
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid biocompatible macrocyclization of peptides with decafluoro-diphenylsulfone†
S.
Kalhor-Monfared
a,
M. R.
Jafari
a,
J. T.
Patterson
b,
P. I.
Kitov
a,
J. J.
Dwyer
b,
J. M.
Nuss
b and
R.
Derda
*a
aDepartment of Chemistry, University of Alberta, Edmonton, AB T6G 2G2, Canada. E-mail: ratmir.derda@ualberta.ca
bFerring Research Institute, San Diego, California 92121, USA
First published on 19th February 2016
Abstract
In this manuscript, we describe modification of Cys-residues in peptides and proteins in aqueous solvents via aromatic nucleophilic substitution (SNAr) with perfluoroarenes (fAr). Biocompatibility of this reaction makes it attractive for derivatization of proteins and peptide libraries comprised of 20 natural amino acids. Measurement of the reaction rates for fAr derivatives by 19F NMR with a model thiol donor (β-mercaptoethanol) in aqueous buffers identified decafluoro-diphenylsulfone (DFS) as the most reactive SNAr electrophile. Reaction of DFS with thiol nucleophiles is >100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 faster than analogous reaction of perfluorobenzene; this increase in reactivity enables application of DFS at low concentrations in aqueous solutions compatible with biomolecules and protein complexes irreversibly degraded by organic solvents (e.g., bacteriophages). DFS forms macrocycles when reacted with peptides of the general structure Xn–Cys–Xm–Cys–Xl, where X is any amino acid and m = 1–15. It formed cyclic peptides with 6 peptide hormones—oxytocin, urotensin II, salmon calcitonin, melanin-concentrating hormone, somatostatin-14, and atrial natriuretic factor (1–28) as well as peptides displayed on M13 phage. Rates up to 180 M−1 s−1 make this reaction one of the fastest Cys-modifications to-date. Long-term stability of macrocycles derived from DFS and their stability toward oxidation further supports DFS as a promising method for modification of peptide-based ligands, cyclization of genetically-encoded peptide libraries, and discovery of bioactive macrocyclic peptides.
000 faster than analogous reaction of perfluorobenzene; this increase in reactivity enables application of DFS at low concentrations in aqueous solutions compatible with biomolecules and protein complexes irreversibly degraded by organic solvents (e.g., bacteriophages). DFS forms macrocycles when reacted with peptides of the general structure Xn–Cys–Xm–Cys–Xl, where X is any amino acid and m = 1–15. It formed cyclic peptides with 6 peptide hormones—oxytocin, urotensin II, salmon calcitonin, melanin-concentrating hormone, somatostatin-14, and atrial natriuretic factor (1–28) as well as peptides displayed on M13 phage. Rates up to 180 M−1 s−1 make this reaction one of the fastest Cys-modifications to-date. Long-term stability of macrocycles derived from DFS and their stability toward oxidation further supports DFS as a promising method for modification of peptide-based ligands, cyclization of genetically-encoded peptide libraries, and discovery of bioactive macrocyclic peptides.
Introduction
Rapid, site-specific modifications of polypeptides are critical for chemical modification of proteins, synthesis of antibody–drug conjugates, and generation of genetically-encoded libraries with unnatural moieties. Biocompatible site-specific cross-linking of two side-chain residues in polypeptides is a rapidly growing field because such modification can increase stability and efficacy of peptide-based therapeutics (for recent reviews see1–6). While such cross-linking or macrocyclization can be performed on amino acid side-chains that contain unnatural reactive groups,7–9 modification of naturally-occurring residues is particularly attractive because it can be used for modification of readily-available peptides/proteins and phage display libraries comprised of 20 natural amino acids.10–16In this article, we report rapid, biocompatible modification of Cys residues via nucleophilic aromatic substitution (SNAr) reaction with perfluoroarenes (fAr). We overcome the deficiencies of previously known SNAr-reagents, such as 1-chloro-2,4-dinitrobenzene (CDNB),17 which are susceptible to background reactivity with nucleophilic residues in proteins, or perfluorobenzene18 and perfluorobiphenyl18,19 which suffer from low reactivity and poor solubility in water. The latter reagents are effective in organic solvents, such as DMF, but are not suitable for modification of biological entities that are irreversibly deactivated by high concentrations of organic solvents (e.g., proteins, bacteriophage, etc.). While glutathione transferase can be used to catalyze SNAr-reactions in water,20 the enzyme acts only on peptides that contain unnatural N-terminal-α-Glu.20,21 Hence, improved fAr cross-linking agents provide the ability to access diverse chemical modalities through native polypeptide sequences.
