR spectroscopy, inhibiting phenol formation. HO2˙ produced by the reaction of [Cu(tmpa)]2+ with H2O2 acts as a chain carrier for the radical chain reactions for formation of phenol. When [Cu(tmpa)]2+ was incorporated into mesoporous silica–alumina (Al-MCM-41) by a cation exchange reaction, the selectivity for production of phenol was much enhanced by prevention of hydroxylation of phenol, which was not adsorbed to Al-MCM-41. The high durability with a turnover number of 4320 for the hydroxylation of benzene to phenol with H2O2 was achieved using [Cu(tmpa)]2+ incorporated into Al-MCM-41 as an efficient and selective catalyst.
Introduction
Phenol is one of the most important basic chemicals, widely used as a source for resins, fibres, and other various organic materials, being produced in industry by the three-step cumene process, which requires high energy consumption due to high-pressure conditions and produces acetone as a coproduct via cumene hydroperoxide.1,2 Thus, much effort has been devoted to develop a one-step hydroxylation of benzene to phenol using different oxidants such as N2O,3 O2 with reductants4–9 and H2O2.10–23 Among these oxidants, H2O2 has a clear advantage from the viewpoint of an environmentally benign green process and economical efficiency because of relatively mild reaction conditions for catalytic hydroxylation of benzene.10–23 However, selective hydroxylation of benzene to phenol with H2O2 has been difficult because phenol is normally easily over-oxidised to yield products such as p-benzoquinone.10–22 Although selective hydroxylation of benzene to phenol without over-oxidation has been achieved in some cases, the catalytic mechanism of hydroxylation of benzene to phenol has yet to be clarified,24–26 except for some cases of photocatalytic hydroxylation of benzene to phenol.23 Extensive efforts have been devoted to enhance the product selectivity and catalytic activity with heterogeneous catalysts, including metal complex catalysts incorporated into a mesoporous material. For instance, it has been reported that a manganese complex incorporated into a mesoporous material Al-MCM-41 exhibits a higher performance compared to that of the complex in a homogeneous solution because of the stabilisation of catalytically active species and prevention of phenol over-oxidation due to the highly acidic nature in Al-MCM-41.26
We report herein the one-step selective hydroxylation of benzene to phenol with H2O2 catalysed by a copper(II) complex ([Cu(tmpa)]2+: tmpa = tris(2-pyridylmethyl)amine) incorporated into mesoporous silica–alumina in acetone at 298 K. The mechanism is clarified by a kinetic study and by detection of a reactive radical intermediate using a spin trap. The incorporation of the [Cu(tmpa)]2+ into mesoporous silica–alumina resulted in significant improvement in the selectivity and the durability of the catalyst.
Results and discussion
Hydroxylation of benzene to phenol with H2O2 catalysed by Cu(II) complexes
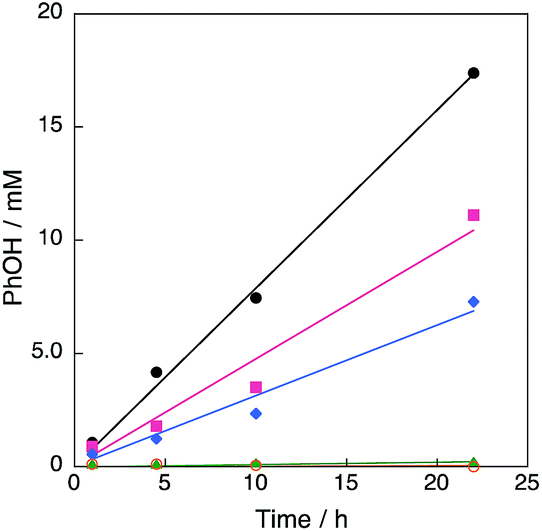
A series of Cu(II) complexes, [CuII(tmpa)(CH3CN)](ClO4)2, [CuII2(N5)(H2O)2](NO3)4 (N5 = –(CH2)5– linked bis[2-(2-pyridyl)ethyl]amine) and [CuII(tepa)(ClO4)]ClO4 (tepa = tris(2-pyridylethyl)amine) were all found to catalyse hydroxylation of benzene with H2O2 to produce phenol (Fig. 1).27 The time profiles for these reactions using a catalytic amount of Cu(II) complexes in acetone at 298 K are shown in Fig. 2. The most reactive Cu(II) complex was [CuII(tmpa)(CH3CN)](ClO4)2, which has the most negative one-electron reduction potential (Ered = −0.01 V vs. SCE)28 compared with those of [CuII2(N5)(H2O)2](NO3)4 (Eredvs. SCE = 0.27 V) and [CuII(tepa)(ClO4)]ClO4 (Eredvs. SCE = 0.44 V),29,30 as shown in Fig. S1 in ESI.† It should be noted that CuII(ClO4)2 with no ligand exhibited virtually no catalytic activity. Thus, the tmpa ligand plays an essential role in the catalytic activity.
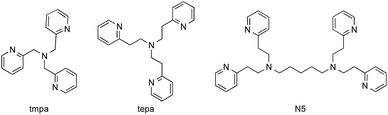 |
| | Fig. 1 Chemical structures of ligands tmpa, tepa and N5. | |
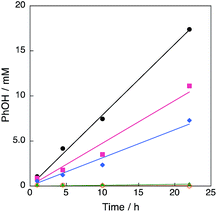 |
| | Fig. 2 Time profiles of formation of phenol in the hydroxylation of benzene with 30 wt% aqueous H2O2 and a catalytic amount of 1 ([[CuII(tmpa)]2+] = 67 μM, black circle), [CuII(tepa)(ClO4)]ClO4 ([[CuII(tepa)]2+] = 74 μM, pink square), [CuII2(N5)(H2O)2](NO3)4 ([[CuII2(N5)]4+] = 65 μM, blue diamond), Cu(ClO4)·6H2O (Cu2+ = 57 μM, green triangle) and no catalyst (orange open circle) in acetone at 303 K; [C6H6] = 2.1 M, [H2O2] = 2.1 M (4.75 mL). | |
When acetone was replaced by strongly coordinating solvents, such as DMF and DMSO, no catalytic hydroxylation of benzene with H2O2 occurred using [CuII(tmpa)(CH3CN)](ClO4)2 (1) as the catalyst (see Fig. S2 in ESI†). Thus, a coordination site is required to exhibit the catalytic activity. In acetonitrile, 1-catalysed hydroxylation of benzene with H2O2 occurred to produce phenol. However, p-benzoquinone was also formed as a byproduct, which may be produced via oxidation of hydroquinone. In MeOH, the catalytic rate of hydroxylation of benzene with H2O2 was slower than those in acetone and acetonitrile. Thus, we have chosen acetone as a suitable solvent for mechanistic studies.
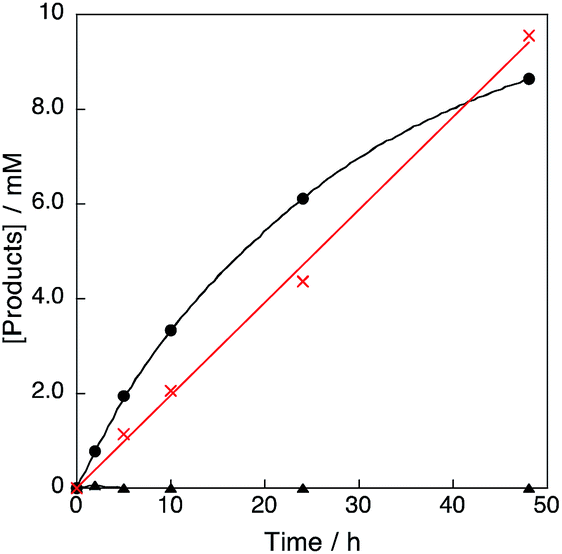
The products in the reaction of benzene with H2O2 and the Cu(II) complex catalyst 1 were examined by GC-MS measurements. When a large concentration of benzene (2.1 M) was used for the 1-catalysed reaction of benzene with H2O2, phenol was selectively produced without formation of p-benzoquinone at the initial stage of the reaction (black circle in Fig. 3). When benzene was replaced by phenol, however, p-benzoquinone was produced with a comparable rate as that for phenol production from benzene (red cross in Fig. 3).
