
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign of potent and highly selective inhibitors for human β-secretase 2 (memapsin 1), a target for type 2 diabetes†
Arun K.
Ghosh
*a,
Bhavanam Sekhara
Reddy
a,
Yu-Chen
Yen
b,
Emilio L.
Cárdenas
a,
Kalapala Venkateswara
Rao
a,
Deborah
Downs
d,
Xiangping
Huang
d,
Jordan
Tang
d and
Andrew D.
Mesecar
abc
aDepartment of Chemistry and Department of Medicinal Chemistry, Purdue University, West Lafayette, IN 47907, USA. E-mail: akghosh@purdue.edu; Fax: +1 765 4961612; Tel: +1 765 4945323
bDepartment of Biological Sciences, Purdue University, West Lafayette, IN 47907, USA
cDepartment of Biochemistry, Purdue University, West Lafayette, IN 47907, USA
dProtein Studies Program, Oklahoma Medical Research Foundation, University of Oklahoma, Oklahoma City, OK 73104, USA
First published on 4th February 2016
Abstract
Design, synthesis and evaluation of very potent and selective β-secretase 2 (memapsin 1, BACE 2) inhibitors are described. The inhibitors were designed specifically to interact with the S2′-site of β-secretase 2 to provide >170![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold selectivity over β-secretase (BACE 1) and >15000-fold selectivity over cathepsin D. BACE 2 is implicated in type 2 diabetes. The studies serve as an important guide to selective BACE 2 inhibitors.
000-fold selectivity over β-secretase (BACE 1) and >15000-fold selectivity over cathepsin D. BACE 2 is implicated in type 2 diabetes. The studies serve as an important guide to selective BACE 2 inhibitors.
Diabetes mellitus is a serious and chronic metabolic disorder that affects nearly 400 million patients worldwide.1,2 Type 2 diabetes is the most common type and as of 2013, an estimated 382 million patients around the world have this disorder. According to the International Diabetes Federation, that number is set to reach 471 million by 2035.3,4 Diabetes is responsible for about 4.5 million deaths each year and it increases the risk of heart disease, stroke, and microvascular complications such as blindness, renal failure, and peripheral neuropathy.5,6 Type 2 diabetes accounts for about 90% of diabetes cases. It is characterized by hyperglycemia resulting from insufficient insulin production because of reduction of the pancreatic beta-cell function and mass and also from an increase in insulin demand associated with insulin resistance.7,8 Current treatments for diabetes have severe limitations. Most current drugs for type 2 diabetes are either insulin sensitizers (e.g., metformin and thiazolidinediones) or insulin secretagogues (e.g., sulfonylureas and glinides).9,10 Therefore, the development of new and effective treatment based upon novel mechanisms of action is of great importance.
Tmem27 (collectrin), a 46 kDa type 1 transmembrane protein plays an important role in the functions of insulin-producing pancreatic beta cells.11,12 It has been shown that over expression of Tmem27 in pancreatic beta cells results in cellular proliferation, increased cell mass and increased insulin production.13 The excess Tmem27 in beta cells is regulated by protease cleavage of Tmem27 which results in the release of ectodomain outside the cells.14,15 The protease, which is responsible for cleavage of Tmem27 ectodomain, has been recently identified as BACE2 or memapsin 1 (Fig. 1).16,17 Furthermore, inhibition of BACE2 in insulin-resistant mice has been shown to increase beta-cell mass and improve insulin levels.16 These results implicate that BACE2 is involved in the maintenance of beta cells. Thus, inhibition of BACE2 may lead to a novel treatment of type 2 diabetes.
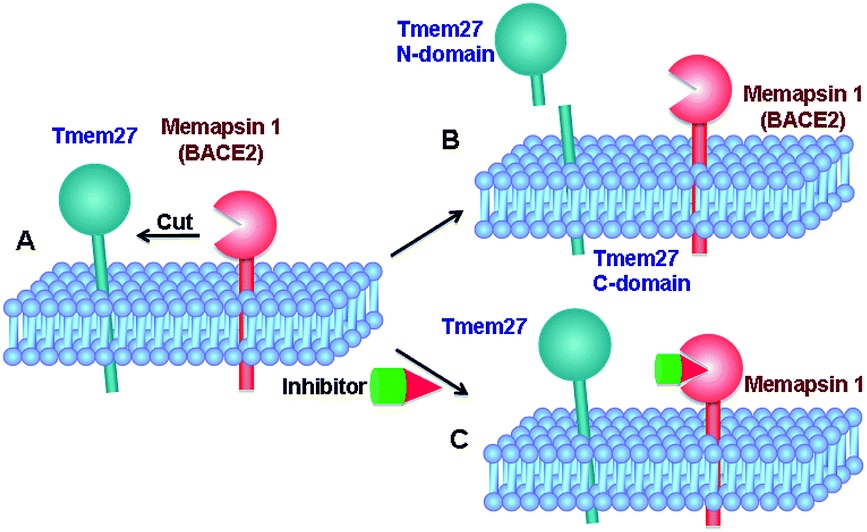 | ||
| Fig. 1 Therapeutic concept of memapsin 1 (BACE2) inhibition for treating type 2 diabetes. (A) The abundance of Tmem27 regulates the function and mass of pancreatic beta-cells. Tmem27 is reduced by memapsin 1 cutting. (B) BACE2 cutting of Tmem27 reduced the number of beta cells and resulted in less insulin production. (C) A BACE2 inhibitor suppresses Tmem27 cutting, increases beta cell function and increases insulin production. | ||
