
Self-assembled structure and relaxation dynamics of diblock copolymers made of polybutadiene and styrene/butadiene rubber†
C. A. da Silvaab,
H. Buddec,
M. Menzeld,
U. Wendlerc,
M. Bartkec,
M. Weyderta and
M. Beiner*bd
aGoodyear Innovation Center Luxembourg, L-7750 Colmar-Berg, Luxembourg
bMartin-Luther-Universität Halle-Wittenberg, Naturwissenschaftliche Fakultät II, D-06099 Halle (Saale), Germany
cFraunhofer PAZ, Value Park A74, D-06258 Schkopau, Germany
dFraunhofer IMWS, Walter-Hülse-Str. 1, D-06120 Halle (Saale), Germany. E-mail: mario.beiner@imws.fraunhofer.de
First published on 18th May 2016
Abstract
Structural features and relaxation dynamics of two series of crosslinked poly(butadiene-block-(styrene-stat-butadiene)) diblock copolymers with systematically varied segregation strength and morphology are studied by atomic force microscopy (AFM) and dynamic shear measurements. Series I contains symmetric diblock copolymers with variable styrene content in the SBR block. In Series II the volume fraction of the SBR block ΦSBR is varied while keeping the styrene content in the SBR block almost constant. AFM results indicate that symmetric diblock copolymers of Series I with styrene contents ≥ 35 mol% in the SBR block are well microphase-separated in accordance with thermodynamic models using an effective interaction parameter χeff. The samples of Series II with volume fractions in the range 30 vol% ≤ ΦSBR ≤ 60 vol% show cylindrical and lamellar morphologies as expected based on thermodynamic equilibrium concepts. This shows that the diblock copolymer morphologies existing at the crosslinking temperature are fixed but not significantly altered by the vulcanization procedure. Depending on the segregation of PB and SBR blocks in the crosslinked state, there are three different relaxation scenarios: (i) two well separated dynamic glass transitions αPB and αSBR for strongly segregated samples consisting mainly of two pure phases, PB and SBR, (ii) significant relaxation modes located between the α relaxation processes of pure PB and SBR for weakly segregated systems with a lot of interfacial material and (iii) one single dynamic glass transition α at intermediate temperatures for practically miscible systems. These three situations are explained based on the chemical composition of subsystems having typical dimensions of about 1–3 nm consistent with the characteristic lengths ξα of the glass transition quantifying the dimensions of cooperative rearranging regions (CRRs).
Introduction
Car tires have been for more than a century a major field of application for industrially used rubbers.1 Rubber composites with optimized relaxation dynamics are, in particular, needed for tire treads. Parameters like wet grip and rolling resistance are strongly related to the relaxation behavior of the used rubber composites. Hence, the viscoelastic properties of these materials are very important for the safety and fuel consumption of cars.2,3 The optimization of such materials remains an issue since it is practically impossible to optimize wet grip and rolling resistance at the same time. Moreover, there is still no predictive understanding of the physical mechanisms influencing the segmental α dynamics of amorphous polymers and their composites determining their application relevant properties.In recent decades polymer blends have been often used in order to improve the performance of tire treads since this allows a better fine-tuning of the relaxation behavior compared to composites with a homogenous polymer matrix. Although this approach is successfully used to improve tire performance, there are still remaining disadvantages. A major drawback of blend-based composites is that the morphology of the polymer matrix is determined by the processing steps. The incompatibility of the blend components leads to a situation where relatively large domains are formed and small modifications in the processing steps can strongly influence the final properties.4
An alternative concept to control the dissipation behavior of rubber composites is to use self-assembled block copolymers5–7 as polymer matrix. Classical examples are diblock copolymers containing two incompatible blocks which self-assemble into nanoscopic domains with typical dimensions in the range 10–40 nm. The morphology of these self-assembled diblock copolymers is usually well reproducible, tunable and weakly affected by the processing conditions. The structural state can be controlled by parameters like volume fraction (Φ) and the order parameter χN, (with χ being the temperature-dependent Flory–Huggins interaction parameter and N the total degree of polymerization). In general, the phase separation behavior of diblock copolymers is well understood and predictable based on established thermodynamic models.8–10 The relaxation behavior of strongly segregated block copolymers is basically a superposition of the contributions of both polymeric components. This opens new routes towards an efficient fine-tuning of the dissipation in those frequency–temperature ranges which are relevant for wet grip (10 Hz, 0 °C) and rolling resistance (10 Hz, 60 °C). However, to make use of this advantage it is urgently required to know as much as possible about the microphase-separated state. This is the basis to understand how structural features like morphology, domain size and interfacial width influence visco-elastic properties, in particular the underlying α relaxation dynamics. This touches fundamental questions in the field of glass transition research, like those for the influence of (i) domain size and geometrical confinement,11–17 (ii) constraints at interfaces18–20 and (iii) local chemical composition21 on the segmental α dynamics in amorphous systems with a complex internal structure.
Main aim of this work is to study the influence of volume fraction (Φ) and compatibility (χN) on microphase-separated state and relaxation behavior of crosslinked diblock copolymers combing a styrene-stat-butadiene rubber (SBR) block with a polybutadiene (PB) block. For that propose two anionically synthesized series of PB–SBR diblock copolymers are used. There is one series of symmetric diblock copolymers where the Flory–Huggins interaction parameter χ is varied by changing the styrene content in the SBR block. In a second series the volume fraction ΦSBR is systematically changed by modifying the SBR and PB block lengths. Changes in the relaxation dynamics dependent on composition are studied and discussed based on interrelations between structural length scales in self-assembled block copolymers and the size of dynamic heterogeneities in glass forming materials.
