DOI:
10.1039/C6RA04916H
(Paper)
RSC Adv., 2016,
6, 48779-48787
Structural, electronic and magnetic properties of metal–organic-framework perovskites [AmH][Mn(HCOO)3]: a first-principles study
Received
24th February 2016
, Accepted 10th May 2016
First published on 11th May 2016
Abstract
We calculate the structural, electronic and magnetic properties of the subgroup of Metal–Organic-Frameworks (MOFs) [AmH][M(HCOO)3] (in which AmH+ = organic ammonium cation, M = divalent metal ion) using density functional theory with GGA+U approximation. The optimized structures and magnetic ground states are in good agreement with available experimental results. The electronic structures of these MOFs are obtained at their magnetic ground states. Using hybrid functional method (HSE06), the band gap is 4.33 eV, 4.12 eV, 4.15 eV and 4.78 eV for NH2NH3+, HONH3+, CH3CH2NH3+ and NH4+ compounds, respectively. The band gap of NH2NH3+ varies from 2.63 eV (−5% compressive strain) to 3.50 eV (+5% tensile strain) at Ueff = 4 eV. It is demonstrated that the band gap of such MOFs can be easily tuned by applying external strain and the AmH+ ligand for the first time. These MOFs all show insulating properties. In addition, such strain engineering may also be useful for enhancing the Neel temperature by changing the distance of magnetic Mn ions. Interestingly, Bader charge analysis indicates that AmH+ is fully ionic suggesting that appropriate arrangement may give rise to polar order associated with the magnetic ordering, these MOFs materials can be considered as potential multiferroics. Finally, this work reveals that both strain and chemical modification are efficient approaches for designing improved and novel MOFs for future applications in photocatalytic, optoelectronic, ferroelectric or multiferroic and electronic device.
1. Introduction
In past decades, metal–organic framework (MOF) materials, consisting of organic ligands, metal ions and the connecting formate, have attracted considerable attention because of their multifunctional properties and potential applications, such as gas storage,1 catalysis,2 photocatalysis,3 luminescence,4,5 magnetism,6 sensors7,8 and electronic devices.9,10 [AmH][M(HCOO)3] (with AmH+ = organic ammonium cation, M = divalent metal ion) is a classical subgroup of MOFs. They are formed into frameworks of metal ions and formate HCOO−, in which cation ligands are located in their channels. Many MOFs of this type have been synthesized, such as [(CH3)2NH2][M(HCOO)3] (M = Mn, Co, Ni, Fe and Zn),11–14 [NH4][M(HCOO)3] (M = Mn, Fe, Co, Ni, Zn and Mg),15–18 [HONH3][M(HCOO)3] (M = Mn, Co, Ni, Zn and Mg),19 [NH2NH3][M(HCOO)3] (M = Mn, Zn, Co and Mg)20 or [AmH][Mn(HCOO)3] (AmH = CH3NH3+, CH3CH2NH3+, (CH3)2NH2+ and (CH2)3NH2+).14 [AmH][M(HCOO)3] have also been investigated experimentally as a new type of multiferroic materials because of their structural diversity and chemical versatility. Jain et al.13 have found that an antiferroelectric order and a weakly ferromagnetic order coexist below 8–36 K in [(CH3)2NH2][M(HCOO)3] with M = Mn, Fe, Co and Ni. Gao et al.18,20 have also reported multiferroic properties in the [NH2NH3][M(HCOO)3] and [NH4][M(HCOO)3] (M = Mn, Fe, Co and Ni) systems. In the NH4+ compound, the ferroelectric or antiferroelectric order is triggered by the order-disorder transition at 191–254 K. Recently, this class of MOFs can be optimized in terms of magnetic, ferroelectric and dielectric properties through mixing ammonium species and/or substituting B-site metal ions. The introduction of nonpolar CH3NH3+ into [NH2NH3][Mn(HCOO)3] decreases framework distortions and phase transition temperature from 355 K to 285 K, depending on CH3NH3+ content.21 On the other hand, the mixed-metal ions of MOFs have been studied mainly in terms of structural, thermal, dielectric properties as well as IR, Raman and luminescence properties by Maczka et al., such as trivalent cations doped-[(CH3)2NH2][M(HCOO)3] (M = Mg, Mn and Co),22 [C2H5NH3][Na0.5Fe0.5(HCOO)3],23 [(CH3)2NH2][FeIIIMII(HCOO)6] (MII = Zn, Ni, Cu, Fe, Mg),24,25 [(CH3)2NH2][Na0.5Cr0.5(HCOO)3].26 Recently, luminescent properties of europium or chromium-doped in such multiferroic MOFs were identified,22,26 which may open a new route for novel potential applications in sensors and solid-state lighting devices. While the spin-canted weak ferromagnetism may originate from antiferromagnetic or ferromagnetic couplings on the corresponding metal sites connected by formate bridge, HCOO−. Unfortunately, the Neel temperature (TN) of these MOFs is often very low. For instance, the highest Neel temperature obtained in [(CH3)2NH2][Ni(HCOO)3] reported so far is only about 36 K, which hinders their multiferroic applications in the future.27
Theoretical simulations are mainly concentrated on ferroelectric polarization in this subclass of MOFs. Stroppa et al.28,29 have investigated that multiferroic properties and strong magnetoelectric coupling do exist in [C(NH2)3][Cu/Cr(HCOO)3] using Berry phase method based on space group analysis. It is shown that Jahn–Teller effect which is active for Cu2+ or Cr2+ ions plays an important role for getting strong magnetoelectric coupling in these MOFs. On the other hand, Sante et al.30,31 have proposed that it is possible to tune the ferroelectric polarization by mediating the magnitude and/or the canting of the organic molecular dipoles as well as epitaxial strain. For instance, 4% compressive strain can enhance the ferroelectric polarization by more than 300% in [C(NH2)3]Cr(HCOO)3. In this series of [NH4][M(HCOO)3], higher phase transition temperature of [NH4][Mg(HCOO)3] is attributed to stronger hydrogen-bonding interactions between NH4+ ligands and oxygen atoms and hardly related to the local environment of the metal center.32 Considering this, the study of electronic structure of these MOFs offer some useful insights for polarization in ferroelectric materials, such as BaTiO3 system.33 The investigation of electronic structure may exploit their other applications in electronic device, sensor, photocatalysis. As a matter of fact, it is thus desirable to modulate the electronic structure of MOFs by modifying the organic linker, the metal center or adding guest molecule as well as external strain. Here, we consider four typical [AmH+][Mn(HCOO)3] with (a) AmH+ = NH2NH3+, (b) AmH+ = HONH3+, (c) AmH+ = CH3CH2NH3+ and (d) AmH+ = NH4+ and investigate their structural, electronic and magnetic properties using density functional theory. Our predictive results on these specific MOFs indicate that strain engineering can modulate the band gap and magnetic interaction which is related to Neel temperature. Bade charge analysis reveals that the polarization mechanism of these MOFs is originated from the ionic properties of the AmH+ ligands.
