DOI:
10.1039/C6RA08724H
(Paper)
RSC Adv., 2016,
6, 52772-52780
Guanidinium rich dendron-appended hydnocarpin exhibits superior anti-neoplastic effects through caspase mediated apoptosis†
Received
5th April 2016
, Accepted 19th May 2016
First published on 20th May 2016
Abstract
Medicinal plants have truly demonstrated their potential as a repository of active biomolecules with promising therapeutic potential and represent an important source for the identification of novel drug leads. Hydnocarpus wightiana Blume is a well known medicinal plant and the acetone extract of its seed demonstrated superior free radical scavenging properties with a high total phenolic and flavonoid content. Hydnocarpin (Hy), which has been isolated and purified from the acetone extract, promotes moderate cytotoxicity in cancer cells. In an attempt to increase the efficiency of Hy as an anticancer agent, chemical coupling with a highly efficient, non-toxic cell-penetrating guanidinium-rich poly(propylene imine) dendron (G8) was attempted. The resultant modified construct (Hy–G8) exhibits superior cytotoxicity preferentially on cancer cells through the induction of apoptosis mediated by caspases. The hybrid construct was also found to be a promising anti-metastatic agent. Therefore, Hy–G8 functioned primarily as a hit compound which requires extensive interdisciplinary approaches and legitimate engineering to accomplish a futuristic lead candidate in cancer chemotherapy.
1. Introduction
A copious amount of natural resources1 for restorative uses exist around the world, of which numerous have not yet been successfully tapped for the development of conceivable drug candidates. Despite extensive research on compounds of natural origin to produce new drug substances occurring, research specifically aimed at naturally derived medicines to optimize dosages for the intended route of administration and to design the most effective dosage forms, has become a greater challenge. Many of the traditional medicines have real, beneficial effects and extracts of these crude combinations lead to the discovery of their active ingredients and eventually to the development of modern chemically pure drugs. In general, phytochemicals identified from traditional medicinal plants are presenting exhilarating opportunities for the development of new drug candidates. Several studies revealed that chemically modified natural compounds, especially from medicinal plants,2 generated better responses as candidate lead molecules in the field of cancer therapy. Cancer3 continues to be one of the major causes of death worldwide with only modest progress being made in reducing the morbidity and mortality of the disease. Phytochemicals from extracts of roots, bulbs, barks, leaves, stems and others parts of plants have shown promising potential as anti-cancer drugs or for serving as lead compounds in the synthesis of new drugs. They are often utilized as traditional medicines in the form of homemade tinctures, teas or crude extracts, but are accompanied by drawbacks, including batch to batch variation in preparation methods and hence in their chemical composition.
Hydnocarpus wightiana Blume3–6 (Flacourtiaceae), is widely cultivated in Southeast Asia, mainly in China, Taiwan, Indonesia, Malaysia and Thailand. Four species occur in India, one of which grows up to 50 feet and is commonly known as Chaulmoogra with globose fruits containing 15–20 seeds. Chaulmoogra oil, extracted from the seeds, has traditionally been effectively used in the treatment of leprosy, chronic skin infections, ophthalmia, and for dressing wounds and ulcers. The plant has also been attributed with anti-obesity activity and the seed3–7 oil is also used in traditional applications in rheumatism, sprains and bruises, sciatica and chest infections. The seeds possess high medicinal value because they are reported to contain an array of therapeutically significant classes of phytochemicals such as triterpenoids, flavones, aglycones, glycosides and a range of fatty acids and esters. Seed extracts yield compounds structurally related to the flavonolignans6,8 namely hydnowightin, hydnocarpin, neohydnocarpin, isohydnocarpin and luteolin. Even though all five flavonolignans demonstrated antidiabetic and antioxidant activities, and moderate cytotoxicity against few cancer cell lines,9,10 hydnocarpin proved to be the most promising agent4,11 with significant cytotoxicity against cancer cells of various origins.
Given their phenomenal concoction of assorted qualities and novel modes of action, natural products keep on working as lead compounds in many drug discovery programs. However, to keep up with the ongoing demand for new therapeutic agents, new strategies for enhancing their therapeutic potential are needed. Semi-synthetic modifications of natural products aimed at enhancing their biological properties or the total synthesis of analogues serve as great starting points for generating molecules with greater bio-availability, bio-distribution and rapid bio-response. Molecular transporters containing octaguanidine residues enhanced the cell penetrating ability most effectively and delivered a variety of cargos.12 To date our group has developed a new guanidinium rich poly(propylene imine) dendron based molecular transporter,13 which was observed to be non-toxic with higher cellular uptake efficiency compared with Arg-8-mer in various cancer cells. The high cell penetrating ability of the G8-dendron encourages us to develop a new hydnocarpin-appended guanidium-rich dendron (Hy–G8). The new synthetic derivative demonstrated excellent water solubility and improved cytotoxicity via caspase-mediated apoptosis in cancer cells. The systematic approach adopted in the current study is illustrated in Fig. 1.
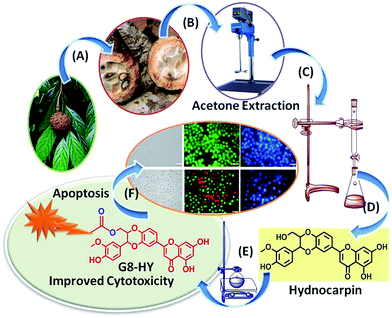 |
| | Fig. 1 Thematic representation of Hy–G8 derivative synthesis. (A) Separation of seeds from Hydnocarpus wightiana Blume, (B) acetone extraction of the seeds, (C) column chromatographic separation, (D) isolation and purification of hydnocarpin, (E) synthetic transformation with G8-PPI dendrons and (F) biological evaluation of Hy–G8. | |
2. Experimental
2.1. Materials and methods
IR spectra were recorded on a Bruker Alpha FT-IR spectrometer. 1H NMR spectra were recorded at 500 MHz using deuterated methanol (CD3OD) as the solvent on a Bruker AV 500 spectrometer. Tetramethylsilane (TMS) was used as the internal standard and chemical shift values are expressed in δ-scale in units of parts per million (ppm) and coupling constants (J) in Hz. Mass spectra were recorded using a Thermo Scientific Exactive mass spectrometer under ESI/HRMS at 61![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 resolution. Analytical thin layer chromatography was performed on a Merck 60 F254 silica gel plate (0.25 mm thickness) and visualization was done with UV light (254 nm and 365 nm) and column chromatography using Merck silica gel (100–200 mesh). Chromatography was carried out using varying polarities of hexane–ethyl acetate mixtures as solvent. Other chemicals were purchased from Sigma Aldrich and Merck.
