
Preparation of graphene oxide by dry planetary ball milling process from natural graphite
Abstract
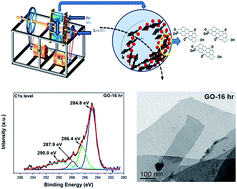
Graphene oxides (GO) with different degrees of oxidation have been prepared by an in-house designed horizontal high energy planetary ball milling process. The prepared graphene oxides have been studied by X-ray diffraction (XRD), field emission scanning electron microscopy (FESEM), transmission electron microscopy (TEM) with selected area electron diffraction (SAED), high resolution transmission electron microscopy (HRTEM), X-ray photoelectron spectroscopy (XPS), micro Raman spectroscopy, Fourier transform infrared (FTIR) spectra, Brunauer–Emmett–Teller (BET) test and thermogravimetric analysis (TGA). XPS study shows an increasing trend of atomic concentration ratio of O/C with increasing ball milling time duration from 2 to 24 h of high purity graphite sample (FEED). This result is attributed to the formation of more oxidation in the graphite sample, produced due to the increasing time duration of milling. From micro Raman analysis it is also noted that ID/IG ratio increases with increasing milling time of FEED, which further supported the preparation of graphene oxide. In this study the graphene oxide prepared by 16 h of milling may be considered as the optimized sample as far as the degree of oxidation, time and energy consumption factors are concerned.
 Please wait while we load your content...
Please wait while we load your content...