2. Computational details
All calculations are implemented using the Vienna Ab Initio Simulation Package (VASP)34,35 with the Perdew–Burke–Ernzerhof (PBE) Gradient Generalized Approximation (GGA) functional36 for the exchange–correlation. The cutoff energy is 400 eV and a 3 × 4 × 2 Monkhorst–Pack grid of k-points is used. Spin-polarized effects are considered in all simulations. Ions are relaxed towards their equilibrium positions until the Hellman–Feynman force is less than 1 meV Å−1. For Mn (3d) MOFs, the strong on-site Coulomb repulsion is modified by considering the Hubbard U correction proposed by Dudarev et al.37 In our simulations, an effective Hubbard U parameter Ueff (Ueff = U − J) is utilized for calculating electronic structure. We test the effects of Mn Ueff values with respect to lattice parameters and compare with the band gap of other MOFs. The total energies are obtained at different magnetic configurations in order to determine the magnetic ground states.
3. Results and discussions
3.1. Structural properties
The structures obtained experimentally using powder X-ray diffraction analysis are used as starting models for bulk [AmH+][Mn(HCOO)3]14,18–20 (8 Mn atoms in NH2NH3+; HONH3+; CH3CH2NH3+; 12 Mn atoms in NH4+) and the unit cell volume and atomic positions are then fully optimized with the GGA+U method. The [NH2NH3+][Mn(HCOO)3], [HONH3+][Mn(HCOO)3] and [CH3CH2NH3+][Mn(HCOO)3] compounds belong to an orthorhombic system. Both NH2NH3+ and CH3CH2NH3+ compounds have a polar space group (Pna21), while space group (P212121) of HONH3+ compound is nonpolar. [NH4+][Mn(HCOO)3] compound crystallizes into a hexagonal symmetry, and belongs to a polar space group (P63). The four unit cells are shown in Fig. 1. NH2NH3+ and CH3CH2NH3+ compounds crystallize into the same anionic perovskite-framework of [Mn(HCOO)3] with the cubic cavities being occupied by NH2NH3+ and CH3CH2NH3+cations, respectively. The optimized equilibrium lattice parameters of these four compounds are listed in Tables 1 and 2, along with the available experimental values published in the literature. The volume of AmH+ cations decreases as following: CH3CH2NH3+ > NH2NH3+ > HONH3+ > NH4+. The calculated lattice constants of the three orthorhombic compounds are all smaller than their corresponding experimental values. The volume of NH2NH3+, HONH3+ and CH3CH2NH3+ are 7.5%, 10.5% and 4.2% smaller than their experimental values, respectively. However, in the hexagonal NH4+ system, the calculated volume is 4.6% bigger than its experimental value. Although the relaxation process induces volume expansion or contraction, these compounds maintain its initial space group system.
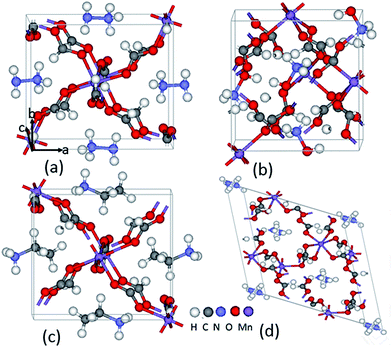 |
| | Fig. 1 Schematic view of the structure of [AmH+][Mn(HCOO)3] (AmH+ = (a) NH2NH3+; (b) HONH3+; (c) CH3CH2NH3+; (d) NH4+). | |
Table 1 Calculated and experimental structural parameters for [AmH+][Mn(HCOO)3] (AmH+ = NH2NH3+; HONH3+; CH3CH2NH3+; NH4+)
| AmH+ |
a (Å) |
b (Å) |
c (Å) |
c/a |
α (°) |
β (°) |
γ (°) |
V (Å3) |
| Ref. 20. Ref. 19. Ref. 14. Ref. 18. |
| NH2NH3+ |
Pna21 |
Cal. |
17.24 (8.62) |
7.72 |
11.36 |
1.32 |
90 |
90 |
90 |
1511.59 (755.80) |
| Exp.a |
8.9319 |
7.82 |
11.70 |
1.31 |
90 |
90 |
90 |
816.61 |
| HONH3+ |
P212121 |
Cal. |
15.30 (7.65) |
7.74 |
12.40 |
1.62 |
90 |
90 |
90 |
1467.13 (733.57) |
| Exp.b |
7.81 |
7.96 |
13.17 |
1.69 |
90 |
90 |
90 |
819.27 |
| CH3CH2NH3+ |
Pna21 |
Cal. |
17.85 (8.93) |
8.14 |
11.84 |
1.33 |
90 |
90 |
90 |
1720.05 (860.03) |
| Exp.c |
9.09 |
8.22 |
12.03 |
1.32 |
90 |
90 |
90 |
897.92 |
| NH4+ |
P63 |
Cal. |
25.68 (12.84) |
12.84 |
8.70 |
0.68 |
90 |
90 |
120 |
2483.02 (1241.51) |
| Exp.d |
12.67 |
12.67 |
8.54 |
0.67 |
90 |
90 |
120 |
1186.61 |
Table 2 Calculated and experimental bond distance and bond angle for [AmH+][Mn(HCOO)3] (AmH+ = NH2NH3+; HONH3+; CH3CH2NH3+; NH4+)
| |
dMn–O (Å) |
cis-O–Mn–O (°) |
trans-O–Mn–O (°) |
dMn–OCHO–Mn (Å) |
dN–O (Å) |
dN–H–O (Å) |
| Ref. 20. Ref. 19. Ref. 14. Ref. 18. |
| NH2NH3+ |
Cal. |
2.16–2.18 |
84.5–96.5 |
178.8 |
5.72–5.77 |
2.75–2.79 |
162.7–166.9 |
| 2.91 |
106.3–113.0 |
| Exp.a |
2.178–2.196 |
86.37–94.43 |
176.1–179.1 |
5.908–5.951 |
2.876–2.926 |
164.9–173.4 |
| 2.990–3.110 |
114.5–121.7 |
| HONH3+ |
Cal. |
2.13–2.21 |
86.0–100.0 |
164.0–169.0 |
5.68–5.73 |
1.42 |
161.6–170.2 |
| 2.72–2.85 |
| Exp.b |
2.15–2.24 |
79.87–97.17 |
167.58–173.80 |
5.945–6.071 |
1.405 |
159.1–168.2 |
| 2.79–3.00 |
| CH3CH2NH3+ |
Cal. |
2.17–2.21 |
87.1–95.7 |
175.1–178.2 |
5.95–6.00 |
2.81 |
121.0; 153.3; 139.1 |
| Exp.c |
2.177–2.218 |
87.68–95.07 |
175.05–178.22 |
6.037–6.155 |
— |
175.6; 124.2; 170.7; 155.6; 139.9 |
| NH4+ |
Cal. |
2.22–2.24 |
81.7–98.7 |
168.5–169.0 |
6.10 |
2.84–2.86 |
176.6 |
| 3.17–3.24 |
100.2 |
| Exp.d |
2.168–2.194 |
81.62–97.95 |
169.26–169.44 |
6.005 |
2.824–2.867 |
171–173 |
| 3.084–3.152 |
104.9–105.8 |
