
Preparation of a novel molecularly imprinted polymer by the sol–gel process for solid phase extraction of vitamin D3
Solmaz
Kia
a,
Mohammad
Fazilati
a,
Hossain
Salavati
b and
Shahab
Bohlooli
*c
aDepartment of Biology, Tehran Payame Noor University, Tehran, Iran
bDepartment of Chemistry, Tehran Payame Noor University, Tehran, Iran
cDepartment of Pharmacology & Toxicology, School of Pharmacy, Ardabil University of Medical Sciences (ArUMS), University Street, 56197, Ardabil, Iran. E-mail: shahab.bohlooli@arums.ac.ir
First published on 24th March 2016
Abstract
A novel molecularly imprinted polymer (MIP) as an artificial receptor for vitamin D3 is prepared and used. A sol–gel process is used to prepare the hybrid MIP. For the organic part (gel) of the MIP, methacrylic acid (MAA), ethylene glycol dimethacrylate (EGDMA) and benzoylperoxide are used as functional monomer, cross-liker and initializer, respectively. As the inorganic part (sol) of the MIP, tetraethyl orthosilicate (TEOS) is used. The main parameters affecting the synthesis of the organic–inorganic hybrid MIP are investigated and optimized. The imprinting efficiency of the MIP is evaluated by comparing the adsorbed amount of vitamin D3 by the MIP with that by the related non-imprinted polymer (NIP). Betamethasone, dexamethasone, 17-beta-stradiol and 25-hydroxy vitamin D3 were chosen to study the selectivity of the MIP. This study revealed that the MIP possesses a remarkable affinity for vitamin D3. A limit of detection of 1 ng mL−1 has been achieved. The linear range and relative standard deviation (RSD%) were 1–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ng mL−1 and 4 (n = 3), respectively. The hybrid MIP provides fast, selective and convenient separation of vitamin D3 from aqueous samples.
000 ng mL−1 and 4 (n = 3), respectively. The hybrid MIP provides fast, selective and convenient separation of vitamin D3 from aqueous samples.
1. Introduction
Vitamin D refers to a group of fat-soluble secosteroids responsible for enhancing intestinal absorption of calcium and phosphate. In addition to the role of vitamin D in the musculoskeletal system, observational studies have shown that vitamin D insufficiency is associated with a wide variety of disorders such as cardiovascular disease, autoimmune diseases and cancer.1 That's why there is an increasing demand for measuring the concentration of vitamin D3 and its metabolites. 25-Hydroxy vitamin D3 (25-OH-D3) is the predominant circulating form of vitamin D and is generally considered to be the best single marker of vitamin D status. Various analytical methods have been reported for the determination of vitamin D concentration.2 The measurement of 25-OH-D can be performed by use of immunoassay, HPLC, and liquid chromatography-tandem mass spectrometry (LC-MS/MS). Most of these methods cooperated with various sample pre-treatment techniques.3,4 Therefore to determine the vitamin D3 concentration in human blood, a material with high selectivity towards vitamin D3 is necessary.Solid phase extraction (SPE) is routinely used for pre-concentration and clean-up in the analysis of biological and environmental samples due to the advantages of simplicity, rapidness and little consumption of organic solvents.5 Common sorbents usually lack selectivity and are easily subjected to interference by non-targeted substances with similar characteristics.6,7 Therefore, new sorbents such as molecular imprinting polymers (MIPs) are increasingly developed to meet the need of high selectivity.8–10 MIPs are made by crosslinked polymers in the presence of “print” molecule (template). After removal of the template, the polymer can be used as a selective binding medium for the template or structurally related compounds. Although, the MIPs synthesized by common methods (organic polymers) exhibit high affinity and selectivity but show poor site accessibility to the target molecules. The kinetics of the extraction/back extraction process is unfavourable and the mass transfer is slow. Moreover, for high performance of polymer we have many limitations in the choice of template molecule and organic polymers are not compatible with aqueous solutions.11 Therefore several challenges remain in bio-macromolecule imprinting, such as those involving proteins, DNAs, and even whole cells and viruses.12
