DOI:
10.1039/C5RA24786A
(Paper)
RSC Adv., 2016,
6, 35815-35824
Polyurethane/58S bioglass nanofibers: synthesis, characterization, and in vitro evaluation
Received
23rd November 2015
, Accepted 29th March 2016
First published on 31st March 2016
Abstract
In this study, polyurethane nanofibers containing 5 and 10 wt% synthesized 58S bioglass were designed and fabricated by the electrospinning process. The physicochemical and mechanical properties and in vitro behavior of the scaffolds were evaluated by scanning electron microscopy (SEM), dynamic mechanical thermal analysis (DMTA), Fourier transform infrared spectroscopy (FTIR), contact angle, phosphate-buffered saline (PBS) uptake, in vitro bioactivity, MTT assay, and cellular response. The FTIR results showed an increase in the urethane bond between the OH (from BG) and NCO (from PU) groups with the increase in BG. When the BG amount increased from 5% to 10%, the contact angle decreased to approximately 20° and the PBS uptake of the scaffold increased because of the hydrophilic property of the BG particles. DMTA showed that the glass transition temperature started to shift slightly to higher temperatures from −17 °C to −15 °C. SEM and X-ray diffraction analysis depicted hydroxyapatite formation on the scaffolds upon immersion in SBF. Cell proliferation and viability also increased significantly with the increase in BG. It is concluded that this composition provides a novel alternative for bone tissue engineering.
Introduction
Tissue engineering is an alternative treatment for repairing and regenerating damaged tissues. In hard tissue engineering, bioceramics, biopolymers, and their composites have been considered suitable candidates for bone tissue engineering.1–4 Bioceramics, such as hydroxyapatite, tricalcium phosphate, glass-ceramics and bioglass (BG), have been used in different forms for bone tissue engineering.5–10 They can be combined with biopolymers to improve the property of scaffolds, including their mechanical properties, biocompatibility, and bioactivity.8,11–13 BG powder has been synthesized using conventional melting methods and the sol–gel process. The sol–gel technique is an economical and technically simple procedure which has many advantages such as low crystallization temperature, high surface area and chemical homogeneity.14,15 Several studies have shown that BG is angiogenic,16 osteoconductive,17,18 and osteoinductive.19 BG also supports osteoblasts, which are vital for adhesion, growth, differentiation, and new bone formation in bone tissue engineering.13,20 Studies demonstrated that bioglass-58S has high bioactivity, biodegradability and osteoconductivity.21 Also recent research have shown degradation of bioglass can active gene expression and stimulate the production of growth factors.22,23 In another point of view, polyurethane (PU) is one the most interesting biopolymers for bone tissue engineering because of its properties and relatively good biocompatibility.24–26 Studies on PU and its potential application as a bone tissue material have increased because of its easy processability, biocompatibility, and mechanical properties.24–26 To our knowledge, only a few studies on the development of PU/BG scaffolds for bone tissue engineering have been reported.27–29
Different methods have been introduced to develop bone tissue engineering scaffolds, including salt leaching,30 3D printing,7,31 and electrospinning.32 Recent studies have shown that electrospinning fibers are suitable candidates due to it can provide many benefits such as suitable conditions for the migration, and differentiation of bone tissue cells, wide range of nanofibers and morphologies, cost-effectiveness and easy to set up.32,33
In the present study, PU/BG scaffolds are prepared by the electrospinning method for the first time to our knowledge. The prepared scaffolds were characterized in terms of physicochemical and mechanical properties. Moreover, apatite formation ability and in vitro biocompatibility were investigated.
Materials and methods
Synthesis and characterization of bioactive glass nanoparticles
Bioactive glass based on the 58SiO2–33CaO–9P2O5 system was synthesized by the sol–gel process. In brief, tetraethyl orthosilicate (Sigma-Aldrich, St. Louis, MO, USA) was added to the mixture of deionized water and hydrochloric acid (mole ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8). After 30 min of mixing, triethyl phosphate (Sigma-Aldrich, St. Louis, MO, USA) was added to the mixture and mixed for 20 min. Then, calcium nitrate (Merck, Darmstadt, Germany) was added and stirred for an additional 1 h. Thereafter, ammonia solution (1.0 mol L−1) was added to the acid sol, with vigorous stirring; then, the sol suddenly gelated. The synthesized gel was heated at 700 °C for 3 h. The synthesized BG was analyzed by X-ray diffraction (XRD; XRD 3003 PTS), Fourier transform infrared spectroscopy (FTIR; Nexus 870), and transmission electron microscopy (TEM; EM208 Philips).
:8). After 30 min of mixing, triethyl phosphate (Sigma-Aldrich, St. Louis, MO, USA) was added to the mixture and mixed for 20 min. Then, calcium nitrate (Merck, Darmstadt, Germany) was added and stirred for an additional 1 h. Thereafter, ammonia solution (1.0 mol L−1) was added to the acid sol, with vigorous stirring; then, the sol suddenly gelated. The synthesized gel was heated at 700 °C for 3 h. The synthesized BG was analyzed by X-ray diffraction (XRD; XRD 3003 PTS), Fourier transform infrared spectroscopy (FTIR; Nexus 870), and transmission electron microscopy (TEM; EM208 Philips).