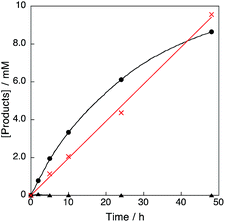 |
| | Fig. 3 Time profiles of formation of phenol (black circle) and p-benzoquinone (black triangle) in the hydroxylation of benzene, and formation of p-benzoquinone (red cross) in the oxidation of phenol with 30 wt% aqueous H2O2 catalysed by 1 in acetone at 298 K; [C6H6] or [PhOH] = 2.1 M, [H2O2] = 2.1 M, [[CuII(tmpa)]2+] = 67 μM in acetone (4.75 mL). | |
The stoichiometry of the catalytic hydroxylation of benzene with H2O2 is given by eqn (1). The amount of reacted H2O2 was determined by the titration with Ti–TPyP (oxo[5,10,15,20-tetra(4-pyridyl)porphyrinato]titanium(IV);31 see Experimental section and Fig. S3 in ESI†). The ratio of the reacted H2O2 to phenol was determined to be nearly 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, indicating that H2O2 decomposed in competition with the catalytic hydroxylation of benzene to phenol.
:1, indicating that H2O2 decomposed in competition with the catalytic hydroxylation of benzene to phenol.
| | | C6H6 + H2O2 → C6H5OH + H2O | (1) |
Kinetics of catalytic hydroxylation of benzene to phenol with H2O2 and 1
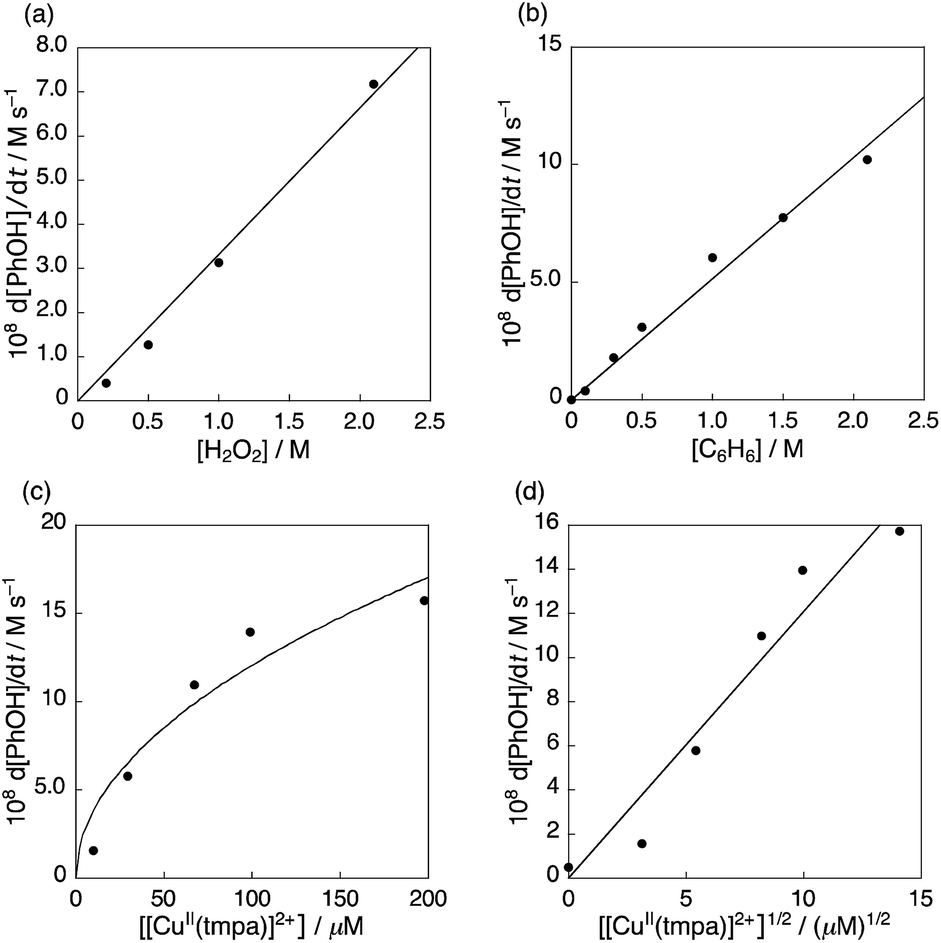
The dependencies of the initial rate of phenol production on the concentrations of 1, H2O2 and benzene were examined to determine a kinetic formulation (Fig. 4). The initial rates of phenol production were determined from the time profiles of production of phenol in the catalytic hydroxylation of benzene with H2O2 (Fig. S4a–c in ESI†). The initial rate of phenol production was proportional to the concentrations of H2O2 and benzene, as shown in Fig. 4a and b, respectively. The dependence of the initial rate of phenol production on the concentration of 1 was not linear (Fig. 4c), but instead was proportional to the square root of the concentration of 1. Thus, the kinetic equation for the catalytic hydroxylation of benzene with H2O2 is given by eqn (2) as follows:| | | d[PhOH]/dt = kcat[1]1/2[C6H6][H2O2] | (2) |
where kcat is the catalytic rate constant. The dependence of the rate on the square root of the concentration of 1 usually suggests that the catalyst may dissociate into two species, which are in equilibrium with 1, and that the concentration of the two catalytically active species are proportional to the square root of the concentration of 1 when the equilibrium lies to the far left-hand side. However, the Cu(II) complex (1) is a monomer complex, which has no way to dissociate into two species. The unusual kinetic formulation in eqn (2) in terms of the dependence of the rate on 1 provides valuable insight into the catalytic mechanism as discussed later.