800 resolution. Analytical thin layer chromatography was performed on a Merck 60 F254 silica gel plate (0.25 mm thickness) and visualization was done with UV light (254 nm and 365 nm) and column chromatography using Merck silica gel (100–200 mesh). Chromatography was carried out using varying polarities of hexane–ethyl acetate mixtures as solvent. Other chemicals were purchased from Sigma Aldrich and Merck.
2.2. Preparation of extracts
The seeds of Hydnocarpus wightiana Blume were thoroughly cleaned and dried in air in an oven maintained at 40 °C for three days. They were then thoroughly powdered and weighed approximately 750 g. The powdered material was first extracted using hexane in a mechanical stirrer (1.5 L) three times at room temperature. It was further extracted using chloroform (1.5 L) three times and finally with acetone (1.5 L) three times at room temperature. The total extracts were concentrated under reduced pressure using a Buchii rotary evaporator. This yielded about 400 g of the crude hexane extract, 25 g of chloroform and 45 g of acetone extract. Based on observation in TLC, the acetone extract was selected for the isolation of compounds.
2.3. Antioxidant activity
Earlier studies revealed that the hexane and chloroform extracts of Hydnocarpus wightiana Blume do not have any appreciable free radical scavenging activity and the acetone extract possesses antioxidant activity against selected free radicals.14 This encouraged us to examine the free radical scavenging properties of the acetone extract against experimentally generated free radicals by established antioxidant assays.
2.3.1. DPPH radical scavenging assay.
The radical scavenging effects of the acetone extract on a 1,1-diphenyl-2-picryl-hydrazil (DPPH) radical were estimated.15 Various concentrations of samples and standards (3 mL) were mixed with 1 mL of 0.1 mM DPPH and the mixture was shaken vigorously and allowed to incubate at room temperature (25 ± 3 °C) for 30 min. The scavenging activity was quantified spectrophotometrically at 507 nm against 95% ethanol as blank. Ascorbic acid was used as the standard and the percent DPPH scavenging effect was expressed as percent inhibition from the given formula:
| % inhibition of DPPH radical = [A0 − A1/A0] × 100 |
where A0 was the absorbance of the control and A1 was the absorbance of the sample/standard.
2.3.2. FRAP (ferric reducing antioxidant power) assay.
This assay measures the total antioxidant capacity of the compound and the procedure was conducted according to Benzie and Strain16 with modification. The working FRAP reagent was prepared by mixing 300 mM acetate buffer (pH 3.6), 10 mM 2,4,6-tripytidyl-s-triazine (TPTZ) solution and 20 mM FeCl3·6H2O in a 10:1:1 ratio prior to use and heated to 37 °C in a water bath. Three millilitres (3 mL) of FRAP reagent was added to 100 μL of various concentrations of the samples and standards. Absorbance was measured at 593 nm at 0 min and again after 90 min. Ascorbic acid was used as the standard and changes in the absorbance after 90 min from the initial reading were compared and percent inhibition was calculated as described.
2.3.3. Hydroxyl radical scavenging assay.
Hydroxyl radical scavenging activity was measured by comparing deoxyribose and the acetone extract for hydroxyl radicals generated by the Fe3+–ascorbate–EDTA–H2O2 system (Fenton reaction).17 The reaction mixture (1 mL), containing 100 μL 2-deoxy-2-ribose (28 mM in 20 mM phosphate buffer, pH 7.4), 500 μL of different concentrations of the samples and controls, 200 μL 1.04 mM FeCl3, 100 μL 1 mM H2O2 and 100 μL ascorbic acid, was incubated at 37 °C for 1 h. Thiobarbituric acid (1%) and trichloroacetic acid (2.8%) were added and incubated at 100 °C for 20 min and after rapid cooling the absorbance was measured at 532 nm. Alpha tocopherol was used as the standard and the percent inhibition was calculated.
2.3.4. Superoxide radical scavenging assay.
The scavenging activity towards superoxide anion radicals was measured by the method of Liu et al.18 Superoxide anions were generated in a non-enzymatic phenazine methosulfate nicotinamide adenine dinucleotide (PMS–NADH) system through the reaction of PMS, NADH and oxygen. It was assayed by the reduction of nitroblue tetrazolium (NBT). Superoxide anions were generated in 3 mL of Tris–HCl buffer (100 mM, pH 7.4) containing 0.75 mL of NBT (300 μM) solution, 0.75 mL of NADH (936 μM) solution and 0.3 mL of various concentrations of samples and standards. The reaction was initiated by adding 0.75 mL of PMS (120 μM) to the mixture. After 5 min of incubation at room temperature (25 ± 3 °C), the absorbance at 560 nm was measured. Alpha tocopherol was used as the positive control and percent inhibition was calculated.
2.4. Total phenolic content (TPC)
Total phenolic content19,20 (TPC) of the acetone extract was determined using the Folin–Ciocalteu reagent. Briefly, 0.5 mL of freshly prepared Folin–Ciocalteu reagent in distilled water (1:2), was added to 1.0 mL extracts of different concentrations (200–1000 μg mL−1) and mixed thoroughly and then neutralized with 1 mL saturated sodium carbonate solution and made up to 10 mL with distilled water. After 2 h, the absorbance of the resulting solution was measured at 764 nm using a spectrophotometer. The TPC was determined by plotting the gallic acid calibration curve (20 to100 μg mL−1) and expressed as gallic acid equivalents (mg GAE/100 g dry weight of sample).