Results and discussion
To identify SNAr derivatives for aqueous bioconjugations, we explored the rate of the reaction of a model thiol donor, β-mercaptoethanol (βME), and twelve fAr reagents using 19F NMR (see ESI Fig. S1–S3,† examples of 19F NMR traces) in aqueous Tris-buffer pH = 8.5 supplemented with CH3CN as co-solvent (Fig. 1A–C). Reactivity of most perfluoroarenes was hampered by both poor solubility and poor reactivity (Fig. 1D); many reactions required 80% or more CH3CN as co-solvent. Similar to studies in organic solvents,22 the reactivity in aqueous conditions correlates with the electronegativity of the substituent on the aromatic ring (Fig. 1C) yielding the fastest reactivity for perfluoronitrobenzene (R = NO2), decafluorobenzophenone (R = –COC6F5), or decafluoro-diphenylsulfone (R = –SO2C6F5, DFS) reagents, or heteroaromatic derivatives such as perfluoropyridine.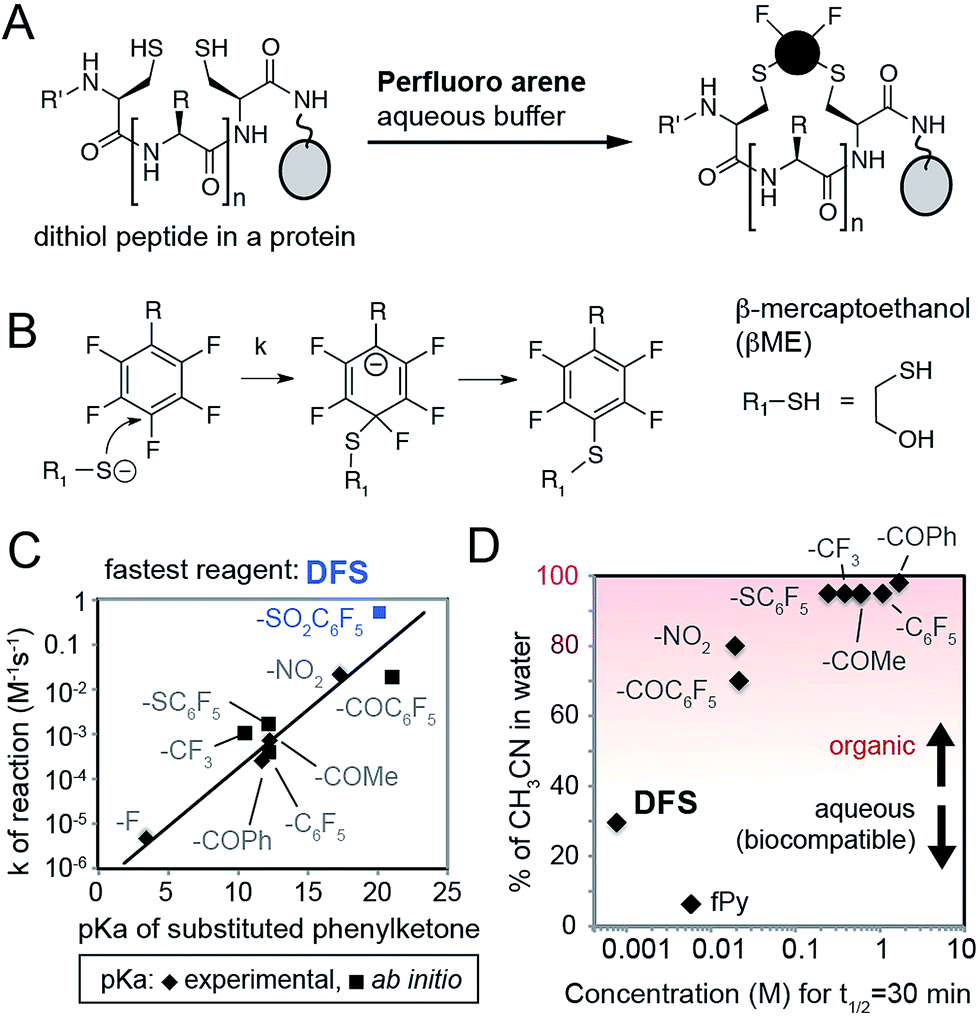 | ||
| Fig. 1 (A) Design of reagents for perfluoroarene macrocyclization by cross-linking two Cys-residues in a protein in aqueous conditions. (B) Mechanism of SNAr reaction. (C) Rate constants (k) of the reaction between substituted fAr and βME, as measured by 19F NMR, increase with the increasing electronegativity of R group. As a measure of electronegativity, we used pKa in a series of phenylketones for which many values are known23 or can be calculated (ESI Fig. S4†). (D) To find the SNAr reagents for rapid modification in water, we plotted molar concentration of fAr necessary to reach 50% conversion in 30 minutes (calculated as ln(2)/(1800 × k), where k is the rate constant) and % of organic co-solvent necessary to dissolve the fAr at this concentration. Most fAr are poorly reactive and/or too insoluble in aqueous solvents; only DFS and perfluoropyridine (fPy) can be used in conditions that require low amount of organic co-solvent. | ||
We then validated the reactivity and specificity of the most reactive substrate (DFS) using model peptides H2N–SWCXC–CONH2, where X = Asp, Ala, Ser, or Arg that contained two types of nucleophiles: N-terminal amine and thiol (Fig. 2A). We were pleased to find that DFS reacted remarkably fast with Cys side-chains (Fig. 2B and ESI Fig. S5 and S6†) in conditions that required as little as 10% of CH3CN co-solvent (Fig. 1) or as little as 5% DMF co-solvent (ESI Fig. S7†). As SNAr reaction alters the aromatic π-system,17,24 we measured rate constants by measuring changes in absorption at 300 nm (ESI Fig. S5 and S6, S8–S11†) and validated these rates by 19F NMR (ESI Fig. S10 and S11†) and LCMS (ESI Fig. S7†). Conveniently, the reaction was accelerated in solvents with high water content, although the rate decreased in water without any CH3CN due to limited solubility of DFS (ESI Fig. S12†). At pH 8.5 and water content of 70–80%, the rate constant was 100–180 M−1 s−1 for positively charged peptide sequence SWCRC and 50–80 M−1 s−1 for neutral analog SWCAC (Fig. 2B).
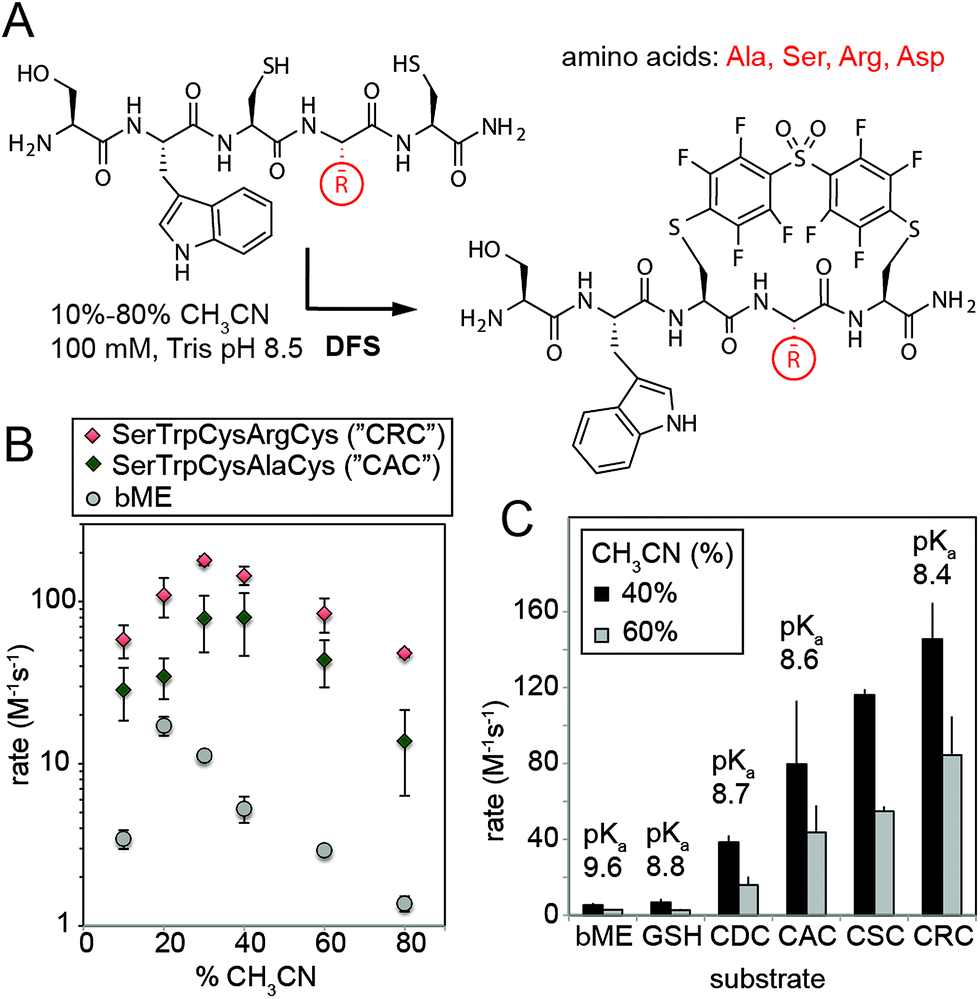 | ||
| Fig. 2 (A) SNAr reaction of DFS with peptides is sequence and solvent-dependent. (B) Rates in all conditions were measured by monitoring absorbance at 320 nm, and identities of the products were confirmed by NMR and Liquid Chromatography Mass-Spectrometry (LCMS, ESI Fig. S7 and S12–S15†). (C) The rate of the reaction correlates with acidity of the thiol; pKa were estimated by measuring the absorbance of peptides at 240 nm as described in ESI Fig. S18† and were in accordance with previously reported values for CXC peptides.25 | ||
