
Simultaneous determination of ciprofloxacin and paracetamol by adsorptive stripping voltammetry using copper zinc ferrite nanoparticles modified carbon paste electrode†
Abstract
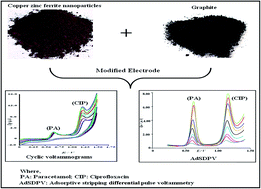
This work presents the development of an electrochemical sensor in the form of a copper zinc ferrite nanoparticle modified carbon paste electrode (CZF-CME) for the simultaneous determination of ciprofloxacin (CIP) and paracetamol (PA) using adsorptive stripping differential pulse voltammetry. The results indicate that the modified electrode exhibited enhancement of the oxidation peak current and shift in the oxidation potential to lower values in comparison with plain carbon paste electrode (CPE) for ciprofloxacin and paracetamol. The electrochemical performances of the modified electrode were investigated by cyclic voltammetry, chronocoulometry and electrochemical impedance spectroscopy, indicating the greater affinity of CIP and PA at the CZF-CME than at PCPE. Under the optimized conditions, the linear working ranges are from 9.09 × 10−7 M to 4.70 × 10−3 M and 1.85 × 10−7 M to 4.76 × 10−4 M for CIP and PA respectively. The detection limits (S/N = 3) of 2.58 × 10−9 M and 8.85 × 10−8 M for CIP and PA respectively were obtained when taken together. The CZF-CME showed relatively high sensitivity, selectivity, stability and the proposed method was successfully used for individual and simultaneous determination of ciprofloxacin and paracetamol in pharmaceutical formulations, serum and urine samples.
 Please wait while we load your content...
Please wait while we load your content...