Electrospinning of scaffolds
Electrospinning was employed to fabricate random nanofiber scaffolds by using PU with and without BG. The polymer was dissolved in DCM/DMF (1:4, v/v) at a concentration of 10 wt%. Then, BG (5 and 10 wt%) was added to the solution dispersed with Dolapix CE64 (Zschimmer & Schwartz GmbH Co., Burgstädt, Germany) for 3 hours. Then, the PU (with and without BG) was electrospun by injecting the solution onto the covered aluminum foil collecting surface by using a syringe at a rate of 0.5 mL h−1, with a distance of 18 cm from the needle tip. A 13 kV difference was applied across the syringe and the collecting surface. All solution preparations and electrospinning studies were conducted at room temperature. The scaffolds were investigated by scanning electron microscopy (SEM; KYKY-EM3200), FTIR (Thermo Nicolet Nexus 870), and contact angle goniometer (OCA 15 plus, Dataphysics). The apatite formation ability of the scaffolds was investigated after immersion in SBF for different days. The structure and apatite formation ability of the scaffolds were monitored by XRD and SEM. In vitro evaluation was conducted by cell culture and MTT assay. The cell attachment was observed by SEM.
Phase structure and morphological characterization
The synthesized BG powders and apatite formation of PU/BG scaffolds were analyzed by X-ray diffractometer (XRD 3003 PTS), with monochromatic Cu-Kα radiation (1.5418 Å). The XRD diagrams were recorded in the interval 0° ≤ 2θ ≤ 40°, with a step size of 0.02° every 1 s. The FTIR spectra were collected by the Thermo Nicolet spectrometer (Nexus 870) in the range of 400 cm−1 to 4000 cm−1, and the spectral resolution was at a minimum of 4 cm−1. TEM (EM208, Philips, Amsterdam, Netherlands) was used to investigate the morphology and the average particle size. After dispersing the powders in methanol with ultrasonication, the samples were collected on carbon-coated gold grids operated at 200 keV. In terms of structure and morphology observation, the samples were sputter-coated with gold prior to scanning electron microscope (KYKY-EM3200) analysis.
Physical properties and mechanical analysis
The sessile drop method was employed to measure the contact angle by using the contact angle goniometer (OCA 15 plus, Dataphysics). The liquid droplet was deposited by a syringe pointed vertically down onto the sample, and the angle was determined by a high-resolution camera and analyzed by analysis software. The phosphate-buffered saline (PBS) uptake of PU and PU/BG nanofibers was evaluated through a simple method. After immersing the samples in PBS solutions in a 37 °C shaking incubator for different days and changing the PBS solution every day, the samples were removed at 3, 6, 9 and 12 days. After drying, the samples were weighed. Then, PBS absorption (%) was calculated as follows:
| PBS absorption = (Wa − Wb)/Wb × 100, |
where Wa and Wb are the weights before and after PBS soaking, respectively.
Dynamic mechanical thermal analysis (DMTA) was conducted by using Teritec 2000 over a temperature range of −50 °C to 200 °C at a heating rate of 10 °C min−1 and frequency of 1 Hz to investigate the effect of BG addition on the bulk material properties of the PU-based composites. The dimension of the samples was 30 × 10 × 1 mm3. The value of tanδ (phase lag) and the elastic and loss moduli were recorded as a function of temperature for each sample.
In vitro studies
L929 mouse fibroblasts were used to assess the biocompatibility of nanofibers, and 4 × 104 cells were used for each sample. The cells were enriched with Dulbecco's modified Eagle's medium supplemented with 4 mM glutamine, 10% fetal bovine serum, and 100 units per μL penicillin/streptomycin and incubated at 37 °C in a humidified atmosphere with 5% CO2. The cell morphologies were observed after 24 and 48 h of cell culture. MTT assay was conducted to determine the cell proliferation and viability rates. After cell seeding on the nanofibers and culturing for 48 h, 100 μL of MTT solution (5 mg mL−1) was added and incubated for 4 h at 37 °C in a humid atmosphere with 5% CO2. Then, the MTT solution was removed, the samples were washed with PBS, and 0.5 mL of dimethyl sulfoxide was added. Finally, the viable cells in the solution were quantified by using a scanning multi-well spectrophotometer (ELISA Microplate Readers, BioTek) at 540 nm. The polystyrene surface of the cell culture plates was used as the control. Samples were prepared by fixing cells with 2.5% glutaraldehyde, rinsing with PBS, and dehydrating in graduated ethanol from 50% to 100% in steps of 10% for 5 min each to observe cell adherence and its morphology on nanofibers by SEM.