The rate of the reaction with DFS was sequence dependent (Fig. 2B and C and ESI Fig. S5†). Positively-charged Arg residue next to Cys accelerated the reaction, negatively charged Asp suppressed it. This observation can be explained by higher basicity of thiol in CRC vs. CDC (Fig. 2C). Additionally, the positive charge on the Arg side-chain can stabilize the intermediate carbanion (Fig. 1B). We anticipate that analogous strong dependence of the rate constant on the pKa of thiol (Fig. 2C) exist in other SNAr reactions between peptides and perfluoroarenes18,19 because SNAr reactions with thiols exhibit large Brønsted coefficients.20 Substrate-dependence was the most pronounced when solvation was poor (e.g., high CH3CN content); increased solvation in solutions with high aqueous content masked the effect (Fig. 2B and ESI Fig. S6†). The pH-dependence of the rate plateaued at pH 8–9 confirming that thiolate is the nucleophile (ESI Fig. S19 and S20†). In acidic pH, the rate dropped exponentially (i.e., decrease of log(k) was linear with pH). Reaction with perfluoropyridine exhibited similar solvent-dependent and substrate-dependent profiles (ESI Fig. S8, S9, and S21†), however, the rates were 10–100 times slower.
LCMS confirmed the identity of the products during the reaction and 24–48 hours after its completion (ESI Fig. S12–S15†). LCMS confirmed that the reaction was selective for Cys in the presence of free N-terminus and large excess of DFS. Side-reactivity was observed only after prolonged exposure to DFS. Specifically, peptides SWCAC or SWCRC incubated with large excess of DFS (10 eq., 2 mM) for 48 hours contained ∼20% of the product with two DFS moieties. Less than 5% of this by-product was visible after 48 hours in the presence of 0.2 mM DFS. In contrast, Cys arylation under analogous conditions was complete in 5–10 minutes (ESI Fig. S6†). We observed this by-product in three different peptide sequences (ESI Fig. S13–S15†) and proposed it to be a product of SNAr reaction between DFS and N-terminus. While the side-reaction of N-terminal amine is possible, it is >104 times slower than Cys-arylation. This data still do not completely rule out the possibility that the cyclization could also happen between the thiol and the terminal amine. To show that cyclization occurs exclusively on Cys residues, we treated peptide with DFS and then with thiol-specific reagent, biotin-PEG2-iodoacetamide (BIA). While BIA reacted readily with thiols in starting material, we detected no reaction between BIA and octafluoro-diphenylsulfone (OFS)-peptide conjugate (Fig. S17†).
To confirm that DFS is useful for modification of complex peptides, we modified peptide CYIQNCPLG (oxytocin, OT). Reaction yielded a single product with expected mass and apparent rate constant of 37.2 M−1 s−1 as measured by LCMS (Fig. 3, ESI Fig. S16†). To stop the reaction before completion, we quenched it with acid and observed mono-arylated intermediates of oxytocin M1 and M2 in a transient open-chain state (Fig. 3A and B). Both intermediates disappeared over the course of the reaction (Fig. 3C). To confirm that DFS can form cyclic peptides regardless of their ring size, we tested reaction between DFS and five other peptide hormones of general structure XN–Cys–XM–Cys–XL, where X is any amino acid other than Cys and M—the number of amino acids flanked by two Cys—ranged from 4 to 15. Urotensin II (UT, M = 4), salmon calcitonin (CT, M = 5), melanin-concentrating hormone (MCH, M = 8), somatostatin-14 (ST, M = 10), and atrial natriuretic factor (1–28) (ANF, M = 15) formed cyclic peptides in reaction with DFS regardless of their ring size. We used HPLC to estimate the rate of cyclization (Fig. 3C, S22–S23†) by quenching the reaction with acid in the presence of 1 mM DFS. After 30 seconds, we detected 10–90% of the final cyclized product and <5–60% of starting material remaining. After 10 min, only urotensin II, which displayed the slowest kinetics, contained ∼5% of the unreacted starting material. Other peptides were completely converted to final cyclic products.
