
Low temperature hydrogenation of pyrolytic lignin over Ru/TiO2: 2D HSQC and 13C NMR study of reactants and products†
Wen
Chen‡
ab,
Daniel J.
McClelland‡
b,
Ali
Azarpira
c,
John
Ralph
c,
Zhongyang
Luo
a and
George W.
Huber
*b
aState Key Laboratory of Clean Energy Utilization, Zhejiang University, Hangzhou, 310027, PR China
bDepartment of Chemical and Biological Engineering, University of Wisconsin-Madison, Madison, WI 53706, USA. E-mail: gwhuber@wisc.edu
cDepartment of Biochemistry, DOE Great Lakes Bioenergy Research Center, and Department of Biological Systems Engineering, University of Wisconsin, Madison, Wisconsin 53726, USA
First published on 15th October 2015
Abstract
Pyrolytic lignin and hydrogenated pyrolytic lignin were characterized by 2D 1H–13C HSQC and quantitative 13C NMR techniques. The pyrolytic lignin was produced from a mixed maple wood feedstock and separated from the bio-oil by water extraction. p-Hydroxyphenyl (H), guaiacyl (G), and syringyl (S) aromatics were the basic units of pyrolytic lignin. The native lignin β-aryl ether, phenylcoumaran and resinol structures were not present in the pyrolytic lignin. The hydrogenation was conducted with a Ru/TiO2 catalyst at temperatures ranging from 25–150 °C with higher temperatures exhibiting higher levels of hydrogenation. Solid coke formed on the catalyst surface (1% coke yield) even for hydrogenation at 25 °C. The carbon yield of pyrolytic lignin to coke increased from 1% to 5% as the hydrogenation temperature increased from 25 to 150 °C. A single-step hydrogenation at 150 °C resulted in a reduction from 65% to 39% aromatic carbons. A three-step hydrogenation scheme at this same temperature resulted in a reduction of aromatic carbons from 65% to 17%. The decrease in the aromatic carbon corresponded with an increase in the aliphatic carbon. Coke formation reduced from a 5% carbon yield of pyrolytic lignin in the first hydrogenation step to a 1% carbon yield in each of the second and third hydrogenation steps. The pyrolytic lignin could be separated into a high and low molecular weight fraction. The coke yield from the high molecular weight fraction was twice as much as that from the low molecular weight fraction.
Introduction
Fast pyrolysis of biomass combined with hydrodeoxygenation (HDO) of the fast pyrolysis produced bio-oil has been proposed as a low cost technology for the production of lignocellulosic fuels.1–5 Techno-economic analyses have shown that pyrolysis-HDO based approaches to biomass conversion have lower capital and operating costs than gasification and fermentation technologies.6–8 However, little is known about the chemistry that occurs during HDO due to the complicated reactions that are occurring in this process.9–12 The HDO approach has several challenges including low liquid product yields, reactor plugging, and high coke yields.10,13,14 This arises from the complexity of the bio-oil or pyrolysis oil which is a complicated emulsion that contains over 400 different compounds.15 Pyrolysis oil can be phase-separated into water-soluble and water-insoluble fractions by extraction with water.16 Upgrading the fractions separately may allow for better understanding of the HDO chemistry of each fraction and the bio-oil as a whole. The water-soluble fraction of bio-oil contains sugars, anhydrosugars, acetic acid, hydroxyacetone, hydroxyacetaldehyde, furfural, and small amounts of phenolics.16,17 We have previously reported on the catalytic chemistry involved in the hydrogenation and HDO of the water-soluble fraction.17–20Less is known about the water-insoluble phase or the pyrolytic lignin. The phase mainly consists of phenolic oligomers derived from thermal depolymerization of the lignin fraction of the biomass source.21 Pyrolytic lignin is difficult to hydrotreat due to rapid polymerization of the complex high molecular weight aromatic structures that comprise it.22 The analysis of the pyrolytic lignin is extremely difficult. Pyrolytic lignin is not volatile which makes GC analysis not possible without functionalizing the pyrolytic lignin.23 Spectroscopic techniques such as FTIR can give qualitative insights into functional groups in pyrolytic lignin.24–26 LC-MS is in principle possible, but remains less effective for the complex array of high molecular weight components. Gel permeation chromatography (GPC) can be used to give the approximate molecular weight of pyrolytic lignin.27–29 In contrast to the techniques mentioned above, NMR has the potential of providing quantitative (or at least comparative) and qualitative insights into pyrolytic lignin. 1H and 13C NMR have been used to identify the primary functionality of the pyrolytic lignin.21,26,27,30,31 2D 1H–13C correlation (HSQC – heteronuclear single-quantum coherence) NMR has been used to provide more detailed information about the substructures in lignin, however this technique has not been used for the analysis of pyrolytic lignin.32–34 As we will show in this publication HSQC NMR can be used to show the absence of β-aryl ether, phenylcoumaran, and resinol structures in pyrolytic lignin as well as the disappearances of G and S units during hydrogenation of pyrolytic lignin. Meier et al.27,35–37 analyzed the structures of pyrolytic lignin using GPC, PY-GC/MS, FTIR, 13C NMR, and MS. They proposed five chemical structures ranging from a tetramer to an octamer with typical oligomers containing biphenyl, phenylcoumaran, diphenyl ether, stilbene, and resinol functionalities in them.
There are a limited number of papers that have reported on the HDO of pyrolytic lignin using Pd/C, Ru/C, or Pt/C in batch reactors.22,26,38,39 Hydrodeoxygenation of lignin and lignin model compounds have been studied; however, pyrolytic lignin has differences in both subunit linkages and functionalities from both the native lignins and the model compounds.40,41 The previous studies focused on the analysis of the final products from hydrotreating of pyrolytic lignin and did not identify the pyrolytic lignin-derived intermediates. Recent studies have focused on stabilization of bio-oil fractions in an effort to reduce the formation of solids during aging as well as prevent coke formation in further hydrotreating reactions.26,42 Rover et al. recently reported that phenolic bio-oil fractions similar to pyrolytic lignin could be hydrogenated at room temperature and 1 bar over a Pd/C catalyst to saturate bio-oil compounds and produce a more stable product. They observed a 25.5% to 350% increase in aliphatic proton resonances through semi-quantitative 1H NMR.26 Elliot et al. also recently reported that a two stage hydrotreating scheme of phenolic bio-oil fractions, a lower temperature Ru/C stabilization followed by a higher temperature Pd/C hydrogenation, removed heteroatoms with up to a 81% carbon yield.42
The objective of this paper is to begin to develop a fundamental understanding of the chemistry that occurs during low temperature hydrogenation of pyrolytic lignin using HSQC and quantitative 13C NMR. Understanding this chemistry will allow us to design more efficient catalytic processes for the production of fuels and chemicals from bio-oils.
