
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Environmental transmission of diarrheal pathogens in low and middle income countries
Timothy R.
Julian
*
Pathogens and Human Health, Department of Environmental Microbiology, Swiss Federal Research Institute of Aquatic Science and Technology, Eawag, BU-F08, Überlandstrasse 133, 8600 Dübendorf, Switzerland. E-mail: tim.julian@eawag.ch; Tel: +41 58 765 5632
First published on 27th June 2016
Abstract
Every year, more than half a million children die due to diarrheal diseases. Recent studies have identified the most important etiologies of diarrheal disease are enterotoxigenic and enteropathogenic E. coli, Shigella spp., rotavirus, norovirus and Cryptosporidium spp. These etiologies are unsurprisingly characterized by a combination of high shedding, high infectivity, and transmissibility through multiple environmental reservoirs. The relative importance of the transmission routes is likely site-specific. So the impact of interventions, which typically target only one or two environmental reservoirs, is likely also site-specific. The factors influencing the transmission routes most important for diarrheal disease are complex, including – at a minimum – etiology of endemic disease; and water, sanitation, and hygiene infrastructure and practices. The site-specific nature – and complexity of transmission – helps explain the observed variation in impacts of water, sanitation, and hygiene interventions. It may also render efforts to estimate or quantify global means for interventions' impacts irrelevant. The theme of this Perspective is that greater reductions in diarrheal disease transmission in LMICs can be achieved by designing interventions to interrupt the most important environmental transmission pathways. Intervention choice should be informed by site-specific conditions, most notably: diarrheal etiology and existing water, sanitation, and hygiene infrastructure and practices. The theme is discussed through the lens of the characteristics of the most important diarrheal diseases (shedding, infectivity, growth, and persistence) and the general characteristics of environmental reservoirs (exposure pathways and fecal contamination). The discussion highlights when interventions – and combinations of interventions – will be most effective at reducing diarrheal disease burden.
Environmental impactGlobally, more than half a million children die every year from diarrheal diseases. Recent studies have identified the diarrheal disease agents most responsible for moderate-to-severe diarrheal disease and diarrhea-related mortality. The agents – enterotoxigenic and enteropathogenic E. coli, Shigella spp., rotavirus, norovirus, and Cryptosporidium spp. – are characterized by high infectivity, high fecal shedding, and transmission through a wide range of environmental reservoirs. This Perspective provides insight into the ecology of the diarrheal disease agents with emphasis on their relationship to environmental reservoirs. Based on this insight, the Perspective advocates for comprehensive interventions targeting exposure reductions across multiple environmental reservoirs. Single interventions are often inadequate. This may partially explain their failure to reduce environmental exposures below thresholds needed to initiate infection. |
Introduction
Every year, there are an estimated 1.7 billion cases of gastrointestinal disease in children under five years old globally.1 Of these, between 5 and 700![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 result in the death of the child.1,2 These deaths are disproportionately experienced by people living in Low and Middle Income Countries (LMICs).1,2 The reason is attributed to a combination of poverty, malnutrition, and living in remote areas with limited access to sufficient safe water, adequate sanitation, and health care.3 Development efforts to reduce these risk factors globally have been largely successful: child deaths due to diarrhea have fallen dramatically – by 70–80% – from an estimated 2.5 million in 2000.4 Despite dramatic reductions in diarrheal disease-related deaths (mortality), reductions in diarrheal episodes (morbidity) have declined only moderately. Between 1990 and 2010, diarrheal disease episodes declined only about 15%: from 3.4 to 2.9 episodes per child year.5 The discrepancy between reductions in morbidity and mortality is due to emphasis on – and effectiveness of – therapeutic treatments such as oral rehydration therapy, zinc, and nutrient supplementation.6 Therapeutic treatments improve recovery from infections but only reduce infection rates indirectly: by reducing pathogen shedding rates and durations. Vaccinations (i.e., rotavirus) are also increasingly important interventions to reduce morbidity and mortality, though they target only a single cause of diarrhea.6
000 result in the death of the child.1,2 These deaths are disproportionately experienced by people living in Low and Middle Income Countries (LMICs).1,2 The reason is attributed to a combination of poverty, malnutrition, and living in remote areas with limited access to sufficient safe water, adequate sanitation, and health care.3 Development efforts to reduce these risk factors globally have been largely successful: child deaths due to diarrhea have fallen dramatically – by 70–80% – from an estimated 2.5 million in 2000.4 Despite dramatic reductions in diarrheal disease-related deaths (mortality), reductions in diarrheal episodes (morbidity) have declined only moderately. Between 1990 and 2010, diarrheal disease episodes declined only about 15%: from 3.4 to 2.9 episodes per child year.5 The discrepancy between reductions in morbidity and mortality is due to emphasis on – and effectiveness of – therapeutic treatments such as oral rehydration therapy, zinc, and nutrient supplementation.6 Therapeutic treatments improve recovery from infections but only reduce infection rates indirectly: by reducing pathogen shedding rates and durations. Vaccinations (i.e., rotavirus) are also increasingly important interventions to reduce morbidity and mortality, though they target only a single cause of diarrhea.6
To improve both morbidity and mortality for all causes of diarrhea, environmental interventions – such as investments to improve water quality and quantity, sanitation, and food and hand hygiene – should be promoted. Public health improvements to interrupt environmental transmission of diarrheal diseases are largely credited as the cause of reductions of diarrheal diseases in developed countries.7,8
Interventions need to consider the importance of transmission through multiple environmental reservoirs.11–13 Although single interventions may reduce exposures, the reduction may be insufficient to impact diarrheal disease prevalence. This is because there are multiple enteric pathogens transmitted via multiple exposure routes. Therefore, the impact of an intervention is dependent on the relative importance of the targeted transmission route.