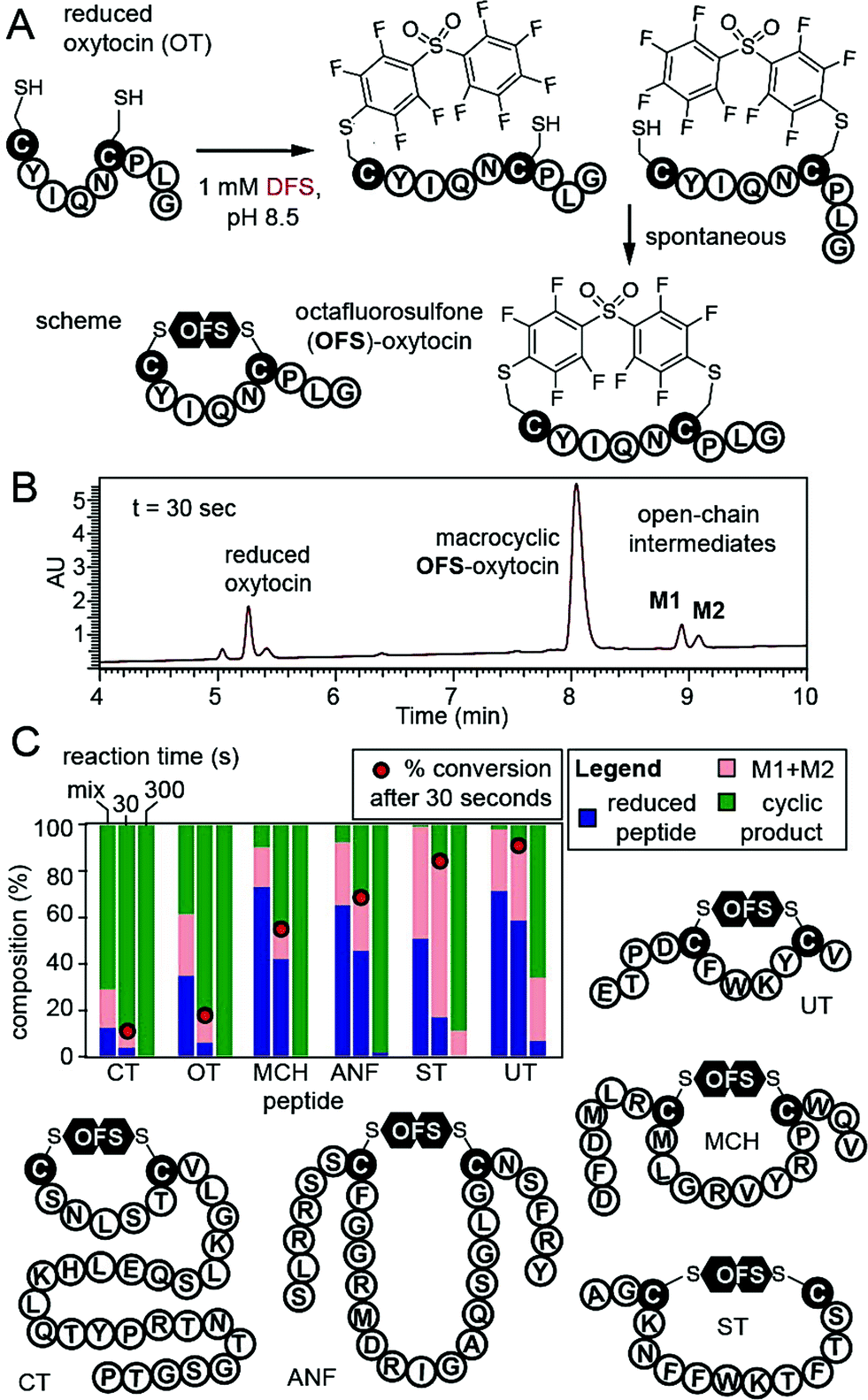 | ||
| Fig. 3 (A and B) Cyclization of reduced oxytocin (OT) by DFS. Quenching the reaction by 2% TFA at 30 s revealed ∼90% consumption of starting material, 80% of product, and 12% of open-chain intermediates M1 and M2. (C) Stacked bar representation of the composition of reaction mixture determined by HPLC shows that both M1 and M2 of OT were completely converted to product by 5 min. Analogous studies with five other peptide hormones in the presence of 1 mM DFS. Fraction of the reactant, sum of M1 + M2 intermediates, and products was determined by integration of the HPLC traces at 215 nm (see ESI Fig. S16, S22, and S23† for raw traces). | ||
We then confirmed reaction of DFS with thiols on intact M13 bacteriophage, which is composed of 5 types of proteins and a single-stranded DNA genome. The phage virion is an ideal model for bioconjugation because (i) it can be engineered to display any peptide sequence, (ii) modification can be detected in individual phage particles,14 (iii) biocompatibility of the modification can be readily quantified by measuring the fraction of M13 particles that can replicate in bacteria; reagents that damage the proteins or DNA disrupt this replication14 (ESI Fig. S24†). We validated the modification on M13 phage using a biotin capture assay developed previously by our group (Fig. 4).14,26 Briefly, a phage displaying a disulfide-peptide ACPARSPLEC was reduced with TCEP and the resulting thiols were alkylated using BIA (Fig. 4B and C). The reaction yielded ∼95% of the biotinylated phage, as determined by titering the phage before and after exposure to streptavidin-coated beads (Fig. 4D). We then used a “pulse-chase” approach13–16,26 to quantify reaction of phage with DFS. “Pulsing” the reduced phage with 0.5 mM DFS in 30% CH3CN–Tris buffer (pH 8.5) for 30 min consumed ∼75% phage-displayed thiols. “Chase” of the reaction with BIA yielded only 20% of biotinylated phage. In contrast, the phage remained reduced and susceptible to alkylation by BIA when it was “pulsed” with buffer that contained no DFS. Thus, disappearance of thiol indicates the reaction with DFS and is not due to side reactions (e.g., oxidation). The same “pulse-chase” quantification confirmed that peptide ACPARSPLEC displayed on phage reacts with perfluoropyridine at much slower rate than DFS (Fig. S25†). We further validated the “pulse-chase” method on purified peptide SWCDYRC and confirmed that the peptide “pulsed” by DFS for 30 minutes forms a product that does not react with BIA (Fig. S17†).