Experimental
Preparation and fractionation of pyrolytic lignin
Pyrolytic lignin was obtained by water extraction of pyrolysis bio-oil obtained from mixed hard woods. The mixed hardwood was obtained from the Cersosimo Lumber Company located in Brattleboro, VT harvested from various parts of the New England region. The bio-oil was collected from the 6th and 7th condenser of a fast pyrolysis Auger reactor.43,44 The bio-oil was stored in the refrigerator to minimize ageing. The bio-oil appeared as one single phase prior to separating it into various components. The procedure used for separation of the pyrolytic lignin from the bio-oil, modified from work by Oasmaa and Kuoppala,28,29 is shown in Fig. 1. In brief, 5 g of bio-oil was added dropwise to 40 g of deionized water in a 50 mL centrifuge tube. The mixture was shaken then centrifuged at 7500 rpm (6603 rcf) in an Eppendorf Centrifuge 5430R v 2.2 for 40 min. After centrifuging, two phases, a light yellow-orange, water-soluble phase and a black, viscous, water-insoluble phase (pyrolytic lignin), were formed and separated by decanting. | ||
| Fig. 1 Fractionation of bio-oil into water-soluble, LMW, and HMW pyrolytic lignin fractions. | ||
The pyrolytic lignin was further fractionated by organic solvent extraction. Dichloromethane was added to the pyrolytic lignin which was shaken and centrifuged with the same method that was used for centrifuging the bio-oil. Two fractions formed, a dichloromethane-soluble fraction and a dichloromethane-insoluble fraction. The dichloromethane-soluble fraction was decanted off and collected. This was repeated two more times for the insoluble fraction using a total of 18 g of dichloromethane for 2.4 g of pyrolytic lignin for the fractionation. Oasmaa et al.29,45 showed that the dichloromethane-insoluble fraction had a 1.5 times higher molecular weight than the dichloromethane-soluble fraction by using GPC. In this paper, the dichloromethane-soluble fraction and the dichloromethane-insoluble fraction were designated as the low molecular weight fraction (LMW fraction) and the high molecular weight fraction (HMW fraction), respectively. After fractionation, the dichloromethane was removed by a 24 h evaporation step at 50 °C for the LMW fraction and at 20 °C for the HMW fraction. The pyrolytic lignin and pyrolytic lignin fraction solutions were kept under refrigeration and hydrogenated or analyzed within two weeks of preparation.
Hydrogenation of pyrolytic lignin and biphenyl activity testing
Pyrolytic lignin is a black viscous material with a smoky odor. It is soluble in ethanol (up to 15 wt%), and hence ethanol was used as the reaction solvent. Reactions were carried out in a 50 mL HEL batch reactor which had a pressure gauge, two inlet/outlet valves, and a pressure relief valve. The HEL batch reactor was placed in an electric heating well/magnetic stirrer with a controller to control the temperature, temperature ramp rate, and stir speed of a magnetic stir bar. The catalyst used in the experiments was Ru/TiO2 prepared by incipient wetness impregnation. This catalyst was chosen due to the high activity of ruthenium for hydrogenation reactions compared to other metals (Pd, Rh, Pt, Ni, Co)20,46 and the high hydrothermal stability of titania.47 Titanium(IV) oxide (Sigma-Aldrich catalog number 718467) and ruthenium(III) nitrosyl nitrate solution (Sigma-Aldrich catalog number 373567) were used as the support and precursor for the preparation of Ru/TiO2. ICP-AES was conducted by Galbraith Laboratories, Inc. The catalyst was determined to be 2.80 wt% Ru. The CO uptake of the catalyst was 16.63 μmol g−1 as measured using CO pulse chemisorption on a Micromeritics Autochem II 2920. Prior to the experiment, 1.50 g of catalyst was reduced in the reactor. The feed, 10 g of 10 wt% pyrolytic lignin in ethanol, was added to the reactor which was then purged with 725 psig of H2 five times and pressurized with H2 to 725 psig. The reactor was then heated at a ramp rate of 10 °C min−1 to 150 °C min−1 and held for a reaction time of 2.5 h. The solution was stirred at 700 rpm for the duration of the heating phase and reaction. After completion of the reaction, the reactor was quenched to room temperature for collection and analysis of the products.Biphenyl was used to test the hydrogenation activity of the fresh and spent catalysts. The activity test comprised of reacting 10 g of 10 wt% biphenyl in n-hexane over a fresh or spent catalyst at 150 °C and 725 psig H2. The procedure was the same as the pyrolytic hydrogenation except the reaction was held for 1.0 h instead of 2.5 h.
Product analysis
The pressure after reaction was recorded to measure the amount of hydrogen consumed. The gaseous products were collected using a gas bag and analyzed using a Shimadzu GC2014 equipped with two parallel lines: (1) a Restek RTX-Alumina column connected to a flame ionization detector (FID) to analyze hydrocarbons (CH4, C2–C5 alkanes and olefins) and (2) a RTX-MS-5A column followed by a RTX-Q-plot with a thermal conductivity detector (TCD) to analyze CO and CO2.After hydrogenation a single phase reddish-brown transparent liquid was obtained. The liquid product and recovered catalyst were collected and separated by filtration. The liquid product was separated by rotary evaporation at 55 °C and 2.5 psi (170 mbar) for 1 h. The distillate and residue are designated as volatile liquids and non-volatile liquids respectively.
The non-volatile liquids were characterized by quantitative 1D 13C and 2D HSQC NMR. The NMR samples were prepared by dissolving the non-volatile liquids in DMSO-d6 with benzaldehyde as an internal standard. The wt% of non-volatile liquids, DMSO-d6, and benzaldehyde were 25%, 72.5%, and 2.5%. All NMR experiments were carried out on a Bruker Biospin (Billerica, MA) AVANCE 500 MHz spectrometer fitted with a cryogenically cooled 5 mm DCH (13C-optimized) gradient probe. Quantitative 13C NMR spectra were obtained by using an inverse-gated decoupling pulse sequence, a 90° pulse, a relaxation delay of 12 s, a sweep width of 350 ppm, a TD of 87![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 714, and 1024 scans. In the quantitative 13C NMR spectra, the relative peak area of the carbonyl group from benzaldehyde was set to 1. The HSQC experiment used the Bruker standard pulse sequence ‘hsqcetgp’ with the following parameters: 16 ppm sweep width (±6 ppm due to automated sweep width adjustment of the spectrometer) in F2 (1H) with a TD2 of 2166, 220 ppm sweep width in F1 (13C) with a TD1 of 352, 24 scans, a 1.5 s relaxation delay, and with the evolution time set for a 1-bond 1H–13C coupling constant of 145 Hz, with a total acquisition time of 3 h. Processing utilized 4096 × 1024 FFT, cosinebell-squared apodization in F1, Gaussian multiplication apodization in F2, and linear prediction in neither dimension.