The relative importance of transmission routes is likely site-specific.13 And the factors influencing that importance are complex, including – at a minimum – etiology of endemic disease; seasonality; and water, sanitation, and hygiene infrastructure and practices.14 This may help explain the observed variation in impacts of water, sanitation, and hygiene interventions.11,15–20 This may also help explain observed multiplicative diarrheal disease reductions due to combined water and sanitation infrastructure.11,19
The theme of this Perspective is that greater reductions in diarrheal disease in LMICs can be achieved when interventions are designed based on knowledge about site-specific conditions – including etiology of disease and existing water, sanitation, and hygiene infrastructure and practices. Interventions should be designed to reduce aggregate exposures by targeting multiple environmental transmission pathways simultaneously. Efforts to identify one single, most effective, intervention – or to quantify a global mean for a single interventions' impact – are flawed in that the effectiveness of interventions varies by site. The theme is discussed through the lens of the characteristics of the most important diarrheal diseases (shedding, infectivity, growth, and persistence) and the general characteristics of environmental reservoirs (exposures, pathogen detection, and fecal contamination). Based on these characteristics, recommendations are made for designing packages of interventions to reduce diarrheal disease burden.
Diarrheal diseases in LMICs
Most diarrheal disease-related deaths are caused by only a handful of pathogens. Although the causes of diarrheal disease are wide and varied – including food allergies or intolerances, chemical or toxin exposures, and microbial infections – the vast majority of cases are caused by pathogens.9,21 A systematic literature review of deaths due to diarrheal disease by Lanata et al. (2013) estimated that 70% (adjusted for age and to sum to 100%) are attributable to 13 pathogens.21 The review highlighted the importance of five: rotavirus (17.8% of all deaths), enteropathogenic E. coli (14.0%), enterotoxigenic E. coli (7.3%), calicivirus (8.2%), and Shigella (6.4%).21The results largely coincide with the Global Enteric Multicenter Study (GEMS), also published in 2013.9 GEMS investigated etiological causes of moderate-to-severe diarrhea in children under five at seven sites across Africa and Asia. The authors were able to detect at least one of the 14 pathogens they tested for in 83% of all diarrheal cases; the cause of the other 17% of cases was not identified. GEMS also highlighted the importance of five – Cryptosporidium spp., E. coli producing heat stable toxin (an enterotoxigenic E. coli), typical enteropathogenic E. coli, rotavirus, and Shigella – as targets of interventions to “substantially reduce the burden of moderate-to-severe diarrhea”.9 Combining evidence from the work of Lanata et al. (2013) and Kotloff et al. (2013) suggests diarrheal disease prevention and treatment efforts should be focused on enterotoxigenic and enteropathogenic E. coli, Shigella spp., rotavirus, norovirus (an important calicivirus), and Cryptosporidium spp. (Table 1).9,21
| Pathogen | Class | Shedding (#/g feces) | Duration (days) | 50% infectious dose (#) | Human feces equivalents for infection (g) | Common detection methods | Hosts |