Experimental section
Synthesis and polymer analytics
Two series of diblock copolymers, PB–SBR, were synthesized by a new living anionic polymerization route previously reported elsewhere.22 The anionic polymerization was initiated by n-BuLi, carried out in cyclohexane at 70 °C and is mainly done in two steps: (1) synthesis of PB block (without modifier) and (2) copolymerization of the SBR block by sequentially adding 1,3-butadiene and styrene monomers to the living polybutadienyllithium in the presence of TMEDA (with molar ratio n-BuLi/TMEDA 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.4). The novelty of this synthesis route is in the step (2) in which 60 wt% of the total 1,3-butadiene needed for the SBR block was metered by means of a pump. It has been performed in this way in order to avoid the formation of PS blockiness. For the considerably high styrene contents in the SBR higher TMEDA concentration would be necessary. However, low 1,2-vinyl contents were desirable for all series in both blocks, PB and SBR. Molecular weight parameters (number average Mn, weight average Mw, polydispersity Mw/Mn) of the PB block and the entire PB–SBR diblock copolymers were taken from gel permeation chromatography (GPC) measurements. These measurements were carried out at 35 °C with a SECcurity GPC System equipped with two columns (PSS SDV linear XL 5 μm) and a Waters 410 refractive index detector. Tetrahydrofuran (THF) was used as mobile phase at a flow rate of 1 ml min−1. Calibration was done using polystyrene standards. Volume fractions (ΦSBR) were calculated based on Mn(SBR), Mn(PB) and Mn(PB–SBR) and on the homopolymer densities (ρPB = 0.89 g cm−3, ρPS = 1.05 g cm−3).23 1,2-vinyl and styrene contents were determined based on 1H nuclear magnetic resonance data (measured on a Varian 400 MHz NMR spectrometer) according to the British Standard – BS ISO 21561:2005 + A1:2010. The samples are dissolved in deuterated chloroform containing 0.03% TMS. The relative amount of styrene sequences longer than six units was estimated based on the ratio of the intensity of the ortho proton peak (6.5 ppm) to that of the peaks representing the entire styrene content (6.1–7.7 ppm).24
:0.4). The novelty of this synthesis route is in the step (2) in which 60 wt% of the total 1,3-butadiene needed for the SBR block was metered by means of a pump. It has been performed in this way in order to avoid the formation of PS blockiness. For the considerably high styrene contents in the SBR higher TMEDA concentration would be necessary. However, low 1,2-vinyl contents were desirable for all series in both blocks, PB and SBR. Molecular weight parameters (number average Mn, weight average Mw, polydispersity Mw/Mn) of the PB block and the entire PB–SBR diblock copolymers were taken from gel permeation chromatography (GPC) measurements. These measurements were carried out at 35 °C with a SECcurity GPC System equipped with two columns (PSS SDV linear XL 5 μm) and a Waters 410 refractive index detector. Tetrahydrofuran (THF) was used as mobile phase at a flow rate of 1 ml min−1. Calibration was done using polystyrene standards. Volume fractions (ΦSBR) were calculated based on Mn(SBR), Mn(PB) and Mn(PB–SBR) and on the homopolymer densities (ρPB = 0.89 g cm−3, ρPS = 1.05 g cm−3).23 1,2-vinyl and styrene contents were determined based on 1H nuclear magnetic resonance data (measured on a Varian 400 MHz NMR spectrometer) according to the British Standard – BS ISO 21561:2005 + A1:2010. The samples are dissolved in deuterated chloroform containing 0.03% TMS. The relative amount of styrene sequences longer than six units was estimated based on the ratio of the intensity of the ortho proton peak (6.5 ppm) to that of the peaks representing the entire styrene content (6.1–7.7 ppm).24
Materials
Series I consists of six symmetric PB–SBR diblock copolymers (ΦPB–ΦSBR ∼ 50 vol%) with variable styrene contents in the SBR block in the range 21 ≤ xS,SBR ≤ 52 mol% (in steps of ∼5 mol%).Series II contains six asymmetric samples with volume fractions in the range 20 ≤ ΦSBR ≤ 69 vol%. The styrene content in the SBR block varies in this series only slightly (xS,SBR = 32 ± 4 mol%).
The diblock copolymers in both series have a common molecular weight of Mn ∼ 200 kg mol−1. Low 1,2-vinyl contents are targeted for all samples in order to minimize known shifts in Tg25,26 and changes in compatibility31 caused by the microstructure of butadiene sequences. Details about the microstructure of the diblock copolymer samples are given in Table 1. The diblock copolymers are labeled as PBΦPB–SxByRΦSBR, where x = xS,SBR and y = xB,SBR are the approximated mole percentages of styrene (S) and butadiene (B) in the random block. A polybutadiene homopolymer (PB, Mw = 86.95 kg mol−1; Mw/Mn = 1.10; c1,2-vinyl = 8.50 mol%) and a random poly(styrene-stat-butadiene) copolymer (S30B70R, Mw = 81.98 kg mol−1; Mw/Mn = 1.04; c1,2-vinyl = 17.63 mol%; xS,SBR = 29.84 mol%) were synthesized in addition as reference materials.
| Label | PB block | SBR block | PB–SBR diblock copolymer | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Mn (kg mol−1) | Mw/Mn | c1,2-vinyl (mol%) | xS,SBRb (mol%) | c1,2-vinyl (mol%) | ΦSBR (vol%) | Mn (kg mol−1) | Mw/Mn | Da (nm) | |
| a Calculated based on room temperature SAXS data for non-crosslinked samples using Bragg's law (details cf. ESI).b Styrene sequences longer than six units were absent in all SBR blocks except for the sample with the highest styrene content in the SBR block (52 mol% S) which contains 8 wt% of such sequences. | |||||||||
| Series I | |||||||||
| PB50–S52B48R50 | 110.75 | 1.06 | 8.39 | 52.15 | 15.74 | 46 | 214.76 | 1.04 | 69.0 |
| PB50–S45B55R50 | 135.44 | 1.06 | 8.33 | 45.50 | 14.57 | 46 | 264.79 | 1.10 | 80.5 |
| PB50–S40B60R50 | 108.85 | 1.06 | 8.72 | 40.41 | 15.05 | 46 | 210.88 | 1.05 | 63.4 |
| PB50–S35B65R50 | 112.38 | 1.06 | 8.40 | 34.81 | 14.22 | 47 | 222.14 | 1.06 | 63.4 |
| PB50–S27B73R50 | 103.24 | 1.06 | 8.60 | 27.38 | 16.15 | 48 | 206.98 | 1.05 | 53.2 |
| PB50–S21B79R50 | 123.19 | 1.07 | 8.60 | 20.76 | 19.40 | 49 | 251.58 | 1.06 | (51.5) |
|
|||||||||
| Series II | |||||||||
| PB80–S30B70R20 | 147.81 | 1.07 | 8.50 | 27.90 | 23.63 | 20 | 188.46 | 1.08 | 47.2 |
| PB70–S30B70R30 | 123.22 | 1.07 | 8.50 | 32.64 | 18.96 | 30 | 179.73 | 1.09 | 62.8 |
| PB62–S30B70R38 | 133.64 | 1.07 | 8.50 | 30.76 | 18.30 | 38 | 221.35 | 1.08 | 66.1 |
| PB50–S35B65R50 | 112.38 | 1.06 | 8.40 | 34.81 | 14.22 | 47 | 222.14 | 1.06 | 63.4 |
| PB40–S35B65R60 | 87.48 | 1.09 | 8.50 | 36.30 | 20.25 | 60 | 233.08 | 1.15 | 68.3 |
| PB31–S35B65R69 | 50.76 | 1.18 | 8.37 | 34.35 | 16.54 | 69 | 173.71 | 1.06 | 49.5 |
Crosslinking
All samples were vulcanized using a standard recipe with 1.5 phr sulfur at 150 °C according to the procedures described elsewhere.22 Samples and crosslinking additives were mixed in an internal mixer (Brabender Plasticorder) of 83 cm3 with filling factor of 0.7 and later the curatives were added to the mixture. Afterwards, they were vulcanized at 150 °C in a frame of 110 × 160 × 2 mm3 applying a hydraulic pressure of 40 bar for 32 min.Atomic force microscopy (AFM)