Statistical analysis
All the measured values were presented as the mean ± standard deviation of at least five experiments. The significance between the mean values was calculated by using one-way analysis of variance and Student's t test. Differences were considered significant at p ≤ 0.05.
Results and discussion
The XRD and TEM data for BG are shown in Fig. 1a and b, respectively. BG exhibited an amorphous phase, and the size of the powders was less than 50 nm. The FTIR spectra of 58S bioactive glass, PU, PU with 5 wt% bioglass (PU5BG), and PU with 10 wt% bioglass (PU10BG) are shown in Fig. 2. The BG spectrum revealed the presence of Si–O and Si–O–Si peaks at 462 cm−1and 725 cm−1, which can be assigned to rocking and bending vibrations, respectively,34 and 1024 cm−1 attributed to the phosphate group.28 The shoulder band at 821 and 1565 cm−1 can be attributed to the bending of the P–O vibrational mode.34 The peak at 1463 cm−1 can be attributed to the O–H bending vibration (O–H absorption of moisture).34 The peak at approximately 3350 cm−1 is attributed to the –OH groups.23 The FTIR spectra of the PU peaks showed N–H stretching vibration at 3427 cm−1,28,35,36 asymmetric CH2 stretching at 2938 cm−1,28,35 symmetric CH2 stretching at 2858 cm−1,35,37 O–CH2 stretching at 2796 cm−1,35 isocyanate group at 2357 cm−1,35 urethane and/or polycaprolactone ester C–O group, urethane linkage at 1711 and 1604 cm−1,35,38 amide II at 1531 cm−1,37 CH2 vibrations at 1459, 1368, and 1309 cm−1,35,39 amide I (ester band) at 1221 cm−1,35,38 ether band at 1107 cm−1,35 C–O–C at 1006 cm−1,35,38,40 amide IV at 767 cm−1,35 amide V at 669 cm−1,35 and δ(N–C–N) at 559 cm−1.35 The FTIR spectra of the composites revealed that the peaks at 3330 and 3302 cm−1 are related to urethane linkage, the peaks at 1719 and 1720 cm−1 are related to carbonyl vibration, and the peaks at 1109 and 1104 cm−1 are related to C–O–C vibration. The peaks at 3733 and 3732 cm−1 are related to the N–H stretching vibration in the composites.35,38 Thus, the NCO intensity decreases with the increase in the BG amount because of the formation of PU and saturated polyester networks.35,38 Simultaneously, (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–O–NH was formed with the decrease in NCO because of interaction between the OH of BG and the NCO of PU (showing the presence of a covalent band).35,38 The peak at 1531 cm−1 shows that the urethane band increases with the increase in the BG amount. PU was linked with BG by hydrogen bonding.6,41 In brief, molecular interactions between PU and BG can be considered the linkage between carboxyl or amino group from PU and hydroxyl group from BG6,42 and the chemical linkage between hydroxyl groups of BG and PU, which causes the disappearance of the peaks at 3550 cm−1 corresponding to the –OH groups in BG.6,41
O)–O–NH was formed with the decrease in NCO because of interaction between the OH of BG and the NCO of PU (showing the presence of a covalent band).35,38 The peak at 1531 cm−1 shows that the urethane band increases with the increase in the BG amount. PU was linked with BG by hydrogen bonding.6,41 In brief, molecular interactions between PU and BG can be considered the linkage between carboxyl or amino group from PU and hydroxyl group from BG6,42 and the chemical linkage between hydroxyl groups of BG and PU, which causes the disappearance of the peaks at 3550 cm−1 corresponding to the –OH groups in BG.6,41
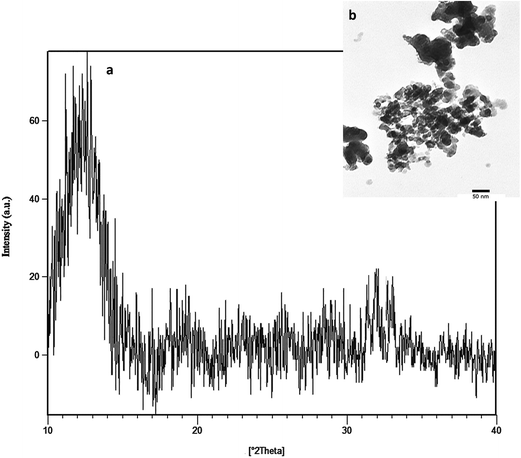 |
| | Fig. 1 XRD pattern (a) and TEM image (b) of the 58S BG powder. | |
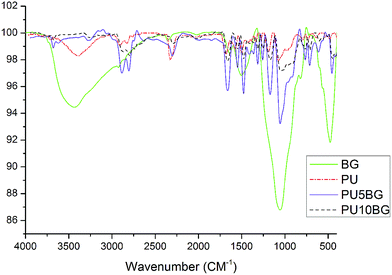 |
| | Fig. 2 FTIR of polyurethane (PU), bioglass (BG), polyurethane containing 5 wt% bioglass (PU5BG), and polyurethane containing 10 wt% bioglass (PU10BG). | |
The storage modulus and glass transition temperature (Tg) of the prepared samples were investigated by DMTA. DMTA detects the type of transitions and relaxations that are related to the structure and morphology of the samples. Glass transition is identified by the decrease in storage modulus and the presence of prominent peaks in tanδ. The DMTA thermographs of the unmodified PU and PU10BG composite are shown in Fig. 3A and B, respectively. The Tg of the unmodified PU is approximately −20 °C, but started to shift slightly to higher temperatures from −20 °C to −14 °C and broadened after being modified with BG. The storage modulus also increased to a higher value in PU modified with BG than pure PU. The presence of inorganic phase BG and its increase to 10% increases the Tg by hindering the molecular and segment rotation with reference to the polymer. This effect also reduces the dipole interaction potential and makes the polymer mechanically stronger.