|---|---|---|---|---|---|---|---|
| Enterotoxigenic E. coli | Bacteria | 107 to 108 | 3–5 | 105 to 108 cells | 10−3 to 101 | PCR | Humans, livestock, dogs |
| Enteropathogenic E. coli | Bacteria | 105, 109 (peak) | >10 | 105 to 107 cells | 100 to 102, 10−4 to 10−2 (peak) | PCR | Humans, livestock, dogs, cats |
| Shigella spp. | Bacteria | 104 to 105, 106 to 1010 (peak) | 7–14 | 103 cells | 10−2 to 100, 10−7 to 10−3 (peak) | Isolation & biochemical profiling | Humans, closely related primates |
| Cryptosporidium spp. | Protozoa | 103 to 107* | 8 (2–35) | 9–160 oocysts | 10−6 to 10−1 | Microscopy & immunoassay | Mammals |
| Norovirus | Virus | 107 to 108, 1012 (peak) | 28 | 1320 genome equivalents | 10−5 to 10−4, 10−9 (peak) | RT-PCR | Humans |
| Rotavirus | Virus | 105 to 1010 | 24 | 6 focal forming units | 10−9 to 10−4 | Immunoassay | Mammals, birds |
Exposure, dose, and infection
For a child to be infected with a diarrheal disease, he must first be exposed. Exposure is describable as a continuous variable, meaning that a child may be exposed to a range of pathogens: from a few to many. Infection, however, is binary: a child is either infected or not. The likelihood that a child will be infected increases with increasing exposure, a pathogen-specific relationship describable by dose–response functions (Fig. 1). Dose–response functions relate dose to probability of infection (seroconversion and illness are also common endpoints). The pathogen dose at which there is a 50% likelihood of infection is known as the human infectious dose 50, or HID50. It is important to note dose–response functions are typically determined using adults, as opposed to children, and so may not accurately reflect infection risks for children.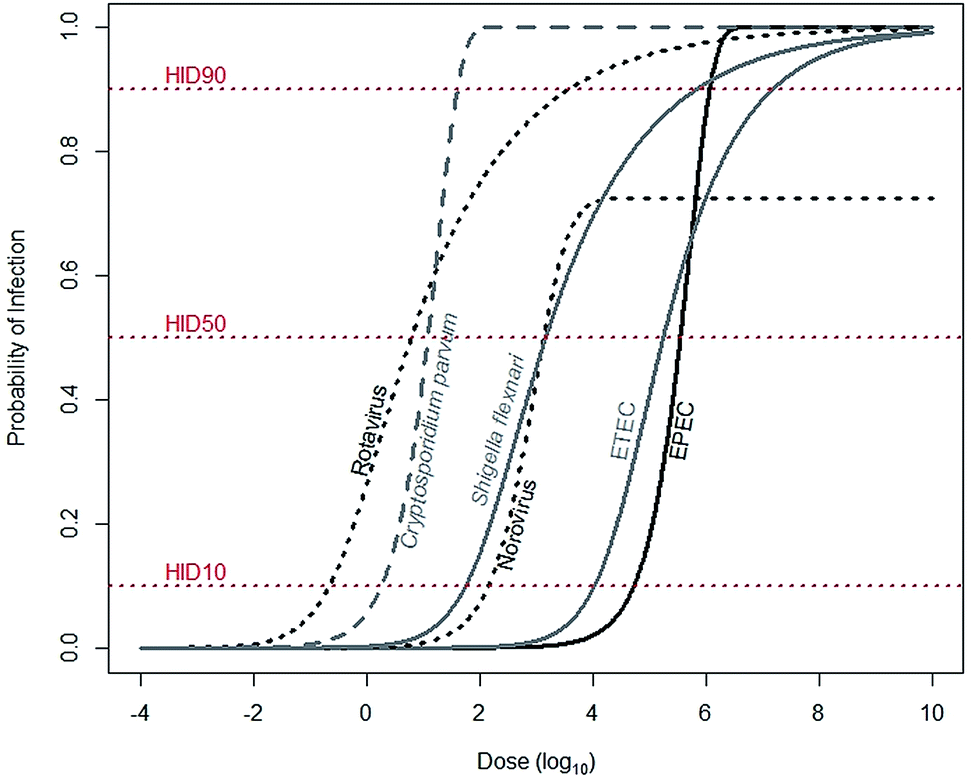 | ||
| Fig. 1 Median estimates for dose–response relationships for common diarrheal diseases. Rotavirus (α = 0.253, N50 = 6.17), Shigella flexnari (α = 0.265, N50 = 1.48 × 103), and ETEC (α = 0.375, N50 = 1.78 × 105) are described by beta-Poisson models, and Cryptosporidium parvum (k = 5.72 × 10−2) and EPEC (k = 1.95 × 10−6) are described by exponential models. Data are from either recommended (rotavirus, S. flexinari, and C. parvum) or most conservative (ETEC, EPEC) models described by the Quantitative Microbial Risk Assessment (QMRA) Wiki, curated and hosted by the Center for Advancing Microbial Risk Assessment (http://qmrawiki.canr.msu.edu). Norovirus (α = 2.910 and β = 2.734) is described by an “aggregated exact beta-Poisson model” with assumed 27.54% immunity, based on Messner et al. (2015).70 The dose at the intersection of the dose–response curves and the HID10, HID50, and HID90 lines corresponds to 10%, 50%, and 90% likelihood of infection, respectively. | ||
To substantially reduce infections, tenfold reductions in exposures are often needed. The dose–response relationship links probability of infection – in arithmetic scale – to dose – in log-scale. So reductions of probability of infection from 50% to, for example, 10%, require a reduction in exposures of anywhere from 0.7 (Cryptosporidium parvum) to 1.5 (rotavirus) orders of magnitude (Fig. 1). Therefore, interventions that reduce exposures may not be sufficient to also reduce infections.