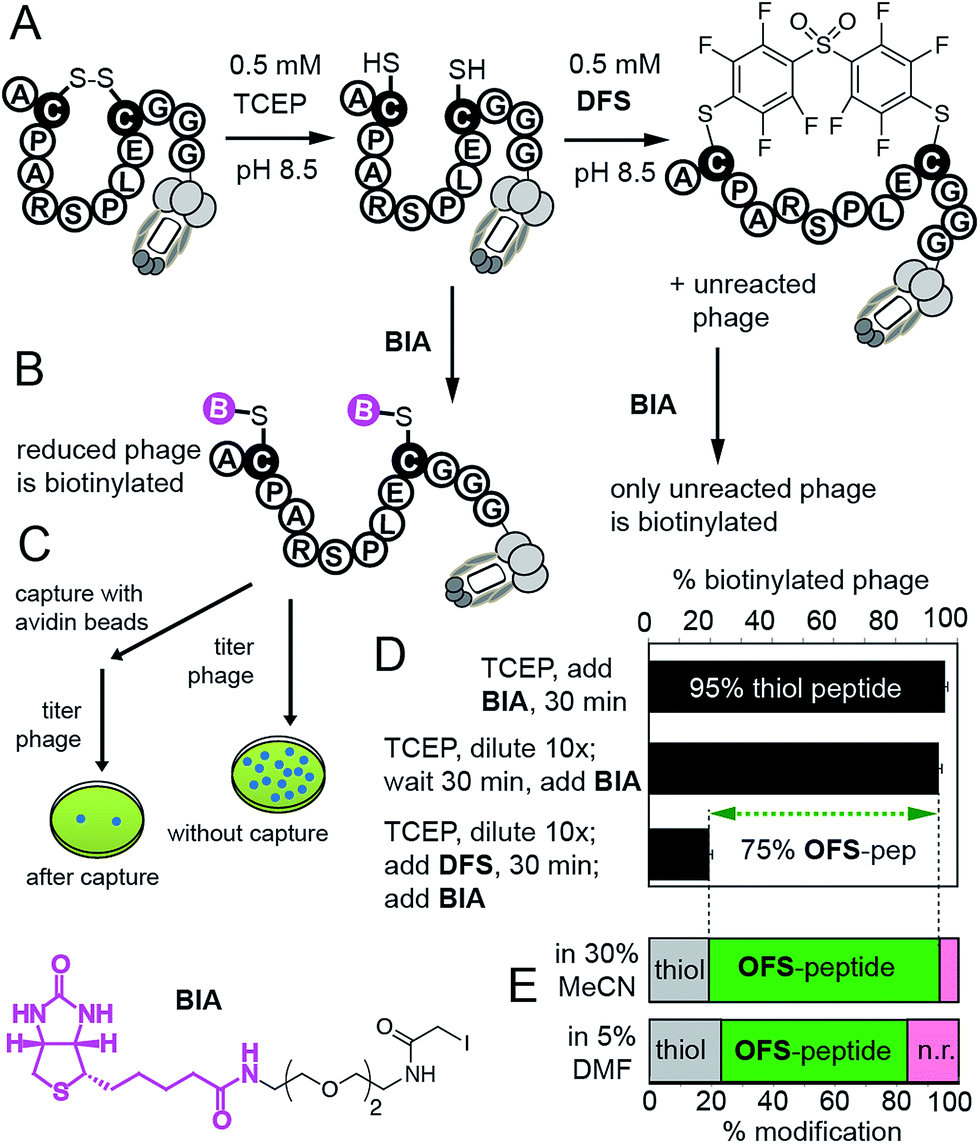 | ||
| Fig. 4 (A) Quantification strategy for the reaction between DFS and M13 phage displaying a peptide ACPARSPLEC.13 (B) Reaction of reduced phage with biotin-iodoacetamide (BIA) biotinylates the phage. (C) Capture assay measures the yield of biotinylation. (D) Capture assay determined that 95% of phage is reduced (reactive with BIA). Diluting the reducing agent 10-fold does not change the number of reduced thiols, but exposing the reduced phage to 0.5 mM DFS in 30% MeCN/Tris buffer for 30 min consumes reactive thiols yielding 80% OFS-modified phage. Error bars represent standard deviations from two experiments. (E) Capture can be summarized as stack bar: bottom bar shows that ∼60% of phage is modified by 0.5 mM solution of DFS in aqueous Tris buffer containing 5% DMF as co-solvent. | ||
While M13 phage is stable in solutions that contain 30% CH3CN, in many applications such as protein modifications, such a high amount of acetonitrile might not be tolerated. To this end, DFS-mediated cyclization can be performed in aqueous solutions containing only 5% DMF as co-solvent. We observed that solubility of DFS in Tris buffer (pH 8.5) containing 5% DMF (v:v) is approximately 500 μM. This concentration is sufficient to perform rapid modifications of synthetic peptides, phage-displayed peptides, and phage-displayed libraries of peptides. For example, reaction between 100 μM DFS and 100 μM peptide SWCDYRC required approximately 100 seconds to reach 50% conversion and 15 min to reach completion (ESI Fig. S7†). LCMS monitoring of this reaction estimated the 2nd order rate constant to be 100–130 M−1 s−1 (ESI Fig. S7†). We then used these conditions to modify a phage clone that contained peptide ACPARSPLEC. The modification efficiency by 0.5 mM DFS in 5% DMF for 30 minutes was ∼60%, (Fig. 4E). Finally, these conditions can also be used to modify commercially available libraries of peptides displayed on phage with 7 random amino acids in between the cysteine residues (New England Biolabs (Ph.D.™-C7C, Fig. 5)). A decrease in chemical modification efficiency of libraries to 40%, when compared to modification of an individual clone (Fig. 4E), is not surprising because library contains 109 peptide sequences with diverse reactivity. This decrease in modification is also consistent with our previous reports.13,15DFS thus can conveniently convert readily available libraries of peptide-disulfides to genetically-encoded macrocyclic libraries that contain unique perfluoroarene cross-linker. We have employed these libraries in selection of peptide-macrocycle ligands for protein targets but results of these investigations extend beyond the scope of this manuscript and will be described in our subsequent report.