2.5. Total flavonoid content (TFC)
About 1 mL of acetone extract (1 mg mL−1) was made up to 5 mL using distilled water and 0.3 mL of 5% (w/v) NaNO2 was added to it and kept for 5 min. After 5 min. 0.3 mL of 10% (w/v) AlCl3 was added to form a flavonoid–aluminum complex. After 6 min, 2 mL of 1 M NaOH was added and the total volume was made up to 10 mL using distilled water. The solution was mixed well again and the absorbance was measured against a reagent blank, at 415 nm. The blank was prepared by adding all reagents except AlCl3. Distilled water was added in place of AlCl3 in the blank. The TFC was determined by plotting the quercetin calibration curve (20 to100 μg mL−1) and expressed as milligrams of quercetin equivalents (mg QE/100 g of dry weight of sample).21
2.6. Isolation of compounds and its chemical modification
Based on the observation in TLC, the acetone extract was used for the isolation of compounds. A fatty acid rich fraction was isolated from the hexane extract. The acetone extract, which showed good anti-oxidant activity, total phenolic and good flavonoid content, was selected for further in depth studies. It was subjected to column chromatography using silica gel (100–200 mesh), the column was eluted with chloroform and an increasing amount of methanol which yielded 574 different fractions. From 354–407 fractions, we have isolated our target compound hydnocarpin in good yield.
2.6.1. Hydnocarpin (Hy).
IR (KBr, νmax, cm−1): 3186, 2961, 1664, 1625, 1518, 1437, 1383, 1271, 1164, 895, 800; 1H NMR (500 MHz, CD3OD): δ 8.65 (s, 1H), 7.55 (d, J = 8.2, 1H), 7.09 (d, J = 8.7, 1H), 7.05, (s, 1H), 6.94 (d, J = 8.7, 1H), 6.88 (d, J = 8.0, 1H), 6.66 (s, 1H), 6.49 (s, 1H), 6.25 (s, 1H), 4.18–4.16 (m, 1H), 3.90 (s, 3H), 3.76 (d, J = 10.8, 1H), 3.54 (dd, J1 = 12.1, J2 = 3.95, 1H), 3.46 (s, 1H) (ESI Fig. 1†); 13C NMR (125 MHz, CD3OD): δ 181.7, 164.2, 162.8, 161.3, 157.3, 147.6, 147.1, 147.0, 143.6, 126.9, 123.6, 120.5, 119.8, 117.5, 115.3, 114.7, 111.6, 103.8, 103.7, 98.9, 94.0, 77.9, 76.3, 60.0, 55.6; HR-MS (m/z): 465.1183 (M + 1 peak).
2.6.2. Hypnocarpin guanidinium derivative (Hy–G8).
To the solution of hydnocarpin (15 mg, 0.3232 mmol) in dry DMF (3 mL), Boc-protected G8-PPI (92 mg, 0.03232 mmol), prepared using a previously reported procedure from our group (ESI Section 2†),13 a catalytic amount of DMAP (base) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) (7.33 mg, 0.03879 mmol) were added. The mixture was stirred at room temperature for 24 h. Completion of the reaction was monitored through TLC and the volatile components were removed under rotary evaporation and the Boc-protected product Hy–Boc–G8 formed was purified by column chromatography. Ethyl acetate (4 mL) saturated with gaseous HCl was added to a solution of Hy–Boc–G8 in ethyl acetate (1 mL). The reaction mixture was stirred for 4 h then the solution was concentrated and the residue was washed with ethyl acetate to remove less polar impurities. The residue was dried and purified using MPLC on Supelclean™ LC-18 reverse-phase silica gel (MeOH/H2O). The purity of the product was checked by HPLC (ESI Fig. 3†) and then dissolved in de-ionised water yielding Hy–G8. The compound was identified by spectroscopic techniques (ESI Fig. 2†). IR (KBr, νmax, cm−1): 3473, 2922, 2817, 2579, 2377, 1747, 1642, 1501, 1260, 1180, 1164; 1H NMR (500 MHz, CD3OD): δ 1.19–1.55 (m, 22H), 2.49 (m, 20H), 2.52 (m, 16H), 3.08 (m, 16H), 3.36 (m, 8H), 3.52 (m, 2H), 3.56 (m, 6H), 3.68 (m, 4H). 3.74 (s, 3H), 4.20 (d, J = 5 Hz, 2H), 4.26 (m, 1H), 5.25 (d, J = 10 Hz, 1H), 6.29–7.57 (m, 9H). MALDI TOF MS calculated 1720.2400 and found 1721.3258.
2.7. Cell lines
The human cancer cell line A549 (lung adenocarcinoma) was obtained from American Type Culture Collection (ATCC, USA). A375 (malignant melanoma) cells were obtained from NCCS, Pune, India. Fibroblast-like mouse preadipocyte cell line 3T3L1 was obtained from the Inter University Centre for Genomics and Gene Technology, University of Kerala, Thiruvananthapuram, India. The cells were maintained in DMEM media with 10% fetal bovine serum and 5% CO2 with incubation at 37 °C.
2.8.
In vitro cytotoxicity assays
The growth inhibition capacity of hydnocarpin (Hy) and hydnocarpin–G8 (Hy–G8) was evaluated in cancer cell lines using the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium (MTT) assay as previously reported.22 This assay measures cell viability by assessing the cleavage of the tetrazolium salt by mitochondrial dehydrogenase. The absorbance was measured at 570 nm using a microplate spectrophotometer (BioTek Power Wave XS). The inhibitory rates on the cells were calculated using the following formulas:
| Proliferation rate (PR)% = [Abs sample/Abs control] × 100; |
| Inhibitory rate (IR)% = 100 − PR. |
MTT assays were performed on A375, A549 and 3T3-L1 with various concentrations of Hy and Hy–G8 ranging from 2.5 μg mL−1 to 80 μM for 24 and 48 h.
The cytotoxicity of Hy–G8 was further confirmed by LDH assay (Pierce LDH Cytotoxicity Assay Kit-88953, USA) as per the protocol given in the kit using the respective controls. Finally the absorbance at 490 nm and 680 nm was measured using a plate-reading spectrophotometer to determine LDH activity.