Of course, diarrhea is not the only consequence of environmental fecal exposures. Environmental bacterial exposures may also contribute to both malnutrition and stunting – which affected as many as 26% of children globally in 2011 – through enteric infections and/or environmental enteric dysfunction (EED).10,22–24 Multiple ongoing research trials are examining links between environmental bacterial exposures and stunting. The Interactions of Malnutrition and Enteric Infections: Consequences for Child Health and Development (MAL-ED, NCT02441426) study is investigating the degree to which enteric infections (with and without diarrhea) contribute to undernutrition as mediated by intestinal inflammation and/or altered intestinal function.10 The Sanitation, Hygiene, Infant Nutrition Efficacy Project in Zimbabwe (SHINE, NCT01824940) is investigating the impacts of water, sanitation, and hygiene (WASH) interventions alone and in combination with nutrition interventions on child health.154 The hypothesis is that WASH interventions reduce fecal bacterial exposures, which reduces EED, thereby increasing intestinal functioning and improving nutrition interventions. Thirdly, the WASH Benefits trials in Bangladesh (NCT01590095) and Kenya (NCT01704105) are also assessing impacts of WASH and nutrition interventions on child health, including enteric infections, undernutrition, and EED.25
This Perspective focuses on enteric infections – as oppose to EED – because the causes and consequences of EED are currently uncertain. However, it is important to note that any reduction in microbial exposures may lead to an improvement in EED, in contrast to the binary outcomes of infection.
Linking environmental contamination to probability of infection using human feces equivalents
Because substantial reductions in exposures are often needed to meaningfully impact infection, interventions should be designed to maximize reductions in pathogen exposures. Intervention impacts could be estimated using data on: (1) pathogen contamination of reservoirs, (2) type, intensity, and frequency of people–reservoir interactions, and (3) impacts of interventions on both (1) and (2). Unfortunately, these data are sparse, especially in LMICs, though ongoing efforts are seeking to remedy this issue (most notably, SaniPath Rapid Assessment Tool by the Center for Global Safe Water at Emory University, http://www.sanipath.org).26,27In the absence of quantitative pathogen and human–environment interaction data, fecal contamination of environmental reservoirs can be linked to probability of infection using human feces equivalents. Human feces equivalents are a proxy measure to estimate exposure risks in the absence of quantitative pathogen data.26,27 Here, this concept is applied to relate risks for multiple diarrheal disease agents to data on environmental fecal contamination. Human feces equivalents are estimated for both infection from diarrheal disease estimates and for environmental fecal contamination. Estimates for infection are estimated by dividing the HID50 – an indicator of a pathogen's infectivity – by the shedding rate – an indicator of pathogen density in feces:
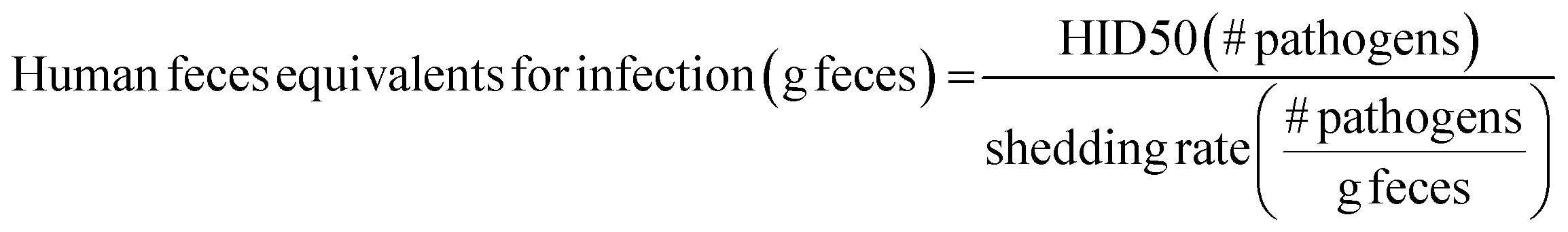
Human feces equivalents for environmental fecal contamination are estimated by dividing E. coli concentrations within reservoirs by the average reported E. coli concentration in feces:
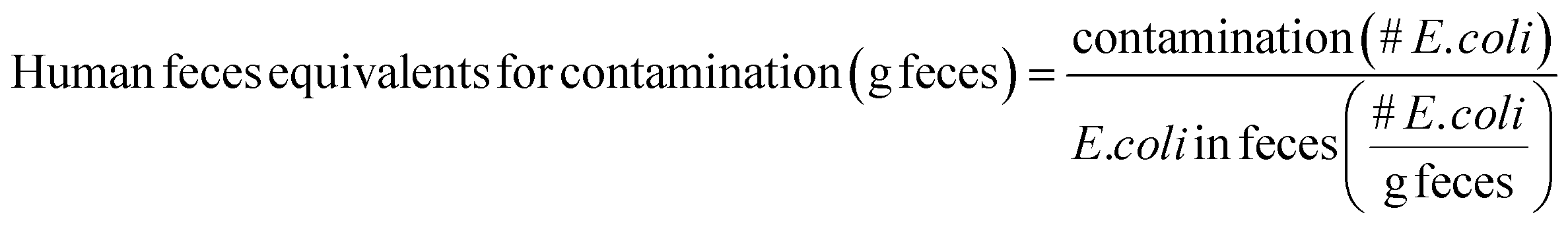
E. coli is typically present in human feces at concentrations of around 105 to 1010 CFU per g feces; the lower end of the range (105 CFU g−1) is used in this Perspective as a conservative choice.28,29 It is important to note that human feces equivalents are intended as approximations. Values for HID50, shedding rates, E. coli contamination, and E. coli in feces are both uncertain and variable, so human feces equivalents will also be uncertain and variable.