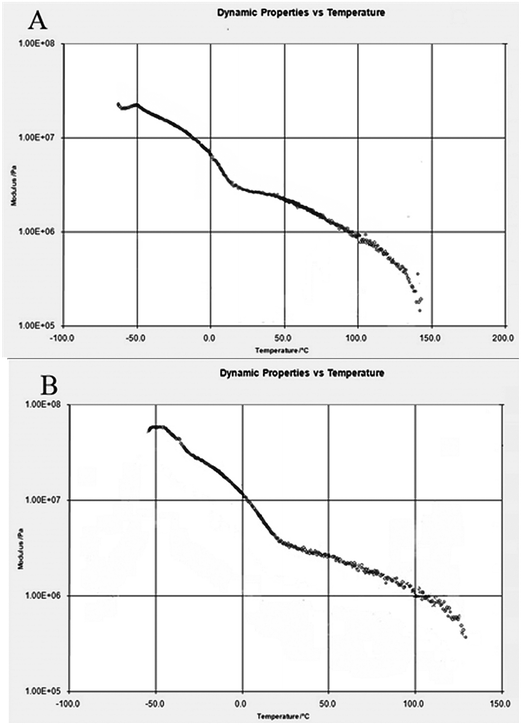 |
| | Fig. 3 DMTA analysis of (A) PU and (B) PU10BG. | |
As shown in Fig. 4A, PU fibers were produced smoothly, continuously, and bead-free. The fiber diameter histogram which determines by Image J software shows in Fig. 4D and nanofibers are mainly ranged below 200 nm. Fig. 4B and C show the SEM of PU5BG and PU10BG as-spun nanofibers, respectively. As shown in Fig. 4A–C, the morphology of the nanofibers did not change with the addition of BG, which was in accordance with a previous research by Khan et al.38 Previous studies have shown that the size of PU nanofibers depends on solution viscosity. With the addition of nanoparticles to the solution, the viscosity increased because of the linkage between PU and nanoparticles or the increase in molecular entanglement.38 Electrospun nanofibrous matrices show morphological similarities to the natural extracellular matrix and are characterized by high surface-to-volume ratio, high porosity, and variable pore size distribution, exhibiting properties that can modulate the cellular behavior.33 Several biopolymers have been electrospun to produce nanofibrous scaffolds for cartilage and bone tissue engineering.43,44 Many bioceramics have been incorporated into biopolymers, and their mineralization, cell adhesion, proliferation, and differentiation have been investigated for use as bone substitutes.45 Given the degradation of biopolymers, the environment would be acidic, which is unsuitable for the cells. Using bioceramics is a suitable method of buffering this environment.9,46 Also adding bioceramics to biopolymers can also increase the mechanical properties of the scaffolds47,48 and prevent the mechanical degradation of polymers.9 In the present study, bioactive glass nanoparticles were incorporated into PU nanofiber to form the nanocomposite nanofibrous web, and its bioactivity was investigated in simulated body fluid (SBF). A significant characteristic of bioactive materials is their ability to bond with bone through hydroxyapatite formation on their surface.49 Previously, the authors observed that the BG-added polymers showed full coverage of the surface with apatite precipitates, biocompatibility, cell growth, and differentiation.48,50,51 The mechanical properties of the composites were also improved.28
 |
| | Fig. 4 SEM images of (A) PU, (B) PU5BG, (C) PU10BG nanofibers, and (D) histograms of the diameter size distribution for PU10BG nanofibers. | |
Fig. 5 shows the SEM of the fibers after SBF immersion for 3, 7, and 14 days under different magnifications. This morphology is typical of hydroxyapatite, which has been reported to grow on the surface of bioactive glass/polymer composite scaffold after incubation in SBF. The difference between the morphology of the surfaces can be evidently observed. Apatite covers the surfaces with BG, and the formation of apatite increased with the increase in the BG amount, as confirmed by XRD. The XRD of the samples after soaking in SBF for seven days showed increase in the formation of the apatite phase with the increase in the BG content (Fig. 6). Hydroxyapatite formation due to the reactivity of BG will stimulate a physiologically relevant environment for the cells to adhere and proliferate. Radev et al.35 showed that nanohydroxyapatite formed on the surface of PU/85S composite after seven days of incubation in SBF and proposed this composite for bone tissue engineering application. The deposition of a hydroxyapatite layer on the surface of the bioactive materials soaked in SBF involves the mechanism proposed by Hench.50 The results indicated the formation of HA that increased gradually with time, consistent with previous research on the immersion of BG and PU/BG composites in SBF.28,52–54 Pores with diameters larger than 150 μm are necessary to accelerate internal mineralized bone formation, whereas pores with diameters smaller than 10 μm inhibit cellular ingrowth. Pores with diameters between 15 μm and 150 μm are suitable for fibrovascular colonization, osteoid growth, cellular penetration, and proliferation.55Fig. 4 shows that the electrospinning method was successful in generating these types of pores.