In these framework structures, each Mn ion connects to six adjacent Mn ions, forming an octahedral spatial arrangement, through six anti–anti formates, thus constructing a topological metal–formate framework, while the ligand cations accommodate the channel sites. For NH2NH3+, the Mn–O distances are equal to 2.16–2.18 Å and the cis-O–Mn–O angles are ranging within 84.5–96.5° interval while trans-O–Mn–O angles are about 178.8°. The framework grid or cavity has Mn–OCHO–Mn edges of 5.72–5.77 Å. For CH3CH2NH3+, the Mn–O distances are in a large range of 2.17–2.21 Å. The longer Mn–OCHO–Mn distances of 5.95–6.00 Å originate from the fact that the CH3CH2NH3+ cation is larger than that of NH2NH3+ cation. The cation forms N–H–O and weak C–H–O hydrogen bonds with the framework. The Mn–O–CH–O–Mn linkages in the framework are parallel to the c axis as well as the two ab diagonal directions in the unit cell. After relaxation, HONH3+ structure remains nonpolar with orthorhombic space group P212121. It possesses a 3D anionic [Mn(HCOO)3] chiral metal–organic framework of rare 49.66 topology, in which HONH3+ cations are arranged, head to tail, in a zigzag manner along the b directional channel. Each octahedral Mn ion is linked to six neighboring Mn ions through the anti–anti formate, forming a trigonal prism. That is different from the octahedral spatial arrangement around Mn metals in polar NH2NH3+ and CH3CH2NH3+ compounds. The MnO6 octahedron has Mn–O distances of 2.13–2.21 Å with cis-O–Mn–O angles and trans-O–Mn–O angles in the interval 86.0–100.0° and 164.0–169.0°, respectively. The channel's wall consists of a bidirectional triple helix of ⋯Mn–OCHO–Mn–OCHO⋯. In the HONH3+ cations, the NH3 end points toward the HO end along the slight tilted b-axis direction with the N–O, H–N and H–O distances being equal to 1.42 Å, 1.05 Å and 1.02 Å, respectively. Each H–O (N–H) donor forms a pair of H–O (N–H)⋯OHCOO, one short and one long distance, to the anionic metal formate framework. The NHONH3–OHCOO and OHONH3–OHCOO distances are 2.89 Å (short) and 3.14 Å (long), and 3.18 Å and 4.09 Å, respectively. The formate-bridged Mn⋯Mn distances are equal to 5.68–5.73 Å. For NH4+ compound, the ab-plane is rhombus, with the NH4+ cations located at three different channels at (0,0,z), (2/3,1/3,z) and (1/3,2/3,z). Each NH4+ cation contains one apical and three basal N–H groups. Each basal N–H group forms stronger H bond to the metal formate framework, with N–O distances of 2.84–2.86 Å and N–H–O angles of 176.6°. The apical N–H group, with the N–H directing to the c axis, still forms a trifurcated acceptor-type H bond, with N–O distances of 3.17–3.24 Å and N–H–O angles of 100.2°. Compared to experimental values, our calculated structures (Tables 1 and 2) show that a good agreement is obtained to support our modeling approach.
3.2. Magnetic properties
The different magnetic configurations are schematized in Fig. 2. We calculate energy differences with respect to the ferromagnetic configuration as well as magnetic exchange interaction energies of Mn atoms for all four compounds at both Mn Ueff = 0 and 4 eV, listed in Table 3. By comparing the total energy of each compound in the four different magnetic configurations considered (i.e. antiferromagnetic (AFM) states with G-type, C-type and A-type arrangements as well as ferromagnetic configuration), it is found that NH2NH3+ and CH3CH2NH3+ compounds are both stable in G-type AFM configuration, while A-type AFM configuration is favored in HONH3+ and NH4+ compounds. All the four compounds keep the same magnetic ground states at Mn Ueff = 0 and 4 eV, although the magnetic interaction strength becomes weaker with increasing of the Mn Ueff value. Previous results using density functional theory calculations obtained by Sante et al.31 have also suggested that CH3CH2NH3+ compounds displays a G-type AFM configuration, which is in good agreement with our finding. Our simulations are also supported by previous experimental results showing that all the compounds exhibit antiferromagnetic properties.14,18–20 Considering the magnetic moment for Mn ion, in case of CH3CH2NH3+ compound, the calculated value is about 4.5 μB using GGA functional with Mn Ueff being zero. Within GGA+U method, the magnetic moment increases linearly with increasing of Ueff value towards the high-spin magnetic moment of Mn2+ ions that is 5.0 μB/Mn, displayed in Fig. 3(b). Similar phenomena38,39 have also been observed in various systems like FeAs or Sr2FeOsO6, indicating that the GGA+U method leads to an enhancement of the magnetic moment through coulomb interactions. In contrast, Kanungo et al.39 have preferred to use GGA functional instead of GGA+U to study the exchange coupling of transition metal ions arguing that it is a better choice for determining the magnetic exchange. In our work, the magnetic ground states at Ueff = 0 and 4 eV are consistent with all the four investigated compounds, see Table 3. Neutron diffraction and further magnetic measurements are desirable to verify the detailed antiferromagnetic configurations.