Diarrheal disease agents
ETEC are a pathotype of E. coli indicated by their mechanism of pathogenicity: colonization targeting intestinal mucosa combined with production of one or more enterotoxins.31 Virulence determinants, which are primarily located on plasmids, are spread via horizontal gene transfer. So ETEC does not describe a “single homogenous group” but rather a cluster of strains from “multiple distinct lineages”.32,33 The high diversity amongst ETEC suggests that estimates of its transmissibility, persistence and growth, and infectivity based on experimental studies of a subset of strains may not be representative of the pathotype as a whole.
Nonetheless, from a handful of strain-specific studies, some consistency in ETEC characteristics is observed. Diarrheal disease caused by ETEC typically lasts 3–5 days though longer bouts have been documented.34 In challenge studies, ETEC is shed in feces at concentrations as high as 107 to 108 CFU per g feces.35–37 Infectivity of ETEC is strain-specific, but for all strains is relatively low: HID50 ranges from around 105 to 108 cells (http://qmrawiki.canr.msu.edu). Notable here is that the HID50 is equivalent to the estimated number of ETEC in 0.001–10 g feces of an infected person. ETEC can also cause infections in livestock (cattle, goats, pigs, and sheep) and other animals (dogs).
EPEC are a pathotype of E. coli characterized by their ability to produce attaching and effacing (A/E) lesions. Diagnosing EPEC relies primarily on molecular methods.38 EPEC infections are indicated by the presence of eae – a gene which encodes the outer membrane protein intimin that mediates intestinal cell attachment – coincident with the absence of stx1 or stx2 – genes which encode Shiga toxin and indicate enterohemorrhagic E. coli (EHEC) infections.38 From there, EPEC infections are classified as either typical or atypical based on the presence (typical) or absence (atypical) of the bundle-forming pilus structural gene bfpA located on the E. coli adherence factor plasmid (EAF).38,39 Occasionally, the presence of the bfpA gene independent of eaeA is sufficient to diagnose typical EPEC, as in the GEMS study.9,40 As EPEC infections can be indicated by the presence of a single gene located on a plasmid, it is not surprising that EPEC isolates also exhibit diversity across lineages.41 EPEC infects other animals besides humans, including cattle, dogs and cats.
EPEC is characterized by substantially longer duration diarrhea than ETEC. EPEC diarrhea frequently lasts >10 days and is often associated with prolonged or persistent diarrhea.42–44 EPEC is shed in feces at concentrations of around 105 cell equivalents per g feces, but can reach as high as 109 cell equivalents per g feces.42 EPEC infectivity, like ETEC, is also strain-specific and relatively low: the HID50 is estimated to, be between 105 to 107 cells (http://qmrawiki.canr.msu.edu). This corresponds to the estimated number of EPEC in 0.01–1 g feces of an infected person.
Shigella serogroups S. flexinari and S. sonnei are responsible for the majority of infections. Within Shigella, there are four serogroups: S. dysentariae, S. flexinari, S. boydii, S. sonnei. In the GEMS study, the majority of Shigella infections (65.9%) were S. flexinari, followed by S. sonnei (23.7%).47 Both S. boydii, and S. dysentarie were responsible for around only 5% of Shigella spp. infections.47 However, S. dysentarie should not be overlooked as it is a causative agent of dysentary outbreaks responsible for both high attack rates and case fatality across all ages.48
Despite similarities to E. coli, Shigella spp. is substantially more infective. For almost all Shigella infections detected in GEMS, children were symptomatic with moderate-to-severe diarrhea.9 The HID50 is estimated to be around 103 cells, though Levine et al. (1973) demonstrated that as few as 10 cells can cause illness (http://qmrawiki.canr.msu.edu).49Shigella infections typically last at least 7, and occasionally longer than 14, days.50Shigella counts per gram of feces typically range from 104 to 105, but reach concentrations of 106 to 1010 per gram at the height of excretion.49,51,52 So the HID50 corresponds to the typical number of Shigella in 0.01–0.1 g of feces of an infected person, though may reach as low as 10−7 g feces at the height of excretion.
Voluntarily infected people shed oocysts at average concentrations of 103 to 107 oocysts per day.56 Shedding continues for 2 to more than 35 days, with a median time of about 8 days.57Cryptosporidium parvum is highly infective, with an HID50 estimated to be as low as 9 oocysts (http://qmrawiki.canr.msu.edu).58 Infectivity may be strain-specific, though, as other strains have HID50s of estimated at around 160 oocysts (http://qmrawiki.canr.msu.edu).59 So the HID50 for Cryptosporidium spp. corresponds to 10−1 to 10−5 of the amount of feces shed in a day during an infection.
High shedding rates combined with high infectivity contribute to rotavirus's ubiquity. Approximately 103 to 1010 genome copies of rotavirus per g feces is shed during infection.62,63 In a study of ten symptomatic children in Vellore, India, viral shedding lasted a median of 24 days.64 Rotavirus infectivity is very high: the HID50 was identified as 6 focus-forming units (FFU) in the most well-known study by Ward et al. (1986).65 Relating FFU to genome copies using a conservative ten-fold estimate, the HID50 is equivalent to a range of 10−3 to 10−9 g feces of an infected person.