Recently, molecularly imprinted sol–gel materials (MISGMs) have been extensively studied.13–15 MISGMs are synthesized by a conventional sol–gel process and incorporation of the template molecules into rigid inorganic or inorganic–organic networks.16 This kind of polymers shows many advantages including high selectivity, more accessible sites, fast mass transfer and binding kinetics. The low temperature processing conditions and versatility of the sol–gel synthesis make these materials very suitable matrices for the molecular imprinting technique.17,18 They are stable against physical and chemical treatments and can be stored in a dry state at room temperature.19,20 Therefore, new sorbents such as MIP based solid phase extraction can improve the efficiency and selectivity of sample preparation techniques. Hence in recent years, the development of MISGMs for solid-phase extraction (SPE) and detection has been extensively reported.21–24
In this study, we developed an inorganic–organic sorbent and vitamin D3 was used as the template compound. We chose cholecalciferol as a template because of the cost-effectiveness of this metabolite of vitamin D3. Up to now, there is no paper for extraction of vitamin D3 using MIP; just a patent has been reported in US at 2013.25 This is perhaps due to the structural characteristics of this analyst. Vitamin D3 has a large molecule with two active double bonds so in common organic molecularly imprinted polymers; it is not suitable as a template. Therefore, sol–gel method for MIP based solid phase extraction of cholecalciferol was chosen. In this procedure, template is captured in SiO2 networks under acid catalysed reactions and then polymerization of the organic polymer around of this network and template is started. Binding sites were formed after the template was removed. The schematic process of the MIP synthesis is shown in Fig. 1. In this work, MIPs and NIPs were synthesized and characterized on the basis of FTIR spectra and SEM image. To confirm the selectivity of the MIP particles, the imprinting factor (IF) and their ability to extract equimolar structural analogs was evaluated.
 | ||
| Fig. 1 Schematic representation of the MIP synthesis. | ||
The purpose of this work is to prepare a new and simple molecularly imprinted sorbent for vitamin D3 by sol–gel polymerization technique and to apply it in SPE coupled with HPLC for the determination of vitamin D3 in real samples. The proposed method presented high selectivity and adsorption capacity for vitamin D3.
2. Experimental
2.1. Chemicals and materials
Methacrylic acid (MAA), ethylene glycol dimethacrylate (EGDMA), benzoyl peroxide, tetraethyl orthosilicate (TEOS), 17-beta estradiol, dexamethasone, betamethasone, 25-hydroxy vitamin D3, vitamin D3 were supplied by Sigma-Aldrich (Munich, Germany). The multiwalled carbon nanotubes (MWCNTs) were obtained from the Research Institute of the Petroleum Industry (Tehran, Iran). The mean diameter of the MWCNTs was 10–15 nm, the length was 50–100 nm and purity > 98%. Methanol, ethanol, dichloromethane, hydrochloric acid and phosphoric acid were purchased from Merck (Schuchardt, Germany). Analytes were analytical grade and solvents all were HPLC grade.2.2. HPLC apparatus
The HPLC system (Jasco liquid chromatography system, Japan) is equipped with a C18, Nucleosil 100 ODS (5 μm) analytical column with a size of 4.6 mm × 150 mm (Teknokroma). An RP-18 guard column was fitted at the upstream side of the analytical column. The mobile phase was methanol with flow rate of 1 mL min−1 and run time in isocratic mode was 10 min. A 20 μL volume of back extracted vitamin D sample was injected onto the HPLC system. The concentration was detected by measuring the absorbance at 265 nm with UV detector and quantified by comparing the peak area produced by back extracted sample with that of vitamin D standard solution.2.3. MIP synthesis
• As inorganic part of the hybrid, 1.35 mL TEOS was mixed with 1.35 mL ethanol for 10 minutes then 50 mg vitamin D3 (the template) was added and stirred at 50 °C. Then 0.8 mL 12.5% (v/v) HCl 37% in de-ionized water was added to the mixture to complete the sol. This mixture was labelled solution (A).
• For organic part of MIP, 10 mmol MAA (functional monomer) was mixed with 9.61 mmol EGDMA (cross-liker) and 0.25 mmol benzoyl peroxide (initializer) was added to the mixture at 50 °C and stirred for 20 min. This was labelled solution (B).