2.9. Apoptotic assays
Evaluation of the mode of cytotoxicity exhibited by Hy–G8 was performed on cancer cells with various apoptotic assays after administration of Hy–G8 (20 μM) for 24 h. Morphological evaluation for apoptotic changes were performed under phase contrast microscope (Olympus 1X51, Singapore) under suitable magnification. Observation of nuclei for any changes was done with Hoechst staining and the cells were observed under an inverted fluorescent microscope using a DAPI filter (Olympus 1X51). Acridine orange–ethidium bromide dual staining is the most commonly used method to detect apoptosis and is based on the differential uptake of two fluorescent DNA binding dyes by viable and nonviable cells. Assessment of apoptosis using the acridine orange–ethidium bromide dual staining procedure was performed as described earlier.23 The cells were observed under an inverted fluorescent microscope, using a FITC filter (Olympus 1X51, Singapore) to view the apoptotic or non-apoptotic cells. Furthermore, TUNEL assay (Dead End™ fluorometric TUNEL system G3250, Promega, USA) was used to detect the incorporation of the fluorescein-12-dUTP in the fragmented DNA of apoptotic cells, using the terminal deoxynucleotidyl transferase recombinant (rTdT) enzyme as per the manufacturer’s instruction using propidium iodide as counter stain.
2.10. Caspase assay
The effect of both initiator caspases (caspases 8, 9 and 2) and executioner caspases (caspase 3) was determined using Apo Alert™ Caspase Profiling kit (Clontech, CA, USA) as per the kit protocol. Cells were treated with 20 μM Hy–G8 for 24 h and finally, samples were transferred to 96-well plates for fluorometric reading (excitation: 380 nm; emission: 460 nm), and the OD obtained was recorded under a spectrofluorometer (FLx800, BioTek).
2.11. Wound healing assay
The scratch wound healing assay was performed to evaluate the anti-metastatic potential of Hy–G8 on A375 and A549 cells, as described previously.24,25 The cancer cells have inherent potential for invasion and metastasis which makes them escape from the cytotoxic agent and establishes a tumor at a new niche. Hence evaluation of the anti-metastasis potential of any cytotoxic agent holds special attention. Briefly, cells were seeded into six-well plates at a density of 1.0 × 105 per well until they reached 80% confluence. A scrape was made through the confluent monolayer with a sterile plastic pipette tip of 1 mm diameter. Afterwards, the dishes were washed twice with PBS and incubated at 37 °C in fresh DMEM complemented with 10% fetal bovine serum in the presence or absence of the indicated concentrations of Hy–G8. At the bottom side of each dish, three arbitrary places were marked where the width of the wound was measured with an inverted microscope (objective × 10). The wells were rinsed with PBS three times to remove floating cells and debris. The culture plates were incubated at 37 °C and in 5% CO2. Wound closure was expressed as the average ± SEM of the difference between the measurements at time zero and the 12–24 h time period considered.
3. Results and discussion
3.1. Isolation of crude extracts from Hydnocarpus wightiana Blume seeds
The seeds were subjected to extraction using hexane and acetone. The acetone extract was selected to screen for antioxidant activity and further fractionalization.
Thin layer chromatography of the acetone extract was carried out using solvents of polarity starting from 100% chloroform to 10% methanol in chloroform. After TLC analysis the extract was subjected to column chromatography. From the acetone extracts 574 fraction pools were collected and a further sub column of 374–407 yielded the isolation of hydnocarpin in good yield (Fig. 2).
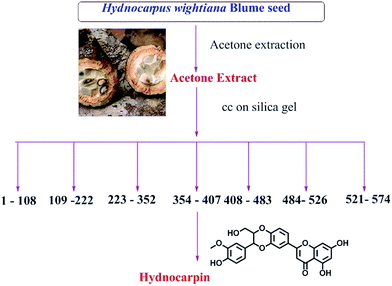 |
| | Fig. 2 Isolation of compounds from acetone extracts of Hydnocarpus wightiana Blume seeds. | |
3.2. Antioxidant action of the acetone extract
Screening of the acetone extract against a wide array of experimentally generated free radicals revealed the astonishing antioxidant properties26 of the extract. The DPPH radical was considered to be a model of lipophilic radicals; a chain reaction in lipophilic radicals was initiated by lipid autoxidation.27 The radical scavenging activity of the acetone extract against experimentally generated DPPH ions is shown in Fig. 3A, which showed excellent scavenging activity even at low concentrations. The scavenging activity increases in a concentration-dependent manner. The IC50 value of the acetone extract was found to be 262.7 ± 1.3 μg mL−1 and for ascorbic acid it was 77.5 ± 1.5 μg mL−1. The FRAP assay directly measures total antioxidants or reductants in a sample. The antioxidant capacity of the acetone extract based on the ability to reduce ferric ions was calculated as percent of inhibition as shown in Fig. 3B. There is a gradual increase in antioxidant activity with an increase in concentration up to 500 μg mL−1 with an IC50 of 70 ± 1.8 μg mL−1, whereas the standard ascorbic acid showed relatively stable activity even from lower concentrations with an IC50 less than even 1 μg mL−1. Hydroxyl radicals attack proteins, DNA, polyunsaturated fatty acids in membranes and most biological molecules they contact, and are known to be capable of abstracting hydrogen atoms from membrane lipids and bring about peroxidation reactions with lipids. The acetone extract was not found to be a good scavenger of hydroxyl radical, with a maximum inhibition of 40% at 500 μg mL−1, whereas alpha tocopherol showed a good scavenging effect with an IC50 at 64 μg mL−1 (Fig. 3C). The scavenging ability of the acetone extract against superoxide ions generated in the PMS–NADH–NBT system was shown in Fig. 3D. The acetone extract exhibited a dose-dependent increase in scavenging activity with an IC50 at 299 ± 2.8 μg mL−1 whereas the positive control alpha tocopherol gave an IC50 of 61.5 ± 1.7 μg mL−1. The radical scavenging activity of the acetone extract is promising and might be helpful in preventing or reducing the progress of various oxidative stress-induced diseases including cancer and thereby be beneficial for human health. From the earlier studies it was proved that the compounds isolated from the acetone extract, hydnocarpin and isohydnocarpin, did not exhibit much antioxidant activity on a DPPH test model; the strong radical scavenging activity of the acetone extract might be due to the presence of luteolin in substantial amounts. Hence the use of the whole extract is preferable to its isolated compounds for free radical scavenging assays.14