BACE2 is a type 1 transmembrane protease which contains an N-terminal ectodomain, a transmembrane domain, and C-terminal cytosolic domain. The ectodomain of BACE2 is a pepsin-like aspartic protease.15 It has a broad substrate specificity similar to its closest homologue BACE1 (memapsin 2).18,19 BACE1 does not directly impact the homeostasis of Tmem27 in the beta-cells of pancreatic islets. However, for inhibitor design, the development of selectivity over BACE1 is important for the following reasons. BACE2 is expressed in the alpha-cells of pancreatic islets and is present in large amounts.16 While its specific function is unknown, the inhibition of BACE1 may indirectly affect glucose metabolism. Also, BACE1 has a physiological function in down-regulating brain neuronal activity through its involvement in β-amyloid peptide production.20 It has been shown that BACE1 gene deletion results in behavior abnormalities in mice.21 In addition, BACE1 is known to process a number of other important protein substrates.22,23 Since type 2 diabetes is a chronic disease, it is important to develop selective inhibitors to reduce off-target toxicity over time. The development of selectivity over cathepsin D is also important as it is abundant in cells and plays a critical role in protein catabolism.24,25 Cathepsin D also plays a significant role in retinal function of the eyes.26 Furthermore, cathepsin D is abundant in human cells and the lack of selectivity would lead to consumption of inhibitor drugs. Thus far, there is no report of selective BACE2 inhibitors. However, a patent application claiming selective BACE2 inhibitors with 2-aminodihydro[1,3]-thiazines has been reported.27
Our design strategies of selective BACE2 inhibitors are based upon hydroxyethylamine isosteres as this scaffold is inherent to several FDA approved HIV-1 protease inhibitors.28,29 Inhibitor 1 (Fig. 2) is a very potent BACE1 inhibitor (Ki = 1.8 nM) designed by us.30 It showed good drug-like properties and low toxicity.31 However, it has shown reduced potency against BACE2 (Ki = 137 nM, 76-fold less potent over BACE1). A high resolution X-ray structure of inhibitor 1-bound BACE1 was determined by us.30 Based upon this X-ray structure and also from the reported inhibitor-bound X-ray structures of BACE2, we speculated that modification of P1′ and P2′ substituent of inhibitor 1 with polar functionalities may lead to improvement in BACE2 activity while reducing BACE1 activity of inhibitor 1.32,33 Herein we report the preliminary results of our investigation which led to the design of highly potent BACE2 inhibitors with exceptional selectivity against BACE1 and cathepsin D.
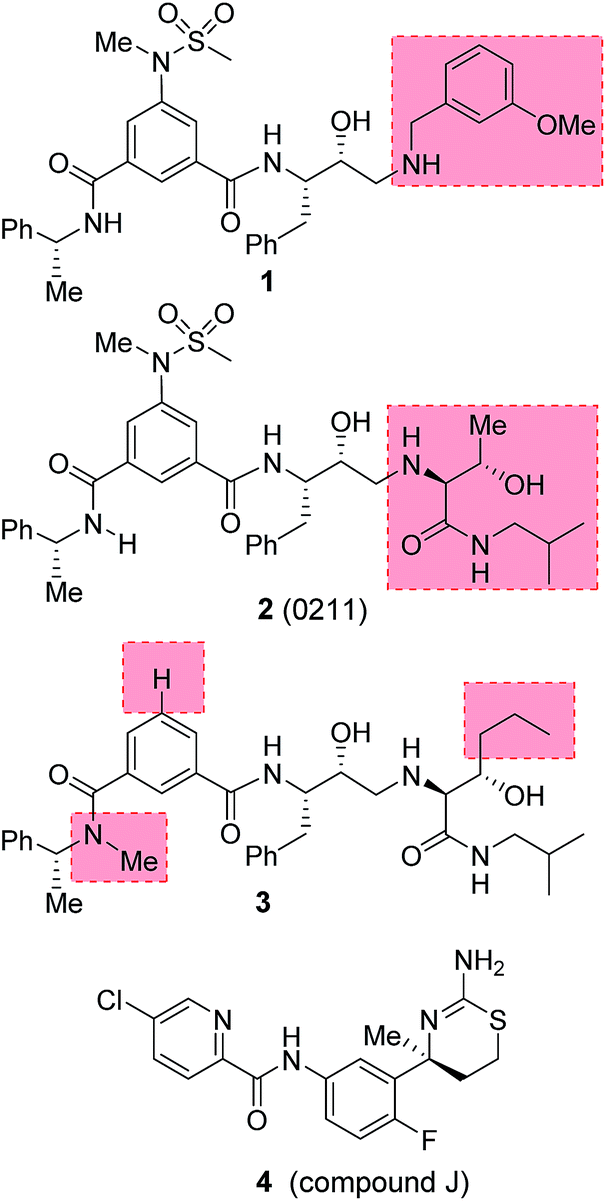 | ||
| Fig. 2 Structures of BACE2 inhibitors 1–4. | ||
Our syntheses of BACE2 inhibitors are shown in Scheme 1. Isophthalic acid derivative 5 was prepared as described by us previously.30 Isophthalic acid derivative 6 was synthesized by coupling of known34 (R)-N-methyl-1-phenylethylamine and 5-(N-methylmethylsulfonamido)-isophthalic acid methyl ester30 followed by saponification of methyl ester with aqueous LiOH at 23 °C to provide carboxylic acid 6. Methyl isophthalic acid derivative 7 was synthesized by saponification of methyl ester with aqueous LiOH at 23 °C.35 The corresponding N-methyl derivative 8 was prepared by N-methylation with NaH and MeI in THF followed by saponification of methyl ester with aqueous LiOH at 23 °C.35 Isophthalic acid derivative 9 was prepared from commercially available 3-(R)-1-(phenylethylcarbamoyl)benzoic acid methyl ester by N-methylation with NaH and MeI in THF followed by saponification of methyl ester with aqueous LiOH at 23 °C to provide the corresponding carboxylic acid. Boc protected allothreonine derivative 10 was synthesized by coupling Boc-allothreonine with isobutylamine using EDC and HOBt as described previously.36 For the synthesis of inhibitor 2, Boc derivative 10 was treated with trifluoroacetic acid (TFA) in CH2Cl2 to effect the deprotection of the Boc group. Reaction of the resulting amine with optically active, commercially available oxirane 11 in the presence of silica gel soaked with CH2Cl2 at 23 °C for 48 h provided Boc-aminoalcohol derivative 12 in excellent yield.37 Exposure of 12 to TFA at 23 °C removed the Boc group. The resulting amine was coupled to substituted aminosulfonamide derivative 5 using EDC and HOBt in the presence of iPr2NEt in CH2Cl2 at 23 °C for 15 h to provide inhibitor 2 in 63% yield. Coupling of the resulting amine with carboxylic acid 6 under similar conditions afforded N-methyl derivative 13 in 35% yield. Coupling of 12-derived amine with carboxylic acids 7 and 8 provided amide derivatives 14 and 15 in 41% and 40% yields, respectively.