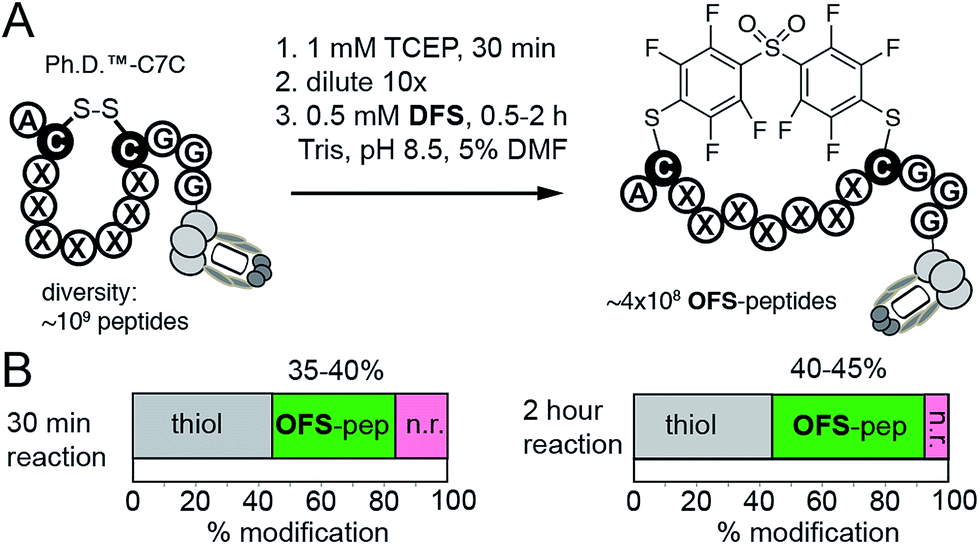 | ||
| Fig. 5 (A) An aqueous solution of DFS containing 5% DMF as co-solvent can conveniently convert commercially available phage displayed library of 109 disulfide heptapeptides—Ph.D. C7C from New England Biolabs—to a library of OFS-macrocyclic peptides. (B) Biotin capture, analogous to that described in Fig. 4C–E, quantified the efficiency of this modification. After 30 minutes to 2 hours reaction with DFS, 35–45% of the library is modified with DFS yielding ∼350–450 million of diverse phage-displayed OFS-macrocycles. Data is averaged from two independent experiments. | ||
Cross-linking of two Cys residues is one of the most common modifications of synthetic peptides and recombinant proteins. To demonstrate advantages of DFS over well-established cysteine cross-linkers, we selected α,α′-dibromo-meta-xylene (DBMX) reagent as a reference (ESI Fig. S26†). Reagents structurally and electronically similar to DBMX are commonly used to stabilize secondary structure of peptides and synthesize genetically-encoded macrocycle libraries.27–29 The major advantage of OFS-peptide conjugates (perfluoroaryl thioethers) is their higher stability to oxidation when compared to conjugates made with DBMX (benzyl thioethers). For example, we routinely use NaIO4 to convert N-terminal Ser to N-terminal aldehyde.14,30,31 Treatment with NaIO4 readily converted SWCDYRC-OFS conjugate to ![[A with combining low line]](https://www.rsc.org/images/entities/i_char_0041_0332.gif)
![[l with combining low line]](https://www.rsc.org/images/entities/i_char_006c_0332.gif)
![[d with combining low line]](https://www.rsc.org/images/entities/i_char_0064_0332.gif) -WCDYRC-OFS, where “-” denotes aldehyde group (Fig. 6), and we observed no oxidation of aryl thioethers. As an example of application, we introduced N-terminal glycosylation14,30,31 in SWCDYRC-OFS conjugate (Fig. 6). One-pot two-step reaction quantitatively converted peptide-OFS conjugate to -peptide-OFS and then to glycopeptide-OFS conjugate. Such transformation would be challenging to achieve in conjugates that contain thioether bonds derived from benzyl or haloacetyl cross-linkers (ESI Fig. S27†). Treatment of SWCDYRC-DMMX conjugate by NaIO4 led to concomitant oxidation of serine and benzyl-thioether (ESI Fig. S27D†), whereas treatment of SWCDYRC-OFS in identical conditions produced no detectable traces of oxidized product (ESI Fig. S27C†).
-WCDYRC-OFS, where “-” denotes aldehyde group (Fig. 6), and we observed no oxidation of aryl thioethers. As an example of application, we introduced N-terminal glycosylation14,30,31 in SWCDYRC-OFS conjugate (Fig. 6). One-pot two-step reaction quantitatively converted peptide-OFS conjugate to -peptide-OFS and then to glycopeptide-OFS conjugate. Such transformation would be challenging to achieve in conjugates that contain thioether bonds derived from benzyl or haloacetyl cross-linkers (ESI Fig. S27†). Treatment of SWCDYRC-DMMX conjugate by NaIO4 led to concomitant oxidation of serine and benzyl-thioether (ESI Fig. S27D†), whereas treatment of SWCDYRC-OFS in identical conditions produced no detectable traces of oxidized product (ESI Fig. S27C†).
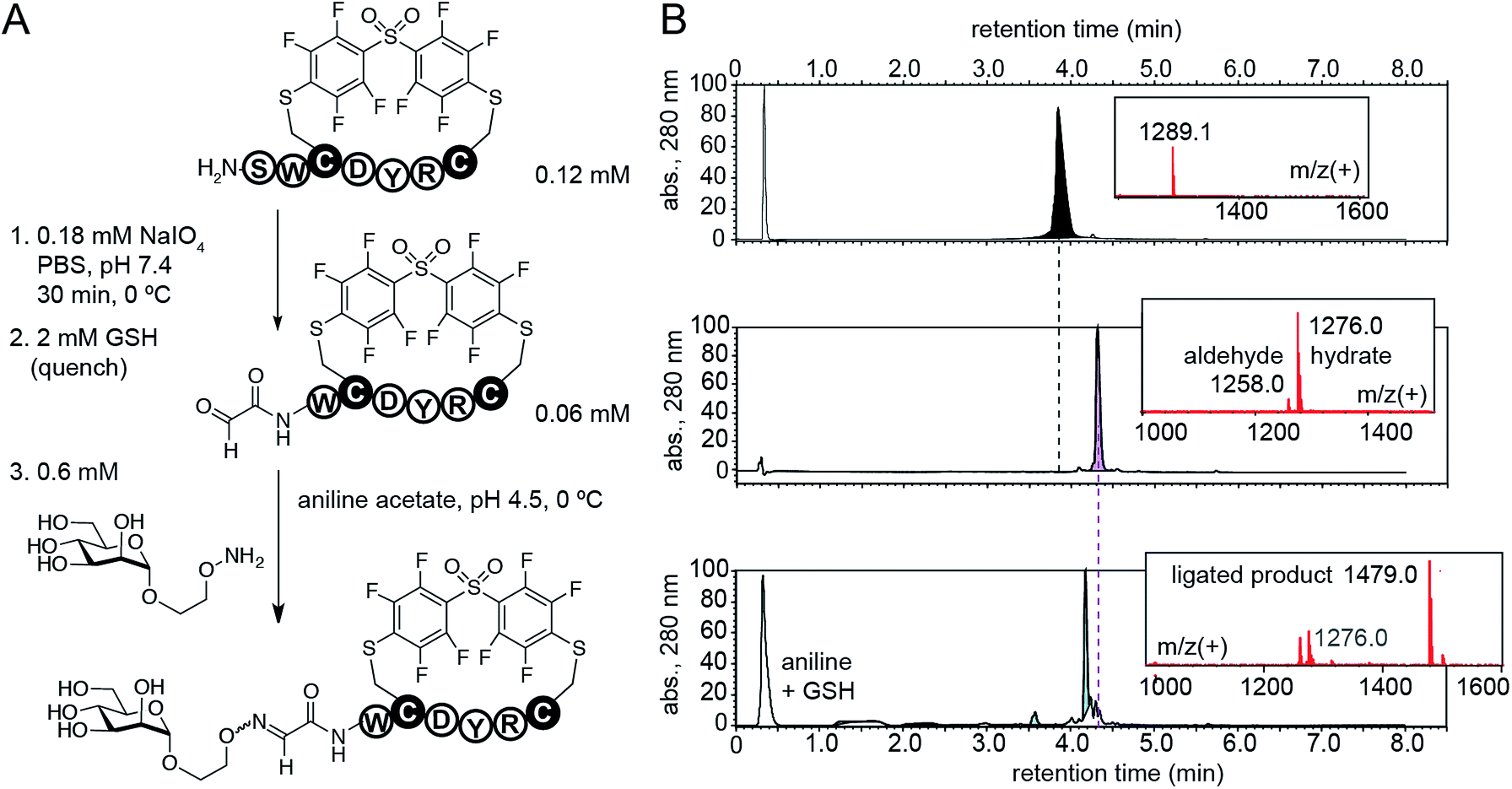 | ||
| Fig. 6 Robust oxidation of SWCDYRC-OFS conjugate to peptide-aldehyde and subsequent one-pot conversion to a cyclic glycopeptide. | ||