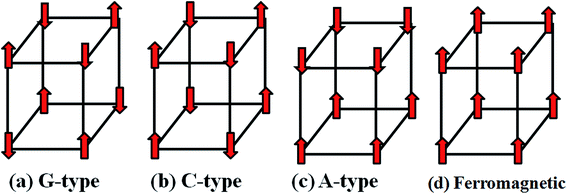 |
| | Fig. 2 Magnetic configurations of [AmH+][Mn(HCOO)3] ((a) NH2NH3+; (b) HONH3+; (c) CH3CH2NH3+; (d) NH4+). | |
Table 3 Calculated energy differences with respect to the ferromagnetic configuration and magnetic interaction strength of these four compounds at Mn Ueff = 0 and 4 eV as well as [NH2NH3][Mn(HCOO)3] under 5% compressive and tensile strain at Mn Ueff = 0 eV. The energy unit is meV
| |
Mn Ueff |
G-type |
C-type |
A-type |
FM |
J1 |
J2 |
| NH2NH3+ |
0 |
−118.674 |
−76.385 |
−26.890 |
0 |
19.0966 |
6.7225 |
| 4 |
−24.820 |
−17.405 |
−8.829 |
0 |
4.3513 |
−2.2073 |
| Comp005 |
0 |
−14.057 |
16.740 |
152.032 |
0 |
−4.185 |
−38.008 |
| Ten005 |
0 |
−106.811 |
−46.537 |
−42.889 |
0 |
11.634 |
10.722 |
| HONH3+ |
0 |
−56.591 |
−42.112 |
−96.159 |
0 |
10.5280 |
24.0398 |
| 4 |
−19.935 |
−18.315 |
−31.554 |
0 |
4.5788 |
7.8885 |
| CH3CH2NH3+ |
0 |
−136.265 |
−105.133 |
−55.257 |
0 |
26.2833 |
13.8143 |
| 4 |
−24.551 |
−16.671 |
−12.985 |
0 |
4.1678 |
3.2463 |
| NH4+ |
0 |
−50.825 |
−78.584 |
−108.824 |
0 |
19.6460 |
27.2060 |
| 4 |
−1.931 |
−16.689 |
−18.150 |
0 |
4.1723 |
4.5375 |
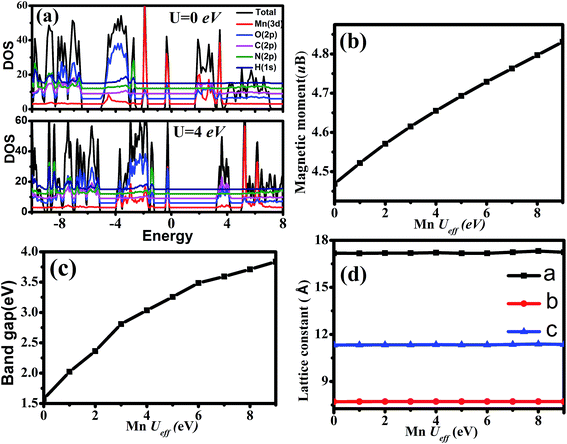 |
| | Fig. 3 (a) The total density of states of [NH3NH2+][Mn(HCOO)3] in G-type antiferromagnetic configuration at Mn Ueff = 0 eV and 4 eV; the relationship between (b) magnetic moment; (c) band gap; (d) lattice constant with increasing of Mn Ueff. | |
According to Goodenough–Kanamori–Anderson rules,40 superexchange interaction of neighboring magnetic ions (Mn) via HCOO and ligand cation plays a leading role in determining their magnetic order. In NH2NH3+, the distance of Mn–HCOO–Mn in ab-plane and c-axis are nearly equal to 5.72–5.77 Å (and weakly affected, if any, by Ueff values, see Fig. 3(d)). The Mn atoms are coupled antiferromagnetically in both the ab-plane and along the c axis. Therefore, for NH2NH3+ and CH3CH2NH3+ compounds, the ground states display a G-type antiferromagnetic (AFM) spin configuration. For NH4+ compound, the Mn–HCOO–Mn along the c-axis direction has a distance of 6.10 Å, while the nearest distance of Mn–Mn in the ab-plane is 7.35 Å. So the superexchange interaction along the c-axis is of AFM type and the magnetic interaction in the ab-plane is ferromagnetic in nature, which satisfies A-type AFM configuration and similarly in HONH3+ compound.
Here, we study the effective exchange interaction among Mn ions with the aim of providing insights about how to increase Neel temperature (TN) of this class of MOFs. Based on the Heisenberg model, the magnetic configurations are usually determined by two parameters J1 and J2; which are the exchange interaction strengths of nearest neighbors and second nearest neighbors, respectively. J1 and J2 are either positive or negative depending on ferromagnetic or antiferromagnetic interactions, respectively. Molecular field theory is utilized to qualitatively estimate Neel temperature of this subgroup of MOFs where TN is approximately given by 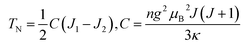 , here n is the number of atoms per unit volume; κ is Boltzmann's constant; μB is Bohr magnetron; g is Lande factor; J is the total angular momentum. According to molecular field theory, TN is related to the difference between J1 and J2. However, whatever the compounds considered with either G-type or A-type magnetic arrangement, TN remains rather modest suggesting that (i) whatever the nature (antiferromagnetic or ferromagnetic) of the interaction for the second nearest neighbors this does not affect TN and thus J2 may be considered as negligible and (ii) J1 remains small in agreement with the low TN temperature reported experimentally. The Mn–OCHO–Mn superexchange magnetic interactions are related to the angles and distances between magnetic Mn sites. In case of NH2NH3+ compound, the distance of nearest neighbor Mn–Mn is 5.7 Å while that of second nearest neighbor is 7.7 Å, and the angle of Mn–OCHO–Mn is 140°. The magnetic interaction strength of NH2NH3+ compound under 5% compressive and tensile strain is displayed in Table 3. It is found that 5% compressive strain enhance the difference of exchange interaction strength of nearest neighbors J1 and second nearest neighbors J2 to get an increase of Néel temperature. This challenge might be addressed by applying a compressive stress by clamping mechanically the MOFs onto a substrate through strain engineering to reinforce the magnetic interactions and finally increase Neel temperature. The Neel temperature of these four MOFs is about 8 K,14,18–20 which indicates that A-ligands offer a slight effect on Neel temperature. In contrast, the introduction of mixed metal atoms may enhance the Neel temperature. For instance, the Neel temperature of [(CH3)2NH2][FeIIINiII(HCOO)6] is up to 42 K.41 As we shall see later on, such strain engineering approach is also powerful for tuning the band gap.