AFM measurements were carried out using a Nanowizard II (JPK-Instruments, Germany) scanning probe microscope with super sharp silicon tips having tip radii of about 2 nm. Phase images were measured in the tapping mode at room temperature. Ultrathin sections (100 nm) of the crosslinked molded sheets were trimmed with a cryo-ultramicrotome (PT-PC Powertome with CR-X cryo unit, RMC products) at a temperature of −120 °C using diamond knives. AFM measurements were carried out on freshly cut samples in order minimize the amount of additives migrating from the bulk to the surface.Differential scanning calorimetry (DSC)
DSC scans were performed on non-crosslinked samples using a METTLER TOLEDO DSC 821 with heating and cooling rates of |dT/dt| = 20 K min−1. The samples were first annealed at 130 °C for 5 min, cooled to −130 °C and held at this temperature for 5 min before a heating scan was measured. Sample masses were 10–20 mg. Glass temperatures Tg were determined using the inflection point method. Further details are given in ref. 22.Dynamic mechanical analysis (DMA)
DMA measurements were performed with an ARES rheometer (TA Instruments) in shear mode. A control strain of 0.1% was applied at low temperatures but increased to 0.3% at temperatures approximately 30 K above Tg,PB of the PB block. Rectangular samples with a size of 30 × 8 × 2 mm3 were used. Temperature sweeps were measured at four frequencies (ω = 0.1, 1, 10, 100 rad s−1) in the temperature range from −120 to Tg,SBR + 30 K with an increment of 3 K and a soak time of 100 s. In addition, isothermal frequency sweeps were measured in the range 0.1–100 rad s−1 with five points per decade at temperatures in the range from −120 °C to Tg,SBR + 30 K to construct master curves. The soak time in this case was 300 s at each temperature and the temperature increment was 2.5 K. All shear measurements were carried out under nitrogen atmosphere.Results
Structural information from atomic force microscopy
AFM phase contrast images of crosslinked symmetric diblock copolymers (Series I) show that all samples with styrene contents in the SBR block larger than 27 mol% do microphase separate. This is an expected finding for symmetric diblock copolymers with incompatible components. Interestingly, long range order and morphology are practically unaffected by the relatively harsh vulcanization procedure. The observed morphologies and periodicities D are comparable to those observed for the corresponding diblock copolymers in the non-crosslinked state, as indicated by small angle X-ray scattering experiments (cf. Table 1 and ESI†). AFM images for samples with lower styrene contents in the SBR block (21 and 27 mol% S) show a completely different behavior. A lamellar morphology is definitively absent in this case. This can be clearly seen in the zoomed insets on their respective AFM images. The zooms indicate that both diblock copolymers are in the disordered state due to increased compatibility of PB and SBR blocks. One can conclude that a transition from a well-ordered microphase-separated state to disorder occurs in this sample series, under the given preparation conditions, somewhere between 35 and 27 mol% S in the SBR block. Note that larger amounts of additives migrating from the bulk and crystallizing on the surface are present in both disordered samples. Zoomed images were made in regions where these migrated additives are practically absent (insets in Fig. 1). | ||
| Fig. 1 AFM phase contrast images for crosslinked symmetric PB–SBR diblock copolymers (Series I) with styrene contents in the SBR block varying from 21 to 52 mol% (upper row) and asymmetric block copolymer samples (Series II) with SBR volume fractions in the range 20 vol% ≤ ΦSBR ≤ 69 vol% (lower row). The image size is commonly 2 × 2 μm2 (except ΦSBR = 69 vol% with 2 × 1 μm2). The scale bar with a length 500 nm is applicable for all images. For the zoomed images the scale bar has a length of 200 nm. The white spots are additives migrating from the bulk to the surface after cryo-microtoming. | ||
AFM images of asymmetric PB–SBR copolymers (Series II) with volume fractions in the range 30 vol% ≤ ΦSBR ≤ 60 vol% show well microphase-separated states with different morphologies (Fig. 1, lower row). Hexagonally packed SBR cylinders embedded in a PB matrix (ΦSBR = 30 vol%), lamellar morphologies (ΦSBR = 38 and 50 vol%), or PB domains dispersed in the SBR phase (ΦSBR = 60 vol%) are observed depending on the volume fraction. Note that in the latter case neither a lamellar nor a cylindrical morphology is seen although microphase separation obviously occurs. One can speculate that a non-equilibrium morphology is frozen during processing at high temperatures (mixing at 70 °C to 110 °C and crosslinking at 150 °C). This might be due to a temperature-dependent transition from lamellae to cylinders during heating combined with kinetic hindrance in a block copolymer with limited mobility under the used preparation conditions. AFM images for the crosslinked diblock copolymer with ΦSBR = 20 vol% indicate that this sample is in the disordered state. Long range order and clear indications for microphase-separated domains are missing. Similar features occur for the diblock copolymer with ΦSBR = 69 vol%. However, in this case certain concentration gradients seems to be also present although clear evidences for long range order are missing. This may be related to concentration fluctuation in the disordered state frozen during the crosslinking step. Morphology studies on the diblock copolymer with ΦSBR = 20 vol% are again impaired by the presence of additives on its surface.
Relaxation data from dynamic mechanical analysis
Temperature-dependent data for shear storage modulus G′(T) and loss modulus G′′(T) measured at a fixed frequency (ω = 10 rad s−1) for crosslinked samples of Series I, Series II and the correspondingly prepared reference samples (PB and S30B70R) are presented in Fig. 2. Symmetric diblock copolymers (Series I) with xS,SBR ≥ 35 mol% show two independent relaxation processes at low and high temperatures corresponding to the dynamic glass transitions of both phases, αPB and αSBR, respectively (Fig. 2a). This can be understood as further evidence for strong segregation of both phases, PB and SBR, for high styrene contents in the SBR block. Expectedly, the αSBR relaxation process shifts systematically to higher temperatures with increasing xS,SBR value like known for random SBR copolymers. Only the symmetric samples with the lowest styrene contents in the SBR block, PB50–S21B79R50 and PB50–S27B73R50, depict different behavior. The only relaxation process observed for the PB50–S21B79R50 sample is slightly shifted towards higher temperatures compared to the αPB processes for strongly segregated samples. A separate αSBR process at higher temperatures is missing in this case. This can be interpreted as an indication for a high degree of miscibility of PB and SBR blocks, i.e. the existence of basically one single phase in accordance with structural information from AFM (Fig. 1). In case of PB50–S27B73R50, the major α relaxation peak in G′′(T) is shifted to higher temperature compared to the αPB peaks for strongly segregated diblock copolymers with larger styrene contents. However, a weak shoulder is observed at higher temperatures (near −45 °C) indicating probably the existence of SBR rich domains. One may understand this relaxation behavior as a consequence of a structure with PB rich domains containing a certain fraction of styrene units combined with a lot of interfacial material with higher concentration of styrene. Consequently, there are pronounced styrene concentration gradients in the sample as indicated also in the AFM image. This should lead to a broader distribution of α relaxation times with two maxima corresponding to PB rich and SBR rich domains where the latter should have a higher Tg corresponding to longer α relaxation times under isothermal conditions. Further details will be considered in the discussion section.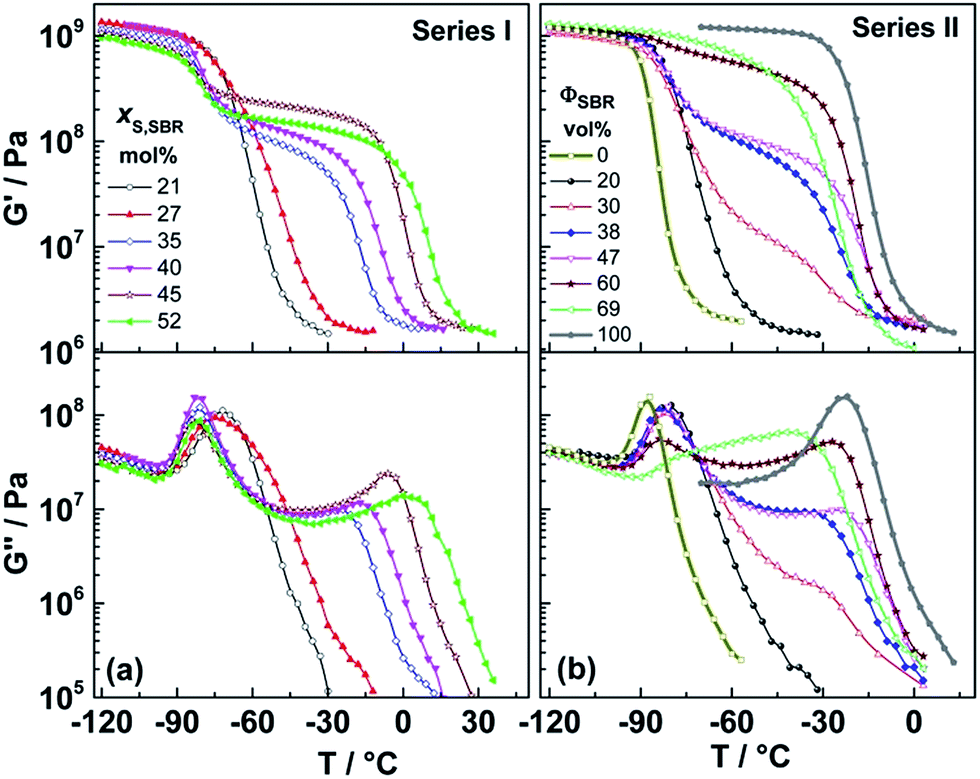 | ||