Norovirus is characterized by high infectivity, but some people are naturally resistant. Norovirus infectivity is unique in that it infects only people with histo-blood group O or A with a functional FUT2 enzyme (“secretor-positive”).68 Based on this finding, a volunteer challenge study estimated that norovirus HID50 is approximately 1320 genome equivalents for susceptible people.69 Subsequent analysis of challenge studies suggested that immunity rates (non-susceptible people) make up about 27% of the study population.70 These values remain debatable, however: Schmidt et al. (2015) contend the HID50 may be overestimated due to limited information on both aggregation of virus and immunity status of volunteers due to other factors, like prior exposures.71
Nevertheless, in those infected with Norwalk virus, the prototype norovirus strain, norovirus shedding in feces ranged from 107 to 1012 genome copies per gram and lasted a median of 28 days.72 Viral loads for natural infections are reportedly within the lower end of the range (107 to 108).73,74 Assuming the HID50 estimate of 1320 genomes, the HID50 is equivalent to approximately 10−4 to 10−5 feces during a natural infection.
Environmental transmission
We can conceptualize the environmental transmission of diarrheal diseases using the F-diagram (Fig. 2). The F-diagram visualizes the role of five or six environmental reservoirs (typically fields, fingers, fluids, flies, food, and sometimes fomites) in diarrheal disease transmission from infected to susceptible people. Pathogens are spread from feces into one or more of the environmental reservoirs through human–environment interactions, animal–environment interactions, and/or natural processes. Subsequent interactions with the reservoirs by susceptible people can result in infection.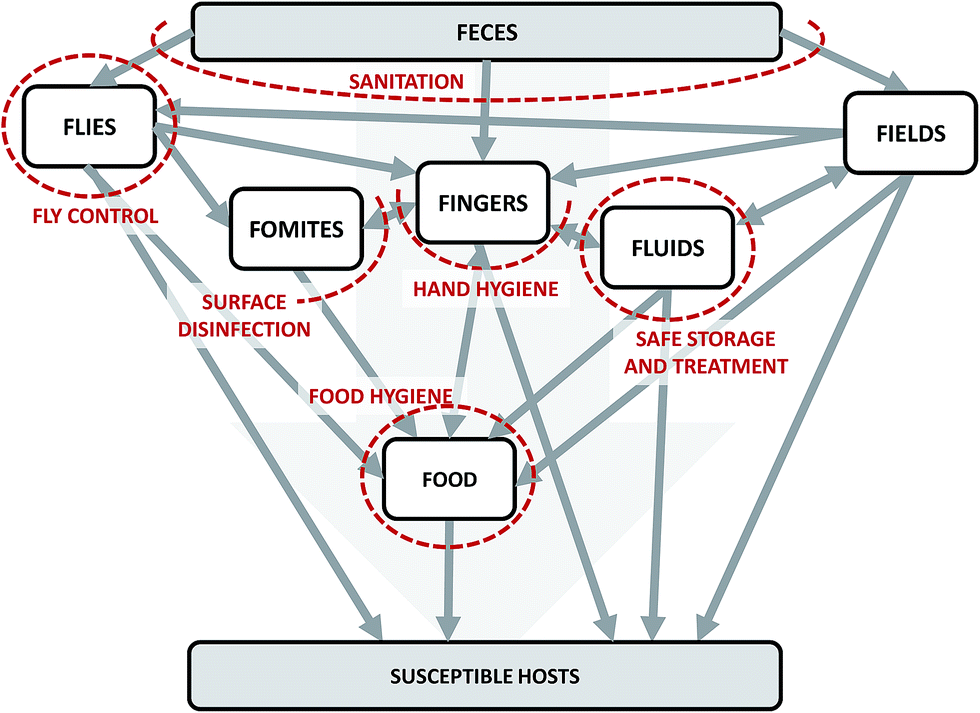 | ||
| Fig. 2 The F-diagram, a conceptual model of the complexity of potential transmission pathways of diarrheal diseases through environmental reservoirs. Adapted from Wagner and Lanoix (1958) and Kawata (1978).12,13 Grey arrows represent potential pathways of pathogen transmission. Red dashed lines represent the impacts of interventions on intersecting arrows. The lines are dashed to imply permeability due to incomplete effectiveness (fly control, food hygiene, hand hygiene, sanitation) and/or imperfect compliance (food hygiene, hand hygiene, safe storage and treatment, and sanitation). | ||
Even if source water is safe, stored water may not be. Contaminated storage containers, hands, drinking cups, and other utensils that come into contact with stored water are responsible for degradation of water quality within a home.77,78 As water is a shared resource, water contaminated by one member of the household puts others at risk. The diarrheal disease agents EPEC, ETEC, Shigella, rotavirus, norovirus, and Cryptosporidium spp. have all been detected in stored drinking water in LMICs.78–85
Drinking water is likely an efficient route of transmission for rotavirus, norovirus, and Cryptosporidium spp.; less so for ETEC, EPEC, and Shigella spp. due to the relatively high HID50s for the bacterial pathogens. In a study of Tanzanian children, Mattioli et al. (2015) estimated median daily fecal consumption through drinking water of 10−4 to 10−6 g feces.26 This range overlaps estimates for feces equivalents of the HID50s for norovirus, rotavirus, Cryptosporidium spp. and Shigella spp. during peak shedding, but is orders of magnitude lower than the HID50 feces equivalent for ETEC, EPEC, and Shigella spp. given typical shedding rates.