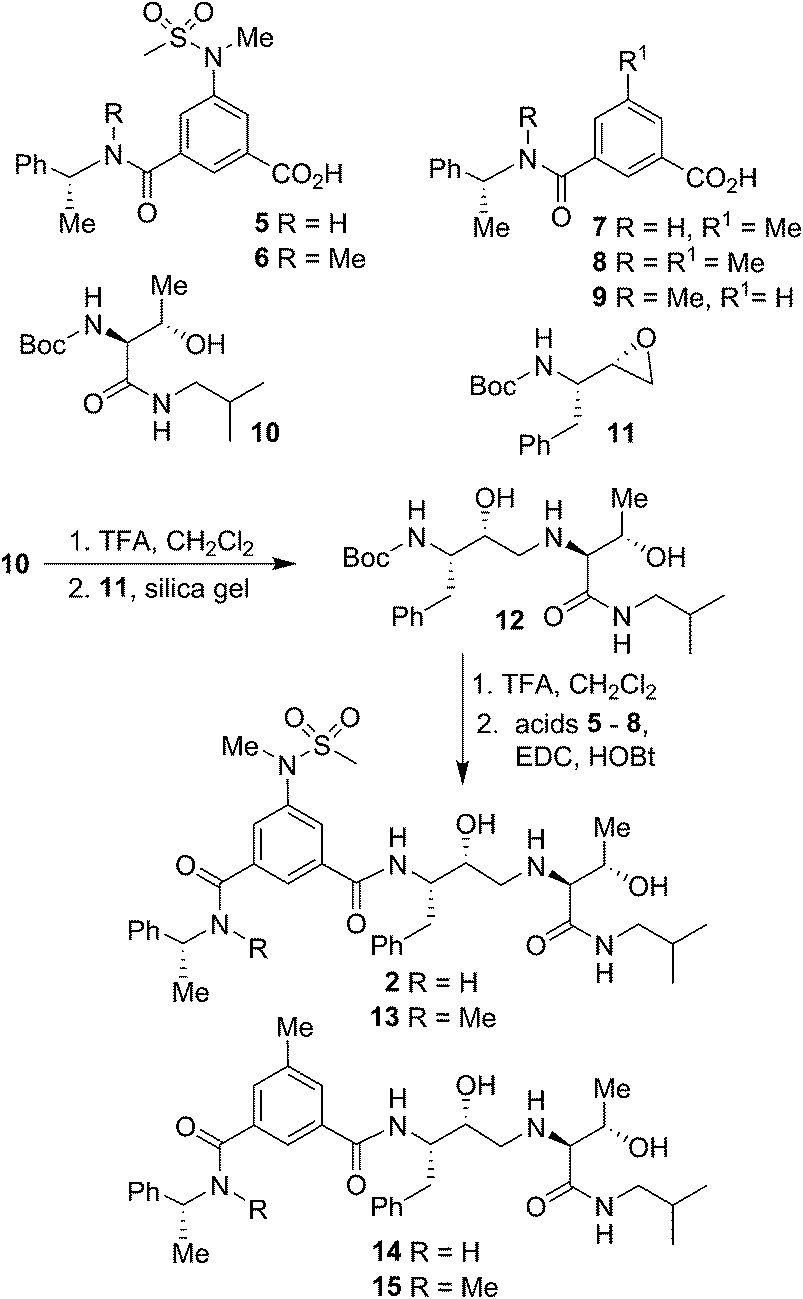 | ||
| Scheme 1 Synthesis of BACE2 inhibitors 2 and 13–15. | ||
The synthesis of BACE2 inhibitors with other structural variations is shown in Scheme 2. For the synthesis of inhibitor 18, the known30 aminoalcohol derivative 16 was coupled with carboxylic acid 8 using EDC and HOBt in the presence of iPr2NEt in CH2Cl2 to provide compound 18 in 65% yield. Boc protected (2S,3S)-3-hydroxyhexanamide derivative 17 was prepared in optically active form using Sharpless asymmetric dihydroxylation as the key reaction.36,38 For the synthesis of inhibitor 20 and 3, hexanamide derivative 17 was treated with TFA in CH2Cl2 at 23 °C for 2 h and the resulting amine was reacted with epoxide 11 to provide Boc-aminoalcohol derivative 19. Boc-derivative 19 was then treated with TFA in CH2Cl2 at 0 °C to 23 °C for 2 h and the resulting amine was reacted with carboxylic acids 8 and 9 providing inhibitors 20 and 3 in 49% and 44% yield, respectively.