| Fig. 2 Temperature-dependence of the shear storage (G′) and loss (G′′) moduli for crosslinked samples of (a) Series I with variable styrene content in the SBR block and (b) Series II with different volume fraction of the SBR block including S30B70R and PB as reference systems. | ||
Shear measurements for diblock copolymers of Series II with volume fractions in the range 30 vol% ≤ ΦSBR ≤ 60 vol% (Fig. 2b) show two well-separated main relaxation processes at about −80 °C (αPB) and at about −25 °C (αSBR). This is the typical relaxation behavior of diblock copolymers in the strong segregation limit. The intensity of both relaxation processes, αPB and αSBR, varies systematically with ΦSBR although the intensity is not expected to be directly proportional to the volume fraction. Note that a certain scatter in the Tα,SBR10 rads−1 values (ESI, Table A1†) corresponding to the maximum position of αSBR peak in G′′(T) is mostly due to a slight variation of xS,SBR during synthesis (Table 1). Quite different behavior is observed for the diblock copolymers of Series II with pronounced asymmetries (PB31–S35B65R69 and PB80–S30B70R20). Sample PB31–S35B65R69 depicts a broad bimodal relaxation process with two maxima in G′′(T) located at temperatures between those of the αPB and αSBR processes in strongly segregated members of this series. This evidences the absence of a really pure PB and SBR domains and instead indicates the existence of a large amount of intermixed material. The relaxation behavior of the PB80–S30B70R20 sample remembers to that of a well miscible diblock copolymer. The peak maximum of the main α relaxation process in G′′(T) is slightly shifted towards higher temperatures compared to that of the αPB process in case of strongly segregated systems. However, weak contributions of SBR rich domains which are only too low to be detectable as a separated peak or shoulder can be hardly excluded.
Finally, we should note that the relaxation temperatures Tα,PB10 rad s−1 of all strongly segregated and crosslinked PB–SBR diblock copolymers are about 7 K higher than the corresponding value of the crosslinked PB reference (Fig. 2). Possibly, this indicates a higher crosslinking density of the PB phase in PB–SBR diblock copolymers since it is well known that Tg,PB of pure PB homopolymers increases with crosslinking density.27 The fact that the DSC glass temperatures Tg,PB of the PB phase in non-crosslinked PB–SBR copolymers are similar to that of PB homopolymer (ESI, Table A1†) may support this hypothesis.
In order to understand further details of the relaxation behavior, isothermal shear experiments in the frequency range 0.1–100 rad s−1 are carried out in a broad temperature interval including both α relaxation processes. Fig. 3 shows the resulting master curves constructed by shifting the isotherms horizontally along the log frequency axis assuming that the shape of the relaxation spectrum is temperature-independent as predicted by the time temperature superposition principle (TTS). The reference temperature is in all cases −60 °C. This shifting procedure neglects differences in the temperature dependence of the relaxation processes αPB and αSBR dominating in different temperature ranges but gives a first overview of the relaxation behavior of the investigated systems. The scatter in the data between both dynamic glass transitions is basically due to the fact that the temperature dependence of αPB and αSBR is significantly different. Thus, the isotherms do not superimpose well. However, the master curves confirm the main trends in the relaxation behavior discussed above based on the isochrones. In Series I the shift of αSBR relaxation towards lower frequencies with increasing styrene content in the SBR block is clearly seen (Fig. 3a) although details should be influenced by the violation of the TTS as discussed below. The relaxation strength of the αSBR processes is very similar for all strongly segregated systems (xS,SBR ≥ 35 mol%). In case of Series II, the most important changes in the relaxation behavior with block copolymer composition are also confirmed by the master curves (Fig. 3b). It is clearly seen that the relaxation strength of the αSBR process (ΔG′SBR) decreases systematically with decreasing volume fraction ΦSBR. The position of both relaxation processes, αPB and αSBR, is weakly influenced for well microphase-separated blocks (38 vol% ≤ ΦSBR ≤ 60 vol%). The strong shift of the αSBR process for ΦSBR = 30 vol% relative to the others is at least partly artificial and a consequence of the violations of the TTS as confirmed by a more detailed evaluation below. A single relaxation process located close to αPB is expectedly observed for the disordered sample PB80–S30B70R20. A bimodal peak in G′′(T) with strong relaxation modes at intermediate frequencies is found for PB31–S35B65R69 supporting the absence of pure PB and SBR phases.
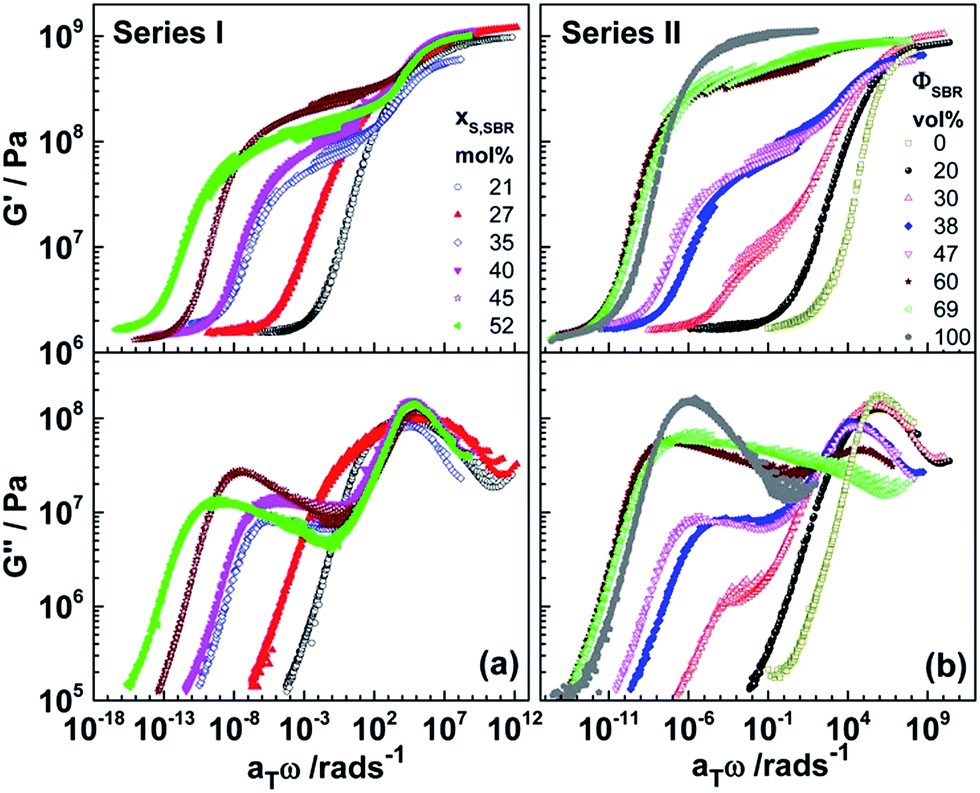 | ||
| Fig. 3 Master curves of the shear storage (G′) and loss (G′′) moduli for crosslinked samples of (a) Series I with variable styrene content xS,SBR and (b) Series II with different volume fraction ΦSBR. Data for S30B70R and PB samples are shown for comparison. All master curves are constructed using −60 °C as reference temperature. | ||
In a final step, the information about the temperature dependence of the average relaxation times (τα = ωα−1) of both segmental relaxation processes, αPB and αSBR, is extracted from shear data for strongly segregated diblock copolymers measured in a broad frequency–temperature range. The shifting behavior of both α relaxation processes was evaluated independently assuming that the segmental dynamics of both phases, PB and SBR, is independent. Individual shift factors aT,PB and aT,SBR are determined by decomposing the isotherms in parts belonging to αPB and αSBR, respectively. These pre-evaluated isotherms are used to construct two independent master curves giving individual shift factors aT,PB and aT,SBR related to the temperature dependence of the average relaxation frequencies ωα,PB and ωα,SBR, respectively. Another approach to learn more about the temperature dependence of ωα,PB and ωα,SBR is to determine the relaxation temperatures, Tα,PBω and Tα,SBRω, corresponding to the maxima of the αPB and αSBR relaxation peaks in G′′(T) isochrones measured at different frequencies in the range 0.1 rad s−1 ≤ ω ≤ 100 rad s−1 (Fig. 2).