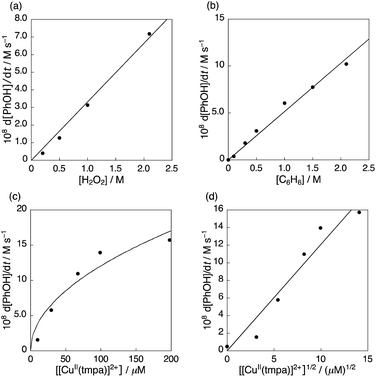 |
| | Fig. 4 (a) Dependence of the initial rate of formation of phenol (d[PhOH]/dt) calculated at 10 h on the concentration of H2O2 ([H2O2] = 0–2.1 M, [C6H6] = 1.0 M). (b) Dependence of d[PhOH]/dt calculated at 14 h on the concentration of benzene ([C6H6] = 0–2.1 M). (c) Dependence of d[PhOH]/dt calculated at the time when ca. 2 mM of phenol was formed on the concentration of 1 ([[CuII(tmpa)]2+] = 0–200 μM). (d) Plot of d[PhOH]/dt vs. [[CuII(tmpa)]2+]1/2. The [PhOH]/dt values were determined from the time profiles of formation of phenol in the hydroxylation of benzene with 30 wt% aqueous H2O2 catalysed by 1 in acetone at 298 K at the same reaction time or when the similar concentration of phenol was produced in the range where the phenol formation rate was constant (Fig. S4†). The concentrations are [C6H6] = 2.1 M, [H2O2] = 2.1 M, [H2O] = 8.4 M and [[CuII(tmpa)]2+] = 67 μM in acetone (4.75 mL) at 298 K unless otherwise noted. | |
A spin trap in the catalytic hydroxylation of benzene with H2O2
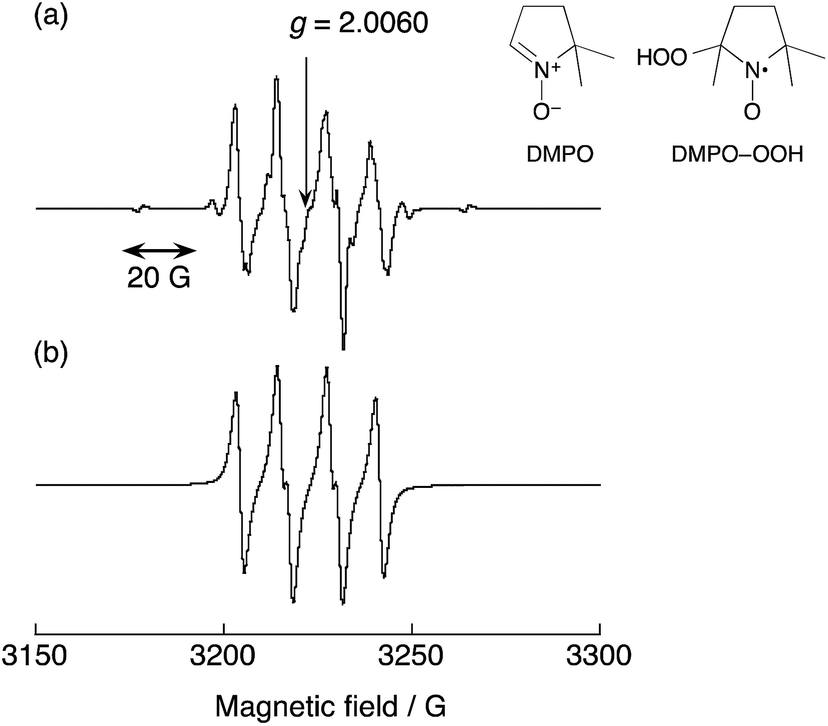
The trapping of a reactive free radical species with a diamagnetic spin trap to generate a persistent spin adduct, which can be characterised by its EPR spectrum constitutes the well known spin trapping technique.32–46 5,5-Dimethyl-1-pyrroline N-oxide (DMPO in Fig. 5a) is one of the most popular spin traps, and EPR features of its spin adducts are well established. For example, the EPR spectrum of the HO˙ radical adduct (DMPO–OH) exhibits signals with a relative peak ratio of 1:2:2:1 with the hyperfine coupling constants of aN = aH = 14.9 G, whereas the EPR spectrum of the HO2˙ radical adduct affords more lines with a hyperfine coupling constant of aN (13–14 G), which is significantly larger than aH (9–11 G).32,37–41,47 Carbon-centred radical adducts afford a larger aH value than the aN value.32,42 Thus, the type of radical species can be readily distinguished based on the hyperfine splitting pattern of the DMPO spin adduct.
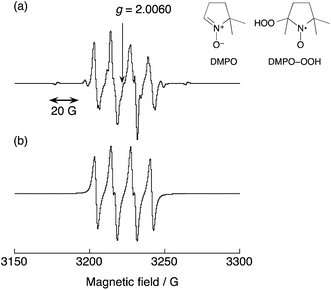 |
| | Fig. 5 (a) An EPR spectrum of DMPO–OOH produced in an acetone solution of benzene, 30 wt% aqueous H2O2, 1 and DMPO at 298 K with structures of DMPO and DMPO–OOH. (b) An EPR simulation spectrum of the DMPO–OOH spin adduct. The parameters obtained by the simulation are as follows: aH = 10.8 G, aN = 13.1 G. The concentrations are [C6H6] = 2.1 M, [H2O2] = 0.10 M, [H2O] = 0.23 M, [[CuII(tmpa)]2+] = 67 μM, [DMPO] = 0.20 M in acetone (2.0 mL). | |
Fig. 5 shows an EPR spectrum observed during the reaction of benzene with H2O2 in the presence of 1 and DMPO. The hyperfine splitting pattern of the observed radical clearly indicates that HO2˙ is produced in the reaction of benzene with H2O2 and 1, and is trapped by DMPO to produce DMPO–OOH, which has the g value of 2.0060 and aN = 13.1 G and aH = 10.8 G, as shown in Fig. 5, wherein the observed spectrum (part a) agrees with the computer simulation spectrum (part b). It was confirmed that the addition of DMPO to an acetone solution of benzene and H2O2 resulted in no observation of an EPR signal. In summary, when 1 was added to the acetone solution of benzene, H2O2 and DMPO, a strong EPR signal assigned to DMPO–OOH was observed.
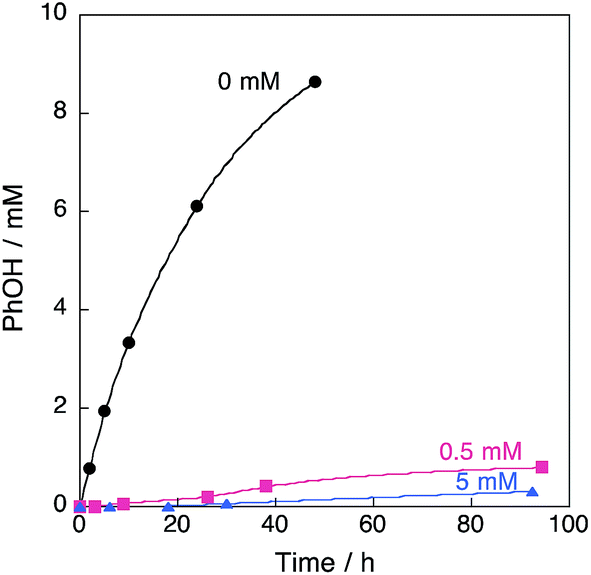
The concentration of DMPO–OOH was determined by comparing the double integration value of the EPR signal of DMPO–OOH with that of a stable reference radical (2,2-diphenyl-1-picrylhydrazyl radical, see Experimental section). The concentration of DMPO–OOH increased with the reaction time, as shown in Fig. S5 (ESI).† More importantly, the addition of a catalytic amount of DMPO to the reaction solution of benzene with H2O2 and 1 resulted in nearly complete inhibition of the reaction, as shown in Fig. 6. This indicates that HO2˙ produced in the reaction of 1 with H2O2 acts as a chain carrier in the catalytic hydroxylation of benzene, which proceeds via radical chain reactions (vide infra). The chain length of the propagation step in Scheme 1 is evaluated as the ratio of the rate of formation of phenol (1.08 × 10−7 M s−1 determined at 5 h) to twice of DMPO–OOH (9.62 × 10−11 M s−1 determined at 6 h), which is 1120, because two HO2˙ radicals are produced in the initiation step (see Scheme 1).48
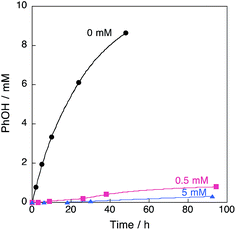 |
| | Fig. 6 Time profiles of formation of phenol in the reaction of benzene with 30 wt% aqueous H2O2 in the presence of 1 and DMPO (0, 0.5, 5 mM) in acetone at 298 K. The concentrations are [C6H6] = 2.1 M, [H2O2] = 2.1 M, [H2O] = 0.4 M and [[CuII(tmpa)]2+] = 67 μM (4.75 mL). | |
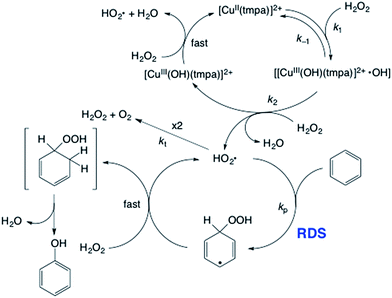 |
| | Scheme 1 Proposed radical chain mechanism of 1-catalysed hydroxylation of benzene with H2O2. | |
A radical chain mechanism of the 1-catalysed hydroxylation of benzene with H2O2
A proposed radical chain mechanism48–50 of the 1-catalysed hydroxylation of benzene with H2O2, which agrees with the kinetic formulation [eqn (2)], is shown in Scheme 1. The catalytic reaction is started by the Fenton-like reaction,43,44,51 in which H2O2 is reduced by [CuII(tmpa)]2+ to produce HO˙ radical and the hydroxide adduct ([CuIII(OH)(tmpa)]2+). In competition with the fast back reaction between HO˙ and [CuIII(OH)(tmpa)]2+, HO˙ reacts with H2O2 to produce HO2˙ and [CuIII(OH)(tmpa)]2+ also reacts with H2O2 to produce HO2˙, accompanied by regeneration of [CuII(tmpa)]2+. HO2˙ reacts with benzene to produce the HO2˙ adduct of benzene as the rate-determining step (RDS). The subsequent fast hydrogen abstraction from H2O2 by the HO2˙ adduct produces phenol and H2O, accompanied by regeneration of HO2˙, thus constituting the radical chain reactions. The termination step is the disproportionation reaction of two HO2˙ to produce H2O2 and O2. Once HO2˙ is trapped by DMPO to produce DMPO–OOH, the radical chain is stopped to inhibit the benzene hydroxylation.