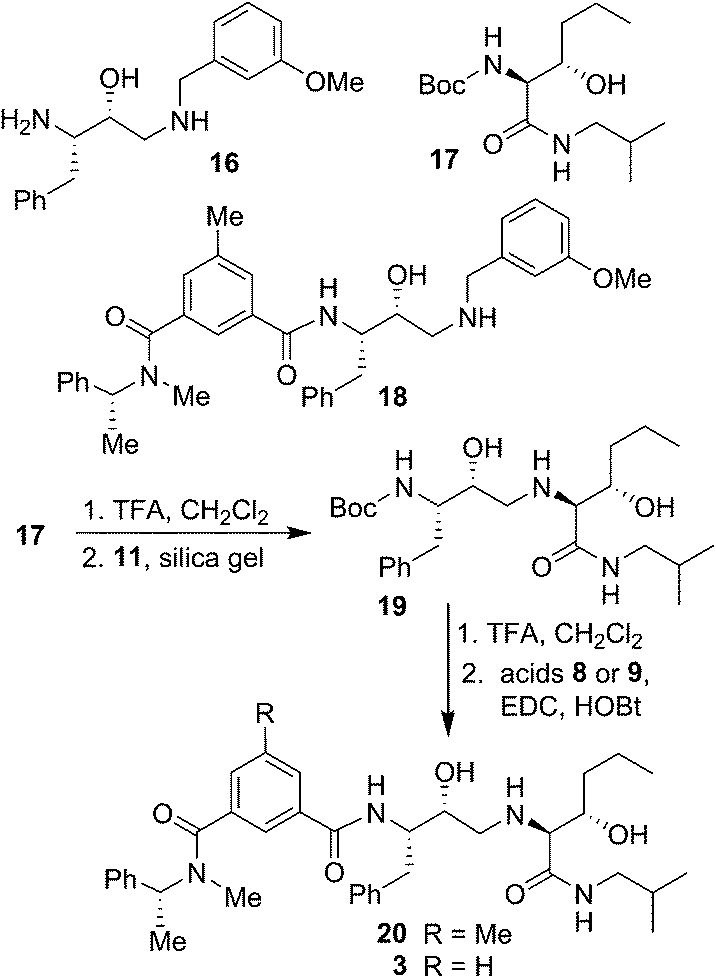 | ||
| Scheme 2 Synthesis of BACE2 inhibitors 3, 18 and 20. | ||
In our preliminary efforts, based upon the X-ray structures of inhibitor-bound BACE2, we have created an active model of inhibitor 1 in the BACE2 active site.30 Our examination of this model indicated that the BACE2 active site contains residues such as Gly50 and Thr88 that can be targeted specifically to improve inhibitor potency and selectivity. Furthermore, it appears that the S2′-subsite of BACE2 can accommodate polar functionalities. In inhibitor 1, the P1′-methoxy benzyl group is mostly involved in non-polar interactions with residues in the hydrophobic S1′–S2′-binding site of BACE1. Therefore, replacement of methoxybenzyl group with polar functionalities could lead to more selective BACE2 inhibitors. We therefore, replaced methoxybenzyl group with an allothreonine isobutylamide derivative in inhibitor 2. This has resulted in significant (∼10-fold) improvement in potency against BACE2 and a significant reduction (∼27-fold) in potency against BACE1 (Ki = 14.6 nM for BACE2; Ki = 48 nM for BACE1).
Our preliminary efforts to obtain a suitable crystal of 2 in complex with BACE2 have been unsuccessful. However, to obtain insight into the inhibitor-BACE2 interactions, we determined the X-ray structure of BACE1 bound to inhibitor 2 to 2.5 Å (Rfree = 23.5% and Rwork = 18.5%.). Using this structural information, we next created a computational model of inhibitor 2 bound to the active site of BACE2. The key interactions between inhibitor 2 and BACE1 and BACE2 are shown in Fig. 3. The interactions between BACE1 and inhibitor 2 determined from the X-ray structure are shown in Fig. 3A and reveal that the P2-sulfonamide functionality fits very well in the S2 subsite and makes extensive interaction with BACE1.39 One of the sulfone oxygens forms a hydrogen bond with Asn233 backbone amide nitrogen and the other oxygen is within proximity to form hydrogen bonds with Ser325 side chains as well as Asn233 side chain carboxamide functionality. There are significant van der Waal interactions of the aromatic ring with Thr231, Thr232, Asn233 and Arg235 in the region. The P3-phenylmethyl group is nicely nestled in the hydrophobic pocket surrounding Thr14, Gln12 and Ser229. The P3-amide nitrogen appears to form a strong hydrogen bond with Gly230 carbonyl and the P3-amide carbonyl is within hydrogen bonding distance to the Thr232 side chain hydroxyl group. Inhibitor 2 also makes extensive interactions in the S2′-subsite, forming a number of hydrogen bonds with Thr72 side chain and backbone atoms of Gly34 and Gln73. Based upon this X-ray structure of compound 2-bound memapsin 2 and the X-ray structure of a known memapsin 1-bound inhibitor, we created a computational model of inhibitor 2 in the memapsin 1 active site.32 The key interactions are shown in Fig. 3B. As can be seen, the P2-sulfonamide functionality forms significantly less polar interactions compared to memapsin 2 active site. In addition, the P3-amide NH does not appear to form any polar interactions in the active site. Both P2 and P3 aromatic rings make significant van der Waals interactions in the memapsin 1 active site. The P2′ and P3′ ligands of inhibitor 2 appear to form extensive interactions in the S2′ and S3′ subsites. Particularly, the hydroxyl group is within proximity to form hydrogen bonds with Lys237 and Tyr211. The carboxamide functionality appears to make polar interactions with the phenolic OH of Tyr211 as well as Thr88 as shown.39 Presumably, these interactions are responsible for enhanced BACE2 activity of inhibitor 2 over BACE1 (Table 1).