 |
| | Fig. 5 Apatite formation on the surfaces of PU, PU5BG, and PU10BG nanofibers after incubation in SBF for seven days. | |
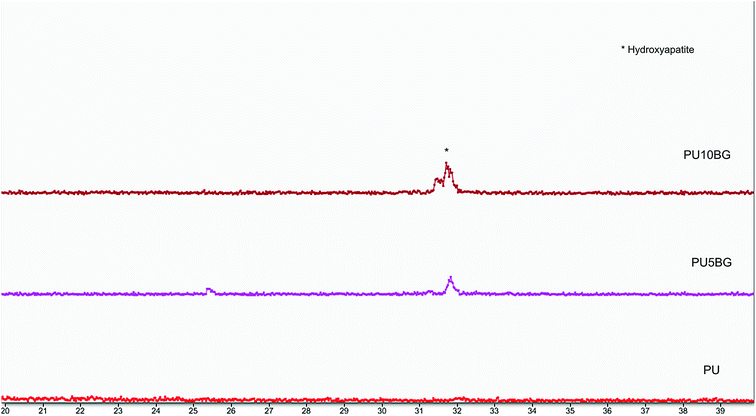 |
| | Fig. 6 XRD pattern of PU, PU5BG, and PU10BG after immersion in SBF for seven days. | |
The contact angle test results (Fig. 7) showed that the pure PU scaffold had the highest contact angle value at 105 ± 3.2, which decreased from 92 ± 2.1 to 72 ± 1.8 with the increase in BG content from 5% to 10%. Fig. 8 shows that scaffolds with a higher BG amount exhibited higher PBS uptake because of BG hydrophilicity.56 When the scaffold is pure PU, the PBS uptake content increased to 16% after six days, which increased to 33% with the increase in BG to the highest amount. These last two tests confirmed the hydrophilicity of BG. The hydrophilic property of the scaffolds can facilitate cellular nutrient supply, cell adhesion and growth, and waste removal and maintain differentiated phenotypic expression.28,57 The hydrophilicity of BG can affect the glass/polymer interface, degradation rate, and pH buffering effect.56,58 Good adhesion between filler and polymer can be obtained by intimate contact between phases, which is a result of suitable wettability.59
 |
| | Fig. 7 Micrograph of the surface contact angle of (a) PU, (b) PU5BG, and (c) PU10BG. | |
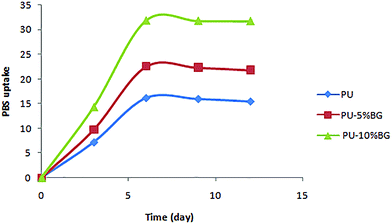 |
| | Fig. 8 PBS uptake of PU, PU5BG, and PU10BG. | |
In this study, L929 mouse fibroblasts cells were seeded on the surface of the scaffolds and cultured for 24 and 48 h. Fig. 9 shows the cell morphology after 24 and 48 h for the control, PU, PU5BG, and PU10BG groups. After culturing for 48 h, the cell morphology was observed by SEM and shown in Fig. 10 with two magnifications. The cells could grow, adhere, and stretch on the surfaces of all scaffolds. The MTT assay showed a low death rate because of the biocompatibility of the scaffold materials, which was enhanced with the increase in the BG amount (Fig. 11). This result indicates that the BG-containing scaffolds may have accelerated cell proliferation. The area covered with cells on the scaffolds increased with the increase in BG concentration. Ionic dissolution in bioactive glasses plays an essential role in such osteoconductivity, angiogenesis,60 and cellular processes as bone cell phenotypes, bone cell differentiation, and gene expression.61–64 For example, the release of Ca2+ depresses osteoclast-mediated bone resorption and addresses osteoblastic proliferation and the presence of a high concentration of phosphorus induces osteoblast apoptosis.41,42 Based on our observations, the incorporation of 58S BG in PU has potential for bone tissue engineering because of its bioactivity, cellular response, and wettability. However, more investigations are necessary to confirm our findings.