Afterwards, 1.5 mL of solution (B) was added very slowly to 3 mL of solution (A) and stirred at 50 °C for 1 hour. The mixture was left for 48 hour until the gel network was formed in situ. The obtained bulk polymers were crushed and ground to obtain regular size particles. Non imprinted polymer (NIP) was prepared and treated with the same method, but in the absence of vitamin D. According to our SEM images size of particles related to MIP and NIP was about 10–50 μm.
2.4. MIP based solid phase extraction and back extraction process for optimization of MIP synthesis
In order to prevent waste of material used for optimization of the organic–inorganic hybrid MIP synthesis the following procedure was used. Vitamin D stock solution was prepared by dissolving 1 mg vitamin D in 1 mL methanol. After serial dilution, 0.5 mL of 1 μg mL−1 vitamin D standard solution was added to 4.5 mL deionised water and pH was adjusted to 3. Then a sample of 5 mg MIP powder was suspended in 5 mL of deionised water containing 100 ng mL−1 of vitamin D. After incubation under stirring at room temperature in speed of 150 RPM for 20 min, the particles were separated from the liquid phase by centrifuging under 4000 RPM for 10 min. The concentration of the vitamin D residue in the liquid phase was determined by UV-Vis absorption in HPLC system. The binding amount of vitamin D was determined as the difference between the initial and residual amounts in the liquid phase before and after the treatment. The recovery of the vitamin D adsorbed by the MIP particles was determined according to the following procedure: the vitamin D adsorbed at MIP particles was disassociated by suspending the particles in 5 mL of ethanol with pH, 3. Afterward; the particles were separated by centrifuging at 4000 RPM for 10 min. The recovered amount of vitamin D was finally determined by UV-Vis absorption in HPLC system. The above procedures were repeated several times to validate the reproducibility and reusability of MIP particles for enrichment and separation.2.5. Preparation of MIP-SPE column
MIP-SPE column were prepared by mixing the dry MIP particles (200 mg) and MWCNT powder (100 mg) which had been acid functionalized.26 Subsequently a black homogeneous powder was packed in a 5 mL empty syringe. The syringe was connected with a stop clock and two sieve plates to the bottom end and the top end of MIP packed particles. The sieve plates were obtained from commercial SPE cartridges. The MIP-SPE columns were washed with 12 mL methanol to remove impurities. Before loading sample solutions, the polymer activated through 5–8 mL deionized water and pH was adjusted to 3.0.3. Results and discussion
3.1. Polymer characterization
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of MAA carboxylic acid, indicating the absence of carbonyl groups in polymer materials. This confirmed the complete polymerization of monomer and cross linker. The sharp band at 1079 cm−1 corresponds to Si–O–Si stretching vibrations, indicating SiO2 existence within matrices.
O stretching vibration of MAA carboxylic acid, indicating the absence of carbonyl groups in polymer materials. This confirmed the complete polymerization of monomer and cross linker. The sharp band at 1079 cm−1 corresponds to Si–O–Si stretching vibrations, indicating SiO2 existence within matrices.
 | ||
| Fig. 2 The scanning electron micrographs images of the MIP (a) and NIP (b) particles produced by sol–gel polymerization. | ||
 | ||