714, and 1024 scans. In the quantitative 13C NMR spectra, the relative peak area of the carbonyl group from benzaldehyde was set to 1. The HSQC experiment used the Bruker standard pulse sequence ‘hsqcetgp’ with the following parameters: 16 ppm sweep width (±6 ppm due to automated sweep width adjustment of the spectrometer) in F2 (1H) with a TD2 of 2166, 220 ppm sweep width in F1 (13C) with a TD1 of 352, 24 scans, a 1.5 s relaxation delay, and with the evolution time set for a 1-bond 1H–13C coupling constant of 145 Hz, with a total acquisition time of 3 h. Processing utilized 4096 × 1024 FFT, cosinebell-squared apodization in F1, Gaussian multiplication apodization in F2, and linear prediction in neither dimension.
A total organic carbon analyzer (TOC; Shimadzu TOC-VCPH) with a solid sample module (Shimadzu SSM-5000A) was utilized to determine the total carbon content of the pyrolytic lignin, its fractions, the non-volatile liquid product, and spent catalyst. Together with the quantitative 13C NMR, TOC was used to determine carbon yields of the hydrogenation products.
The liquid products were qualitatively analyzed on a GCMS-QP2010S (Shimadzu) equipped with a Restek-VMS column. The operating conditions were as follows: (1) hold for 5 min at 35 °C, (2) heat to 140 °C at 5 °C min−1, (3) heat to 230 °C at 50 °C min−1, and (4) hold for 20 min at 230 °C. Less than 2% of the carbon in the liquid products and non-volatile liquids of pyrolytic lignin, its fractions, and hydrogenated pyrolytic lignin was detected with GCMS.
The liquid products from the biphenyl hydrogenation were analyzed by Shimadzu GC2010 system with an Agilent HP INNOWax column (60 m, 0.32 mm, 0.5 μm) and a FID. The following GC conditions were used: (1) hold for 10 min at 70 °C, (2) heat to 95 °C at 2 °C min−1, (3) heat to 240 °C at 15 °C min−1 and (4) hold for 10 min at 240 °C.
The recovered catalyst was washed with acetone until the filtrate was colorless. Prior to coke analysis, the washed catalyst was rinsed three times with water and dried in the oven overnight at 110 °C. The dried catalyst was then run through the TOC to determine the mass of carbon as coke per mass of the spent catalyst. This allowed for the total moles of carbon as coke to be determined and with eqn (4), the carbon yield to coke.
The mass and moles of carbon in volatile liquids from pyrolytic lignin were estimated according to eqn (1) and (2).
| Mass of volatile liquids = MEL − MLP × Weth | (1) |
 | (2) |
The mass and carbon yields of volatile liquids, non-volatile liquids, and coke from pyrolytic lignin were calculated according to eqn (3) and (4).
 | (3) |
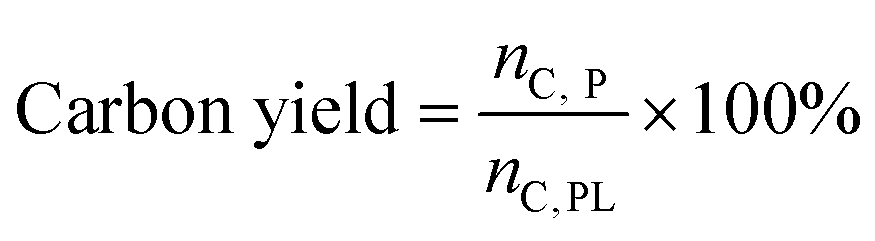 | (4) |
Results and discussion
Characterization of pyrolytic lignin and its fractions by NMR
Two dimensional HSQC NMR experiments have the ability to resolve otherwise overlapping resonances observed in either of the 1D 1H or 13C NMR spectra.33,34 The HSQC spectra of the pyrolytic lignin is shown in Fig. 2. This figure can be divided up into three regions, (1) the aromatic region (δC/δH 95–145/6–8.25), (2) the aliphatic C–O region (δC/δH 50–95/2.75–6.0), and (3) the aliphatic C–C region (δC/δH 5–50/0.5–3.0). These three regions are displayed in more detail in Fig. 3–5. | ||
| Fig. 2 HSQC spectra of pyrolytic lignin highlighting the aliphatic C–C, aliphatic C–O, and aromatic regions. | ||
 | ||
| Fig. 3 Aromatic regions of HSQC spectra from (A) pyrolytic lignin, (B) LMW pyrolytic lignin fraction, (C) HMW pyrolytic lignin fraction, and (D) three-step hydrogenated pyrolytic lignin. Dotted lines represent positions of the syringyl and guaiacyl signals. | ||
 | ||
| Fig. 4 Aliphatic C–O regions of HSQC spectra from (A) pyrolytic lignin, (B) LMW pyrolytic lignin fraction, (C) HMW pyrolytic lignin fraction, and (D) three-step hydrogenated pyrolytic lignin. Positions of absent resinol, phenylcoumaran, and β-O-4 unit correlation peaks are shown by dotted circles. | ||
 | ||
| Fig. 5 Aliphatic C–C regions of HSQC spectra from (A) pyrolytic lignin, (B) LMW pyrolytic lignin fraction, (C) HMW pyrolytic lignin fraction, and (D) three-step hydrogenated pyrolytic lignin. | ||
HSQC correlation signals from the pyrolytic lignin were compared with published data for lignin structures.48–53 The pyrolytic lignin, LMW fraction, HMW fraction, and three-step hydrogenated pyrolytic lignin are characterized by 2D 1H–13C HSQC NMR in combination with quantitative 13C NMR as shown in the ESI (Fig. S1†). Fig. 3 shows the aromatic region for the pyrolytic lignin, the LMW fraction, the HMW fraction, and the three-step hydrogenated pyrolytic lignin. The intense correlation signals between δC/δH 128.0–136.0/7.40–8.00 derive from the protonated aromatic carbons of benzaldehyde that was added as an internal standard. The cross-signals at δC/δH 105.5/6.42 are attributed to C2,6/H2,6 correlations of syringyl (S) units. From previous literature,32–34,54,55δC/δH 103.8/6.69 and 106.2/7.27 are attributed to C2,6/H2,6 positions of syringyl units in native lignin. Strong correlation signals at δC/δH 112.8/6.70, 115.4/6.61, and 119.3/6.61 correspond to C2/H2, C5/H5, and C6/H6 correlations of guaiacyl (G) units. If present small correlations at δC/δH 129.3/6.91–7.15 would correlate to C2,6/H2,6 from p-hydroxyphenyl (H) units. The correlations of the H3,5 position overlap with those from G5/6 position. Slight changes in the chemical shifts in the aromatic region from previously reported signals32–34,54,55 to our observed signals might be due to interactions of other functional groups that are formed in the pyrolysis process, i.e., on the altered sidechain on the H, G, and S aromatic rings. The aromatic regions from pyrolytic lignin and LMW fraction are quite similar. The HMW spectra has a higher ratio of G to S units than the LMW and pyrolytic fractions. Signals in the HMW fraction are much broader than those in the LMW fraction, consistent with its higher molecular weight and slower molecular motion.21 Only S groups are observed in the aromatic region of the three-step hydrogenated pyrolytic lignin.