The rate of formation of phenol in Scheme 1 is given by eqn (3) as follows:
| | | d[PhOH]/dt = kp[C6H6][HO2˙] | (3) |
where
kp is the propagation rate constant of the reaction of HO
2˙ with benzene. The rate of formation and decay of HO
2˙ is given by
eqn (4) as follows:
| | | d[HO2˙]/dt = 2k2[([CuIII(OH)(tmpa)]2+ HO˙)][H2O2] − 2kt[HO2˙]2 | (4) |
where
k2 is the rate constant of the reaction of the caged radical pair ([Cu
III(OH)(tmpa)]
2+ HO˙) with H
2O
2 to produce HO
2˙ and
kt is the rate constant of disproportionation of HO
2˙. The rate of decay of HO
2˙ by the reaction with benzene is the same as the rate of formation of HO
2˙ by the reaction of the HO
2˙ adduct of benzene with H
2O
2. The rate of formation and decay of the caged radical pair ([Cu
III(OH)(tmpa)]
2+ HO˙) is given by
eqn (5) as follows:
| | | d[([CuIII(OH)(tmpa)]2+ HO˙)]/dt = k1[1][H2O2] − (k−1 + k2[H2O2])[([CuIII(OH)(tmpa)]2+ HO˙)] | (5) |
where
k1 is the rate constant of the Fenton-like reduction of H
2O
2 by
1 and
k−1 is the rate constant of the back reaction. Under the steady-state conditions (d[HO
2˙]/d
t = 0), [HO
2˙] is given from
eqn (4) by
eqn (6), whereas [([Cu
III(OH)(tmpa)]
2+HO˙)] is given by equation as follows:
| | | [HO2˙] = (k2/kt)1/2([([CuIII(OH)(tmpa)]2+ HO˙)][H2O2])1/2 | (6) |
Under the conditions that k2[H2O2] ≪ k−1, eqn (7) is
| | | [([CuIII(OH)(tmpa)]2+ HO˙)] = k1[1][H2O2]/(k−1 + k2[H2O2]) | (7) |
reduced to
eqn (8). From
eqn (3),
(6) and
(8), the kinetic
| | | [([CuIII(OH)(tmpa)]2+ HO˙)] = k1[1][H2O2]/k−1 | (8) |
formulation is derived as given by
eqn (9), which agrees with
| | | d[PhOH]/dt = kp(k1/k−1)1/2(k2/kt)1/2[1]1/2[C6H6][H2O2] | (9) |
the experimental observations (
eqn (2)). Such agreement, including the unusual dependence of the rate on [
1] strongly supports the validity of
Scheme 1. The feasibility of the chain reactions in
Scheme 1 is also supported by DFT calculations (see Fig. S6, Tables S1 and S2 in ESI
†).
52 The rate-determining propagation step of the reaction of HO
2˙ with benzene to produce the HO
2˙ adduct of benzene is calculated to be uphill by 11.1 kcal mol
−1, whereas the reaction of the HO
2˙ adduct of benzene with HO
2˙ to produce phenol and H
2O is downhill by 72.3 kcal mol
−1. The formation of the intermediate prior to phenol and H
2O is uphill by 5.6 kcal mol
−1. Thus, the intermediate is given in parenthesis in
Scheme 1.
Incorporation of Cu(II) complex catalyst 1 in a mesoporous silica–alumina
Mesoporous silica and silica–alumina have often been used to improve the catalytic performance of metal complexes in a role as catalyst supports whose pores provide broad reaction fields with efficient incorporation.15,16,21,26,53–57 To improve the reaction selectivity of the benzene hydroxylation by H2O2, the catalyst 1 was incorporated into a mesoporous silica–alumina Al-MCM-41 (ref. 53a, 54–56) (the Brunauer–Emmett–Teller (BET) surface area: 1200 m2 g−1, the external surface area: 32 m2 g−1, the pore diameter: 3.5 nm) to prepare [CuII(tmpa)]2+@Al-MCM-41 (see Experimental section).26,53a,58 [CuII(tmpa)]2+@Al-MCM-41 was characterised by a UV-Vis diffuse reflectance spectrum (Fig. S7 in ESI†) and the amount of incorporated 1 was determined to be 2.50 × 10−5 mol g−1 by the absorption spectral change of the mother liquid according to the literature (Fig. S8 in ESI†).53a The much larger BET surface area compared to the external surface suggests that most of the incorporated [Cu(tmpa)]2+ is placed inside the mesopore, which is large enough (diameter 3.5 nm) for incorporation of [Cu(tmpa)]2+ species smaller than 1.2 nm.