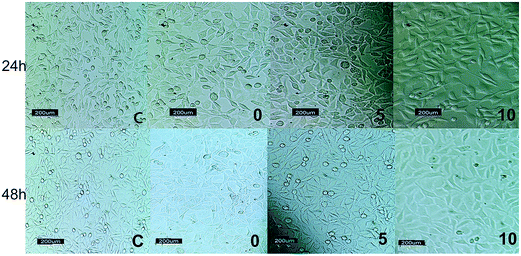 |
| | Fig. 9 Cell morphology after cell cultures with the control group (c), PU (0), PU5BG (5), and PU10BG (10) after 24 and 48 h. | |
 |
| | Fig. 10 Morphology of the cells on the surfaces of PU, PU5BG, and PU10BG nanofibers and the control group after two days of culture with two magnifications. | |
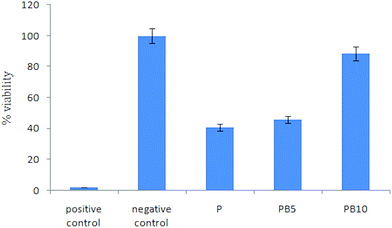 |
| | Fig. 11 MTT assay results. | |
Conclusion
In this study, highly in vitro bioactive and biocompatible materials, in the system of SiO2–CaO–P2O5, have been obtained by the sol–gel technique. Moreover, a novel porous scaffold based on PU and 58S BG was fabricated by electrospinning. This study shows that covalently linked PU/BG composite and the nanofibres of this composite were successfully collected by the electrospinning process and their diameter is below 200 nm. The morphological appearance of the nanofibres was smooth and uniform. The effect of different amounts of BG (5 and 10 wt%) on the physical and chemical properties, bioactivity, wettability, PBS uptake, cellular response, and MTT assay was investigated. The incorporation of the BG in the PU/BG scaffold significantly enhanced the bioactivity, biocompatibility, hydrophilicity and storage modulus of composite. L929 fibroblast cells adhered to and grew well on the bioglass-added PU scaffold, with a higher level of growth than on the pure PU. In brief, the results showed that the prepared scaffolds represent an interesting candidate for bone tissue engineering because of their ease of fabrication and suitable properties.
Acknowledgements
The authors would like to thanks High Impact Research Grant UM.C/HIR/MOHE/ENG D000014-16001 from the University of Malaya and Iranian Council for Stem Cell Sciences and Technologies for financial supports. The technical assistance of Dr Sumit Pramanik for the Image J analysis is appreciated.
References
- A. R. Shrivats, M. C. McDermott and J. O. Hollinger, Drug Discovery Today, 2014, 19, 781–786 CrossRef CAS PubMed.
- S. Wu, X. Liu, K. W. K. Yeung, C. Liu and X. Yang, Mater. Sci. Eng., R, 2014, 80, 1–36 CrossRef.
- P. Gentile, V. Chiono, I. Carmagnola and P. V. Hatton, Int. J. Mol. Sci., 2014, 15, 3640–3659 CrossRef CAS PubMed.
- J. Venkatesan and S.-K. Kim, J. Biomed. Nanotechnol., 2014, 10, 3124–3140 CrossRef CAS PubMed.
- S. Adzilaa, I. Sopyanc, T. C. Yonga, R. Singha, W. Y. Hoonga, J. Purbolaksonoa and M. Hamdia, Indian J. Chem., 2013, 52, 1570–1575 Search PubMed.
- A. Asefnejad, A. Behnamghader, M. T. Khorasani and B. Farsadzadeh, Int. J. Nanomed., 2011, 6, 93–100 CrossRef CAS PubMed.
- S. C. Cox, J. A. Thornby, G. J. Gibbons, M. A. Williams and K. K. Mallick, Mater. Sci. Eng., C, 2015, 47, 237–247 CrossRef CAS PubMed.
- Z. Fereshteh, P. Nooeaid, M. Fathi, A. Bagri and A. R. Boccaccini, Data in Brief, 2015, 4, 524–528 CrossRef PubMed.
- M. Kikuchi, Y. Koyama, T. Yamada, Y. Imamura, T. Okada, N. Shirahama, K. Akita, K. Takakuda and J. Tanaka, Biomaterials, 2004, 25, 5979–5986 CrossRef CAS PubMed.
- M. Hafezi, A. Reza Talebi, S. Mohsen Miresmaeili, F. Sadeghian and F. Fesahat, Ceram. Int., 2013, 39, 4575–4580 CrossRef CAS.
- M. K. Narbat, F. Orang, M. S. Hashtjin and A. Goudarzi, Iran. Biomed. J., 2006, 10, 215–223 CAS.
- M. Dziadek, E. Menaszek, B. Zagrajczuk, J. Pawlik and K. Cholewa-Kowalska, Mater. Sci. Eng., C, 2015, 56, 9–21 CrossRef CAS PubMed.