The aliphatic C–O regions in the HSQC spectra are presented in Fig. 4. Signals from methoxyls (δC/δH 55.7/3.75) and ethanol (δC/δH 56.0/3.50) are the most prominent in this region. Lignin is a complex macromolecule synthesized from two main precursors, coniferyl, and sinapyl alcohols (with low levels from p-coumaryl alcohol),56 in which the monomer-derived units are connected by several types of C–C or C–O–C linkages, including β-5, 5–5, β–β, β-O-4, and 4-O-5, forming phenylcoumaran, biphenyl, resinol, alkyl–aryl ether, and biphenyl ether substructures. β-O-4-linkages are involved in the predominant β-aryl ether units in lignin.56 The β-aryl ether units have characteristic signals at δC/δH 72.2/4.86 from Cα/Hα, 85.8/4.12, 86.8/3.99, or 83.9/4.29 from Cβ/Hβ, and δC/δH 59.5–59.7/3.41–3.64 from Cγ/Hγ.32–34,54 These signals, along with those corresponding to the characteristic signals from resinols or phenylcoumarans, are not observed in the spectra shown in Fig. 4. This is in accordance with previous pyrolysis studies that indicate that ether bonds in cyclic resinols and phenylcoumarans, and β-O-4-linkages in β-ethers are broken during pyrolysis.57,58 Our results differ from the work of Meier who did not use HSQC analysis but claimed that resinols and phenylcoumarans were present in their pyrolytic lignin.36,37 In this region, the HMW fraction presents signals at δC/δH 71.8/3.32, 73.8/3.48, 76.6/4.40, and 89.1/5.24, whereas the LMW fraction, does not have these signals. This demonstrates that the C–O linkages in LMW and HMW are different. The three-step hydrogenated pyrolytic lignin shows increases in both the number of signals and signal intensities. This is expected as G, H, and S aromatic units are converted to aliphatics during hydrogenation.
The aliphatic C–C regions of HSQC spectra are shown in Fig. 5. The signal intensity in this region is higher than in the C–O region. Similar results have been reported with the 13C NMR spectra of pyrolytic lignin from different feedstocks obtained by Scholze et al.27 and Mullen et al.21 Pyrolytic lignin, the LMW fraction, and the HMW fraction show similar spectra in this region. The three-step hydrogenated pyrolytic lignin spectra has many more signals in the aliphatic C–C region than the non-hydrogenated fractions.
Based on these results, it can be concluded that the aromatic units in pyrolytic lignin are primarily S and G with minor amounts of H. No evidence can be found for the formation of polynuclear aromatics such as naphthalenes indicating that such condensations are not prominent during lignin pyrolysis. Meier proposed several different dimeric substructures including biphenyl, phenylcoumaran, resinol, and diphenyl ether while Boateng et al.21 did not detect phenylcoumaran or resinol structures in their pyrolytic lignin. In this work, characteristic HSQC signals corresponding to phenylcoumaran and resinol structures are not evident. Both Meier and Boateng suggested the existence of biphenyl structures in which the two aromatics are linked by a 5–5′-bond. This structure was also identified in lignin and has the highest bonding dissociation energy among the various inter-unit linkages. This implies that this structure might be retained during pyrolysis although it could not be confirmed from 2D-HSQC or 1D 13C NMR. Biphenyls are not easily detectable in 2D-HSQC due to the tertiary nature of the diagnostic 5-carbons (without a bonded hydrogen); in native lignins they are readily identified as they exist as dibenzodioxocins that has diagnostic α- and β-C/H correlations.59,60 Biphenyls could also not be identified in 13C NMR spectra due to the large number of overlapping peaks in that region.
Two contrasting hypotheses have been proposed for the formation of high molecular weight pyrolytic lignin molecules, one by reactive boiling ejection61 and the other by depolymerisation to volatile monomers and repolymerization after escaping primary pyrolysis.58 Teixeira et al. has shown reactive boiling ejection to be a mechanism for the production of anhydro-oligomers during pyrolysis of cellulose61 and involves an intermediate liquid cellulose which violently boils expelling liquid aerosols. The liquid aerosols become entrained in the gas phase allowing for transportation of the larger non-volatile molecules out of the pyrolysis bed. Further investigation of this method with lignin showed the formation of a lignin liquid phase but the ejection of particles could not be confirmed.61 Contrasting this, Patwardhan et al. has shown that pyrolysis of lignin yields up to 30.5% of monolignols with a 17% yield of CO and CO2 and 37% yield of char. The accompanying bio-oil condensation conditions were shown to facilitate polymerization of the monolignols.58 The absence of β-aryl ether, phenylcoumaran, and resinol structures in the pyrolytic lignin supports that depolymerisation occurs during pyrolysis.
Hydrogenation of pyrolytic lignin
| Reaction temperature (°C) | Gas products | Carbon balance (Carbon%) | |||||
|---|---|---|---|---|---|---|---|
| CH4a (mmol) |
C2H6a (mmol) |
H2a (mmol) |
Volatile liquidsb | Non-volatile liquidsc | Coke | Total | |
| a Calculated by multiplying the volume percentage of a specific gas by total moles of gas product assuming that all gases obey the ideal gas law. b Volatile liquids: distillates after rotary evaporation of liquid product at 55 °C and 2.47 psi (170 mbar) for 1 h. c Non-volatile liquids: distillation residue after rotor evaporation of liquid product at 55 °C and 2.47 psi (170 mbar) for 1 h. | |||||||
| 25 | 0 | 0 | 93.6 | 15.0 | 71.8 | 1.1 | 87.9 |
| 50 | 0 | 0 | 92.6 | 15.0 | 74.3 | 1.4 | 90.7 |
| 100 | 0.1 | 0 | 90.6 | 16.3 | 72.1 | 1.5 | 90.0 |
| 150 | 1.5 | 0.2 | 84.2 | 15.9 | 76.5 | 4.9 | 97.3 |
Rotary evaporation was used to remove the solvent after reaction. As shown in Table 1, over 85% of the carbon from the pyrolytic lignin remained in the volatile or non-volatile liquid phases after hydrogenation for all the temperatures tested. The hydrogenation of pyrolytic lignin does not have a dramatic effect on the change of the product volatility. The coke yield slightly increased from 1.1 to 1.5% as the temperature increases from 25 to 100 °C. Furthermore, the coke yield increased to 4.9% as the temperature increases to 150 °C. As no coke formed during the reaction of the pure ethanol, ethanol was not the cause for coke formation.