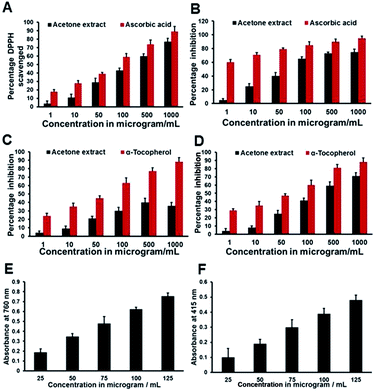 |
| | Fig. 3 The antioxidant activity of the acetone extract. (A) DPPH radical scavenging assay using ascorbic acid as the standard, (B) FRAP assay using ascorbic acid as the standard, (C) hydroxyl radical scavenging assay using alpha tocopherol as the standard and (D) superoxide radical scavenging assay using alpha tocopherol as the standard. (E) Total phenolic content (absorbance value of gallic acid at 760 nm, expressed as gallic acid equivalent in mg g−1 dry weight of sample) and (F) total flavonoid content (absorbance value of quercetin at 415 nm), expressed as quercetin equivalent in mg g−1 dry weight of sample. | |
3.3. Total phenolic and flavonoid content
The estimation of total phenolic content revealed that the acetone extract contains 642 ± 27 mg gallic acid equivalents in 100 g of extract. Total flavonoid content was determined by plotting the quercetin calibration curve (20 to100 μg mL−1) and is expressed as milligrams of quercetin equivalents (mg QE/100 grams of dry weight of sample). The study revealed that the acetone extract of Hydnocarpus wightiana Blume seeds contains 135 ± 9 mg quercetin equivalents in 100 g acetone extract. The concentration and type of phenolic substances are mainly responsible for the biological activities of the extract and high phenolic contents often attributes to higher free radical scavenging properties.28 Flavonoids are stable and effective scavengers of most oxidizing molecules, including singlet oxygen and various free radicals29 implicated in several diseases. The higher phenolic and flavonoid contents of the acetone extract could be highly responsible for the observed superior free radical scavenging effects.
3.4. Synthetic modification of hydnocarpin
The flavonolignan hydnocarpin (Hy) was isolated from acetone extract. Its structure is mostly similar to silybin and it can be characterized as a small, highly functionalized molecule with alternating carbo- and hetero-cycles. Preliminary screening for the cytotoxicity of Hy revealed moderate effects on cancer cells. The cytotoxic nature of Hy prompted us to investigate further structural modification in order to improve its bio-availability, water solubility and to consider transformed Hy as an excellent anti-cancer agent. Our group developed an excellent cell penetrating carrier with an octaguanidinium scaffold which shows higher cellular uptake compared to Arg-8-mer, the well-known cell penetrating peptide. It has excellent selectivity towards the lysosome and the transporter alone (G8–PPI–FL) is found to be non-toxic.13 The primary alcoholic group at the C-23 position of Hy can be readily esterified and oxidized to a carboxylic acid. So, we have conjugated the primary OH of Hy with the acid terminal of the molecular transporter octaguanidinium–poly(propylene imine) hybrid dendron (G8–Boc–PPI) by ester coupling using carboxyl activating agent EDC in combination with DMAP (Scheme 1). Finally, all the Boc groups of the guanidine moieties were deprotected by treatment with ethyl acetate saturated with HCl gas reagent. The final compound Hy–G8 was characterized by HPLC, NMR spectroscopy and MALDI-TOF mass spectrometry.
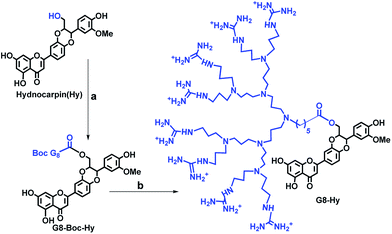 |
| | Scheme 1 Synthesis of hydnocarpin–G8 derivative; (a) EDC, DMAP, dry DMF, N2 atm, 36 h; (b) EtOAc saturated with gaseous HCl, EtOAc. | |
3.5. Evaluation of cytotoxic potential
Evaluation of cytotoxicity is the major step for any lead compounds aimed at human therapeutic applications. The cytotoxic potential was evaluated for Hy, Hy–G8 and the carrier G8 on A375, A549 and 3T3-L1 for 24 and 48 h from a wide dosage range of 2.5 to 80 μM by MTT assay. Hy–G8 executed superior cytotoxicity than Hy in all dosages and incubation times towards A375 cells. Hy also executed significant cytotoxicity whereas the carrier G8 was found to be non-toxic (Fig. 4A, ESI Fig. 4A and B†). A375 cells were affected by Hy–G8 with an IC50 value of 22.7 ± 0.9 and 14.4 ± 0.7 at 24 and 48 h, respectively, but Hy gave an IC50 at relatively higher concentrations of 57.6 ± 1.3 and 41.5 ± 1.7 at 24 and 48 h, respectively. A similar trend in cytotoxicity was observed for A549 cells, where Hy produced an IC50 of 72.7 ± 1.4 at 48 h but failed to generate an IC50 at 24 h. A marked increase in cytotoxic potential was shown by Hy–G8 (Fig. 4B, ESI Fig. 4C and D†) with IC50s of 33.7 ± 0.9 and 24 ± 1.2 at 24 and 48 h, respectively. The cytotoxic potential of Hy and Hy–G8 increased in a dose-dependent manner in both of the cancer cell lines. Absence of an IC50 in the normal fibroblast cell line for both Hy and Hy–G8 highlights the cancer cell-oriented cytotoxicity of the agents. The enhanced cytotoxic potential of Hy–G8 in comparison to Hy demonstrated on cancer cells was not observed on 3T3-LI (Fig. 4C, ESI Fig. 4E and F†). Even though both Hy and Hy–G8 produced a maximum cytotoxicity of 18 and 17% on 3T3-L1 at 80 μM at 48 h, lower dosages produced only negligible cytotoxicity; G8 proved to be non-toxic towards non-cancer cells also. The increased cytotoxicity of Hy–G8 than Hy on cancer cells was further tested by quantification of lactate dehydrogenase (LDH) with 24 and 48 h of compound administration. Hy–G8 exhibited a higher cytotoxicity towards A375 (Fig. 4D) and A549 (Fig. 4E) cells, with marginal cytotoxicity towards 3T3-L1 cells (Fig. 4F). The results obtained from the LDH assay highly correlated with the cytotoxicity evaluation from the MTT assay.