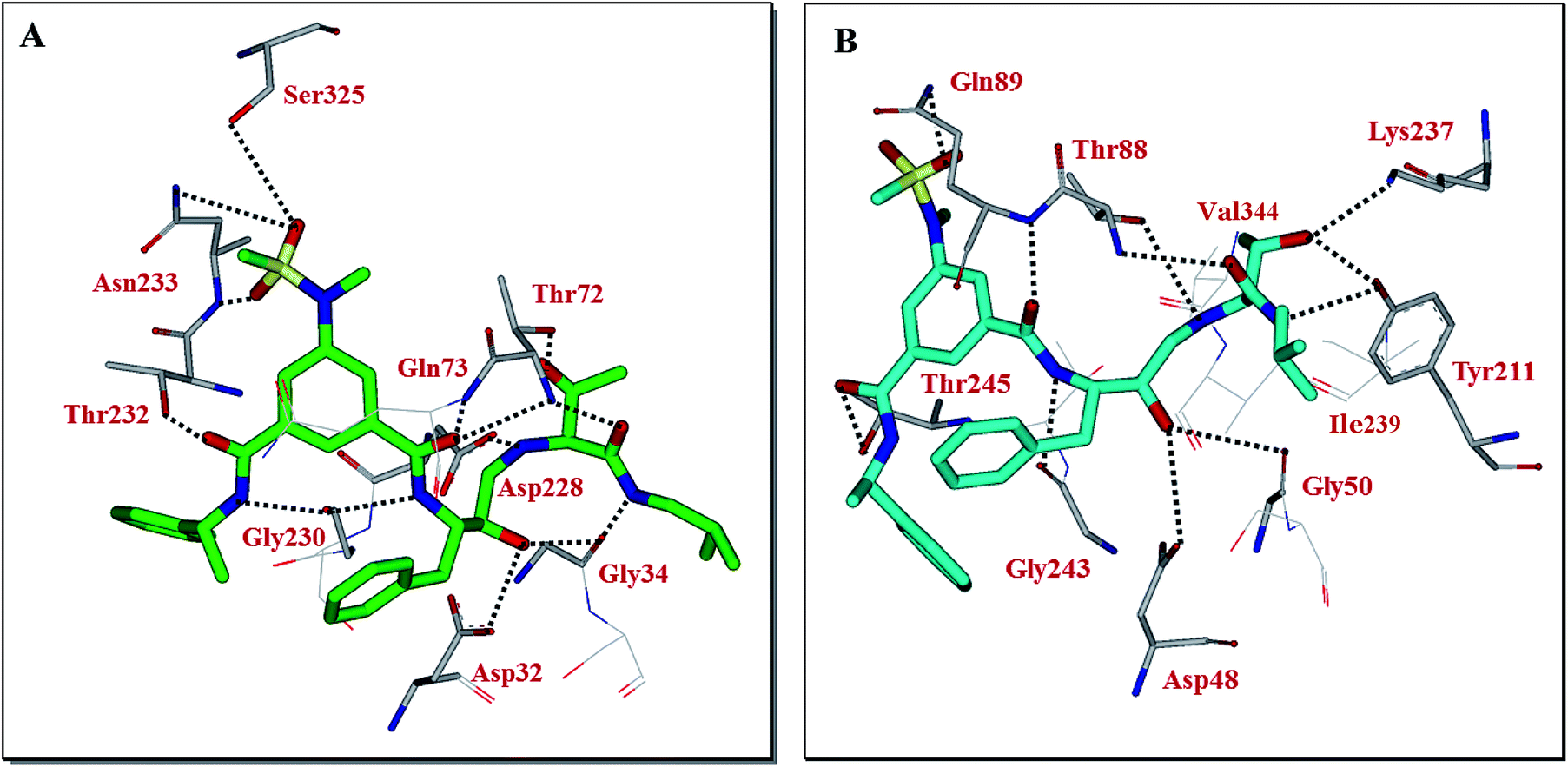 | ||
| Fig. 3 (A) X-ray structure of inhibitor 2 (green color) in complex with BACE 1 (memapsin 2) (PDB code: 5DQC). (B) An energy-minimized model of 2 (turquoise) bound within the BACE2 (memapsin 1) active site. All strong hydrogen bonding interactions are shown as dotted lines. | ||
| Compound | BACE2 Ki (nM) | BACE1 Ki (nM) |
|---|---|---|
| 2 | 14.6 ± 2.9 | 45.6 ± 15.3 |
| 3 | 0.031 ± 0.021 | 5244 ± 1711 |
| 13 | 80.5 ± 4.7 | 1812 ± 194 |
| 14 | 182.8 ± 22.7 | 731 ± 65 |
| 15 | 1.0 ± 0.15 | 1657 ± 44.6 |
| 18 | 14.02 ± 6.6 | 837.3 ± 25.1 |
| 20 | 0.038 ± 0.014 | 563.5 ± 18.5 |
We have evaluated the ability of inhibitor 2 to process Tmem27 in pancreatic β-cell line MIN6.40,41 MIN6 cells were grown in the presence of various inhibitors, lysed and then subjected to Western blot using a monoclonal antibody vs. Tmem27 C-terminal region. Fig. 4 shows that the 22 kDa C-terminal fragment of Tmem27 (lane 1) was reduced to less than 5% (lane 3) and completely abolished (lane 4) by 0.4 μM and 0.9 μM of inhibitor 2 respectively. Inhibitor 4, which is known as compound J (4, BACE2 Ki = 6 nM; BACE1 Ki = 18 nM) was used as positive control.16 The C-terminal fragment of Tmem27 was preserved from degradation (lane 5) by the presence of gamma-secretase inhibitor DAPT in lanes 1–4. These results indicated that inhibitor 2 penetrated MIN6 cells and effectively inhibited the processing of Tmem27 by BACE2. This assumption is due to the fact that BACE2 processes its protein substrate PMEL in endosomes.42 It is therefore expected that BACE2 and its all protein substrates, including Tmem27 are endocytosed to endosomes for processing.