The non-volatile liquids of the hydrogenated pyrolytic lignin were analyzed by quantitative 13C NMR with the results shown in Fig. 6. The percentages of carbonyl (including carboxyl), and aromatic carbons decrease by 31.9% and 11.9% respectively after hydrogenation at 25 °C demonstrating that hydrogenation begins at a low temperature. Increasing the reaction temperature to 50 °C, causes a 34.1% and 14.5% decrease from the feed in the carbonyl and aromatic carbons. More prominent decreases in carbonyl and aromatic carbons and increases in aliphatic C–O and C–C carbons are observed at the reaction temperatures of 100 °C and 150 °C. The percentage of aromatic carbon decreases from 65% in the pyrolytic lignin feed to 39% in the non-volatile liquids at 150 °C. The carbonyl and carboxyl group decrease from 6.5% to 2.0% and the percentage of aliphatic C–O and C–C bonds increase from 5.6% and 23.3% to 16.5% and 43.0% at the same reaction temperature. This indicates that unsaturated compounds are hydrogenated to saturated aliphatics.
 | ||
| Fig. 6 Quantitative 13C NMR results of non-volatile liquids from hydrogenation of pyrolytic lignin at various temperatures. Carbonyl and carboxyl region (□), aromatic region (○), aliphatic C–O region (△), and aliphatic C–C region (∇). | ||
The quantitative 13C NMR spectra of the pyrolytic lignin feed and hydrogenated pyrolytic lignin at 150 °C was further analyzed to compare the carboxyl and other carbonyl functionality. In the quantitative 13C NMR spectra, 165–180 ppm corresponds to carboxyl group, while 180–215 ppm are attributed to other carbonyl groups.62 The peak area ratio of carboxyl groups to carbonyl groups changes from 0.78:1 in pyrolytic lignin to 24.5:1 in hydrogenated pyrolytic lignin. This suggests that carbonyl groups are easier to hydrogenate than carboxyl functionalities. This is in agreement with literature results which shows that the rate of hydrogenation of carbonyl groups is about 80 times higher than the rate of hydrogenation of carboxyl groups with Ru catalysts.19,46,63
The hydrogen balance for the run at 150 °C was calculated through NMR, pressure change, and GC gas product analyses. The hydrogen consumption was estimated to be 11.3 mmol by the change in pressure and gas composition. The stoichiometric hydrogen consumption calculated from the 13C NMR results and gas product analysis was calculated to be 9.3 mmol, within which 7.62 mmol was used to hydrogenate the non-volatile liquids. The overall hydrogen balance was 81.8%.
Though a pressure drop is observed after hydrogenation at 150 °C and cooling, the reactor pressure did not decrease with reaction time during the actual reaction (ESI Fig. S2†). This suggests that the catalytic reactions occurred at very short time periods and the coke poisoned the catalysts during the initial heating period.
Multistep hydrogenation of pyrolytic lignin
Hydrogenation of pyrolytic lignin was investigated using a multistep approach to understand the potential limitations of single-step hydrogenations. Table 2 highlights the carbon balances of the three-step hydrogenation of pyrolytic lignin. In each of these experiments the product is separated from the spent catalyst and the hydrogenation reaction is repeated with fresh catalyst.| Hydrogenation step | Gas products | Carbon balance (carbon%) | |||||
|---|---|---|---|---|---|---|---|
| CH4a (mmol) |
C2H6a (mmol) |
H2a (mmol) |
Volatile liquidsb | Non-volatile liquidsc | Coke | Total | |
| a Calculated by multiplying the volume percentage of a specific gas by total moles of gaseous product assuming that all gases obey the ideal gas law. b Volatile liquids: distillates after rotor evaporation of liquid product at 55 °C and 170 mbar for 1 h. c Non-volatile liquids: distillation residue after rotor evaporation of liquid product at 55 °C and 170 mbar for 1 h. | |||||||
| 1st hydrogenation | 1.5 | 0.2 | 84.2 | 15.9 | 76.5 | 4.9 | 97.3 |
| 2nd hydrogenation | 4.0 | 0.3 | 85.1 | 21.1 | 72.6 | 1.0 | 94.7 |
| 3rd hydrogenation | 4.2 | 0.4 | 84.8 | 26.7 | 71.8 | 1.0 | 99.4 |
The amount of CH4 and C2H6 produced increases with the number of hydrogenation steps. The carbon yield to coke is 1% after the second and the third step of the hydrogenation, which is nearly five times lower than after the first step. The carbon yield to volatile liquids increases from 15.9% in the first step to 26.7% in the third step, which implies that the pyrolytic lignin can undergo C–C bond hydrogenolysis to create more volatile compounds during this low temperature hydrogenation.
The quantitative 13C NMR results of the multistep hydrogenation are displayed in Fig. 7. The percentages of carbonyl (including carboxyl), and aromatic carbons decrease and the percentages of aliphatic C–O and C–C moieties increase with additional hydrogenation steps. The percentage of carboxyl groups decrease from 1.9% after the first step hydrogenation to 1.6% after the second hydrogenation step and further decrease to 1.1% after the third hydrogenation. The S/G ratio increases from 1:2.4 to 1:1.8 after the second hydrogenation step. After the third hydrogenation step, almost no signals remained in the G or H regions, whereas signals in the S region were still present. Zhang et al.64 carried out aqueous-phase hydrodeoxygenation of monomeric lignin model compounds including anisole, catechol, guaiacol and syringol over Ru/HZM-5 and observed the rate of hydrogenation of guaiacol was greater than that of syringol which is consistent with our results.
 | ||
| Fig. 7 Quantitative 13C NMR results of non-volatile liquids from multistep hydrogenation of pyrolytic lignin. Carbonyl and carboxyl region (□), aromatic region (○), aliphatic C–O region (△), aliphatic C–C region (∇). | ||
The ratio of C–O:C–C aliphatics is 0.24:1.00 in the pyrolytic lignin and changes to 0.38:1.00, 0.37:1.00, and 0.35:1.00 after the first, second, and third 150 °C hydrogenation respectively. The C–O:C–C aliphatic ratio increases with temperature from 0.24:1.00 in the feed to 0.29:1.00, 0.32:1.00, 0.37:1.00, and 0.38:1.00 at 25, 50, 100, and 150 °C respectively.
The catalysts were recovered and the activity for hydrogenation of biphenyl was measured to compare the activity of the spent and fresh catalyst as shown in Table 3. The conversion of biphenyl was 69.3% over a fresh Ru/TiO2 catalyst, whereas the conversion decreased to 7.4% over the Ru/TiO2 after hydrogenation of pyrolytic lignin at 150 °C.
| Catalyst | Conversion of Bipa (%) | Selectivity to Bica (%) | Selectivity to Phca (%) | Selectivity to unidentified (%) |
|---|---|---|---|---|
| a Bip: Biphenyl; Bic: Bicyclohexyl; Phc: Phenylcyclohexyl. b 1st Ru/TiO2: recovered Ru/TiO2 from the 1st hydrogenation step of pyrolytic lignin. c 2nd Ru/TiO2: recovered Ru/TiO2 from the 2nd hydrogenation step of pyrolytic lignin. d 3rd Ru/TiO2: recovered Ru/TiO2 from the 3rd hydrogenation step of pyrolytic lignin. | ||||
| Fresh Ru/TiO2 | 69.3 | 24.3 | 69.9 | 5.9 |
| 1st Ru/TiO2b |
7.4 | 7.2 | 35.2 | 57.7 |
| 2nd Ru/TiO2c |
25.7 | 17.6 | 68.6 | 13.8 |
| 3rd Ru/TiO2d |
69.5 | 25.2 | 68.6 | 6.2 |
Furthermore, the selectivity to the fully hydrogenated product, bicyclohexyl, decreases over the recovered Ru/TiO2. This result shows that Ru/TiO2 deactivates after hydrogenation of pyrolytic lignin at 150 °C most likely due to coke forming on the catalyst surface. As the number of hydrogenation steps increases from 1 to 3, the conversion increases from 7.4% to 69.5%. This indicates that further hydrogenation steps decreases the coke formation. It also demonstrates that the remaining aromatics were harder to hydrogenate as the catalyst was still active towards biphenyl hydrogenation yet did not hydrogenate more of the aromatics.