- W. Lu, K. Ji, J. Kirkham, Y. Yan, A. R. Boccaccini, M. Kellett, Y. Jin and X. B. Yang, Cell Tissue Res., 2014, 356, 97–107 CrossRef CAS PubMed.
- R. Ravarian, F. Moztarzadeh, M. S. Hashjin, S. M. Rabiee, P. Khoshakhlagh and M. Tahriri, Ceram. Int., 2010, 36, 291–297 CrossRef CAS.
- M. Yamamoto, G. Nishikawa, A. M. Afifi, I. Wataoka, J.-c. Lee and H. Yamane, American Journal of Macromolecular Science, 2015, 2, 11–24 Search PubMed.
- I. D. Xynos, A. J. Edgar, L. D. Buttery, L. L. Hench and J. M. Polak, J. Biomed. Mater. Res., 2001, 55, 151–157 CrossRef CAS PubMed.
- L. Drago, E. D. Vecchi, M. Bortolin, M. Toscano, R. Mattina and C. L. Romanò, Future Microbiol., 2015, 1–7 Search PubMed.
- M. Stevanović, N. Filipović, J. Djurdjević, M. Lukić, M. Milenković and A. Boccaccini, Colloids Surf., B, 2015, 132, 208–215 CrossRef PubMed.
- R. Detsch, S. Alles, J. Hum, P. Westenberger, F. Sieker, D. Heusinger, C. Kasper and A. R. Boccaccini, J. Biomed. Mater. Res., Part A, 2015, 103, 1029–1037 CrossRef PubMed.
- I. A. Silver, J. Deas and M. Erecińska, Biomaterials, 2001, 22, 175–185 CrossRef CAS PubMed.
- L. Hench, Curr. Orthop., 2000, 14, 7–15 CrossRef.
- P. Valerio, M. Pereira, A. Goes and M. F. Leite, Biomed. Mater., 2009, 4, 045011 CrossRef CAS PubMed.
- S. Joughehdoust and S. Manafi, Mater. Sci., 2012, 30, 45–52 CAS.
- H. Y. Mi, S. Palumbo, X. Jing, L. S. Turng, W. J. Li and X. F. Peng, J. Biomed. Mater. Res., Part B, 2014, 102, 1434–1444 CrossRef PubMed.
- G. Tetteh, A. Khan, R. Delaine-Smith, G. Reilly and I. Rehman, J. Mech. Behav. Biomed. Mater., 2014, 39, 95–110 CrossRef CAS PubMed.
- W. Yang, S. K. Both, Y. Zuo, Z. T. Birgani, P. Habibovic, Y. Li, J. A. Jansen and F. Yang, J. Biomed. Mater. Res., Part A, 2015, 103(7), 2251–2259 CrossRef CAS PubMed.
- F. Baino, E. Verné and C. Vitale-Brovarone, J. Mater. Sci.: Mater. Med., 2009, 20, 2189–2195 CrossRef CAS PubMed.
- J. L. Ryszkowska, M. Auguścik, A. Sheikh and A. R. Boccaccini, Compos. Sci. Technol., 2010, 70, 1894–1908 CrossRef CAS.
- M. Bil, J. Ryszkowska, J. Roether, O. Bretcanu and A. Boccaccini, Biomed. Mater., 2007, 2, 93 CrossRef CAS PubMed.
- W. Yang, S. K. Both, Y. Zuo, Z. T. Birgani, P. Habibovic, Y. Li, J. A. Jansen and F. Yang, J. Biomed. Mater. Res., Part A, 2015, 103(7), 2251–2259 CrossRef CAS PubMed.
- M. O. Wang, C. E. Vorwald, M. L. Dreher, E. J. Mott, M. H. Cheng, A. Cinar, H. Mehdizadeh, S. Somo, D. Dean and E. M. Brey, Adv. Mater., 2015, 27, 138–144 CrossRef CAS PubMed.
- S.-H. Shin, O. Purevdorj, O. Castano, J. A. Planell and H.-W. Kim, J. Tissue Eng., 2012, 3, 2041731412443530 CrossRef PubMed.
- T. Subbiah, G. S. Bhat, R. W. Tock, S. Parameswaran and S. S. Ramkumar, J. Appl. Polym. Sci., 2005, 96, 557–569 CrossRef CAS.
- M. R. Ahsan, M. A. Uddin and M. G. Mortuza, Indian J. Pure Appl. Phys., 2005, 43, 89–99 CAS.
- L. Radev, D. Zheleva and I. Michailova, Open Chem., 2013, 11, 1439–1446 CrossRef CAS.
- E. S. Jang, S. B. Khan, J. Seo, Y. H. Nam, W. J. Choi, K. Akhtar and H. Han, Prog. Org. Coat., 2011, 71(1), 36–42 CrossRef CAS.
- A. S. Khan, Z. Ahmed, M. J. Edirisinghe, F. S. L. Wong and I. U. Rehman, Acta Biomater., 2008, 4, 1275–1287 CrossRef CAS PubMed.