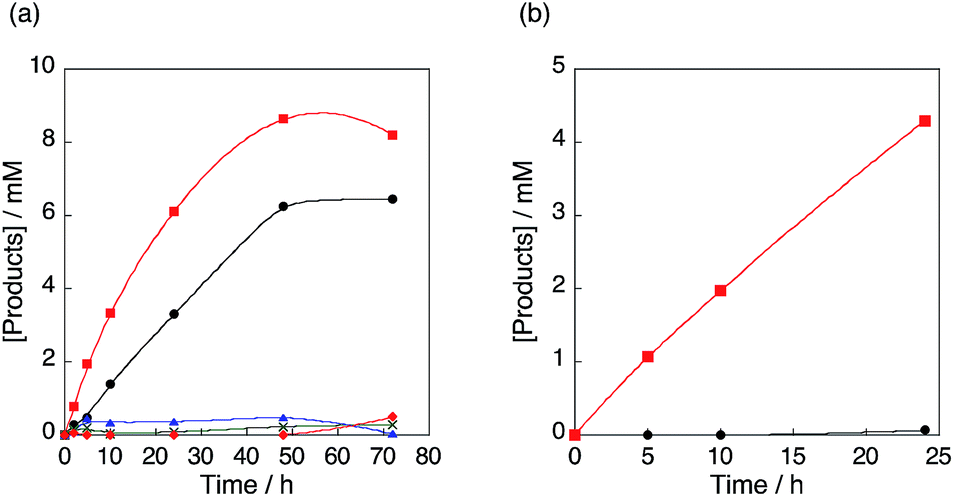
When [CuII(tmpa)]2+@Al-MCM-41 was used as a catalyst for the hydroxylation of benzene with H2O2, benzene was oxidised to phenol as in the case of the homogeneous system (Fig. 7a).59 A slower phenol formation with [CuII(tmpa)]2+@Al-MCM-41 compared to that with [CuII(tmpa)]2+ can be explained by the smaller diffusion rate in the pore. The unchanged blue colour of [CuII(tmpa)]2+@Al-MCM-41 after the reaction suggests that [CuII(tmpa)]2+ cations are encapsulated in Al-MCM-41 without major leaching. The selectivity to produce phenol was significantly improved as p-benzoquinone was hardly produced from phenol, as shown in Fig. 7b. It was confirmed that no phenol was produced using Al-MCM-41 without 1, indicating that the reaction was catalysed by [CuII(tmpa)]2+ incorporated into Al-MCM-41 (Fig. 7a). It was found that benzene does adsorb to Al-MCM-41, whereas phenol was hardly adsorbed (Fig. S9 and S10 in the ESI†). The difference in the adsorption behaviours can be explained by the solvophobic surface present in Al-MCM-41 compared to the environment in solution.60 This selective adsorption of benzene and lack of adsorption of phenol results in the selective hydroxylation of benzene with H2O2 catalysed by the [CuII(tmpa)]2+ sites in Al-MCM-41 to produce only phenol, which is desorbed from Al-MCM-41. The solution was not coloured after the reaction, suggesting that leaching of the Cu2+ species is not the major reason for the saturation of phenol formation. This result shows the potential for recyclability of [CuII(tmpa)]2+@Al-MCM-41 after recovery and washing with acetonitrile.
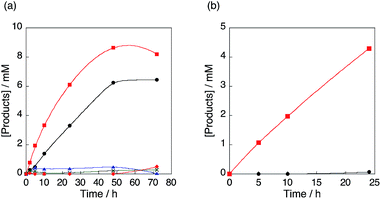 |
| | Fig. 7 (a) Time profiles of formation of phenol in the hydroxylation of benzene with 30 wt% aqueous H2O2 and a catalytic amount of 1 (red square), [CuII(tmpa)]2+@Al-MCM-41 (12.5 mg, black circle), Al-MCM-41 (12.5 mg, blue triangle) or no Cu(II) species nor Al-MCM-41 (green cross), and formation of p-benzoquinone in the hydroxylation of benzene with 1 (red diamond) in acetone at 298 K. (b) Time profiles of formation of p-benzoquinone in hydroxylation of phenol with 30 wt% aqueous H2O2 and a catalytic amount of [CuII(tmpa)]2+@Al-MCM-41 (12.5 mg, black circle) or [CuII(tmpa)]2+ (red square) in acetone at 298 K. The concentrations are [C6H6] or [PhOH] = 2.1 M, [H2O2] = 2.1 M and [[CuII(tmpa)]2+] = 67 μM in acetone (4.75 mL). | |
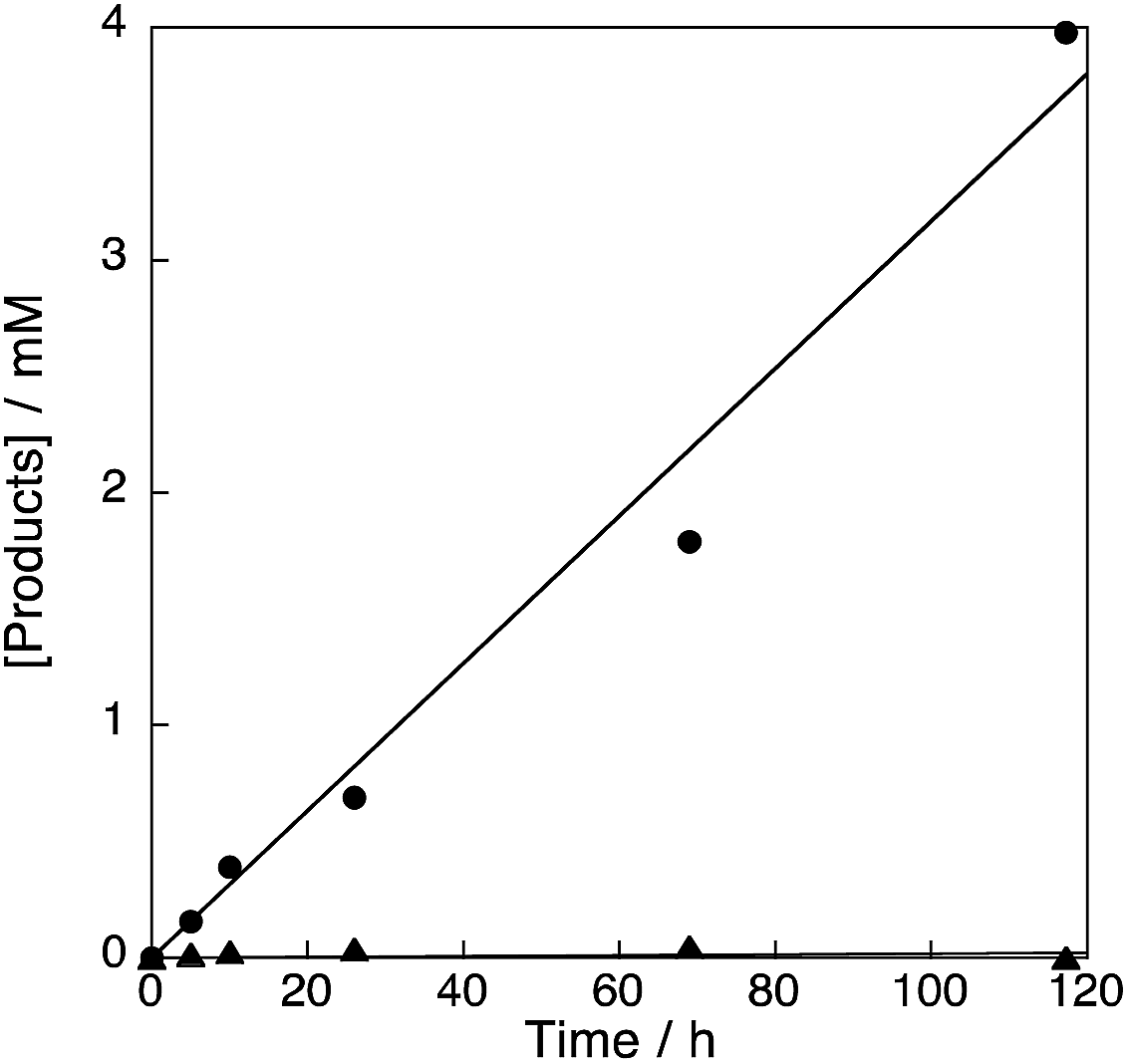
The reaction of 1 mM benzene with 2.1 M H2O2 and 200 μM of [CuII(tmpa)]2+@Al-MCM-41 resulted in formation of phenol in 17% yield with 100% selectivity after 118 h (Fig. S11 in ESI†). The durability of the catalyst [CuII(tmpa)]2+@Al-MCM-41 was also examined by the reaction of 2.1 M benzene with 2.1 M H2O2 and 1 μM of the catalyst. The turnover number was determined to be 4320 after 118 h, demonstrating a high durability for this catalyst (Fig. 8).