To compare the stability of OFS-thioethers towards oxidation with stability of other conjugates, we exposed them to conditions recently used to evaluate stability of diverse thioethers (1 mM H5IO6 at 37 °C).32 Consistent with the published results,32 we observed that H5IO6 rapidly oxidized benzyl-thioether bonds and converted SWCDYRC-DMMX conjugate to four oxidized products in 10 minutes (ESI Fig. S27F†). In contrast, half-life of oxidation for thioethers in SWCDYRC-OFS by H5IO6 was 16 hours (ESI Fig. S27E†). This value exceeds the reported oxidation half-lives for tolyl- or 4-cyanophenyl-thioether (1 and 6 h, see ESI Fig. S4† in ref. 32). As the rate of oxidation of aryl thioether scales with electronegativity of the aryl substituent,32 it is not surprising that the peptide-OFS macrocycles exhibit high resistance to oxidation. We believe that fast reactivity of DFS combined with exceptional oxidative stability of resulting OFS-thioethers will make it possible to conduct robust multi-step modification of peptides, which combines Cys cross-linking and N-terminal ligations to functionalize natural proteins and diversify genetically-encoded libraries.
Conclusions
The DFS framework combines fast reactivity up to 180 M−1 s−1, specificity to Cys-residues, biocompatibility with complex multiprotein complexes (here, bacteriophage), and maximum performance at neutral to mildly-basic pH. Its reactivity significantly exceeds the rates of substitution reactions of Cys with CDNB (k = 1 M−1 s−1),17 haloacetamide cross-linkers (k = 10–20 M−1 s−1 at pH 8.5),13,33 bis-bromobenzyl cross-linkers (k = 30 M−1 s−1 at pH 8.8),34 bis-methyl-maleimide cross-linkers (k = 1–3 M−1 s−1),35 and allenamides36 (k = 16–30 M−1 s−1).16 Recent summaries from the Bode37 and Prescher38 groups highlight that in modern bio-orthogonal reactions, very few reach rates of >100 M−1 s−1 as exhibited by DFS. To date only two Cys-reactive reagents, dichlorotetrazene39 and maleimide (rates up to 800 M−1 s−1),37 can rival DFS in reactivity, but the products are susceptible to long-term hydrolytic instability.39–42 In contrast, DFS adducts of oxytocin are stable for over 2 weeks at pH = 7.4 (ESI Fig. S28†). We note that the size of the DFS is compatible to the size of many previously reported thiol crosslinkers such as bisbromoxylene,27–29 decafluorobenzene,18,19 benzophenone,32 bismaleimide, chloroacetamide-azobenzene,13 and others. Introduction of DFS into proteins or peptides will be context dependent: it can provide favourable stabilization of secondary structure but it can also interfere with biological function. In contrast to many known Cys cross-linkers, DFS produces conjugates that are stable to oxidation; this stability will enable development of multi-step transformations that convert peptides made of natural amino acids to functionality-rich macrocyclic derivatives. We anticipate that this reaction will serve as a platform for development of new bioconjugation and macrocyclization strategies as well as post-translational diversification of genetically-encoded libraries.Acknowledgements
This research was supported by a research contract from the Ferring Research Institute. We thank Cindy Wu for the help in measurement of viability of phage, Dr Randy Whittal for assistance with LCMS, and Mark Miskolzie for assistance with 19F NMR kinetics. We thank Prof. Todd Lowary for critical review of this manuscript as well as Prof. Hans Reich and Dr Simon Ng for helpful discussions. M. R. J. acknowledges Alberta Innovated Technology Futures for graduate fellowship, and P. K. acknowledges Alberta Glycomics Centre for salary support and access to computational cluster.References
- A. Bhat, L. R. Roberts and J. J. Dwyer, Eur. J. Med. Chem., 2015, 94, 471–479 CrossRef CAS PubMed.
- C. P. R. Hackenberger and D. Schwarzer, Angew. Chem., Int. Ed., 2008, 47, 10030–10074 CrossRef CAS PubMed.
- N. Tsomaia, Eur. J. Med. Chem., 2015, 94, 459–470 CrossRef CAS PubMed.
- C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509–524 CrossRef CAS PubMed.
- V. Azzarito, K. Long, N. S. Murphy and A. J. Wilson, Nat. Chem., 2013, 5, 161–173 CrossRef CAS PubMed.
- M. Pelay-Gimeno, A. Glas, O. Koch and T. N. Grossmann, Angew. Chem., Int. Ed., 2015, 54, 8896–8927 CrossRef CAS PubMed.
- H. E. Blackwell and R. H. Grubbs, Angew. Chem., Int. Ed., 1998, 37, 3281–3284 CrossRef CAS.