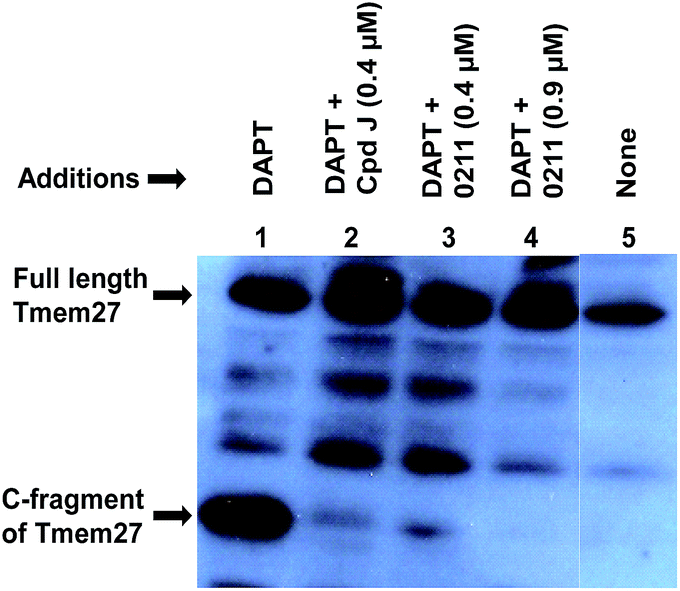 | ||
| Fig. 4 Inhibition of BACE2 processing of Tmem27 by inhibitor 2 in MIN6 cells. | ||
We then utilized the molecular insight into the ligand-binding site interactions of inhibitor 2 in the BACE2 active site to further improve inhibitor properties. We speculated that N-alkylation of P2-amide may lead to further improvement of BACE2 potency and selectivity as the NH-group was not involved in hydrogen bonding interactions in the active site. Also, it appeared that a shallow hydrophobic pocket adjacent to this group can be filled with a small alkyl group. We synthesized the corresponding N-methyl derivative 13. As shown in Table 1, it showed loss of potency against both BACE1 and BACE2 over compound 2. However, compound 13 displayed >20-fold BACE2 selectivity. Furthermore, we planned to remove the sulfonamide functionality from the P2-ligand as this group appears to form nice polar interactions with BACE1 active site residues such as Ser325 but not so much with BACE2 active site. Thus, the sulfonamide group on the P2-isophthalamide ligand was replaced with a methyl group in compound 14. This has also resulted in significant loss of potency against both BACE1 and BACE2. To examine the effect of both N-methylation of the amide NH group and removal of sulfonamide group, we synthesized compound 15. This has resulted in a very potent BACE2 inhibitor (Ki = 1 nM for BACE2; Ki = 1657 nM for BACE1) and showed >1500-fold selectivity over BACE1. Incorporation of this modified P2-ligand in inhibitor 1 provided derivative 18. Interestingly, these changes also provided potent selective BACE2 inhibitor.
Furthermore, based upon the inhibitor 2 model in BACE2 active site, we incorporated a propyl chain in place of P1′-methyl side chain of inhibitor as it appeared that a larger alkyl chain can make more effective van der Waals interactions with surrounding residues such as Ile239 and Val344 (Fig. 3B). These changes resulted in inhibitor 20 which exhibited a marked improvement in BACE2 inhibitory activity (Ki = 0.038 nM), significant reduction in BACE1 activity (Ki = 563 nM, BACE1) and a selectivity over 14000-fold. Further modification of P2-ligand by removal of the ring methyl group resulted in inhibitor 3 which also showed marked BACE2 inhibitory activity. Inhibitor 3 exhibited significant reduction in BACE1 activity (Ki = 5.24 μM, BACE1), showing more than 174000-fold selectivity over BACE1. Furthermore, inhibitor 3 showed excellent selectivity against the lysosomal aspartic protease, cathepsin D, having a Ki = 473 nM which results in a selectivity of >15000-fold over BACE2. The molecular size of inhibitor 3 (Mw = 630 Da) is smaller compared to inhibitor 2 (Mw 695 Da). Also, it has favorable lipophilicity and polar surface area (clogP 4.6; tPSA 131) compared to compound 2 (clogP 2.54; tPSA 177).