| Fig. 3 FT-IR analysis of vitamin D3-imprinted MIP (a) and NIP (b) nanoparticles. Characteristic IR absorption band for the SiO2 group is at 1079 cm−1. | ||
3.2. Optimization of the organic–inorganic hybrid MIP synthesis
The polymer synthesis conditions strongly affect the efficiency of extraction and back extraction procedure. Therefore, essential conditions were studied and optimized carefully.:2 ratios. The scanning electron micrograph images of the MIP and NIP particles are shown in Fig. 2. The control polymer (NIP) was observed to have smoother surface than the MIP, while the MIP after the template removal on the other side had rough surfaces which can be attributed to the formation of cavities during the synthesis process. It has been reported that the roughness of MIP particles can lead to high surface area than that of NIP and thus MIP can adsorb analyte of interest much better than the NIP.27
 | ||
| Fig. 4 Influence of volume ratios between (A) and (B) solutions. Extraction condition: amount of MIP powder 5 mg; volume and concentration of donor phase 5 mL, 100 ng mL−1; pH of sample, 3. | ||
 | ||
| Fig. 5 The effect of MAA to EGDMA in polymer efficiency. Extraction condition: amount of MIP powder 5 mg; volume and concentration of donor phase 5 mL, 100 ng mL−1; pH of sample, 3. | ||
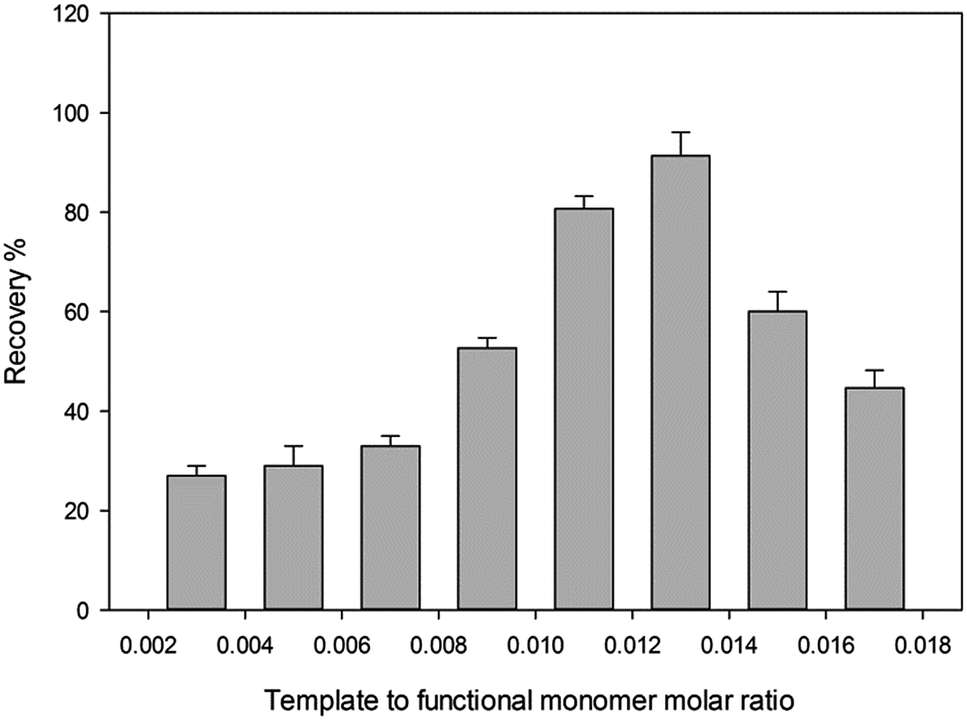 | ||
| Fig. 6 Influence of molar ratios between template and functional monomer. Extraction condition: amount of MIP powder 5 mg; volume and concentration of donor phase 5 mL, 100 ng mL−1; pH of sample, 3. | ||
3.3. Optimization of extraction and back extraction conditions
In this study removing the template and back extraction condition of the analyte were studied and optimized. Thus the effect of pH (2.5–8.5), different eluents in a wide range of polarity (CH3OH, ACN, CH2Cl2, C3H7OH, C6H14), flow rate of sample solution (0.5–4 mL min−1) and flow rate of eluent (0.5–4 mL min−1) were studied. | ||
| Fig. 7 Selection of back extraction solvent. Extraction condition: experimental conditions: pH of sample, 3, sample volume; 10 mL, sample flow rate; 1 mL min−1, amount of absorbent; 0.2 g. | ||
 | ||
| Fig. 8 The effect of sample flow rate on the recovery of vitamin D3; experimental conditions: pH of sample, 3, sample volume; 10 mL, amount of absorbent; 0.2 g. | ||

Moreover, the imprinting factor (IF) was considered to calculate the recognition capabilities of the MIP-SPE.

| Sample | ER | IF | |
|---|---|---|---|
| MIP-SPE | NIP-SPE | ||
| Water | 0.95 | 0.30 | 3.16 |
| Plasma | 0.80 | 0.32 | 2.5 |
Moreover, the effect of the salt on the recovery of vitamin D3 extraction by MIP-SPE was considered. For this aim, a different range of NaCl (10–100 mM) concentrations was added to aliquots with same concentration of vitamin D3 solution using the same extraction process and conditions. The results are shown in Fig. 9. The percentage recovery of vitamin D3 in the plasma sample shows that the extraction recovery decreases with increasing NaCl concentration within range of 10–100 mM in the buffer (pH 3).
 | ||
| Fig. 9 The effect of ionic strength on the recovery of vitamin D; experimental conditions: pH 3, sample volume; 10 mL, sample flow rate; 1 mL min−1, amount of adsorbent; 0.2 g. | ||
3.3.7.1. Application to real sample. The proposed method has been applied for the determination of vitamin D3 in plasma sample. The listed results in Table 1, indicate the suitability of the present polymer for pre concentration and separation of vitamin D3 from natural sample.