- C. E. Schafmeister, J. Po and G. L. Verdine, J. Am. Chem. Soc., 2000, 122, 5891–5892 CrossRef CAS.
- L. D. Walensky, A. L. Kung, I. Escher, T. J. Malia, S. Barbuto, R. D. Wright, G. Wagner, G. L. Verdine and S. J. Korsmeyer, Science, 2004, 305, 1466–1470 CrossRef CAS PubMed.
- A. Angelini and C. Heinis, Curr. Opin. Chem. Biol., 2011, 15, 355–361 CrossRef CAS PubMed.
- E. Baslé, N. Joubert and M. Pucheault, Chem. Biol., 2010, 17, 213–227 CrossRef PubMed.
- J. M. Smith, J. R. Frost and R. Fasan, J. Org. Chem., 2013, 78, 3525–3531 CrossRef CAS PubMed.
- M. R. Jafari, L. Deng, P. I. Kitov, S. Ng, W. L. Matochko, K. F. Tjhung, A. Zeberoff, A. Elias, J. S. Klassen and R. Derda, ACS Chem. Biol., 2014, 9, 443–450 CrossRef CAS PubMed.
- S. Ng, M. R. Jafari, W. L. Matochko and R. Derda, ACS Chem. Biol., 2012, 7, 1482–1487 CrossRef CAS PubMed.
- S. Ng and R. Derda, Org. Biomol. Chem., 2016 10.1039/c5ob02646f.
- M. R. Jafari, J. Lakusta, R. J. Lundgren and R. Derda, Bioconjugate Chem., 2016 DOI:10.1021/acs.bioconjchem.6b00026.
- A. M. Gold, Biochemistry, 1968, 7, 2106–2115 CrossRef CAS PubMed.
- A. M. Spokoyny, Y. Zou, J. J. Ling, H. Yu, Y.-S. Lin and B. L. Pentelute, J. Am. Chem. Soc., 2013, 135, 5946–5949 CrossRef CAS PubMed.
- Y. Zou, A. M. Spokoyny, C. Zhang, M. D. Simon, H. Yu, Y.-S. Lin and B. L. Pentelute, Org. Biomol. Chem., 2014, 12, 566–573 CAS.
- B. S. Nieslanik and W. M. Atkins, J. Am. Chem. Soc., 1998, 120, 6651–6660 CrossRef CAS.
- C. Zhang, A. M. Spokoyny, Y. Zou, M. D. Simon and B. L. Pentelute, Angew. Chem., Int. Ed., 2013, 52, 14001–14005 CrossRef CAS PubMed.
- R. D. Chambers, J. A. Cunningh and D. J. Spring, J. Chem. Soc. C, 1968, 1560–1565 RSC.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS.
- L. Ekladius and H. K. King, Biochem. J., 1957, 65, 128–131 CrossRef CAS PubMed.
- L. D. Lavis, T. J. Rutkoski and R. T. Raines, Anal. Chem., 2007, 79, 6775–6782 CrossRef CAS PubMed.
- P. I. Kitov, D. F. Vinals, S. Ng, K. F. Tjhung and R. Derda, J. Am. Chem. Soc., 2014, 136, 8149–8152 CrossRef CAS PubMed.
- N. K. Bashiruddin and H. Suga, Curr. Opin. Chem. Biol., 2015, 24, 131–138 CrossRef CAS PubMed.
- Y. V. Guillen Schlippe, M. C. T. Hartman, K. Josephson and J. W. Szostak, J. Am. Chem. Soc., 2012, 134, 10469–10477 CrossRef CAS PubMed.
- P. Timmerman, J. Beld, W. C. Puijk and R. H. Meloen, ChemBioChem, 2005, 6, 821–824 CrossRef CAS PubMed.
- S. Ng, E. Lin, P. I. Kitov, K. F. Tjhung, O. O. Gerlits, L. Deng, B. Kasper, A. Sood, B. M. Paschal, P. Zhang, C.-C. Ling, J. S. Klassen, C. J. Noren, L. K. Mahal, R. J. Woods, L. Coates and R. Derda, J. Am. Chem. Soc., 2015, 137, 5248–5251 CrossRef CAS PubMed.
- K. F. Tjhung, P. I. Kitov, S. Ng, E. N. Kitova, L. Deng, J. S. Klassen and R. Derda, J. Am. Chem. Soc., 2016, 138, 32–35 CrossRef CAS PubMed.
- E. V. Vinogradova, C. Zhang, A. M. Spokoyny, B. L. Pentelute and S. L. Buchwald, Nature, 2015, 526, 687–691 CrossRef CAS PubMed.
- R. L. Lundblad, in Chemical Reagents For Protein Modification, Taylor and Francis Group, LLC, New York, 2014, ch. 7, pp. 217–337 Search PubMed.
- L. E. J. Smeenk, N. Dailly, H. Hiemstra, J. H. van Maarseveen and P. Timmerman, Org. Lett., 2012, 14, 1194–1197 CrossRef CAS PubMed.
- Y. Chen, C. M. Clouthier, K. Tsao, M. Strmiskova, H. Lachance and J. W. Keillor, Angew. Chem., Int. Ed., 2014, 53, 13785–13788 CrossRef CAS PubMed.
- A. Abbas, B. Xing and T.-P. Loh, Angew. Chem., Int. Ed., 2014, 53, 7491–7494 CrossRef CAS PubMed.
- F. Saito, H. Noda and J. W. Bode, ACS Chem. Biol., 2015, 10, 1026–1033 CrossRef CAS PubMed.
- D. M. Patterson, L. A. Nazarova and J. A. Prescher, ACS Chem. Biol., 2014, 9, 592–605 CrossRef CAS PubMed.
- S. P. Brown and A. B. Smith III, J. Am. Chem. Soc., 2015, 137, 4034–4037 CrossRef CAS PubMed.
- S. C. Alley, D. R. Benjamin, S. C. Jeffrey, N. M. Okeley, D. L. Meyer, R. J. Sanderson and P. D. Senter, Bioconjugate Chem., 2008, 19, 759–765 CrossRef CAS PubMed.
- A. D. Baldwin and K. L. Kiick, Bioconjugate Chem., 2011, 22, 1946–1953 CrossRef CAS PubMed.
- N. Toda, S. Asano and C. F. Barbas, Angew. Chem., Int. Ed., 2013, 52, 12592–12596 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc03856a |
| This journal is © The Royal Society of Chemistry 2016 |