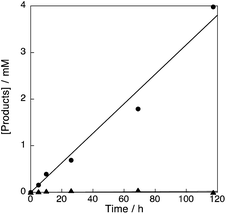 |
| | Fig. 8 Time profiles of formation of phenol (circle) and p-benzoquinone (triangle) in the hydroxylation of benzene (2.1 M) with 30 wt% aqueous H2O2 (2.1 M) catalysed by [CuII(tmpa)]2+@Al-MCM-41 (1.0 μM) in acetone (4.75 mL) at 298 K. | |
Conclusions
In conclusion, one-step hydroxylation of benzene with H2O2 to produce phenol with a high turnover number (TON = 4320) and high selectivity (>99%) has been achieved using a copper(II) complex, ([Cu(tmpa)]2+), incorporated into mesoporous silica–alumina (Al-MCM-41) in acetone at 298 K. The catalytic hydroxylation of benzene with H2O2 proceeds via a radical chain mechanism in which HO2˙ produced via the Fenton-like reduction of H2O2 with [Cu(tmpa)]2+ acts as the chain carrier radical. Spin trapping of the chain carrier radical resulted in the complete inhibition of the catalytic benzene hydroxylation. The present study on the selective catalytic hydroxylation of benzene to phenol using metal complex-incorporated mesoporous silica–alumina as a catalyst provides a general strategy that may be applied to other selective catalytic reactions.
Experimental section
Materials
Chemicals were purchased from commercial sources and used without further purification, unless otherwise noted. Tetraethyl orthosilicate, cetyltrimethylammonium bromide, sodium aluminate, sodium hydroxide, aqueous solutions of hydrogen peroxide (30 wt%) and perchloric acid (60%) were purchased from Wako Pure Chemical Industries. Tris(2-pyridylmethyl)amine and oxo[5,10,15,20-tetra(4-pyridyl)porphyrinato]titanium(IV) (Ti–TPyP) were supplied from Tokyo Chemical Industry. Copper(II) perchlorate hexahydrate was supplied by Sigma-Aldrich. Acetone and acetonitrile were purchased from Nacalai tesque as a spectral grade and used as received. Ultra-pure water was provided by a water purification system, Millipore Direct-Q3 UV, wherein the electronic conductance was 18.2 MΩ cm. Benzene was purchased from Wako Pure Chemical Industries and purified by washing with sulphuric acid and water following distillation. [Cu(tmpa)(CH3CN)](ClO4)2, [Cu(tepa)(ClO4)]ClO4, [CuII2(N5)(H2O)2](NO3)4 and Al-MCM-41 were synthesised by literature methods.53a,61–64
Synthesis of a mesoporous silica–alumina Al-MCM-41
Al-MCM-41 was synthesised by a reported method.64 Cetyltrimethylammonium bromide (18.3 g, 50.4 mmol) was dissolved in an aqueous solution (1.0 L) of NaOH (8.39 g, 210 mmol). Tetraethylorthosilicate (87.5 g, 94.0 mL, 420 mmol) was added dropwise to the aqueous solution during 3 h at 35–40 °C with stirring in water bath and stirred for a further 30 min. Then, sodium aluminate (0.484 g, 5.90 mmol) was added to the reaction vessel and stirred for 4 h at 298 K. The white precipitate obtained was filtered under reduced pressure and washed with distilled water. After drying at 333 K in an oven for 10 h, the solid obtained was calcined at 873 K for 6 h at a rate of 1 K min−1 of temperature increase. The Si/Al ratio of this solid is 60,65 which was calculated from the amount of the precursor materials. The Brunauer–Emmett–Teller (BET) surface area and the external surface area of Al-MCM-41 were determined to be 1200 m2 g−1 and 32 m2 g−1, respectively, by the N2 isotherm and the t-plot. The pore diameter was also determined to be 3.5 nm from the powder XRD pattern. Al-MCM-41 with the ratio of Si/Al = 20 was prepared by the same method with the appropriate amount of sodium aluminate.
Preparation of [CuII(tmpa)]2+@Al-MCM-41
[CuII(tmpa)]2+@Al-MCM-41 was prepared by a cation exchange method in an acetonitrile solution according to the literature method.26 Al-MCM-41 (300 mg) was suspended in an acetonitrile solution (15 mL) of [CuII(tmpa)(CH3CN)](ClO4)2 (16.5 mg, 27.8 μmol, [[CuII(tmpa)]2+]: 1.85 mM) and stirred for 2 h at 298 K. The suspension was centrifuged to collect solid, which was washed with acetonitrile (5.0 mL × 2) and dried in vacuo. Incorporation of [CuII(tmpa)]2+ was confirmed by the diffuse reflectance spectrum of the solid obtained resembling the absorption spectrum of [CuII(tmpa)]2+ in acetonitrile (Fig. S7†),26,53a which could also be observed by clear colour change of the solid from white to blue. The amount of incorporated [CuII(tmpa)]2+ was determined to be 2.50 × 10−5 mol g−1 by the decrease in the UV-Vis absorption band around 860 nm due to [CuII(tmpa)]2+ in the mother liquid according to the literature (Fig. S8†).53a
The catalytic hydroxylation reaction
A typical procedure for catalytic hydroxylation is as follows: 4.75 mL of an acetone solution containing benzene (2.1 M) and 30 wt% aqueous H2O2 (2.1 M) was added to [CuII(tmpa)(CH3CN)](ClO4)2 (0.31 μmol) and vigorously stirred at 298 K. The sample solutions for GC-MS measurements were prepared from the reaction solution by dilution with acetonitrile as needed to analyse products. The Al-MCM-41 and [CuII(tmpa)]2+@Al-MCM-41 were removed by filtration before the measurement. GC-MS measurements were performed with a Shimadzu QP-2010 Ultra instrument.
Electrochemical measurements
Cyclic voltammetry (CV) measurements were performed with an ALS630B electrochemical analyser in deuterated acetonitrile containing 0.1 M Bu4NPF6 (TBAPF6) as a supporting electrolyte at 298 K. The platinum working electrode (BAS, surface i.d. 1.6 mm) was polished with BAS polishing alumina suspension and rinsed with Milli-Q ultra-pure water, acetone and acetonitrile before use. The counter electrode was a platinum wire (0.5 mm dia.). The measured potentials were recorded with respect to an Ag/AgNO3 (0.01 M) reference electrode. The values of redox potentials (vs. Ag/AgNO3) are converted into those vs. SCE by addition of 0.29 V.66
Titanium(IV)–porphyrin method
The concentration of hydrogen peroxide was determined using the Ti–TPyP reagent according to the literature as follows:31 aqueous solutions of 50 μM Ti–TPyP with 50 mM hydrochloric acid and 5 M perchloric acid were prepared. Sample solutions were prepared by 5000 times dilution of 50 μL of the reaction mixture (the concentrations are [C6H6] = 2.1 M, [H2O2] = 0.6 M and [[CuII(tmpa)]2+] = 200 μM in acetone (4.75 mL)) with acetonitrile. The sample solution (50 μL) was added to the mixed solution of Ti–TPyP (250 μL) and perchloric acid (250 μL), shaken and diluted with H2O (1950 μL). The concentration of hydrogen peroxide was calculated from the absorbance of the resulting peroxo complex at 435 nm observed by UV-Vis measurements (Hewlett-Packard 8453 diode array spectrophotometer).