Of note, the WHO guidelines for drinking water do not necessarily indicate an absence of pathogens. The current guideline states that E. coli or thermotolerant coliform must not be detectable in 100 ml samples.86 This standard (<1 CFU E. coli per 100 ml) is equivalent to approximately 10−5 to 10−10 g human feces. Water in households where there is shedding of rotavirus, norovirus, Cryptosporidium spp., or Shigella spp. could be sufficiently contaminated to cause disease even when there are no E. coli detectable.
Although contamination of water needs to be fairly high to risk exposure to the HID50 for the bacterial pathogens EPEC, ETEC, and Shigella, bacteria can increase during storage through growth. E. coli, including enterotoxigenic E. coli, can grow in drinking water distribution systems and persist in freshwater for up to 3 months.87–89 In stored drinking water containers, risk factors for growth of total coliform – of which E. coli is a member – include hand contacts, presence of biofilms, high temperature, and high assimilable organic carbon (AOC).79,90 So bacterial pathogen growth is primarily a concern when water with high AOC is stored at warm temperatures for long periods of time.90
The greatest risk factor for foodborne transmission of pathogens is the ability of bacterial pathogens to grow. Both Shigella spp. and E. coli are capable of growing rapidly in various food stuffs, including cheese, rice, milk, and beef.98Shigella flexinari and EPEC for example, reached concentrations between 105 to 108 CFU per g or CFU per ml in these matrices when incubated at 25 °C.98,99 Lower temperature storage via refrigeration reduces growth, but refrigerator ownership is generally low in LMICs.100 Accounting for growth, food is an efficient carrier of the equivalent of 1–100 g feces for bacterial pathogens.
log10CFU/2 hands E. coli and enterococci, for example.101,102 Studies that have looked for diarrheagenic pathogens have noted the presence of multiple E. coli pathotypes including ETEC and EPEC, Shigella spp., rotavirus, and norovirus.83,101,103–106 Once on hands, pathogens can survive for long periods of time. Rotavirus, for example, survives for more than 260 minutes.107
Hand contamination poses both direct and indirect diarrheal disease risks. Here, direct risks refer to direct exposure from hand-to-mouth contacts. Children and adults touch their mouths approximately 3–28 and 8 times per hour, respectively.75 The frequency of contacts drives risks because anywhere from 33–41% of microbial contamination can be transferred from hands-to-mouth on a single contact.108 Based on E. coli and enterococci contamination on hands in Tanzania, Mattioli et al. (2015) estimate that children consume the equivalent of a median 10−3 to 10−4 g feces per day due to hand–mouth contacts.26 Hands are also responsible for indirect risks. Drinking water, food, and fomites are often contaminated due to contacts with contaminated hands.
Hands are likely an efficient route of rotavirus, norovirus, Shigella spp., and Cryptosporidium spp. transmission due to the high infectivity of these diarrheal pathogens. This is also observed in developed countries, where epidemiological surveys have suspected outbreaks were due to poor hand hygiene, often during food preparation.109–112 Direct transmission of ETEC and EPEC by hands is likely only when hands are heavily contaminated. During outbreaks, for example. The exposure estimates by Mattioli et al. (2015) border the estimated HID50 feces equivalents for these pathogens.26 However, the role of hands in indirect bacterial pathogen transmission is likely very important. Transfer of EPEC, ETEC, and Shigella spp. to environmental reservoirs where they can grow (like food), may contribute substantially to diarrheal disease burden.
Quantitative data on pathogen contamination of flies is scarce, making it difficult to determine the role of flies in disease transmission. On a United States cattle farm in Kansas – admittedly very different than households in LMICs – Alam et al. (2004) estimated that approximately 1–3% of flies carried 101 to 105 CFU E. coli 0157:H7, an enterohemorrhagic E. coli strain.117 De Jesús et al. (2004) estimated that flies are capable of contaminating surfaces with bacteria from the equivalent of about 10−4 g of food.118 Extrapolating these results to feces suggests that flies may be effective carriers of infectious doses of rotavirus, norovirus, Cryptosporidium spp., and Shigella spp. but that the doses may be lower than would be needed for effective transmission of EPEC or ETEC.
But even though flies may not be carriers of infectious doses of EPEC or ETEC, flies provide opportunities for these bacteria to grow. Studies demonstrated that E. coli could grow on the surface of flies and in regurgitation spots, though the latter phenomenon was only observed for E. coli artificially deposited on regurgitation spots.116,119 Similarly, E. coli and Shigella transferred to food are capable of growing, as discussed earlier.