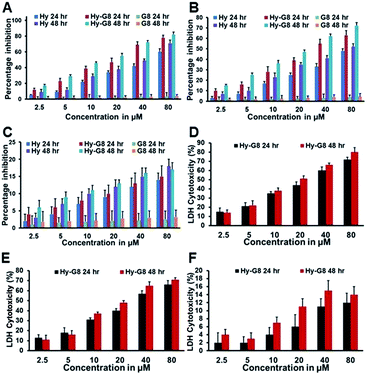 |
| | Fig. 4 Assay for cytotoxicity in cancer cells and normal cells treated with Hy, Hy–G8 and G8 after 24 and 48 h of MTT assay. (A) A375 cells; (B) A549 cells; (C) 3T3-L1 cells. Assessment of the cytotoxicity of Hy–G8 by LDH assay towards (D) A375 cells; (E) A549 cells and (F) 3T3-L1 cells. | |
The cytotoxicity of Hy and Hy–G8 evaluated for lung adenocarcinoma, malignant melanoma and fibroblast-like mouse preadipocyte cell line indicates a cancer cell-oriented cytotoxicity profile. Even though Hy alone was found to be a good cytotoxic agent, the G8 coupled construct (Hy–G8) shows superior cytotoxicity towards both the cancer cells, but a similar increase in cytotoxic potential was not found for normal cells. Plasma membrane damage due to cytotoxic agents releases LDH into the cell culture media which could be quantified for determining the percentage of cytotoxicity caused by the agent. The improved cytotoxicity of Hy–G8, which could be attributed to the enhanced water solubility of Hy–G8 in contrast to Hy, was confirmed by MTT and LDH assays.
3.6. Hy–G8 induced anticancer effects through apoptosis
To investigate the cell death mechanism induced by Hy–G8 in cancer cells, various apoptotic assays were conducted on A375 and A549 cells after the administration of 20 μM of the compound for 24 h. Morphological changes evaluated with phase contrast microscopy revealed a decrease in cell number, which was accompanied by salient morphological features of apoptosis, such as distorted shape, membrane blebbing, and the presence of apoptotic bodies compared to the control group on both A375 (Fig. 5Aa) and A549 (Fig. 5Ba) cells. In comparison with the respective controls, acridine orange–ethidium bromide staining displayed a change in colour from green to yellow/red, which is associated with other apoptotic features such as the presence of apoptotic bodies, damaged cell membrane and nuclear condensation in Hy–G8-treated cells. Significant changes in fluorescence were observed for A375 (Fig. 5Ab) and A549 (Fig. 5Bb) cells. Cells undergoing apoptosis demonstrated nuclear condensation and DNA fragmentation, which was detected by Hoechst 33342 nuclear staining. The percentage of chromatin condensation after Hy–G8 treatment was significantly higher in A375 (Fig. 5Ac) and A549 (Fig. 5Bc) cells compared to the respective controls. The mechanism of cell death induced by Hy-G8 was further confirmed with the TUNEL assay in which propidium iodide was used as counter stain. Hy–G8-treated cells displayed a green colour indicating TUNEL positivity, but control cells are largely TUNEL negative, indicating the targeted apoptotic induction of the compound on both A375 (Fig. 6A) and A549 cells (Fig. 6B). One of the mechanistic features of apoptotic cell death is the activation of caspases; a class of cysteine proteases and many anticancer drugs was reported to execute apoptosis through a caspase dependent pathway.30 Since caspases are the effective mediators of apoptosis, the expression of caspases 3, 8, 9 and 2 was estimated using fluorimetry to thoroughly substantiate the cell death mechanisms induced by Hy–G8. Both A375 and A549 presented a significant (P < 0.001) increase in the expression of caspases 2, 3 and 8 (Fig. 6C). Caspase 3 was the most over-expressed with 2.4- and 2.6-fold increases within A375 and A549 cells, respectively, in comparison with their controls. The expression pattern of caspase 9 was not that significant in comparison with other caspases in both of the cells.