Arrhenius plots combining such relaxation temperatures, Tα,PBω and Tα,SBRω, with shift factors, aT,PB and aT,SBR, obtained from a horizontal shift of decomposed isotherms are shown in Fig. 4. One can clearly see that the temperature dependencies of ωα,PB and ωα,SBR are quite different. The temperature dependence of the shift factors is commonly well approximated by the Vogel–Fulcher–Tammann (VFT) equation.28,29 The fit parameters are given in Table A1 in the ESI.† Considering the temperature-dependent data for the αPB relaxations for all strongly segregated samples it can be directly seen in Fig. 4 that the traces (more or less) coincide. This holds for Series I as well as Series II. Thus, one can conclude that the softening behavior of the butadiene segments in PB domains of well microphase-separated block copolymers is basically domain shape and domain size independent. Note that the obtained ωα,PB traces are also in reasonable agreement with those for the corresponding PB homopolymer with similar microstructure although a certain vertical shift is obvious in the Arrhenius plot. The temperature dependencies are obviously identical although the average relaxation frequencies are significantly different. This finding corresponds to the already mentioned fact that the Tα,PB10 rad s−1 values for crosslinked block copolymers are commonly a bit higher compared to the value for the crosslinked PB homopolymer.
 | ||
| Fig. 4 Arrhenius diagram showing shift factors aT (open symbols) and relaxation temperatures Tαω taken from G′′(T) isochrones (full symbols) for the relaxation processes, αPB and αSBR, of crosslinked diblock copolymers of (a) Series I with variable styrene content xS,SBR and (b) Series II with different volume fractions ΦSBR. Data for S30B70R and PB homopolymer are shown for comparison. The symbols and colors correspond to those used in Fig. 2 and 3. The reference temperature was adapted in such a way that the aT values coincide with Tαω. The lines are VFT fit curves for the shift factors. | ||
One can also see in Fig. 4 that the relaxation temperatures Tα,SBR10 rad s−1 in Series I systematically increase with increasing styrene content as long as the samples are well microphase-separated. A systematic shift of the individual αSBR traces to higher temperatures appears in the Arrhenius diagram as the styrene content increases. This is expected based on the findings for random SBR copolymers. This effect is accompanied by a certain change in the overall temperature dependence of the relaxation frequencies. The Tα,SBR10 rad s−1 values, the αSBR traces as well as the VFT fitting parameters (cf. ESI, Table A1†) for the SBR phase of strongly segregated asymmetric samples (Series II) in the range 30 vol% ≤ ΦSBR ≤ 60 vol% are quite comparable (although weak differences due to variation of styrene content in the SBR block are indicated). The data for the αSBR process in these diblock copolymers are also comparable with the results for the S30B70R copolymer used as reference.
Discussion
PS–SBR diblock copolymer phase behavior
The influence of the styrene content in the SBR block on miscibility can be systematically studied based on Series I. AFM images and shear data clearly demonstrate that symmetric PB–SBR diblock copolymers with xS,SBR ≥ 35 mol% styrene are in the microphase separated state showing long-range order even after vulcanization. Despite of high shear forces applied at processing temperatures between 70 °C and 150 °C, these diblock copolymers show a nice lamellar morphology like expected for incompatible symmetric diblock copolymers (in equilibrium). On the other hand, the samples of Series I with xS,SBR ≤ 27 mol% are obviously not microphase-separated but in a disordered state. Thus, one can conclude that depending on the styrene content in the SBR block an order–disorder–transition (ODT) occurs in the range 27 mol% ≤ xS,SBR ≤ 35 mol%. This seems to be reasonable since the incompatibility of both blocks, PB and SBR, is reduced if xS,SBR decreases. However, it is interesting and highly application relevant to predict at which styrene content in the SBR block the ODT occurs for processed and crosslinked PB–SBR diblock copolymers.Equilibrium predictions for non-crosslinked diblock copolymers can be made based on conventional thermodynamic approaches.30 Leibler's weak segregation theory predicts that the ODT occurs for symmetric diblock copolymers at (χN)c = 10.5.8 In order to describe the equilibrium phase behavior of PS–SBR diblock copolymers with variable styrene contents in the SBR block an effective interaction parameter χeff has to be introduced. A thermodynamic model taking into account the individual pair interactions between styrene (S), 1,2-vinyl (V) and 1,4-cis/trans (B) units has been reported by Sakurai et al.31 based on experimental data for SBR/PB blends. According to their phase separation studies on blends an effective interaction parameter
| χeff = kφS,SBRχVS + (φS,SBR − k)φS,SBRχBS − k(φS,SBR − k)χVB | (1) |
Based on eqn (1) as well as on χVS, χBS and χVB, the segregation strengths χeffN of the symmetric diblock copolymer samples of Series I has been calculated at 25 °C and 150 °C (Fig. 5). As expected there is a strong increase of the segregation strength χeffN with increasing styrene volume fraction in the SBR block φS,SBR. An interpolation of the calculated χeffN data at 150 °C predicts an ODT (χeffN = 10.5) at about φS,SBR = 0.31 corresponding to xS,SBR ∼ 21 mol%. This prediction is in a reasonable agreement with AFM results for Series I (Fig. 1) where clear indications of disorder and a trend towards miscibility are seen for the samples B50–S21B79R50 (xS,SBR = 21 mol%) and B50–S27B73R50 (xS,SBR = 27 mol%). Based on this model predictions the sample B50–S27B73R50 (χeffN = 15.6) should be close to the order–disorder transition during the vulcanization process at 150 °C while the sample B50–S21B79R50 (χeffN = 9.8) should be disordered under identical conditions. Considering further details one should note that the true value of (χN)c might be slightly higher than 10.5 if fluctuation corrections are taken into account.32 This shows that the model used to calculate the effective interaction parameter χeff (eqn (1)) is describing the phase separation behavior for symmetric diblock copolymers with a random SBR block relatively well. Note that we assume here that the microphase-separated state occurring at 150 °C is fixed without further changes during the vulcanization process. This is understandable if we consider that the crosslinking reaction is much faster than reorganization processes in a block copolymer at temperatures below the TODT.