- A. Khan, Z. Ahmed, M. Edirisinghe, F. Wong and I. Rehman, Acta Biomater., 2008, 4, 1275–1287 CrossRef CAS PubMed.
- K. Gorna and S. Gogolewski, Polym. Degrad. Stab., 2002, 75, 113–122 CrossRef CAS.
- A. K. Mishra, D. K. Chattopadhyay, B. Sreedhar and K. V. S. N. Raju, Prog. Org. Coat., 2006, 55, 231–243 CrossRef CAS.
- L. Wang, Y. Li, Y. Zuo, L. Zhang, Q. Zou, L. Cheng and H. Jiang, Biomed. Mater., 2009, 4, 025003 CrossRef PubMed.
- M.-N. Huang, Y.-L. Wang and Y.-F. Luo, J. Biomed. Sci. Eng., 2009, 2, 36 CrossRef CAS.
- C. Li, C. Vepari, H.-J. Jin, H. J. Kim and D. L. Kaplan, Biomaterials, 2006, 27, 3115–3124 CrossRef CAS PubMed.
- H. Yoshimoto, Y. M. Shin, H. Terai and J. P. Vacanti, Biomaterials, 2003, 24, 2077–2082 CrossRef CAS PubMed.
- D. Verma, K. Katti and D. Katti, J. Biomed. Mater. Res., Part A, 2006, 78, 772–780 CrossRef PubMed.
- M. Ara, M. Watanabe and Y. Imai, Biomaterials, 2002, 23, 2479–2483 CrossRef CAS PubMed.
-
S. Dasgupta and K. Maji, Comparative study on mechanical strength of macroporous hydroxyapatite-biopolymer based composite scaffold, International Conference on Advances in Engineering and Technology (ICAET’2014), Singapore, 2014 Search PubMed.
- H. H. Lu, S. F. El-Amin, K. D. Scott and C. T. Laurencin, J. Biomed. Mater. Res., Part A, 2003, 64, 465–474 CrossRef PubMed.
- G. Ciardelli, A. Rechichi, S. Sartori, M. D'Acunto, A. Caporale, E. Peggion, G. Vozzi and P. Giusti, Polym. Adv. Technol., 2006, 17, 786–789 CrossRef CAS.
- L. L. Hench, J. Am. Ceram. Soc., 1991, 74, 1487–1510 CrossRef CAS.
- A. R. Boccaccini and V. Maquet, Compos. Sci. Technol., 2003, 63(16), 2417–2429 CrossRef CAS.
- A. R. Boccaccini and V. Maquet, Compos. Sci. Technol., 2003, 63, 2417–2429 CrossRef CAS.
- K. Rezwan, Q. Chen, J. Blaker and A. R. Boccaccini, Biomaterials, 2006, 27, 3413–3431 CrossRef CAS PubMed.
- C. Y. Kim, A. E. Clark and L. L. Hench, J. Non-Cryst. Solids, 1989, 113, 195–202 CrossRef CAS.
- R. C. M. Dias, A. M. Góes, R. Serakides, E. Ayres and R. L. Oréfice, Mater. Res., 2010, 13, 211–218 CrossRef CAS.
- H.-W. Kim, E.-J. Lee, I.-K. Jun, H.-E. Kim and J. C. Knowles, J. Biomed. Mater. Res., Part B, 2005, 75, 34–41 CrossRef PubMed.
- P. Van Wachem, T. Beugeling, J. Feijen, A. Bantjes, J. Detmers and W. Van Aken, Biomaterials, 1985, 6, 403–408 CrossRef CAS PubMed.
- C. Wu, Y. Ramaswamy, A. Soeparto and H. Zreiqat, J. Biomed. Mater. Res., Part A, 2008, 86, 402–410 CrossRef PubMed.
- J. J. Blaker, V. Maquet, A. R. Boccaccini, R. Jérôme and A. Bismarck, E-Polymers, 2005, 5, 248–260 Search PubMed.
- R. M. Day, Tissue Eng., 2005, 11, 768–777 CrossRef CAS PubMed.
- S. Hattar, S. Loty, D. Gaisser, A. Berdal and J. M. Sautier, J. Biomed. Mater. Res., Part A, 2006, 76, 811–819 CrossRef CAS PubMed.
- I. D. Xynos, A. J. Edgar, L. D. K. Buttery, L. L. Hench and J. M. Polak, Biochem. Biophys. Res. Commun., 2000, 276, 461–465 CrossRef CAS PubMed.
- I. D. Xynos, A. J. Edgar, L. D. K. Buttery, L. L. Hench and J. M. Polak, J. Biomed. Mater. Res., 2001, 55, 151–157 CrossRef CAS PubMed.
- J. Yao, S. Radin, G. Reilly, P. S. Leboy and P. Ducheyne, J. Biomed. Mater. Res., Part A, 2005, 75, 794–801 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a 