, here n is the number of atoms per unit volume; κ is Boltzmann's constant; μB is Bohr magnetron; g is Lande factor; J is the total angular momentum. According to molecular field theory, TN is related to the difference between J1 and J2. However, whatever the compounds considered with either G-type or A-type magnetic arrangement, TN remains rather modest suggesting that (i) whatever the nature (antiferromagnetic or ferromagnetic) of the interaction for the second nearest neighbors this does not affect TN and thus J2 may be considered as negligible and (ii) J1 remains small in agreement with the low TN temperature reported experimentally. The Mn–OCHO–Mn superexchange magnetic interactions are related to the angles and distances between magnetic Mn sites. In case of NH2NH3+ compound, the distance of nearest neighbor Mn–Mn is 5.7 Å while that of second nearest neighbor is 7.7 Å, and the angle of Mn–OCHO–Mn is 140°. The magnetic interaction strength of NH2NH3+ compound under 5% compressive and tensile strain is displayed in Table 3. It is found that 5% compressive strain enhance the difference of exchange interaction strength of nearest neighbors J1 and second nearest neighbors J2 to get an increase of Néel temperature. This challenge might be addressed by applying a compressive stress by clamping mechanically the MOFs onto a substrate through strain engineering to reinforce the magnetic interactions and finally increase Neel temperature. The Neel temperature of these four MOFs is about 8 K,14,18–20 which indicates that A-ligands offer a slight effect on Neel temperature. In contrast, the introduction of mixed metal atoms may enhance the Neel temperature. For instance, the Neel temperature of [(CH3)2NH2][FeIIINiII(HCOO)6] is up to 42 K.41 As we shall see later on, such strain engineering approach is also powerful for tuning the band gap.
3.3. Electronic properties
3.3.1 Density of states. We firstly study the effect of the different A-site ligands (NH2NH3+, HONH3+, CH3CH2NH3+ and NH4+) on the electronic structure of these MOFs. As traditional GGA functional neglects the on-site Coulomb interactions of transition metal (Mn) and underestimates seriously the band gap, the GGA+U method is performed in this work. As the band gap of these four MOFs are experimentally unavailable, we considered other classic MOFs (i.e. MOF-5) to obtain a suitable Mn Ueff correction for our calculations. Taking Ueff = 4 eV, the band gap of the four MOFs ranges from 3.12 eV to 3.56 eV (see Table 4) which are close to those of MOF-5 references.42–44 Let us take the NH2NH3+ compound as an example to describe the relationship between band gap, magnetic moment, lattice constants and the effective Hubbard Mn Ueff correction, as shown in Fig. 3. In Fig. 3(b)–(d), it is shown that both band gap and magnetic moment of NH2NH3+ compound increase almost linearly with increasing of the Mn Ueff value. For examples, the band gap increases from 1.64 eV (with Ueff = 0 eV) to 3.12 eV (with Ueff = 4 eV) and to 3.84 eV (with Ueff = 9 eV); the magnetic moment reaches 4.84 μB with Mn Ueff = 9 eV while it is 4.66 μB with Ueff = 4 eV and 4.47 μB with Ueff = 0 eV. Similar magnetic features were observed in MnO by Franchini et al.45 the magnetic moment of MnO is 4.67 μB with Mn Ueff = 6 eV, which is comparable with our results. Interestingly, the lattice constants are only slightly, if any, affected with increasing of Mn Ueff value, presented in Fig. 3(d). Fig. 3(a) displays the total density of states (TDOS) of NH2NH3+ compound at Mn Ueff = 0 eV and 4 eV, respectively. The bottom of conduction band is pushed away from the Fermi level in the TDOS when Mn (Ueff) is increased from 0 eV to 4 eV, which induces a larger band gap. In case of Mn Ueff = 0 eV, the conduction band consists of Mn 3d, C 2p and O 2p orbitals, while the valance band mainly originates from Mn 3d and O 2p orbitals. For Mn Ueff = 4 eV, the big difference in TDOS shows that Mn 3d electrons become more localized and are pushed away from the bottom of the conduction band.
Table 4 Band gap of [AmH+][Mn(HCOO)3] (AmH+ = (a) NH2NH3+; (b) HONH3+; (c) CH3CH2NH3+; (d) NH4+) at Mn Ueff = 0 eV and 4 eV as well as hybrid functional (HSE06)
| Mn Ueff |
NH2NH3+ |
HONH3+ |
CH3CH2NH3+ |
NH4+ |
| 0 |
1.64 |
1.23 |
1.80 |
1.71 |
| 4 |
3.12 |
3.03 |
3.27 |
3.56 |
| HSE06 |
4.33 |
4.12 |
4.15 |
4.78 |
Total density of states (TDOS) and partial density of states (PDOS) of the four MOFs in their antiferromagnetic configuration with Ueff = 4 eV are displayed in Fig. 4 and 5, respectively. The band gap is 3.12 eV, 3.03 eV, 3.27 eV and 3.56 eV for NH2NH3+, HONH3+, CH3CH2NH3+ and NH4+ compounds, respectively. In order to obtain accurate band gap, hybrid functional method (HSE06) is performed, as shown in Table 4. In NH2NH3+, the band gap is 4.33 eV. For the case of IRMOF-1,46 the band gap using HSE06 is 4.66 eV, larger than the experimental band gap of 3.4 eV. For MOFs, hybrid functional may overestimate the band gap. In summary, these MOFs are good insulators. Observing partial density of states, a small bandwidth in the lower part of the conduction band is observed for all MOFs, except NH4+ compound. At Mn Ueff = 4 eV, these four compounds exhibit similar density of states. The Mn 3d and O 2p orbitals contribute mainly to the valence band, while the conduction band consists of C 3d and N 2p orbitals. In Fig. 5(d) of NH4+, the H 1s and N 2p orbitals are weakly hybridized with the C 2p and O 2p orbitals near Fermi level, which indicates that ammonium exist weak interaction with the Mn–OCHO framework. It is clear that the C 2p orbitals have stronger hybridization of O 2p orbitals at the bottom of valance band. So HCOO− is more stable than ammonium like NH4+, which is easily rotated, distorted and moved under external stimulus such as high temperature, high pressure. Such results offer some insights to the mechanism of polarization and phase transitions originating from A-site ligands. Two possible methods are suggested to modulate the band gap of these MOFs: (1) our results show that the A-site ligands induce the change of electronic structure, suggesting that it is also possible to modifying the band gap through shifting the conduction band through implantation of foreign molecule.47 (2) Doping with metal atoms may shift the valence band to the Fermi level.