In conclusion, our structure-based design strategies led to the discovery of very potent and highly selective BACE2 inhibitors. The X-ray structural analysis provided potentially important molecular interactions useful in the design of selectivity. We have shown that inhibitor 2 completely abolished the processing of Tmem27 in pancreatic β-cell line MIN6. Since the therapeutic principle of BACE2 inhibitors is to enhance the beta cell function and mass, it represents a potentially significant new type of therapeutic target for diabetes treatment. Based upon X-ray structural analysis, we have further designed BACE2 inhibitors such as 3 and 20 by removing the P2-sulfonamide, incorporating N-methyl amide in P3-ligand and appending 3-carbon propyl chain at P1′-ligand. This has resulted in inhibitors with extraordinary BACE2 potency and marked improvement in selectivity against BACE1 and cathepsin D. Further studies using X-ray crystallography combined with computational docking of inhibitors with BACE2 and elucidation of the role of interactions for selectivity and molecular modifications are in progress.
Acknowledgements
Financial support by the National Institutes of Health and Purdue University is gratefully acknowledged. A.D.M also wishes to acknowledge partial support from the Walther Cancer Foundation. NMR, mass spectrometry and protein crystallization were supported in part by the Purdue Center for Cancer Research Shared Resources, which are supported by NIH grant (P30 CA023168). Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).Notes and references
- Centers for Disease Control and Prevention, National Diabetes Statistics Report: Estimates of Diabetes and Its Burden in the United States, 2014, Dept of Health and Human Services, Atlanta, GA, U.S., 2014, accessed http://www.cdc.gov/diabetes/pubs/factsheet11.htm on 20 August 2014 Search PubMed.
- American Diabetes Association, Diagnosis and classification of diabetes mellitus, Diabetes Care, 2013, 36(1), S67–S74 CrossRef PubMed.
- International Diabetes Federation, Sixth edn, 2013, http://www.idf.org/sites.
- Facts about Type 2 diabetes: American Diabetes Association, 2014, http://www.diabetes.org/diabetes-basics.
- W. T. Cade, Phys. Ther., 2008, 88, 1322–1335 CrossRef PubMed.
- A. M. J. Boulton, A. I. Vinik, J. C. Arezzo, V. Bril, E. L. Feldman, R. Freeman, R. A. Malik, R. E. Maser, J. M. Sosenko and D. Ziegler, Diabetes Care, 2005, 28, 956–962 CrossRef PubMed.
- N. Mazzola, Am. J. Manag. Care, 2012, 18, S17–S26 Search PubMed.
- C. M. Ripsin, H. Kang and R. J. Urban, Am. Fam. Physician, 2009, 79, 29–36 Search PubMed.
- S. Bolen, L. Feldman, J. Vassy, L. Wilson, H.-C. Yeh, S. Marinopoulos, C. Wiley, E. Selvin, R. Wilson, E. B. Bass and F. L. Brancati, Ann. Intern. Med., 2007, 147, 386–399 CrossRef PubMed.
- R. R. Holman, S. K. Paul, M. A. Bethel, D. R. Matthews and H. A. Neil, N. Engl. J. Med., 2008, 359, 1577–1589 CrossRef CAS PubMed.
- P. Akpinar, S. Kuwajima, J. Krutzfeldt and M. Stoffel, Cell Metab., 2005, 2, 385–397 CrossRef CAS PubMed.
- J. Altirriba, R. Gasa, S. Casas, M. J. Ramirez-Bajo, S. Ros, S. Gutierrez-Casas, P. Casini, S. Piquer, J. Altirriba, M. Soty, L. Cadavez, R. Gomis and A. Novials, Am. J. Physiol.: Endocrinol. Metab., 2010, 299, E1087–1095 CrossRef PubMed.
- R. Yan, M. J. Bienkowski, M. E. Shuck, H. Miao, M. C. Tory, A. M. Pauley, J. R. Brashler, N. C. Stratman, W. R. Mathews, A. E. Buhl, D. B. Carter, A. G. Tomasselli, L. A. Parodi, R. L. Heinrikson and M. E. Gurney, Nature, 1999, 402, 533–537 CrossRef CAS PubMed.
- P. Akpinar, S. Kuwajima, J. Krutzfeldt and M. Stoffel, Cell Metab., 2005, 2, 385–397 CrossRef CAS PubMed.
- X. Lin, G. Koelsch, S. Wu, D. Downs, A. Dashti and J. Tang, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 1456–1460 CrossRef CAS.
- D. Esterhazy, I. Stutzer, H. Wang, M. P. Rechsteiner, J. Beauchamp, H. Dobeli, H. Hilpert, H. Matile, M. Prummer, A. Schmidt, N. Lieske, B. Boehm, L. Marselli, D. Bosco, J. Kerr-Conte, R. Aebersold, G. A. Spinas, H. Moch, C. Migliorini and M. Stoffel, Cell Metab., 2011, 14, 365–377 CrossRef CAS PubMed.
- C. Southan, Expert Opin. Ther. Pat., 2013, 23, 649–663 CrossRef CAS PubMed.
- R. T. Turner III, J. A. Loy, C. Nguyen, T. Devasamudram, A. K. Ghosh, G. Koelsch and J. Tang, Biochemistry, 2002, 41, 8742–8746 CrossRef.
- R. T. Turner III, G. Koelsch, L. Hong, P. Castanheira, J. Ermolieff, A. K. Ghosh and J. Tang, Biochemistry, 2001, 40, 10001–10006 CrossRef.