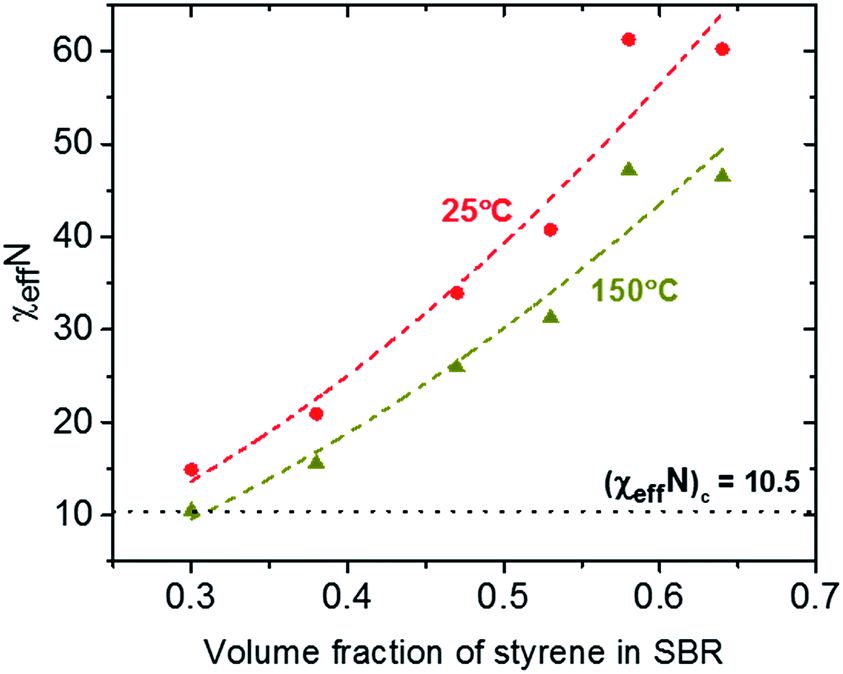 | ||
| Fig. 5 Segregation strength χeffN as a function of the volume fraction of styrene in the SBR block for the diblock copolymers of Series I. The total chain length N was determined based on the total molecular weight values and the effective interaction parameter χeff was calculated at 25 °C (triangles) and 150 °C (circles) using eqn (1). The dashed lines are an interpolation of the data using a quadratic fit. The dotted line represents the critical value (χeffN)c = 10.5 at the ODT. | ||
The influence of volume fraction ΦSBR on the morphology can be seen considering the results for asymmetric PB–SBR diblock copolymers of Series II. The samples with volume fractions in the range 30 vol% ≤ ΦSBR ≤ 60 vol% show morphologies similar to those obtained for ordinary non-crosslinked diblock copolymers in the strong segregation limit under equilibrium conditions. For ΦSBR = 38 vol% and 50 vol% lamellar morphologies are obtained, for ΦSBR = 30 vol% a cylindrical morphology is observed and for ΦSBR = 60 vol% a transition from lamellae to cylinders seems to occur after heating the sample to the vulcanization temperature (150 °C). The sample PB80–S30B70R20 with ΦSBR = 20 vol% seems to be disordered with quasi-miscible PB and SBR blocks. The sample PB31–S35B65R69 with ΦSBR = 69 vol%, shows also disorder but concentration fluctuations seem to occur which are frozen during the crosslinking process at 150 °C.
In general, the microphase-separated states obtained for the investigated crosslinked diblock copolymers with random SBR block seems to be well in-line with phase diagrams known for conventional non-crosslinked diblock copolymers composed of two homopolymer blocks. Thermodynamic equilibrium concepts can be obviously applied successfully since the state occurring at the vulcanization temperature is probably fixed by fast crosslinking without significant changes in the morphology caused by previous processing steps or due to crosslinking.
Influence of microstructure and microphase-separated morphology on the segmental α dynamics
Styrene content and butadiene microstructure of SBR are known to influence the glass temperature and α relaxation dynamics systematically.33 Since the styrene content in the SBR block is systematically varied in Series I while the butadiene microstructure is basically kept constant, corresponding behavior is expected in this case. The dependence of the thermal (non-crosslinked samples) and dynamic (crosslinked samples) glass transition temperatures of the SBR block, Tg,SBR and Tα,SBR10 rad s−1, on its styrene content xS,SBR is shown in Fig. 6 for strongly segregated systems of Series I. Both data sets were fitted to the phenomenological Gordon–Taylor equation describing the softening behavior of random copolymers depending on composition33| Tg,SBR = (Tg,PSwS,SBR + ATg,PB(1 − wS,SBR)/(wS,SBR + A(1 − wS,SBR)) | (2) |
 | ||
| Fig. 6 Tg,SBR of non-crosslinked samples (open symbol) and Tα,SBR10 rad s−1 of crosslinked samples (full symbols) depending on the styrene content wS,SBR of the SBR block for well microphase separated samples of Series I (circles) and Series II (stars). The dashed and dotted lines are independent Gordon–Taylor fits for the Tg,SBR and Tα,SBR10 rad s−1 data of Series I. The inset shows a zoomed version of the central part of the plot. | ||
Tg,SBR values for non-crosslinked samples as well as Tα,SBR10 rad s−1 for crosslinked samples of well microphase-separated members of Series II with styrene contents in the SBR block of about 32 ± 4 mol% are shown for comparison in Fig. 6. The values are in-line with the findings for Series I. Tg,SBR and Tα,SBR10 rad s−1 scatter within a small window what is mainly due to the slight variation of styrene concentration in the SBR block, as already mentioned before. In general, the softening behavior of the SBR blocks for strongly segregated PB–SBR diblock copolymers nicely confirms the trends known from corresponding random copolymers depending on the styrene content.
The influence of the block copolymer morphology on the segmental α dynamics can be nicely studied in the investigated PB–SBR diblock copolymers showing a large variety of structural states fixed during the vulcanization step. There are (i) various samples in the strongly segregated state with different morphologies and domain sizes, there are (ii) samples close to the order–disorder transition (weakly segregated or disordered systems with pronounced concentration gradients frozen during vulcanization) with a large fraction of interfacial material and there are (iii) quasi miscible diblock copolymers. Moreover, there is information about (iv) the relaxation dynamics of a corresponding random S30B70R copolymer. This is an excellent opportunity to analyze the influence of domain size, domain shape, and local chemical composition in domains and interphases on the cooperative dynamics seen in particular as segmental α relaxation processes.
Considering case (i), i.e., strongly segregated PB–SBR diblock copolymers, we see clear evidence for α relaxation dynamics of the blocks which is quite similar to that of pure bulk materials with the same chemical composition. This holds for the PB block if compared to PB homopolymer of identical microstructure. This observation is also valid for the SBR blocks if compared with random SBR copolymers with identical styrene and 1,2-vinyl contents. Interestingly, the influence of domain shape (layer-like or cylindrical) seems to be weak. Materials in well defined, long range ordered systems and somehow disturbed, less perfect morphologies (e.g. PB40–S35B65R60) behave identical as long as the phases are strongly segregated. Moreover, there is no obvious influence of domain size on the cooperative α dynamics as long as the domains have dimensions of about 20–30 nm like in the investigated samples. Although there is a certain Tg difference compared to bulk samples there is no evidence for significant effects due to geometrical confinement. This finding is not really new but important in the light of the ongoing controversial debate about changes in the α relaxation dynamics and Tg in ultrathin films.34–37 Our data indicate, that significant size-dependent changes in Tg are basically absent in self-assembled block copolymers with domain sizes > 20 nm while larger changes have been reported in self-assembled alkyl nanodomains having sizes in the range 1–3 nm.38,39 This observation is somehow also in-line with many reports on the softening behavior of glass-forming guest systems in nanoporous hosts40–42 and supports the observation that purely geometrical confinement effects can change the α dynamics only in extremely small domains of size of a few nanometers.