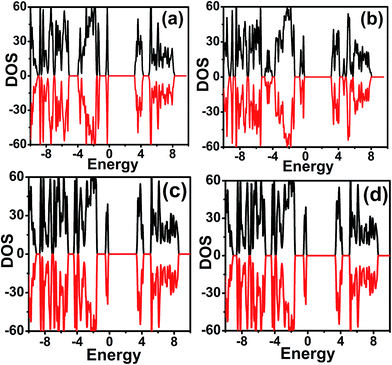 |
| | Fig. 4 Total density of states of [AmH+][Mn(HCOO)3] at Ueff = 4 eV ((a) NH2NH3+; (b) HONH3+; (c) CH3CH2NH3+; (d) NH4+). | |
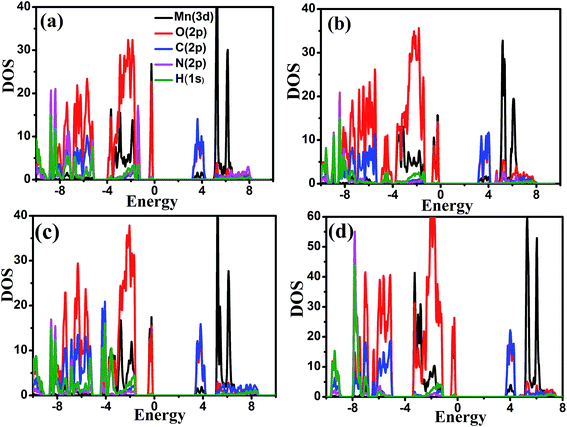 |
| | Fig. 5 Partial density of states of [AmH+][Mn(HCOO)3] at Ueff = 4 eV ((a) NH2NH3+; (b) HONH3+; (c) CH3CH2NH3+; (d) NH4+). | |
3.3.2 Band gap modulation with strain engineering. Next, we investigate another strategy to tune the electronic properties of NH2NH3+ compound, i.e. the application of compressive or tensile strain. Recently, thin films of MOFs48–53 have been fabricated to study their multifunctional properties. The mismatch of the lattices between MOFs and substrates can indeed affect the mechanical, electronic, and magnetic properties of the materials. In this work, in-plane strain is applied biaxially on the orthorhombic and hexagonal unit cell of NH2NH3+ compound. The in-plane strain is defined as ε = Δa/a0, where a0 and Δa + a0 are the unit cell parameters without and with external strain, respectively (ε < 0 stands for compressive strain, ε > 0 represents tensile strain). The effect of strain (from −5% compressive strain to +5% tensile strain) on the band gap at Ueff = 4 eV is plotted in Fig. 6. The Fermi energy level is marked at zero. The band gap of NH2NH3+ compound increases from 2.63 eV under −5% compressive strain to 3.50 eV under +5% tensile strain at Ueff = 4 eV. The modulation of band gap arises mainly from the shift of the conduction band (CBM), the valence band (VBM) is influenced slightly. Compared to previous studies,54,55 the band gap of MOFs such as MIL-53, MOF-5 can be modulated through hydrostatic pressure or internal (external) pressure. In MIL-53-Al materials,55 the bang gap increases from 3.6 eV to 4.6 eV under tensile pressure, which also indicates that tensile strain increases the band gap. Therefore, strain engineering is revealed to be an powerful tool for modifying the electronic structure by shifting the conduction band in these MOFs.
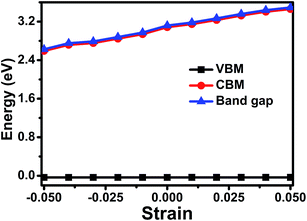 |
| | Fig. 6 Band gap, valence band (VBM), conduction band (CBM) of [NH2NH3][Mn(HCOO)3] at Ueff = 4 eV under external strain. | |
3.4. Bader charge analysis
The investigation of Bader charges help us obtain information on electrons arrange of the compounds by revealing charge transfer, chemical state and so on. Bader charges are calculated using Bader's atoms-in-molecules theory.56 For the four compounds (see Table 5), both the Bader charges of Mn atoms and strength of the bond are nearly same whatever the AmH+ ligands, which is consistent with the close distances of Mn–O bond in these four compounds. The Bader charges of H in HCOO− and AmH+ are completely different for all the four compounds. The Bader charge of H is 0.07–0.26 e, 0.074–0.088 e, −0.03–0.21 e and 0.02–0.175 e in HCOO− for NH2NH3+, HONH3+, CH3CH2NH3+ and NH4+ compounds, respectively. As a result, the H atom transfers its charge to the C atom in HCOO−, regardless of the type of the compounds giving rise to a more covalent C–H bond. However, all the charge of H atoms in AmH+ are 1 e, implying that there is no charge transfer. The complete ionic bond in AmH+ indicates that the AmH+ cation is very soft and suggesting that the cations can be ordered and/or disordered using external parameters like temperature, pressure and electric field. As a consequence, the AmH+ cations by creating electrical dipoles may arrange into a polar state that might exhibit ferroelectric and/or antiferroelectric behavior associated with magnetic ordering, which makes these MOFs become novel multiferroic materials.
Table 5 Bader charges of [AmH+][Mn(HCOO)3] (AmH+ = (a) NH2NH3+; (b) HONH3+; (c) CH3CH2NH3+; (d) NH4+)
| |
HCOO− |
AmH+ |
| Mn |
H |
C |
O |
N |
C |
O |
H |
| NH2NH3+ |
1.64 |
0.07–0.26 |
2.52–2.73 |
−1.79 to −1.82 |
−1.88 |
— |
— |
1.00 |
| −2.33 |
| HONH3+ |
1.66 |
0.074–0.088 |
2.70–2.74 |
−1.77 to −1.82 |
−2.12 |
— |
−1.12 |
1.00 |
| |
| CH3CH2NH3+ |
1.64 |
0.12, 0.21, 0.04, −0.05, −0.03 |
2.66–2.71 |
−1.77 to −1.85 |
−2.96 |
0.12 |
— |
1.00 |
| 0.60 |
| NH4+ |
1.63 |
0.02–0.175 |
2.67–2.78 |
−1.79 to −1.86 |
−3.178 |
— |
— |
1.00 |
| −3.183 |
4. Conclusions
In summary, a comprehensive study is conducted to investigate structural, electronic and magnetic properties of a subgroup of MOFs using density functional theory with GGA+U method. The calculated lattice constants are in good agreement with previous results. The magnetic ground states are determined through comparing the energy difference of different magnetic configurations. Using hybrid functional method (HSE06), the band gap is 4.33 eV in NH2NH3+, 4.12 eV in HONH3+, 4.15 eV in CH3CH2NH3+ and 4.78 eV in NH4+, respectively. The band gap of NH2NH3+ at Ueff = 4 eV decreases from 2.63 eV to 3.50 eV with increasing external strain (from −5% compressive to +5% tensile strain). The band gap in these four insulating MOFs is modulated via changing the AmH+ ligands or external strain. Bader charge analysis shows that the AmH+ ligands are purely ionic, which is helpful to elucidate the physical mechanism of ferroelectric polarization. We analyze magnetic interaction of Mn ions, it is expected that the low Neel temperature of these MOFs may be increased by mediating the distance of magnetic metal atoms using external strain or changing the metal atoms. Our results suggest thus that appropriate design of these MOFs may make them potential applications in novel multifunctional devices.