Spin trap by DMPO and EPR measurements
A typical procedure for a spin trap experiment is as follows: 4.75 mL of an acetone solution containing benzene (2.1 M), 30 wt% aqueous H2O2 (0.1 M), 1 (0.31 μmol, 67 μM), and DMPO (0.2 M) was vigorously stirred at 298 K. The sample for EPR was prepared by Ar bubbling (15 min) with a Teflon tube of 200 μL of the reaction solution in a quartz EPR tube (2.0 mm i.d.). EPR spectra of solutions were obtained on a JEOL X-band spectrometer (JES-RE1XE) at 298 K. The g value was calibrated using a Mn2+ marker. The EPR spectra were obtained under non-saturating microwave power conditions. The magnitude of modulation was chosen to optimise the resolution and the signal-to-noise (S/N) ratio of the observed spectra. The double integrated first derivative of a stable radical, 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical (10 μM), was obtained from an EPR spectrum in a mixed solvent benzene/acetone/H2O (1:1:5, v/v/v) similar to the reaction conditions and used as a reference. The spin concentrations of DMPO–OOH signals in the reaction solutions observed by EPR were calculated by comparing with the value obtained from DPPH. The GC-MS (Shimadzu QP-2010 Ultra) measurements were performed as needed to examine the relationship between the spin trap and the hydroxylation.
UV-Vis absorption spectra measurements
UV-Vis absorption spectra were obtained on a Hewlett-Packard 8453 diode array spectrophotometer for UV-Vis at 298 K. UV-Vis diffuse reflectance spectra were obtained on a JASCO V-670 spectrophotometer equipped with an SIN-768 attachment.
Theoretical calculations
Density functional theory (DFT) calculations were performed on a 32CPU workstation (PQS, Quantum Cube QS8-2400C-064). Geometry optimisations were carried out using the B3LYP/6-31++G(d) level of theory for compounds and intermediates relating to the reaction as implemented in the Gaussian 09 program, Revision A.02.52
Acknowledgements
This study was supported by an Advanced Low Carbon Technology Research and Development (ALCA) and SENTAN programs from Japan Science Technology Agency (JST) (to S. F.) of the Ministry of Education, Culture, Sports, Science and Technology, Japan. Support from the USA National Institutes of Health (NIH) for K. D. K. is also acknowledged.
Notes and references
- R. J. Schmidt, Appl. Catal., A, 2005, 280, 89 CrossRef CAS.
- R. Molinari and T. Poerio, Asia-Pac. J. Chem. Eng., 2010, 5, 191 CrossRef CAS.
- H. Xin, A. Koekkoek, Q. Yang, R. Van Santen, C. Li and E. J. M. Hensen, Chem. Commun., 2009, 7590 RSC.
- T. Kusakari, T. Sasaki and Y. Iwasawa, Chem. Commun., 2004, 992 RSC.
- K. Ohkubo, T. Kobayashi and S. Fukuzumi, Angew. Chem., Int. Ed., 2011, 50, 8652 CrossRef CAS PubMed.
- K. Ohkubo, A. Fujimoto and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 5368 CrossRef CAS PubMed.
- W. Ge, Z. Long, X. Cai, Q. Wang, Y. Zhou, Y. Xu and J. Wang, RSC Adv., 2014, 4, 45816 RSC.
- Y. Guo, X. Zhang, H. Zou, H. Liu, J. Wang and K. L. Yeung, Chem. Commun., 2009, 5898 RSC.
- T. D. Bui, A. Kimura, S. Ikeda and M. Matsumura, J. Am. Chem. Soc., 2010, 132, 8453 CrossRef CAS PubMed.
- K. Ohkubo, K. Hirose and S. Fukuzumi, Chem.–Eur. J., 2015, 21, 2855 CrossRef CAS PubMed.
- L. Balducci, D. Bianchi, R. Bortolo, R. D'Aloisio, M. Ricci, R. Tassinari and R. Ungarelli, Angew. Chem., Int. Ed., 2003, 42, 4937 CrossRef CAS PubMed.
- J. Yang, G. Sun, Y. Gao, H. Zhao, P. Tang, J. Tan, A. H. Lu and D. Ma, Energy Environ. Sci., 2013, 6, 793 CAS.
- X. Wang, X. Zhang, H. Liu, J. Qiu, W. Han and K. L. Yeung, Catal. Today, 2012, 193, 151 CrossRef CAS.
- C. A. Antonyraj and K. Srinivasan, Catal. Surv. Asia, 2013, 17, 47 CrossRef CAS.
- P. Borah, X. Ma, K. T. Nguyen and Y. Zhao, Angew. Chem., Int. Ed., 2012, 51, 7756 CrossRef CAS PubMed.
- P. K. Khatri, B. Singh, S. L. Jain, B. Sain and A. K. Sinha, Chem. Commun., 2011, 47, 1610 RSC.
- Y. Leng, J. Liu, P. Jiang and J. Wang, Chem. Eng. J., 2014, 239, 1 CrossRef CAS.
- B. Xu, W. Zhong, Z. Wei, H. Wang, J. Liu, L. Wu, Y. Feng and X. Liu, Dalton Trans., 2014, 43, 15337 RSC.
- M. Goto, Y. Kajita and H. Masuda, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2011, 50, 459 Search PubMed.
- C. Detoni, N. M. F. Carvalho, R. O. M. A. De Souza, D. A. G. Aranda and O. A. C. Antunes, Catal. Lett., 2009, 129, 79 CrossRef CAS.
- Y. Kong, X. Xu, Y. Wu, R. Zhang and J. Wang, Chin. J. Catal., 2008, 29, 385 CrossRef CAS.
- L. Wu, W. Zhong, B. Xu, Z. Wei and X. Liu, Dalton Trans., 2015, 44, 8013 RSC.
- S. Fukuzumi and K. Ohkubo, Asian J. Org. Chem., 2015, 4, 836 CrossRef CAS.
- O. Shoji, T. Kunimatsu, N. Kawakami and Y. Watanabe, Angew. Chem., Int. Ed., 2013, 52, 6606 CrossRef CAS PubMed.
- Y. Morimoto, S. Bunno, N. Fujieda, H. Sugimoto and S. Itoh, J. Am. Chem. Soc., 2015, 137, 5867 CrossRef CAS PubMed.
- Y. Aratani, Y. Yamada and S. Fukuzumi, Chem. Commun., 2015, 51, 4662 RSC.
- A series of Cu(II) complexes have been chosen because of the expectation that the higher electron donating ability of the tri- or tetra-dentate ligands employed will improve catalytic activity, according to previous works.
-
(a) S. Fukuzumi, H. Kotani, H. R. Lucas, K. Doi, T. Suenobu, R. Peterson and K. D. Karlin, J. Am. Chem. Soc., 2010, 132, 6874 CrossRef CAS PubMed;
(b) S. Kakuda, R. L. Peterson, K. Ohkubo, K. D. Karlin and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 6513 CrossRef CAS PubMed;
(c) S. Kakuda, C. Rolle, K. Ohkubo, K. D. Karlin and S. Fukuzumi, J. Am. Chem. Soc., 2015, 137, 3330 CrossRef CAS PubMed.
- D. Das, Y.-M. Lee, K. Ohkubo, W. Nam, K. D. Karlin and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 2825 CrossRef CAS PubMed.
- L. Tahsini, H. Kotani, Y.-M. Lee, J. Cho, W. Nam, K. D. Karlin and S. Fukuzumi, Chem.–Eur. J., 2012, 18, 1084 CrossRef CAS PubMed.