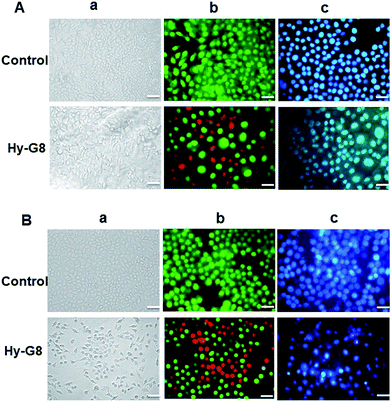 |
| | Fig. 5 Morphological evaluation of apoptosis by the 24 h administration of Hy–G8 (20 μM) on (A) A375 and (B) A549 cells; (a) represents phase contrast images, (b) represents acridine orange–ethidium bromide and (c) represents Hoechst staining. The scale bar corresponds to 50 μm. | |
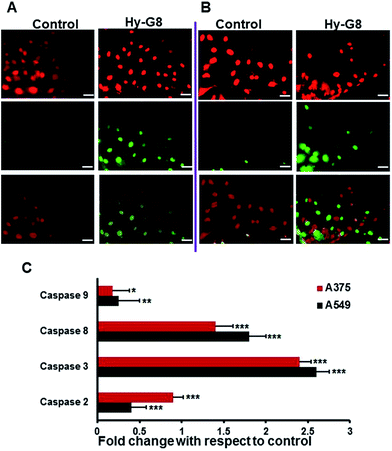 |
| | Fig. 6 Morphological evaluation of apoptosis by TUNEL assay after 24 h administration of Hy–G8 (20 μM) on (A) A375 and (B) A549 cells. The top panel represents images with PI filter, the middle panel represents the FITC filter and the bottom panel shows the merged images. The scale bar corresponds to 50 μm. (C) Caspase activity profiling after 24 h administration of Hy–G8 (20 μM) on A375 and A549 cells. Results are expressed as mean ± SD. | |
Apoptosis is the most predominant mode of cell death, and several naturally occurring compounds have shown to induce apoptosis in cancer cell lines.31 The presence of irregular bulges in the plasma membrane of the cell due to localized decoupling of the cytoskeleton from the plasma membrane is a hallmark of apoptosis. Although the budding phenomenon of apoptotic cells lasts for only a few minutes, the formation of apoptotic bodies remains visible for a few hours.32 The enhanced cytotoxicity of Hy–G8 compared with Hy is found to be effected through caspase-mediated apoptosis.
3.7. Anti-metastatic potential
The wound healing assay allows the researcher to study cell migration, cell interactions and metastatic potential of any agent under investigation. The anti-metastatic potential of Hy–G8 was evaluated for both A375 and A549 cells. It was clearly observed that Hy–G8 could hinder the migration of cells in a time-dependent manner for both A375 (Fig. 7A) and A549 cells (Fig. 7B) whereas the untreated cells attain confluency within 24 h. The cell-free scratch zone of the treated wells are large even after 24 h but the control wells displayed almost complete closure of the zone even after 12 h in both of the cell lines. The in vitro scratch assay is an easy, low-cost and well-developed method to measure cell migration. Compared to other methods, the in vitro scratch assay is particularly suitable for studies on the effects of cell-matrix and cell–cell interactions on cell migration, mimicking cell migration during wound healing in vivo, and are compatible with imaging of live cells during migration to monitor intracellular events if desired.33 Tumor invasion and metastasis is a multistep process involving cell adhesion, proteolytic degradation and migration through the extra-cellular membrane, angiogenesis and many more. Common chemotherapeutic agents involved in cancer treatment aim at blocking cell cycle progression, inducing cell death and inhibiting tumor migration and thereby metastasis. Several natural compounds34,35 have been reported to exhibit cancer chemoprevention through blocking cell migration.
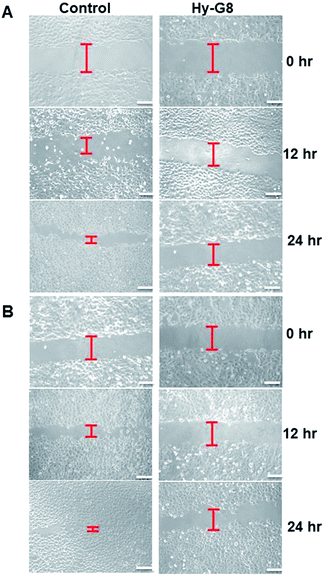 |
| | Fig. 7 Representative wound healing images at 0, 12, and 24 hours. Wounds were made with a pipette tip in confluent monolayers on (A) A375 and (B) A549 cells after administration of Hy–G8 (20 μM). Red lines indicate the cell-free scratch zone. The scale bar corresponds to 50 μm. | |
4. Conclusions
Phytochemical investigations on the popularly used medicinal plant Hydnocarpus wightiana Blume (seed) acetone extract decipher its excellent free radical scavenging property with high total phenolic and flavonoid content. The most abundant compound hydnocarpin (Hy), which was isolated, purified and characterised from the extract demonstrates moderate cytotoxic potential towards cancer cells. In an attempt to increase the anticancer potential of Hy, chemical conjugation with a highly efficient, non-toxic cell-penetrating guanidinium-rich poly(propylene imine) dendron (G8) was carried out via EDC coupling. The hybrid molecule (Hy–G8) exhibited excellent cytotoxicity preferentially towards lung adenocarcinoma and malignant melanoma cells mediated through caspase-dependent programmed cell death and also demonstrated anti-metastatic potential. The superior anticancer potential of Hy–G8 over hydnocarpin warrants the need for detailed investigations into its molecular mechanistic mode of action both in cell lines and in animal models.
Acknowledgements
Author KKM wishes to thank Science and Engineering Research Board (SERB), Govt. of India (DST No. SR/S1/OC-67/2012) and Council of Scientific and Industrial Research (CSIR), Govt. of India, network project (NaPHA: CSC-0130), (M2D: CSC-0134), (NanoSHE: BSC-0112) for research funding. Author MMJ wishes to thank Kerala Biotechnology Commission, Govt. of Kerala (KBC-KSCSTE) for the Post-Doctoral research fellowship. AcSIR PhD student SMG acknowledges CSIR for research fellowship. AcSIR PhD student JBN acknowledges CSIR network project (M2D) for the research fellowship. The authors also thank CSIR-NIIST for the instrumental facilities and Division of Cancer Research, Regional Cancer Centre (RCC), Thiruvananthapuram.
Notes and references
- O. Prakash, A. Kumar, P. Kumar and A. Ajeet, Am. J. Pharmacol. Sci., 2013, 1, 104–115 Search PubMed
 .
.
- B. V. Raman, N. V. Krishna, B. N. Rao, M. P. Saradhi and M. V. B. Rao, Int. Res. J. Pharm., 2012, 3, 11–15 Search PubMed .
-
(a) A. Bhanot, R. Sharma and M. N. Noolvi, Int. J. Phytomed., 2011, 3, 09–26 Search PubMed ;
(b) A. Saha, S. Mohapatra, P. Kurkute, B. Jana, J. Sarkar, P. Mondal and S. Ghosh, RSC Adv., 2015, 5, 92596–92601 RSC .