Considering case (ii), i.e., PB–SBR copolymers close to the order–disorder transition, interfacial material becomes much more important. In the relaxation spectra this is indicated by a higher intensity of the relaxation modes between both segmental relaxation processes, αPB and αSBR. Obviously, there is a certain fraction of material having intermediate Tg's corresponding to α relaxation times which are in between those of the pure phases. In this respect the material in interphases is behaving like random copolymers with different styrene contents indicating that the chemical composition is important for the α dynamics. Very pronounced effects of the same type are seen for so called gradient copolymers containing practically only interfacial material without pure phases.43
In case (iii), i.e., for quasi miscible block copolymers like the sample B50–S21B79R50, we observe a softening behavior which is similar to that in case (iv), i.e., for a random copolymer with identical content of co-units. The position of the single peak at Tα10 rad s−1 = −71.9 °C for B50–S21B79R50 containing in total 9.68 mol% styrene is almost the same than that of a random S10B90R copolymer with styrene content of 10 mol% having a Tα,SBR10 rad s−1 = −71.8 °C according to Gordon–Taylor approximation shown in Fig. 5. This can be understood as a clear hint for miscibility and a practically homogeneous distribution of the SBR chains in the PB matrix. However, the question remains what “homogenous” means in this context and at which length scale the α relaxation dynamics is influenced by a non-homogenous distribution of the styrene units. From our point of view, this is a special way to ask the question for a characteristic length of the glass transition as proposed in the related literatures.44–46 If there are cooperatively rearranging regions (CRRs) with a typical dimension ξα in the 2–3 nanometer range as proposed for example by the fluctuation approach to the glass transition,46,47 the α relaxation dynamics should change as soon as the average chemical composition (co-unit contents) in a volume Vα = ξα3 is varying in different spatial regions.21 Hence, this should be the criterion to discriminate whether or not a system is homogeneous from the viewpoint of the α relaxation dynamics. If this is not the case, domains with different softening behavior start to appear and α relaxation process is significantly affected. Based on this idea the interference of static length scales (domain size, interfacial width) and dynamic length scales (CRR sizes) is of major importance for an understanding of the cooperative α dynamics in block copolymers in different states (Fig. 7).
 | ||
| Fig. 7 Sketches showing snapshots of the chemical composition of differently structured copolymers containing butadiene (red) and styrene (blue) monomers together with the cooperatively rearranging regions (CRR) (having a characteristic length ξα) related to a fluctuating density pattern. The three presented situations are (a) a strongly and (b) a weakly segregated PB–SBR diblock copolymer as well as (c) a random SBR copolymer with similar overall composition. | ||
Considering the cases discussed above, one can conclude that the observed behavior is at least qualitatively in agreement with the predictions of this physical picture and with ξα values in the range of a few nanometers. For very strongly segregated systems the contributions of the interfacial material to the relaxation spectrum should be small (of the order of 20 vol% for the domain sizes considered here) since the number of subsystems of size Vα where the chemical average composition is different from that of both pure phases are rare (Fig. 7a). Hence, we observe in this case basically two α relaxations corresponding to those of both pure phases and only weak contributions to the relaxation spectrum in-between these processes. For weakly segregated block copolymers and systems close to the ODT, however, the contributions of material in interphases are very important. In that case there are more subsystems with a size Vα having different chemical average compositions and intermediate α relaxation times (Fig. 7b). Hence, we find strong relaxation modes related to the interphase between both dynamic glass transition processes, αPB and αSBR, representing the segmental dynamics of the pure phases. Finally, one single α relaxation process is observed if the block copolymer system is well miscible or if we consider random copolymers, where each subsystem of size Vα has the same chemical average composition (Fig. 7c). Consequently, only one α relaxation peak corresponding to this composition is found. Note that this is a criterion defining an internal length scale at which a multi component system must be chemically homogenous before a spatially uniform α dynamics can be expected. This length scale is related to ξα and this criterion is not (a priory) identical with that what is defining the ODT in block copolymers from the thermodynamic and structural point of view. From this perspective it is also not surprising and understandable that in several “miscible” blends two α relaxations have been observed above the ODT where concentration fluctuations and gradients are still present.48–51
In summary, we understand the experimental findings for differently structured PB–SBR diblock copolymers reported in this work as a certain evidence for the existence of a characteristic lengths scale ξα of the glass transition in the 1–3 nm range. Without such a length scale it is hard to explain the dynamically different behavior of the investigated diblock copolymers containing different amounts of interfacial material. This insight can be further used to fine-tune relaxation behavior and dissipation in complex composite materials like those used for tire treads.
Conclusions
The influence of local chemical composition and interfacial material as well as structural parameters like domain size and domain shape on the cooperative α dynamics has been studied based on two series of PB–SBR diblock copolymers with different composition. In Series I, the styrene content in the SBR block is varied in the range 21–52 mol% for a given volume fraction of ΦSBR ≈ 50 vol%, comparable total number molecular weight of 200 kg mol−1 and fixed 1,2-vinyl content in the butadiene sequences. This leads for higher styrene contents (35–52 mol% styrene) to systems with lamellar morphology but different segregation strength χeffN and variable amount of interfacial material. Disorder and increasing miscibility are only indicated for the lowest styrene contents under consideration (21 and 27 mol% styrene). This confirms the predictions of a thermodynamic model describing the PB–SBR interaction based on an effective Flory–Huggins interaction parameter χeff derived for PB/SBR blends in the literature. Series II contains PB–SBR diblock copolymers with variable volume fraction (20 vol% ≤ ΦSBR ≤ 69 vol%) and fixed styrene content in the SBR block (32 ± 4 mol% styrene) resulting in well microphase-separated systems with different morphologies as well as quasi miscible states. Interestingly, the morphology is practically in all investigated cases weakly influenced by the processing and vulcanization procedures at 150 °C showing the robustness of the self-assembling process in block copolymers.A careful inspection of the relaxation behavior shows that (i) the relaxation behavior of strongly segregated diblock copolymers is dominated by the α relaxations of the pure phases being similar to those of the corresponding bulk polymers, (ii) interfacial material majorly contributes at temperatures and frequencies between αPB and αSBR relaxations in weakly segregated or disordered systems close to the ODT, while (iii) quasi-miscible diblock copolymers show like random copolymers only one α relaxation process at intermediate temperatures and frequencies. These findings and the transitions between the different model situations can be explained by the variation of the average chemical composition of small subsystems determining the segmental α dynamics. The size of the relevant subsystems is defined by the volume of a cooperatively rearranging region Vα = ξα3 with ξα = 1–3 nm being the characteristic length scale for the cooperative segmental dynamics related to the dynamic glass transition α. The presented results indicate how the relaxation behavior of self-assembled diblock polymers can be efficiently influenced and fine-tuned towards the requirements for tire tread applications.
Acknowledgements
The authors thank Goodyear S. A. for allowing us to publish the data shown, Dr Benoit Duez for the technical help and the Fonds National de la Recherche in Luxembourg (FNR) for financial support.Notes and references
- A. N. Gent and J. D. Walter, Mechanics of pneumatic tires, in National Highway Traffic Safety Administration, Washington D.C., 2005 Search PubMed.