3.3.7.2. Separation of vitamin D3 from blood serum. In blood the strongly hydrophobic 25-OH-D is largely bound to vitamin D-binding protein (VDBP) (14), to dissociate vitamin D from VDBP and precipitate protein, to 150 μL of plasma sample, 150 μL of 0.1 mol L−1 NaOH, was added and then 500 μL of 100% methanol was added for proteins precipitation. After centrifugation the supernatant was diluted with 8–10 mL buffer solution at pH 3 and then transferred to the column for solid-phase extraction. For determination of calibration curve and limit of detection, pool serum samples were spiked with vitamin D3 at different concentration levels between 10 and 100 ng mL−1. The recoveries and reproducibility of the method were calculated and summarized in Table 2. As can be seen, the average recovery of the MISPE method was 87% at the studied levels. These results demonstrated that the MIP based sorbent had a good recovery. However, the recovery of vitamin D3 was decreased when the concentration of vitamin D3 solution was increased.
| Sample | Total volume (μL) | Adding (ng mL−1) | Found (ng mL−1) | Recovery (%) | RSD (%) (n = 3) |
|---|---|---|---|---|---|
| a Experimental conditions: pH 3, sample flow rate and eluent; 1 mL min−1, amount of absorbent; 0.2 g. | |||||
| 150 μL plasma | 10 | 0 | 0 | — | — |
| 10 | 8.9 | 89.06 | 3.1 | ||
| 20 | 18.13 | 90.65 | 2.8 | ||
| 40 | 34.74 | 86.86 | 3.5 | ||
| 60 | 53.55 | 89.25 | 3.9 | ||
| 80 | 68.01 | 85.02 | 3.7 | ||
| 100 | 78.82 | 78.82 | 4.0 | ||
3.4. Analytical consideration of the method
 | ||
| Fig. 10 HPLC chromatogram for selectivity consideration. (a) Before MIP-solid phase extraction and (b) after MIP solid phase extraction. HPLC condition: gradient mode, pump (A) de-ionized water/acetonitrile (70/30% v/v), pump (B) methanol/de-ionized water (95/5% v/v), 0–10 min 100% pump A, 10–30 min 100% pump B, flow rate 1.0 mL min−1, detection wavelength 265 nm. | ||
Moreover, the differences in the rebinding capacities for the MIP and NIP (MIP is three times more), can reflect the specific adsorption of cholecalciferol at its imprinted sites. It is clear that the imprinted sites exhibit a high selectivity rebinding to the cholecalciferol target. Despite the 25-hydroxy-vitamin D3 and vitamin D3 have high structural similarity to each other, synthesized polymer in a solution containing both components, extract vitamin D3 with higher binding affinity than 25-hydroxyl-D3. This efficient uptake of the main analyte by the hybrid MIP was acceptable and adsorption capacity for the method.
000 ng mL−1 of cholecalciferol. The equation for the line was y = 3913.7x + 117348 (R2 = 0.9951). The limit of detection (LOD) under optimal experimental conditions was 1 ng mL−1 (n = 3). The relative standard deviation (RSD) for 100 ng mL−1 of cholecalciferol was 4. The relative standard deviation (RSD) was lower than 5%, showing a good precision. The MIP adsorbent can be reused at least 30 times. The results obtained from the present study were revealed that with applying this method the LOD improved 10 times. It means we can use this method for concentration and extraction of 1,25-dihydroxyvitamin D3 (calcitriol). Calcitriol exists in a trace amount (pg) in serum and we cannot determine it by UV-Vis absorption in HPLC System. The accuracy, precision and selectivity of the method encourage us to develop a MIP-SPE for calcitriol in our future work.