- M. R. Sahoo, S. P. Dhanabal, A. N. Jadhav, V. Reddy, G. Muguli, U. V. Babu and P. Rangesh, J. Ethnopharmacol., 2014, 154, 17–25 CrossRef CAS PubMed .
- V. Vimberg, M. Kuzma, E. Stodu°lková, P. Novák, L. Bednárová, M. Šulc and R. Gažák, J. Nat. Prod., 2015, 78, 2095–2103 CrossRef CAS PubMed .
- M. R. Sahoo, S. P. Dhanabal, A. N. Jadhav, V. Reddy, G. Muguli, U. V Babu and P. Rangesh, J. Ethnopharmacol., 2014, 154, 17–25 CrossRef CAS PubMed .
- D. K. Sharma and I. H. Hall, J. Nat. Prod., 1991, 54, 1298–1302 CrossRef CAS .
- V. Vimberg, M. Kuzma, E. Stodůlková, P. Novák, L. Bednárová, M. Šulc and R. Gažák, J. Nat. Prod., 2015, 78, 2095–2103 CrossRef CAS PubMed .
- D. K. Sharma and I. H. Hall, J. Nat. Prod., 1991, 54, 1298–1302 CrossRef CAS .
- M. R. Sahoo, S. P. Dhanabal, A. N. Jadhav, V. Reddy, G. Muguli, U. V. Babu and P. Rangesh, J. Ethnopharmacol., 2014, 154, 17–25 CrossRef CAS PubMed .
- K. A. L. Pan, Y.-W. Chin, H.-B. Chai, T. N. Ninh and D. D. Soejarto, Bioorg. Med. Chem., 2009, 17, 2219–2224 CrossRef PubMed .
- E. G. Stanzl, B. M. Trantow, J. R. Vargas and P. A. Wender, Acc. Chem. Res., 2013, 46, 2944–2954 CrossRef CAS PubMed .
- J. B. Nair, S. Mohapatra, S. Ghosh and K. K. Maiti, Chem. Commun., 2015, 51, 2403–2406 RSC .
-
(a) S. V. Reddy, A. K. Tiwari, U. S. Kumar, R. J. Rao and J. M. Rao, Phys. Ther., 2005, 19, 277–281 CAS ;
(b) S. B. Nimse and D. Pal, R. Soc. Chem., 2015, 5, 27986–28006 CAS .
- P. Molyneux, Songklanakarin J. Sci. Technol., 2004, 26, 211–219 CAS .
- I. F. Benzie and J. J. Strain, Anal. Biochem., 1996, 239, 70–76 CrossRef CAS PubMed .
- R. Singh, N. Singh, B. S. Saini and H. S. Rao, Indian J. Pharmacol., 2008, 40, 147–151 CrossRef PubMed .
- F. Liu, V. E. C. Ooi and S. T. Chang, Life Sci., 1997, 60, 763–771 CrossRef CAS PubMed .
- L. A. Shaver, S. H. Leung, A. Puderbaugh and S. Angel, J. Chem. Educ., 2011, 88, 492–495 CrossRef CAS .
- M. S. Stankovi, Kragujev. J. Sci., 2011, 33, 63–72 Search PubMed .
- R. Sahu and J. Saxena, J. Pharmacogn. Phytochem., 2013, 2, 176–179 Search PubMed .
- M. M. Joseph, S. R. Aravind, S. Varghese, S. Mini and T. T. Sreelekha, Colloids Surf., B, 2013, 104, 32–39 CrossRef CAS PubMed .
- M. M. Joseph, S. R. Aravind, S. K. George, K. Raveendran Pillai, S. Mini and T. T. Sreelekha, J. Biomed. Nanotechnol., 2014, 10, 3253–3268 CrossRef CAS PubMed .
- F.-Y. Zhang, Y. Hu, Z.-Y. Que, P. Wang, Y.-H. Liu, Z.-H. Wang and Y.-X. Xue, Int. J. Mol. Sci., 2015, 16, 23823–23848 CrossRef CAS PubMed .
- A. Shiozaki, X. Bai, G. Shen-Tu, S. Moodley, H. Takeshita, S.-Y. Fung, Y. Wang, S. Keshavjee and M. Liu, PLoS One, 2012, 7, e38049 CAS .
- M. N. Alam, N. J. Bristi and M. Rafiquzzaman, Saudi Pharm. J., 2013, 21, 143–152 CrossRef PubMed .
- E. J. Garcia, T. L. Cadorin Oldoni, S. M. de Alencar, A. Reis, A. D. Loguercio and R. H. Miranda Grande, Braz. Dent. J., 2012, 23, 22–27 CrossRef PubMed .
- B. H. A. Meziti, H. Meziti, K. Boudiaf and B. Mustapha, World. Acad. Sci. Eng. Tech., 2012, 64, 24–32 Search PubMed .
- L. Bravo, D. Sources and N. Significance, Nutr. Rev., 1998, 56, 317–333 CrossRef CAS PubMed .
- A. Vaculova, V. Kaminskyy, E. Jalalvand, O. Surova and B. Zhivotovsky, Mol. Cancer, 2010, 9, 87 CrossRef PubMed .
- Y. Dong, C. Y. Kwan, Z. N. Chen and M. M. Yang, Res. Commun. Mol. Pathol. Pharmacol., 1996, 92, 140–148 CAS .
- B. A. Barres, I. K. Hart, H. S. Coles, J. F. Burne, J. T. Voyvodic, W. D. Richardson and M. C. Raff, Cell, 1992, 70, 31–46 CrossRef CAS PubMed .
- C.-C. Liang, A. Y. Park and J.-L. Guan, Nat. Protoc., 2007, 2, 329–333 CrossRef CAS PubMed .
- T. Kuno, T. Tsukamoto, A. Hara and T. Tanaka, J. Biophys. Chem., 2012, 3, 156–173 CrossRef CAS .
- L. S. Adams, S. Phung, N. Yee, N. P. Seeram, L. Li and S. Chen, Cancer Res., 2010, 70, 3594–3605 CrossRef CAS PubMed .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra08724h |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a 