Hydrogenation of pyrolytic lignin fractions
The LMW and HMW fractions of the pyrolytic lignin were hydrogenated with Ru/TiO2 at 150 °C as shown in Table 4 and Fig. 8. Methane and ethane were produced in both experiments. According to the changes in pressure and gas composition, the hydrogen consumed during LMW fraction hydrogenation was 10.1 mmol, while that during HMW fraction hydrogenation was 6.4 mmol. | ||
| Fig. 8 Quantitative 13C NMR results of non-volatile liquids from low molecular weight fraction (LMW fraction), high molecular weight fraction (HMW fraction), hydrogenated low molecular weight fraction (Hydro LMW fraction), and hydrogenated high molecular weight fraction (Hydro HMW fraction). Carbonyl and carboxyl bond (□), aromatic bond (○), aliphatic C–O bond (△), and aliphatic C–C bond (∇). | ||
| Reaction feedstock | Gas products | Carbon balance (Carbon%) | |||||
|---|---|---|---|---|---|---|---|
| CH4a (mmol) |
C2H6a (mmol) |
H2a (mmol) |
Volatile liquidsb | Non-volatile liquidsc | Coke | Total | |
| a Calculated by multiplying the volume percentage of a specific gas by total moles of gaseous product assuming that all gases obey the ideal gas law. b Volatile liquids: distillates from rotor evaporation of liquid product at 55 °C and 170 mbar for 1 h. c Non-volatile liquids: distillation residue from rotor evaporation of liquid product at 55 °C and 170 mbar for 1 h. d LMW fraction: low molecular weight fraction. e HMW fraction: high molecular weight fraction. | |||||||
| LMW fractiond | 0.5 | 0.05 | 85.4 | N/A | 87.4 | 3.9 | 91.3 |
| HMW fractione | 0.6 | 0.04 | 89.0 | 25.4 | 63.5 | 7.7 | 96.6 |
Hydrogenation of the LMW and HMW fractions did not change the distribution of volatile liquids and non-volatile liquids from the feedstock (Table S1†). The coke yield from hydrogenation of the HMW fraction was 7.7%, which was nearly two times higher than the coke yield from the LMW fraction.
The quantitative 13C NMR results of the non-volatile liquids from the LMW fraction, the HMW fraction, and their respective hydrogenated products are shown in Fig. 8. Compared with non-volatile liquids from HMW fraction, those from the LMW fraction contained more carbonyl, carboxyl, and aliphatic carbons but less aromatic carbons. After hydrogenation, the percentages of carbonyl, carboxyl, and aromatics decreased whereas the aliphatic carbons increased for the non-volatile liquids from both fractions. The percentage of aromatic carbons in the non-volatile liquids of LMW fraction decreased from 60.4% to 43.5% whereas in the non-volatile liquids of HMW fraction, decreased from 72.8% to 59.4%.
Pyrolytic lignin can be hydrogenated over a Ru/TiO2 catalyst although catalyst deactivation due to coke formation is a major issue. The higher molecular weight compounds in pyrolytic lignin contribute more to the formation of coke than the lower molecular weight compounds. Along with the reduction in the formation of coke from further hydrogenation steps, this eludes to certain molecules or functionalities in the molecules being responsible for, or more prone to, coke formation. To reduce the formation of coke, a filtration or mild reaction may be necessary to remove or stabilize these compounds. Rover et al. demonstrated low pressure, low temperature hydrogenation over Pd/C to stabilize phenolic oligomers, a bio-oil fraction similar to pyrolytic lignin.26 Another method to reduce coke formation during the hydrotreating would be to use a catalytic pyrolysis approach to produce the pyrolysis oil. Catalytic pyrolysis approaches may convert the most reactive part of the pyrolytic lignin thereby producing a pyrolysis oil that has a lower coke yield during hydrotreating than conventional pyrolysis oils produced with a non-catalytic approach.65–76
In this paper we start to understand some of the key classes of reactions that are occurring during hydrotreating of pyrolysis oils, but more advanced analytical approaches are needed to understand the complicated fundamental chemistry that is occurring during hydrogenation of pyrolytic lignin. These analytical approaches should be combined with theoretical and additional kinetic experiments to develop a molecular based model for hydrotreating of pyrolytic lignin. Free-radical reactions during the hydrotreating may be responsible for forming coke and, with this in mind, in situ electron paramagnetic resonance spectroscopy (EPR) could be an important technique to study the chemistry involved. More advanced mass spectrometric (MS) techniques capable of analyzing non-volatile substances could also be beneficial in understanding the chemistry of non-volatile pyrolytic lignin. Techniques such as Fourier transform ion cyclotron resonance mass spectrometry (FTICR-MS) and ion mobility mass spectrometry (IM-MS) are two MS techniques that allow for accurate detection of compounds without the need for volatilization that is required for other MS techniques.
Conclusions
Characterizations of pyrolytic lignin and hydrogenated pyrolytic lignin were performed utilizing 2D HSQC and quantitative 13C NMR analyses. The aromatic regions contained mainly guaiacyl and syringyl units, and low levels of p-hydroxyphenyl units whereas the aliphatic C–O region contained mainly methoxyl group peaks. β-aryl ether, phenylcoumaran, and resinol structures were not found in the pyrolytic lignin or products.The hydrogenation of pyrolytic lignin over a Ru/TiO2 catalyst converted a large amount of carboxyl/carbonyl and aromatic carbons into C–O and C–C aliphatic carbons. The G aromatics were easier to hydrogenate than the S aromatics. Hydrogenation was able to be carried out as low as 25 °C although coking still occurred at this temperature. Higher reaction temperatures increased both the degree of hydrogenation as well as formation of coke. Hydrogenation decreased the aromatic functionality 40.5% for a single-step and 74.2% for the three-step process. Hydrogenation of the HMW pyrolytic lignin fraction produced almost twice the coke yield as hydrogenation of the LMW pyrolytic lignin fraction. Understanding the cause of coke formation and developing techniques for mitigation of coke will be necessary for upgrading of pyrolytic lignin and pyrolysis oil as a whole.