Acknowledgements
This work was supported by the National Science Foundation of China (NSFC No. 51372195, 41372055, 11204230 and 51390472), the Ministry of Science and Technology of China through a 973-Project (No. 2012CB619401, No. 2012CB619402, No. 2014CB644003 and No. 2015CB654903), the Fundamental Research Funds for the Central Universities (2013JDGZ03), and Grant No. IRT13034. X. J. Lou would like to thank the “One Thousand Youth Talents” program for support. Y. Liu and B. Dkhil acknowledge the China Scholarship Council (CSC) for funding Y. Liu's stay in France.
References
- L. J. Murray, M. Dincă and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- M. Nasalevich, M. Vander Veen, F. Kapteijn and J. Gascon, CrystEngComm, 2014, 16, 4919–4926 RSC.
- M. Maczka, A. Sieradzki, B. Bondzior, P. Deren, J. Hanuza and K. Hermanowicz, J. Mater. Chem. C, 2015, 3, 9337–9345 RSC.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353–1379 RSC.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2011, 112, 1105–1125 CrossRef PubMed.
- M. D. Allendorf, R. J. Houk, L. Andruszkiewicz, A. A. Talin, J. Pikarsky, A. Choudhury, K. A. Gall and P. J. Hesketh, J. Am. Chem. Soc., 2008, 130, 14404–14405 CrossRef CAS PubMed.
- M. D. Allendorf, A. Schwartzberg, V. Stavila and A. A. Talin, Chem.–Eur. J., 2011, 17, 11372–11388 CrossRef CAS PubMed.
- V. Stavila, A. Talin and M. Allendorf, Chem. Soc. Rev., 2014, 43, 5994–6010 RSC.
- X. Wang, L. Gan, S. W. Zhang and S. Gao, Inorg. Chem., 2004, 43, 4615–4625 CrossRef CAS PubMed.
- P. Jain, N. S. Dalal, B. H. Toby, H. W. Kroto and A. K. Cheetham, J. Am. Chem. Soc., 2008, 130, 10450–10451 CrossRef CAS PubMed.
- P. Jain, V. Ramachandran, R. J. Clark, H. D. Zhou, B. H. Toby, N. S. Dalal, H. W. Kroto and A. K. Cheetham, J. Am. Chem. Soc., 2009, 131, 13625–13627 CrossRef CAS PubMed.
- Z. Wang, B. Zhang, T. Otsuka, K. Inoue, H. Kobayashi and M. Kurmoo, Dalton Trans., 2004, 2209–2216 RSC.
- M. Mączka, A. Pietraszko, B. Macalik and K. Hermanowicz, Inorg. Chem., 2014, 53, 787–794 CrossRef PubMed.
- Z. Wang, B. Zhang, K. Inoue, H. Fujiwara, T. Otsuka, H. Kobayashi and M. Kurmoo, Inorg. Chem., 2007, 46, 437–445 CrossRef CAS PubMed.
- G. C. Xu, X. M. Ma, L. Zhang, Z. M. Wang and S. Gao, J. Am. Chem. Soc., 2010, 132, 9588–9590 CrossRef CAS PubMed.
- G. C. Xu, W. Zhang, X. M. Ma, Y. H. Chen, L. Zhang, H. L. Cai, Z. M. Wang, R. G. Xiong and S. Gao, J. Am. Chem. Soc., 2011, 133, 14948–14951 CrossRef CAS PubMed.
- B. Liu, R. Shang, K. L. Hu, Z. M. Wang and S. Gao, Inorg. Chem., 2012, 51, 13363–13372 CrossRef CAS PubMed.
- S. Chen, R. Shang, K. L. Hu, Z. M. Wang and S. Gao, Inorg. Chem. Front., 2014, 1, 83–98 RSC.
- S. Chen, R. Shang, B. W. Wang, Z. M. Wang and S. Gao, Angew. Chem., Int. Ed., 2015, 127, 11245–11248 CrossRef.
- M. Maczka, A. Sieradzki, B. Bondzior, P. Deren, J. Hanuza and K. Hermanowicz, J. Mater. Chem. C, 2015, 3, 9337–9345 RSC.
- M. Ptak, M. Mączka, A. Gągor, A. Sieradzki, A. Stroppa, D. Di Sante, J. M. Perez-Mato and L. Macalik, Dalton Trans., 2016, 45, 2574–2583 RSC.
- A. Ciupa, M. Maczka, A. Gagor, A. Sieradzki, J. Trzmiel, A. Pikul and M. Ptak, Dalton Trans., 2015, 44, 8846–8854 RSC.
- A. Ciupa, M. Maczka, A. Gagor, A. Pikul and M. Ptak, Dalton Trans., 2015, 44, 13234–13241 RSC.
- M. Maczka, B. Bondzior, P. Deren, A. Sieradzki, J. Trzmiel, A. Pietraszko and J. Hanuza, Dalton Trans., 2015, 44, 6871–6879 RSC.
- G. Rogez, N. Viart and M. Drillon, Angew. Chem., Int. Ed., 2010, 49, 1921–1923 CrossRef CAS PubMed.
- A. Stroppa, P. Barone, P. Jain, J. PerezMato and S. Picozzi, Adv. Mater., 2013, 25, 2284–2290 CrossRef CAS PubMed.
- A. Stroppa, P. Jain, P. Barone, M. Marsman, J. M. PerezMato, A. K. Cheetham, H. W. Kroto and S. Picozzi, Angew. Chem., Int. Ed., 2011, 123, 5969–5972 CrossRef.
- S. Ghosh, D. Di Sante and A. Stroppa, J. Phys. Chem. Lett., 2015, 6, 4553–4559 CrossRef CAS PubMed.
- D. Di Sante, A. Stroppa, P. Jain and S. Picozzi, J. Am. Chem. Soc., 2013, 135, 18126–18130 CrossRef CAS PubMed.
- J. Xu, B. E. G. Lucier, R. Sinelnikov, V. V. Terskikh, V. N. Staroverov and Y. Huang, Chem.–Eur. J., 2015, 21, 14348–14361 CrossRef CAS PubMed.