It appears that the unique selectivity of the HPLC by separation of isobaric compounds are able to meet this requirement. Recent technical developments have centred on reducing the manual component of sample preparation prior to chromatography and elimination of liquid–liquid extraction using MIP based solid-phase extraction. The presented approach demonstrates the selectivity, in both chromatographic and sample preparation, allows for efficient analysis. All of these method changes contribute to increased throughput, improved reliability of HPLC assay for 25-OH-D. Table 3 demonstrate a comparison between reported methods.
| Method | Extraction method | Linear range nmol L−1 | LOD nmol L−1 | Recovery (%) | RSD (%) | Cross-reactivity | Ref. |
|---|---|---|---|---|---|---|---|
| a n.d. = not determined. | |||||||
| Chemiluminescent immunoassay | None | 17.5–375 | ≤5.0 | 101 | ±13 | Vitamin D2 1.4%, vitamin D3 1.1%, 25-OH vitamin D2 100.0%, 25-OH vitamin D3 100.0%, 1,25-(OH)2 vitamin D2 7.1%, 1,25-(OH)2 vitamin D3 21.7% | 28 |
| HPLC-UV | Liquid–liquid extraction | n.d. | 1.1 | 86.6–101 | 0.6–5 | None | 29 |
| HPLC-MS/MS | None | 4.0–265.3 | 4.0 | 89.6–98.7 | n.d. | None | 30 |
| HPLC-UV | MIP-SPE | 2.5–2500 | 2.5 | 78.8–89.0 | 4 | None | Present work |
4. Conclusion
In this paper, a simple procedure was developed to synthesize vitamin D-imprinted sorbent with sol–gel polymerization technique. The present study is the first work to use imprinting process for vitamin D extraction. Various effective parameters, including the template, monomer and cross linker molar ratios, time, pH and desorption solution were investigated and optimized. The selectivity of MIP was studied, and the results revealed that MIP possesses a remarkable affinity for cholecalciferol. Promising mechanical and thermal stability, excellent durability, wide linear range and good extraction efficiency suggest that the developed MIP is an attractive tool as a solid-phase extraction sorbent to determine vitamin D in real samples. The validity data demonstrated that the method is accurate, selective and sensitive. It is the first step and we hope to use these artificial receptors for developing pseudo immunoassays.Acknowledgements
The authors would like to thank Dr Shahryar Bahar for his generous guidance in this project.References
- P. Pludowski, M. F. Holick, S. Pilz, C. L. Wagner, B. W. Hollis, W. B. Grant, Y. Shoenfeld, E. Lerchbaum, D. J. Llewellyn, K. Kienreich and M. Soni, Autoimmun. Rev., 2013, 12, 976–989 CrossRef CAS PubMed.
- C.-J. L. Farrell, S. Martin, B. McWhinney, M. Herrmann, I. Straub and P. Williams, Clin. Chem., 2012, 58, 531–542 CAS.
- A. M. Wallacea, S. A. Gibsonb, D. l. Hunty, C. L. Allardt and M. Ashwell, Steroids, 2010, 75, 477–488 CrossRef PubMed.
- E. W. Holme, J. Garbincius and K. M. McKenna, American Society for Clinical Pathology, 2013, 140, 550–560 CrossRef PubMed.
- M. M. Moein, A. Abdel-Rehim and M. Abdel-Rehim, TrAC, Trends Anal. Chem., 2015, 67, 34–44 CrossRef CAS.
- L. Vidal, M. L. Riekkola and A. Canals, Anal. Chim. Acta, 2012, 715, 19–41 CrossRef CAS PubMed.
- M. M. Moein, R. Said, F. Bassyouni and M. Abdel-Rehim, J. Anal. Methods Chem., 2014, 2014, 1–24 CrossRef PubMed.
- F. Augusto, L. W. Hantao, N. G. S. Mogollón and S. C. G. N. Braga, TrAC, Trends Anal. Chem., 2013, 43, 14–23 CrossRef CAS.
- G. Vasapollo, R. D. Sole, L. Mergola, M. R. Lazzoi, A. Scardino, S. Scorrano and G. Mele, Int. J. Mol. Sci., 2011, 12, 5908–5945 CrossRef CAS PubMed.
- M. Zhao, C. Zhang, Y. Zhang, X. Guo, H. Yan and H. Zhang, Chem. Commun., 2014, 50, 2208–2210 RSC.