Acknowledgements
The authors appreciate the financial support from China Scholarship Council. This work was also supported by the National Natural Science Foundation of China (51336008) and the Major State Basic Research Development Program of China (2013CB228100). JR and AA were funded by the DOE Great Lakes Bioenergy Research Center (DOE BER Office of Science DE-FC02-07ER64494). The authors would like to thank the Magnetic Resonance Facility in the Chemistry Department of the University of Wisconsin-Madison for use of the Bruker Advance III 500 gifted by Paul J. Bender.References
- A. V. Bridgwater, Biomass Bioenergy, 2012, 38, 68–94 CrossRef CAS.
- D. Meier, B. van de Beld, A. V. Bridgwater, D. C. Elliott, A. Oasmaa and F. Preto, Renewable Sustainable Energy Rev., 2013, 20, 619–641 CrossRef.
- C. Zhao and J. A. Lercher, Angew. Chem., Int. Ed., 2012, 51, 5935–5940 CrossRef CAS PubMed.
- M. Saidi, F. Samimi, D. Karimipourfard, T. Nimmanwudipong, B. C. Gates and M. R. Rahimpour, Energy Environ. Sci., 2014, 7, 103–129 CAS.
- M. Ringer, V. Putsche and J. Scahill, Large-scale pyrolysis oil production: A technology assessment and economic analysis, Report NREL/TP-510–37779, Midwest Research Institute, 2006 Search PubMed.
- M. M. Wright, D. E. Daugaard, J. A. Satrio and R. C. Brown, Fuel, 2010, 89, S11–S19 CrossRef.
- R. M. Swanson, A. Platon, J. A. Satrio and R. C. Brown, Fuel, 2010, 89, S2–S10 CrossRef.
- R. P. Anex, A. Aden, F. K. Kazi, J. Fortman, R. M. Swanson, M. M. Wright, J. A. Satrio, R. C. Brown, D. E. Daugaard, A. Platon, G. Kothandaraman, D. D. Hsu and A. Dutta, Fuel, 2010, 89, S29–S35 CrossRef CAS.
- E. Furimsky, Appl. Catal., A, 2000, 199, 147–190 CrossRef CAS.
- D. C. Elliott, Energy Fuels, 2007, 21, 1792–1815 CrossRef CAS.
- J. Wildschut, F. H. Mahfud, R. H. Venderbosch and H. J. Heeres, Ind. Eng. Chem. Res., 2009, 48, 10324–10334 CrossRef CAS.
- J. Wildschut, I. Melian-Cabrera and H. J. Heeres, Appl. Catal., B, 2010, 99, 298–306 Search PubMed.
- E. Butler, G. Devlin, D. Meier and K. McDonnell, Renewable Sustainable Energy Rev., 2011, 15, 4171–4186 CrossRef CAS.
- P. M. Mortensen, J. D. Grunwaldt, P. A. Jensen, K. G. Knudsen and A. D. Jensen, Appl. Catal., A, 2011, 407, 1–19 Search PubMed.
- D. Mohan, C. U. Pittman and P. H. Steele, Energy Fuels, 2006, 20, 848–889 CrossRef CAS.
- C. R. Vitasari, G. W. Meindersma and A. B. de Haan, Bioresour. Technol., 2011, 102, 7204–7210 CrossRef CAS PubMed.
- T. P. Vispute and G. W. Huber, Green Chem., 2009, 11, 1433–1445 RSC.
- T. P. Vispute, H. Y. Zhang, A. Sanna, R. Xiao and G. W. Huber, Science, 2010, 330, 1222–1227 CrossRef CAS PubMed.
- J. C. Lee, Y. Xu and G. W. Huber, Appl. Catal., B, 2013, 140, 98–107 CrossRef.
- J. Lee, Y. T. Kim and G. W. Huber, Green Chem., 2014, 16, 708–718 RSC.
- C. A. Mullen and A. A. Boateng, J. Anal. Appl. Pyrolysis, 2011, 90, 197–203 CrossRef CAS.
- H. X. Ben, W. Mu, Y. L. Deng and A. J. Ragauskas, Fuel, 2013, 103, 1148–1153 CrossRef CAS.
- M. Garcia-Perez, A. Chaala, H. Pakdel, D. Kretschmer and C. Roy, Biomass Bioenergy, 2007, 31, 222–242 CrossRef CAS.
- N. Ozbay, A. E. Putun and E. Putun, Int. J. Energy Res., 2006, 30, 501–510 CrossRef CAS.
- A. E. Putun, B. B. Uzun, E. Apaydin and E. Putun, Fuel Process. Technol., 2005, 87, 25–32 CrossRef.
- M. R. Rover, P. H. Hall, P. A. Johnston, R. G. Smith and R. C. Brown, Fuel, 2015, 153, 224–230 CrossRef CAS.
- B. Scholze, C. Hanser and D. Meier, J. Anal. Appl. Pyrolysis, 2001, 58–59, 387–400 CrossRef CAS.
- A. Oasmaa, E. Kuoppala, S. Gust and Y. Solantausta, Energy Fuels, 2003, 17, 1–12 CrossRef CAS.
- A. Oasmaa, E. Kuoppala and Y. Solantausta, Energy Fuels, 2003, 17, 433–443 CrossRef CAS.
- L. Ingram, D. Mohan, M. Bricka, P. Steele, D. Strobel, D. Crocker, B. Mitchell, J. Mohammad, K. Cantrell and C. U. Pittman, Energy Fuels, 2008, 22, 614–625 CrossRef CAS.
- H. Ben and A. J. Ragauskas, Energy Fuels, 2011, 25, 2322–2332 CrossRef CAS.
- S. D. Mansfield, H. Kim, F. Lu and J. Ralph, Nat. Protoc., 2012, 7, 1579–1589 CrossRef CAS PubMed.
- T. Q. Yuan, S. N. Sun, F. Xu and R. C. Sun, J. Agric. Food Chem., 2011, 59, 10604–10614 CrossRef CAS PubMed.
- H. Kim and J. Ralph, Org. Biomol. Chem., 2010, 8, 576–591 CAS.
- B. Scholze and D. Meier, J. Anal. Appl. Pyrolysis, 2001, 60, 41–54 CrossRef CAS.
- R. Bayerbach, V. D. Nguyen, U. Schurr and D. Meier, J. Anal. Appl. Pyrolysis, 2006, 77, 95–101 CrossRef CAS.
- R. Bayerbach and D. Meier, J. Anal. Appl. Pyrolysis, 2009, 85, 98–107 CrossRef CAS.
- T. L. Marker and J. A. Petri, US Pat, 7578927, 2009 Search PubMed.
- T. Zhe, Z. Ying and G. Qingxiang, Ind. Eng. Chem. Res., 2010, 49, 2040–2046 CrossRef.