-
(a) C. Matsubara, N. Kawamoto and K. Takamura, Analyst, 1992, 117, 1781 RSC;
(b) K. Takamura and C. Matsubara, Bull. Chem. Soc. Jpn., 2003, 76, 1873 CrossRef CAS.
-
(a) M. A. Grela, M. E. J. Coronel and A. J. Colussi, J. Phys. Chem., 1996, 100, 16940 CrossRef CAS;
(b) G. R. Buettner, Free Radical Biol. Med., 1987, 3, 259 CrossRef CAS.
- H. Karoui, C. Nsanzumuhire, F. Le Moigne, M. Hardy, D. Siri, E. Derat, A. Rockenbauer, O. Ouari and P. Tordo, Chem.–Eur. J., 2014, 20, 4064 CrossRef CAS PubMed.
- O. V. Zalomaeva, N. N. Trukhan, I. D. Ivanchikova, A. A. Panchenko, E. Roduner, E. P. Talsi, A. B. Sorokin, V. A. Rogov and O. A. Kholdeeva, J. Mol. Catal. A: Chem., 2007, 277, 185 CrossRef CAS.
- M. Mrowetz and E. Selli, Phys. Chem. Chem. Phys., 2005, 7, 1100 RSC.
- R. Li, H. Kobayashi, J. Tong, X. Yan, Y. Tang, S. Zou, J. Jin, W. Yi and J. Fan, J. Am. Chem. Soc., 2012, 134, 18286 CrossRef CAS PubMed.
- G. Rosen, A. Beselman, P. Tsai, S. Pou, C. Mailer, K. Ichikawa, B. H. Robinson, R. Nielsen, H. J. Halpern and A. D. MacKerell Jr, J. Org. Chem., 2004, 69, 1321 CrossRef CAS PubMed.
- F. A. Villamena, Y. Liu and J. L. Zweier, J. Phys. Chem. A, 2008, 112, 12607 CrossRef CAS PubMed.
- C. Houriez, N. Ferré, D. Siri, P. Tordo and M. Masella, J. Phys. Chem. B, 2010, 114, 11793 CrossRef CAS PubMed.
- J. R. Harbour, V. Chow and J. R. Bolton, Can. J. Chem., 1974, 52, 3549 CrossRef CAS.
- P. L. Zamora, A. Rockenbauer and F. A. Villamena, Chem. Res. Toxicol., 2014, 27, 765 CrossRef CAS PubMed.
- A. Molinari, M. Montoncello, H. Rezala and A. Maldotti, Photochem. Photobiol. Sci., 2009, 8, 613 CAS.
- J. P. Kehrer, Toxicology, 2000, 149, 43 CrossRef CAS PubMed.
- L. Lyu, L. Zhang, Q. Wang, Y. Nie and C. Hu, Environ. Sci. Technol., 2015, 49, 8639 CrossRef PubMed.
- J. R. Harbour and J. R. Bolton, Biochem. Biophys. Res. Commun., 1975, 64, 803 CrossRef CAS PubMed.
- J. R. Harbour and M. L. Hair, J. Phys. Chem., 1978, 82, 1397 CrossRef CAS.
- The aN and aH values are somewhat dependent on the solvent.
- S. Fukuzumi and J. K. Kochi, J. Org. Chem., 1980, 45, 2654 CrossRef CAS.
- Y. Morimoto, Y.-M. Lee, W. Nam and S. Fukuzumi, Chem. Commun., 2013, 49, 2500 RSC.
- F. Haber and J. Weiss, Proc. R. Soc. London, Ser. A, 1934, 147, 332 CrossRef CAS.
- S. I. Liochev, Met. Ions Biol. Syst., 1999, 36, 1 CAS.
-
(a)
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin. V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009;
(b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS;
(c) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS.
-
(a) S. Fukuzumi, K. Doi, T. Suenobu, K. Ohkubo, Y. Yamada and K. D. Karlin, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15572 CrossRef CAS PubMed;
(b) Y. Yamada, K. Maeda, K. Ohkubo, K. D. Karlin and S. Fukuzumi, Phys. Chem. Chem. Phys., 2012, 14, 9654 RSC.
-
(a) C.-C. Liu, T.-S. Lin, S. I. Chan and C.-Y. Mou, J. Catal., 2015, 322, 139 CrossRef CAS;
(b) C.-H. Lee, S.-T. Wong, T.-S. Lin and C.-Y. Mou, J. Phys. Chem. B, 2005, 109, 775 CrossRef CAS PubMed.
- C. Suspéne, S. Brandés and R. Guilard, Chem.–Eur. J., 2010, 16, 6352 CrossRef PubMed.
- K. Zhang, K.-F. Lam, B. Albela, T. Xue, L. Khrouz, Q.-W. Hou, E.-H. Yuan, M.-Y. He and L. Bonneviot, Chem.–Eur. J., 2011, 17, 14258 CrossRef CAS PubMed.
- L. Fu, X. Li, M. Liu and H. Yang, J. Mater. Chem. A, 2013, 1, 14592 CAS.
- [CuII(tmpa)]2+ can be incorporated into Al-MCM-41 by a cation exchange with Na+ at the cationic exchange site on Al in the silica framework. This method prevents decomposition of the metal complex by calcination.
- The radical trap experiment with [CuII(tmpa)]2+@Al-MCM-41 was consistent with the reaction mechanism which is the same as that with [CuII(tmpa)]2+.
- Using acetonitrile instead of acetone, the reaction with [CuII(tmpa)]2+@Al-MCM-41 showed formation of p-benzoquinone with the loss of reaction selectivity; this observation supports the importance of environmental differences between the inside and outside of Al-MCM-41, for selectivity.
- J. Wang, M. P. Schopfer, S. C. Puiu, A. A. N. Sarjeant and K. D. Karlin, Inorg. Chem., 2010, 49, 1404 CrossRef CAS PubMed.
-
(a) K. D. Karlin, J. C. Hayes, S. Juen, J. P. Hutchinson and J. Zubieta, Inorg. Chem., 1982, 21, 4106 CrossRef CAS;
(b) H. K. Baek, K. D. Karlin and R. A. Holwerda, Inorg. Chem., 1986, 25, 2347 CrossRef CAS.
- S. Thyagarajan, N. N. Murthy, A. A. N. Sarjeant, K. D. Karlin and S. E. Rokita, J. Am. Chem. Soc., 2006, 128, 7003 CrossRef CAS PubMed.
- E. G. Vaschetto, G. A. Monti, E. R. Herrero, S. G. Casuscelli and G. A. Eimer, Appl. Catal., A, 2013, 453, 391 CrossRef CAS.
- The reaction using [CuII(tmpa)]2+@Al-MCM-41 with the same concentration of [CuII(tmpa)]2+ prepared from Al-MCM-41 with the Si/Al = 20 ratio results in the similar phenol production regardless of the Si/Al ratio. This result indicates that the amount of Al has little influence on the catalytic activity under the present reaction conditions.
-
C. K. Mann and K. K. Barnes, in Electrochemical Reactions in Non-aqueous Systems; Mercel Dekker, New York, 1970 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Cyclic voltammogram (Fig. S1), time profiles of phenol, p-benzoquinone, H2O2, spin or benzene (Fig. S2–S5 and S9 and S10), DFT results (Fig. S6 and Tables S1 and S2), UV-Vis absorption spectra (Fig. S7) and UV-Vis DRS (Fig. S8). See DOI: 10.1039/c5sc04312c |
|
| This journal is © The Royal Society of Chemistry 2016 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a