- R. E. Cohen, Nature, 1992, 358, 136–138 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- S. Dudarev, G. Botton, S. Savrasov, C. Humphreys and A. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505 CrossRef CAS.
- S. M. Griffin and N. A. Spaldin, arXiv preprint arXiv:1401.2277, 2014.
- S. Kanungo, B. Yan, M. Jansen and C. Felser, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 214414 CrossRef.
- J. B. Goodenough, Magnetism and chemical bond, Interscience publishers, 1963 Search PubMed.
- A. Ciupa, M. Maczka, A. Gagor, A. Pikul and M. Ptak, Dalton Trans., 2015, 44, 13234–13241 RSC.
- L. M. Yang, P. Vajeeston, P. Ravindran, H. Fjellvåg and M. Tilset, Inorg. Chem., 2010, 49, 10283–10290 CrossRef CAS PubMed.
- M. Alvaro, E. Carbonell, B. Ferrer, F. X. L. i. Xamena and H. Garcia, Chem.–Eur. J., 2007, 13, 5106–5112 CrossRef CAS PubMed.
- S. Bordiga, C. Lamberti, G. Ricchiardi, L. Regli, F. Bonino, A. Damin, K. P. Lillerud, M. Bjorgen and A. Zecchina, Chem. Commun., 2004, 2300–2301 RSC.
- C. Franchini, R. Podloucky, J. Paier, M. Marsman and G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 195128 CrossRef.
- R. Warmbier, A. Quandt and G. Seifert, J. Phys. Chem. C, 2014, 118, 11799–11805 CAS.
- A. A. Talin, A. Centrone, A. C. Ford, M. E. Foster, V. Stavila, P. Haney, R. A. Kinney, V. Szalai, F. El Gabaly and H. P. Yoon, Science, 2014, 343, 66–69 CrossRef CAS PubMed.
- K. Otsubo, T. Haraguchi, O. Sakata, A. Fujiwara and H. Kitagawa, J. Am. Chem. Soc., 2012, 134, 9605–9608 CrossRef CAS PubMed.
- M. Van deráAuweraer and D. E. áDe Vos, Chem. Commun., 2010, 46, 3735–3737 RSC.
- A. Bétard and R. A. Fischer, Chem. Rev., 2011, 112, 1055–1083 CrossRef PubMed.
- M. C. So, S. Jin, H. J. Son, G. P. Wiederrecht, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 15698–15701 CrossRef CAS PubMed.
- H. Fei, S. Pullen, A. Wagner, S. Ott and S. M. Cohen, Chem. Commun., 2015, 51, 66–69 RSC.
- H. Guo, Y. Zhu, S. Qiu, J. A. Lercher and H. Zhang, Adv. Mater., 2010, 22, 4190–4192 CrossRef CAS PubMed.
- K. T. Butler, C. H. Hendon and A. Walsh, ACS Appl. Mater. Interfaces, 2014, 6, 22044–22050 CAS.
- S. Ling and B. Slater, J. Phys. Chem. C, 2015, 119, 16667–16677 CAS.
- R. F. Bader, Atoms in molecules: a quantum theory, Clarendon Press, 1994 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 



 , here n is the number of atoms per unit volume; κ is Boltzmann's constant; μB is Bohr magnetron; g is Lande factor; J is the total angular momentum. According to molecular field theory, TN is related to the difference between J1 and J2. However, whatever the compounds considered with either G-type or A-type magnetic arrangement, TN remains rather modest suggesting that (i) whatever the nature (antiferromagnetic or ferromagnetic) of the interaction for the second nearest neighbors this does not affect TN and thus J2 may be considered as negligible and (ii) J1 remains small in agreement with the low TN temperature reported experimentally. The Mn–OCHO–Mn superexchange magnetic interactions are related to the angles and distances between magnetic Mn sites. In case of NH2NH3+ compound, the distance of nearest neighbor Mn–Mn is 5.7 Å while that of second nearest neighbor is 7.7 Å, and the angle of Mn–OCHO–Mn is 140°. The magnetic interaction strength of NH2NH3+ compound under 5% compressive and tensile strain is displayed in Table 3. It is found that 5% compressive strain enhance the difference of exchange interaction strength of nearest neighbors J1 and second nearest neighbors J2 to get an increase of Néel temperature. This challenge might be addressed by applying a compressive stress by clamping mechanically the MOFs onto a substrate through strain engineering to reinforce the magnetic interactions and finally increase Neel temperature. The Neel temperature of these four MOFs is about 8 K,14,18–20 which indicates that A-ligands offer a slight effect on Neel temperature. In contrast, the introduction of mixed metal atoms may enhance the Neel temperature. For instance, the Neel temperature of [(CH3)2NH2][FeIIINiII(HCOO)6] is up to 42 K.41 As we shall see later on, such strain engineering approach is also powerful for tuning the band gap.
, here n is the number of atoms per unit volume; κ is Boltzmann's constant; μB is Bohr magnetron; g is Lande factor; J is the total angular momentum. According to molecular field theory, TN is related to the difference between J1 and J2. However, whatever the compounds considered with either G-type or A-type magnetic arrangement, TN remains rather modest suggesting that (i) whatever the nature (antiferromagnetic or ferromagnetic) of the interaction for the second nearest neighbors this does not affect TN and thus J2 may be considered as negligible and (ii) J1 remains small in agreement with the low TN temperature reported experimentally. The Mn–OCHO–Mn superexchange magnetic interactions are related to the angles and distances between magnetic Mn sites. In case of NH2NH3+ compound, the distance of nearest neighbor Mn–Mn is 5.7 Å while that of second nearest neighbor is 7.7 Å, and the angle of Mn–OCHO–Mn is 140°. The magnetic interaction strength of NH2NH3+ compound under 5% compressive and tensile strain is displayed in Table 3. It is found that 5% compressive strain enhance the difference of exchange interaction strength of nearest neighbors J1 and second nearest neighbors J2 to get an increase of Néel temperature. This challenge might be addressed by applying a compressive stress by clamping mechanically the MOFs onto a substrate through strain engineering to reinforce the magnetic interactions and finally increase Neel temperature. The Neel temperature of these four MOFs is about 8 K,14,18–20 which indicates that A-ligands offer a slight effect on Neel temperature. In contrast, the introduction of mixed metal atoms may enhance the Neel temperature. For instance, the Neel temperature of [(CH3)2NH2][FeIIINiII(HCOO)6] is up to 42 K.41 As we shall see later on, such strain engineering approach is also powerful for tuning the band gap.