- M. Wang, KGK, Kautsch. Gummi Kunstst., 2007, 60, 438–443 CAS.
- M. Wang, KGK, Kautsch. Gummi Kunstst., 2008, 61, 33–42 Search PubMed.
- G. Thielen, KGK, Kautsch. Gummi Kunstst., 2007, 60, 389–392 CAS.
- T. Kawazura, M. Kawazoe, Y. Kikuchi, T. Nakamura, M. Nakamura and T. Karato, US Patent 6,355,728, 2002.
- T. Yasumasa, S. Mitsuhiko, T. Fumio, T. Akio and H. Iwakazu, European Patent 0134909, 1988.
- H. Feng, X. Zhang and S. Zhao, J. Appl. Polym. Sci., 2008, 110, 228–236 CrossRef CAS.
- L. Leibler, Macromolecules, 1980, 13, 1602–1617 CrossRef CAS.
- F. S. Bates and G. H. Fredrickson, Annu. Rev. Phys. Chem., 1990, 41, 525–557 CrossRef CAS PubMed.
- I. W. Hamley, The physics of block copolymers, Oxford University Press, 1998 Search PubMed.
- M. Alcoutlabi and G. B. McKenna, J. Phys.: Condens. Matter, 2005, 17, R461–R524 CrossRef CAS.
- M. Arndt, R. Stannarius, H. Groothues, E. Hempel and F. Kremer, Phys. Rev. Lett., 1997, 79, 2077–2080 CrossRef CAS.
- F. Kremer, A. Huwe, M. Arndt, P. Behrens and W. Schwieger, J. Phys.: Condens. Matter, 1999, 11, A175–A188 CrossRef CAS.
- C. Schick and E. Donth, Phys. Scr., 1991, 43, 423–429 CrossRef CAS.
- J. A. Forrest and K. Dalnoki-Veress, Adv. Colloid Interface Sci., 2001, 94, 167–196 CrossRef CAS.
- C. J. Ellison, M. K. Mundra and J. M. Torkelson, Macromolecules, 2005, 38, 1767–1778 CrossRef CAS.
- S. F. Swallen, P. A. Bonvallet, R. J. McMahon and M. D. Ediger, Phys. Rev. Lett., 2003, 90, 015901 CrossRef PubMed.
- A. Serghei, M. Tress and F. Kremer, J. Chem. Phys., 2009, 131, 154904 CrossRef CAS PubMed.
- V. M. Boucher, D. Cangialosi, A. Alegria, J. Colmenero, J. Gonzalez-Irun and L. M. Liz-Marzan, Soft Matter, 2010, 6, 3306–3317 RSC.
- A. Papon, K. Saalwächter, K. Schäler, L. Guy, F. Lequeux and H. Montes, Macromolecules, 2011, 44, 913–922 CrossRef CAS.
- E. Donth, The Glass Transition. Relaxation Dynamics in Liquids and Disordered Materials, Springer, Berlin, 2001 Search PubMed.
- C. A. da Silva, M. Weydert, M. Menzel, U. Wendler, M. Bartke and M. Beiner, Rubber Chem. Technol., 2016 DOI:10.5254/rct.16.84842.
- J. E. Mark, Physical properties of polymers handbook, Springer, 2007 Search PubMed.
- Y. Tanaka, H. Sato, K. Saito and K. Miyashita, Rubber Chem. Technol., 1981, 54, 685–691 CrossRef CAS.
- A. Hofmann, A. Alegria, J. Colmenero, L. Willner, E. Buscaglia and N. Hadjichristidis, Macromolecules, 1996, 29, 129–134 CrossRef CAS.
- R. Zorn, F. I. Mopsik, G. B. McKenna, L. Willner and D. Richter, J. Chem. Phys., 1997, 107, 3645–3655 CrossRef CAS.
- M. Mansilla, A. R. Garraza, L. Silva, W. Salgueiro, C. Macchi, A. Marzocca and A. Somoza, Polym. Test., 2013, 32, 686–690 CrossRef CAS.
- M. L. Williams, R. F. Landel and J. D. Ferry, J. Am. Chem. Soc., 1955, 77, 3701–3707 CrossRef CAS.
- H. Vogel, Phys. Z., 1921, 22, 645–646 CAS.
- F. S. Bates, Science, 1991, 251, 898–905 CrossRef CAS PubMed.
- S. Sakurai, T. Izumitani, H. Hasegawa, T. Hashimoto and C. C. Han, Macromolecules, 1991, 24, 4844–4851 CrossRef CAS.
- G. H. Fredrickson and E. Helfand, J. Phys. Chem., 1987, 87, 697–705 CrossRef CAS.
- M. Gordon and J. S. Taylor, J. Appl. Chem., 1952, 2, 493 CrossRef CAS.
- J. Forrest and K. Dalnoki-Veress, Adv. Colloid Interface Sci., 2001, 94, 167–196 CrossRef CAS.
- A. Serghei and F. Kremer, Macromol. Chem. Phys., 2008, 209, 810–817 CrossRef CAS.
- C. Ellison and J. Torkelson, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 2745–2758 CrossRef CAS.
- H. Yin, S. Napolitano and A. Schönhals, Macromolecules, 2012, 45, 1652–1662 CrossRef CAS.
- M. Beiner and H. Huth, Nat. Mater., 2003, 2, 595–599 CrossRef CAS PubMed.
- K. L. Ngai and M. Beiner, Macromolecules, 2004, 37, 8123–8127 CrossRef CAS.
- C. L. Jackson and G. B. McKenna, J. Non-Cryst. Solids, 1991, 131, 221–224 CrossRef.
- M. Arndt, R. Stannarius, H. Groothues, E. Hempel and F. Kremer, Phys. Rev. Lett., 1997, 79, 2077–2080 CrossRef CAS.
- A. Schönhals, H. Goering, C. Schick, B. Frick and R. Zorn, Eur. Phys. J. E, 2003, 12, 173–178 CrossRef PubMed.
- M. M. Mok, S. Pujari, W. R. Burghardt, C. M. Dettmer, S. T. Nguyen, C. J. Ellison and J. M. Torkelson, Macromolecules, 2008, 41, 5818–5829 CrossRef CAS.
- G. Adam and J. H. Gibbs, J. Chem. Phys., 1965, 43, 139–146 CrossRef CAS.
- L. Berthier, G. Biroli, J. P. Bouchaud, L. Cipelletti, D. El Masri, D. L'Hote, F. Ladieu and M. Pierno, Science, 2005, 310, 1797–1800 CrossRef CAS PubMed.
- E. Donth, J. Non-Cryst. Solids, 1982, 53, 325–330 CrossRef CAS.
- E. Hempel, G. Hempel, A. Hensel, C. Schick and E. Donth, J. Phys. Chem. B, 2000, 104, 2460–2466 CrossRef CAS.
- J. Dudowicz, J. F. Douglas and K. F. Freed, J. Chem. Phys., 2014, 140, 244905 CrossRef PubMed.
- J. Colmenero and A. Arbe, Soft Matter, 2007, 3, 1474–1485 RSC.
- D. Cangialosi, A. Alegria and J. Colmenero, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2007, 76, 011514 CrossRef CAS PubMed.
- D. Cangialosi, G. A. Schwartz, A. Alegria and J. Colmenero, J. Chem. Phys., 2005, 123, 144908 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Small angle X-ray scattering (SAXS) data for non-crosslinked diblock copolymer samples are provided and results from a detailed analysis of temperature-dependent α relaxation times are presented. See DOI: 10.1039/c6ra06786g |
| This journal is © The Royal Society of Chemistry 2016 |