- T. Prasomsri, M. Shetty, K. Murugappan and Y. Román-Leshkov, Energy Environ. Sci., 2014, 7, 2660 CAS.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- D. C. Elliott, H. Wang, M. Rover, L. Whitmer, R. Smith and R. Brown, ACS Sustainable Chem. Eng., 2015, 3, 892–902 CrossRef CAS.
- P. C. Badger, US Pat, 20080006519, 2008 Search PubMed.
- P. C. Badger and P. B. Fransham, US Pat, 20080006520, 2008 Search PubMed.
- J. Wildschut, M. Iqbal, F. H. Mahfud, I. M. Cabrera, R. H. Venderbosch and H. J. Heeres, Energy Environ. Sci., 2010, 3, 962 CAS.
- H. Olcay, L. J. Xu, Y. Xu and G. W. Huber, ChemCatChem, 2010, 2, 1420–1424 CrossRef CAS.
- J. Duan, Y. T. Kim, H. Lou and G. W. Huber, Catal. Today, 2014, 234, 66–74 CrossRef CAS.
- J. C. del Rio, J. Rencoret, G. Marques, A. Gutierrez, D. Ibarra, J. I. Santos, J. Jimenez-Barbero, L. M. Zhang and A. T. Martinez, J. Agric. Food Chem., 2008, 56, 9525–9534 CrossRef CAS PubMed.
- J. C. del Rio, J. Rencoret, G. Marques, J. B. Li, G. Gellerstedt, J. Jimenez-Barbero, A. T. Martinez and A. Gutierrez, J. Agric. Food Chem., 2009, 57, 10271–10281 CrossRef CAS PubMed.
- J. J. Villaverde, J. B. Li, M. Ek, P. Ligero and A. de Vega, J. Agric. Food Chem., 2009, 57, 6262–6270 CrossRef CAS PubMed.
- F. C. Lu and J. Ralph, Plant J., 2003, 35, 535–544 CrossRef CAS PubMed.
- F. C. Lu, J. Ralph, K. Morreel, E. Messens and W. Boerjan, Org. Biomol. Chem., 2004, 2, 2888–2890 CAS.
- J. Rencoret, G. Marques, A. Gutierrez, L. Nieto, J. Jimenez-Barbero, A. T. Martinez and J. C. del Rio, Ind. Crops Prod., 2009, 30, 137–143 CrossRef CAS.
- T. Q. Yuan, S. N. Sun, F. Xu and R. C. Sun, J. Agric. Food Chem., 2011, 59, 6605–6615 CrossRef CAS PubMed.
- J. C. del Rio, J. Rencoret, P. Prinsen, A. T. Martinez, J. Ralph and A. Gutierrez, J. Agric. Food Chem., 2012, 60, 5922–5935 CrossRef CAS PubMed.
- M. P. Pandey and C. S. Kim, Chem. Eng. Technol., 2011, 34, 29–41 CrossRef CAS.
- S. Chu, A. V. Subrahmanyam and G. W. Huber, Green Chem., 2013, 15, 125–136 RSC.
- P. R. Patwardhan, R. C. Brown and B. H. Shanks, ChemSusChem, 2011, 4, 1629–1636 CrossRef CAS PubMed.
- P. Karhunen, P. Rummakko, J. Sipilä, G. Brunow and I. Kilpeläinen, Tetrahedron Lett., 1995, 36, 169–170 CrossRef CAS.
- J. Ralph and L. L. Landucci, in Lignin and Lignans; Advances in Chemistry, ed. C. Heitner, D. R. Dimmel and J. A. Schmidt, CRC Press, Taylor & Francis Group, Boca Raton, FL, 2010, ch. 5, pp. 137–234 Search PubMed.
- A. R. Teixeira, K. G. Mooney, J. S. Kruger, C. L. Williams, W. J. Suszynski, L. D. Schmidt, D. P. Schmidt and P. J. Dauenhauer, Energy Environ. Sci., 2011, 4, 4306 CAS.
- C. A. Mullen, G. D. Strahan and A. A. Boateng, Energy Fuels, 2009, 23, 2707–2718 CrossRef CAS.
- H. Olcay, Y. Xu and G. W. Huber, Green Chem., 2014, 16, 911–924 RSC.
- W. Zhang, J. Chen, R. Liu, S. Wang, L. Chen and K. Li, ACS Sustainable Chem. Eng., 2014, 2, 683–691 CrossRef CAS.
- V. Paasikallio, C. Lindfors, E. Kuoppala, Y. Solantausta, A. Oasmaa, J. Lehto and J. Lehtonen, Green Chem., 2014, 16, 3549 RSC.
- E. F. Iliopoulou, S. Stefanidis, K. Kalogiannis, A. C. Psarras, A. Delimitis, K. S. Triantafyllidis and A. A. Lappas, Green Chem., 2014, 16, 662–674 RSC.
- E. F. Iliopoulou, E. V. Antonakou, S. A. Karakoulia, I. A. Vasalos, A. A. Lappas and K. S. Triantafyllidis, Chem. Eng. J., 2007, 134, 51–57 CrossRef CAS.
- F. A. Agblevor, S. Beis, O. Mante and N. Abdoulmoumine, Ind. Eng. Chem. Res., 2010, 49, 3533–3538 CrossRef CAS.
- F. A. Agblevor, O. Mante, N. Abdoulmoumine and R. McClung, Energy Fuels, 2010, 24, 4087–4089 CrossRef CAS.
- M. M. R. Corredores, V. Sanchez and X. Tong, US Pat, 8,669,405, 2014 Search PubMed.
- M. M. R. Corredores, J. Sorrells and C. Zhang, US Pat, 8,377,152, 2013 Search PubMed.
- J. A. Kocal and T. A. Brandvold, US Pat, 8,927,793, 2015 Search PubMed.
- R. Marinangeli, J. A. Kocal and T. A. Brandvold, US Pat, 8,519,203, 2013 Search PubMed.
- M. Biddy, A. Dutta, S. Jones and A. Meyer, Ex situ catalytic fast pyrolysis technology pathway, Report PNNL-22317, National Renewable Energy Laboratory and Pacific Northwest National Laboratory, 2013 Search PubMed.
- M. Biddy, A. Dutta, S. Jones and A. Meyer, In situ catalytic fast pyrolysis technology pathway, Report PNNL-22320, National Renewable Energy Laboratory and Pacific Northwest National Laboratory, 2013 Search PubMed.
- A. Dutta, A. Sahir, E. Tan, D. Humbird, L. J. Snowden-Swan, P. Meyer, J. Ross, D. Sexton, R. Yap and J. Lukas, Process design and economics for the conversion of lignocellulosic biomas to hydrocarbon fuels: Thermochemical research pathways with In Situ and Ex Situ upgrading of fast pyrolysis vapors, Report PNNL-23823, National Renewable Energy Laboratory and Pacific Northwest National Laboratory, 2015 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional figures and tables. See DOI: 10.1039/C5GC02286J |
| ‡ Authors contributed equally to the production of this paper. |
| This journal is © The Royal Society of Chemistry 2016 |