- H. Yan and K. H. Row, Int. J. Mol. Sci., 2006, 7, 155–178 CrossRef CAS.
- L. Feng, B. Kan, K. Zhao, J. Wei, D. Zhu and L. Zhang, J. Sol-Gel Sci. Technol., 2014, 71, 428–436 CrossRef CAS.
- M. A. Golsefidi, Z. Es'haghia and A. Sarafraz-Yazdic, J. Chromatogr. A, 2012, 1229, 24–29 CrossRef CAS PubMed.
- M. M. Moeina, A. El-Beqqalia, A. Abdel-Rehimb, A. Jeppsson-Dadouna and M. Abdel-Rehima, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 967, 168–173 CrossRef PubMed.
- M. M. Moein, D. Jabbar, A. Colmsjö and M. Abdel-Rehim, J. Chromatogr. A, 2014, 1366, 15–23 CrossRef CAS PubMed.
- M. Díaz-Bao, R. Barreiro, J. Miranda, A. Cepeda and P. Regal, Chromatography, 2015, 2, 79–95 CrossRef.
- K. Zhao, L. Feng, H. Lin, Y. Fu, B. Lin, W. Cui, S. Li and J. Wei, Catal. Today, 2014, 236, 127–134 CrossRef CAS.
- K. Zhao, B. Lin, W. Cui, L. Feng, T. Chen and J. Wei, Talanta, 2014, 121, 256–262 CrossRef CAS PubMed.
- B. H. Fumes, M. R. Silva, F. N. Andrade, C. E. D. Nazario and F. M. Lanças, TrAC, Trends Anal. Chem., 2015, 71, 9–25 CrossRef CAS.
- J. Ou, Z. Liu, H. Wang, H. Lin, J. Dong and H. Zou, Electrophoresis, 2015, 36, 62–75 CrossRef CAS PubMed.
- M. M. Moein, M. Javanbakht, M. Karimi and B. Akbari-adergani, Talanta, 2015, 134, 340–347 CrossRef CAS PubMed.
- F. Bates and M. del Valle, Microchim. Acta, 2014, 182, 933–942 CrossRef.
- G. D. Giustina, A. Sonato, E. Gazzola, G. Ruffato, S. Brusa and F. Romanato, Mater. Lett., 2016, 152, 44–47 CrossRef.
- S. Liao, W. Zhang, W. Long, D. Hou, X. Yang and N. Tan, Appl. Surf. Sci., 2015, 12, 184 Search PubMed.
- W. A. Jensen, J. J. Belbruno and Y. Liu, US Pat., 20130288385 A1, 2013.
- M. A. Golsefidi, Z. Es'haghi and A. Sarafraz-Yazdi, Sample Preparation, 2013, 1, 1–9 CrossRef.
- P. Sikiti, T. A. Msagati, B. B. Mamba and A. K. Mishra, J. Environ. Health Sci. Eng., 2014, 12, 82 CrossRef PubMed.
- D. L. Ersfeld, D. S. Rao, J. J. Body, J. L. Sackrison Jr, A. B. Miller, N. Parikh, T. L. Eskridge, A. Polinske, G. T. Olson and G. D. MacFarlane, Clin. Biochem., 2004, 37, 867–874 CrossRef CAS PubMed.
- L. Hymoller and S. K. Jensen, J. Chromatogr. A, 2011, 1218, 1835–1841 CrossRef CAS PubMed.
- S. Baecher, A. Leinenbach, J. A. Wright, S. Pongratz, U. Kobold and R. Thiele, Clin. Biochem., 2012, 45, 1491–1496 CrossRef CAS PubMed.
- M. Pal, S. Datta, A. K. Pradhan, L. Biswas, J. Ghosh, P. Mondal, R. Rahut, A. R. Chaudhuri, S. Sau and S. Das, Adv. Biol. Chem., 2013, 03, 501–504 CrossRef CAS.
- W. D. Fraser and A. M. Milan, Calcif. Tissue Int., 2013, 92, 118–127 CrossRef CAS PubMed.
- D. Enko, G. Kriegshauser, R. Stolba, E. Worf and G. Halwachs-Baumann, Biochem. Med., 2015, 25, 203–212 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2016 |