- F. Kamenetz, T. Tomita, H. Hsieh, G. Seabrook, D. Borchelt, T. Iwatsubo, S. Sisodia and R. Malinow, Neuron, 2003, 37, 925–937 CrossRef CAS PubMed.
- D. Kobayashi, M. Zeller, T. Cole, M. Buttini, L. McConlogue, S. Sinha, S. Freedman, R. G. Morris and K. S. Chen, Neurobiol. Aging, 2008, 29, 861–873 CrossRef CAS PubMed.
- S. M. Harrison, A. J. Harper, J. Hawkins, G. Duddy, E. Grau, P. L. Pugh, P. H. Winter, C. S. Shilliam, Z. A. Hughes, L. A. Dawson, M. I. Gonzalez, N. Upton, M. N. Pangalos and C. Dingwall, Mol. Cell. Neurosci., 2003, 24, 646–655 CrossRef CAS PubMed.
- M. Willem, A. N. Garratt, B. Novak, M. Citron, S. Kaufmann, A. Rittger, B. DeStrooper, P. Saftig, C. Birchmeier and C. Haass, Science, 2006, 314, 664–666 CrossRef CAS PubMed.
- S. Diment, M. S. Leech and P. D. Stahl, J. Biol. Chem., 1988, 263, 6901–6907 CAS.
- E. Liaudet-Coopman, M. Beaujouin, D. Derocq, M. Garcia, M. Glondu-Lassis, V. Laurent-Matha, D. Prebois, H. Rochefort and F. Vignon, Cancer Lett., 2006, 237, 167–179 CrossRef CAS PubMed.
- R. Steinfeld, K. Reinhardt, K. Schreiber, M. Hillebrand, R. Kraetzner, W. Bruck, P. Saftig and J. Gartner, Am. J. Hum. Genet., 2006, 78, 988–998 CrossRef CAS PubMed.
- Patent Application: J. Beauchamp, A. Benardeau, H. Hilpert, C. Migliorini, W. Riboulet and H. Wang, 2-aminodihydro[1,3] thiazines as BACE2 inhibitors for the treatment of diabetes, WO2011029803A1, 2011.
- A. K. Ghosh, D. D. Anderson and H. Mitsuya, Burger's Medicinal Chemistry, Drug Discovery and Development, 2010, vol. 7, pp. 1–72 Search PubMed.
- A. K. Ghosh, B. D. Chapsal and H. Mitsuya, Methods Princ. Med. Chem., 2010, 45, 205–235 CAS.
- A. K. Ghosh, N. Kumaragurubaran, L. Hong, S. Kulkarni, X. Xu, H. B. Miller, S. D. Reddy, V. Weerasena, R. Turner, W. P. Chang, G. Koelsch and J. Tang, Bioorg. Med. Chem. Lett., 2008, 18, 1031–1036 CrossRef CAS PubMed.
- W. P. Chang, X. Huang, D. Downs, J. R. Cirrito, G. Koelsch, D. M. Holtzman, A. K. Ghosh and J. Tang, FASEB J., 2011, 25, 775–784 CrossRef CAS PubMed.
- N. Ostermann, J. Eder, U. Eidhoff, F. Zink, U. Hassiepen, S. Worpenberg, J. Maibaum, O. Simic, U. Hommel and B. Gerhartz, J. Mol. Biol., 2006, 355, 249–261 CrossRef CAS PubMed.
- I. Stutzer, N. Selevsek, D. Esterhazy, A. Schmidt, R. Aebersold and M. Stoffel, J. Biol. Chem., 2013, 288, 10536–10547 CrossRef CAS PubMed.
- T. Wirth, K. J. Kulicke and G. Fragale, Helv. Chim. Acta, 1996, 79, 1957–1966 CrossRef CAS.
- Patent Application: S. Lundberg, S. Ayesa, O. Belda, I. Dorange, K. Ersmark, K. Hammer, P.-O. Johansson, S. Lindstroem, A. Rosenquist, B. Samuelsson, et al., Benzamides and pyridinecarboxamides as aspartyl protease inhibitors and their preparation and use in the treatment of Alzheimer's disease, WO2010042030, 2010.
- A. K. Ghosh, K. V. Rao, N. D. Yadav, D. D. Anderson, N. Gavande, X. Huang, S. Terzyan and J. Tang, J. Med. Chem., 2012, 55, 9195–9207 CrossRef CAS PubMed.
- A. K. Chakraborti, S. Rudrawar and A. Kondaskar, Org. Biomol. Chem., 2004, 2, 1277–1280 CAS.
- P. R. Fleming and K. B. Sharpless, J. Org. Chem., 1991, 56, 2869–2875 CrossRef CAS.
- All key hydrogen bonding distances are shown in Fig. D (ESI†).
- M. Rupnik and A. Cencic, ALTEX, 2010, 27, 105–113 Search PubMed.
- H. Ishihara, T. Asano, K. Tsukuda, H. Katagiri, K. Inukai, M. Anai, M. Kikuchi, Y. Yazaki, J. Miyazaki and Y. Oka, Diabetologia, 1993, 36, 1139–1145 CrossRef CAS PubMed.
- L. Rochin, I. Hurbain, L. Serneels, C. Fort, B. Watt, P. Leblanc, M. S. Marks, B. De Strooper, G. Raposo and G. van Niel, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10658–10664 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure and characterization data of new compounds. See DOI: 10.1039/c5sc03718b |
| This journal is © The Royal Society of Chemistry 2016 |