Recent evidence, however, has suggested that fields should be expanded to include flooring inside and near households. E. coli concentrations ranged from a mean of 10 to 103 CFU per g soil in household plots in Zimbabwe and Tanzania.92,121 In Tanzania, E. coli pathotypes including EPEC and ETEC were also detected. Though the fecal source is uncertain, suggestions include inadequate management of animal feces, child feces, and/or wastewater, as well as off-plot fecal bacteria sources.92 Growth is also a risk for bacterial pathogens, as E. coli have been shown to grow in soils.122
Fecal contamination of soil in the household is a transmission concern for all diarrheal pathogens. In Zimbabwe, Ngure et al. (2012) estimated that infants consume the equivalent of 1 g of chicken feces (coprapaghy) and 20 g of soil (geophagy) daily.121 Copraphagy alone would ensure infection with any zoonotic diarrheal diseases – including ETEC, EPEC, and rotavirus – if the animal is shedding. Under the assumption of proper animal fecal management, children would still be exposed to the equivalent of 10−3 to 10−1 g of feces per day through geophagy.
It is important to note, however, that the role of soil around households in disease transmission is also site-specific. For example, Ngure et al. (2012) estimates of soil ingestion are 500 times greater than estimates for children in the United States.75 Similarly, there was a ten-fold difference in E. coli contamination of household soils between Tanzania and Zimbabwe; there is likely greater variation at other sites. Nevertheless, soil contamination is a transmission concern given infant and young child interactions with flooring.
Once contaminated, fomites readily transfer pathogens to other surfaces. Transfer events move a fraction of the pathogen (typically from <0.01–50%) between the fomite and other reservoirs.130 The magnitude of the fraction is dependent on pathogen, fomite, hand or other surface characteristics, and environmental (i.e., temperature, humidity) characteristics.130,131
Compounding risks of fomite transmission is the ability of pathogens to persist on surfaces for extended periods of time. Persistence of Shigella spp., and E. coli ranges from 1.5 days up to 16 months.132 Norovirus and rotavirus persist for at least 2 months.133 In contrast, Cryptosporidium parvum persists on surfaces for less than two hours.134 Factors that influence persistence are similar to those that influence transfer: material type, humidity, and temperature.132–134
Given the modest fecal contamination levels observed on fomites, transmission concerns are primarily for rotavirus and norovirus, and to a lesser extent Shigella spp. following peak shedding. Given Cryptosporidium spp. susceptibility to desiccation, it is unsurprising that evidence of Cryptosporidium spp. transmission by fomites is sparse. Fomite-mediated transmission for bacterial pathogens is likely relevant primarily as an intermediate prior to transfer to other reservoirs where subsequent growth is a concern (like food).
Discussion
This Perspective argues that greater reductions in diarrheal disease in LMICs can be achieved when interventions are designed based on site-specific conditions to interrupt multiple transmission routes. Systems-based approaches to interventions are needed to further reduce diarrheal disease burden in LMICs.11 The diarrheal diseases most important for child health – enterotoxigenic and enteropathogenic E. coli, Shigella spp., rotavirus, norovirus, and Cryptosporidium spp. – are characterized by high infectivity, high fecal shedding, and transmission through a wide range of environmental reservoirs (Table 1, Fig. 2). There is likely no single intervention that will universally and uniformly reduce diarrheal diseases globally. Interrupting a single transmission route may reduce total exposure, but other pathways may still contribute sufficient exposure to cause infection. Packages of interventions should be designed to interrupt simultaneously all of the relevant transmission pathways to sufficiently reduce infections. The most effective intervention packages are site-specific. Characteristics that influence intervention effectiveness include diarrheal disease etiology and existing water, sanitation, and hygiene infrastructure and practices.Investment in combined interventions does not necessarily lead to interruption of multiple exposure pathways. Previous reviews have failed to demonstrate additive impacts of combined interventions on diarrheal disease.15,135,136 There are several hypotheses as to why combined interventions have failed to show health improvements. One review – by Fewtrell et al. (2005) – suggested the lack of additive impacts of combined interventions on health may be due to incomplete or inconsistent implementation.15 Related to incomplete implementation, Enger et al. (2013) showed that imperfect user compliance strongly influences intervention effectiveness.137 This Perspective contributes an alternative explanation, that standard interventions may be redundant because they impact similar transmission pathways while others are neglected.
To avoid redundancies, the transmission pathways for the main pathogens responsible for diarrheal diseases should be considered. For example, this Perspective suggests that bacterial pathogens (EPEC, ETEC, and Shigella spp.) can be controlled through reducing geophagy (consumption of contaminated soil),121 prevention of growth in food,138 and – especially in the context of Shigella spp. – fly control.139,140 Given reported high rates of geophagy for soil flooring, it should be unsurprising that the upgrade of concrete flooring alone reduced diarrheal disease incidence by 13% during the Piso Firme Program in Mexico.121,141 Hand hygiene is also important for control of bacterial pathogens due to its reduction of both direct hand-to-hand contact transmission and – perhaps more importantly – physical transfer of bacteria to reservoirs where growth is possible.
Cryptosporidium spp. and norovirus are more difficult to control than bacterial pathogens due to the combination of low infectious doses and high shedding rates (Table 1, Fig. 3). Interventions targeting Cryptosporidium spp. or norovirus need to interrupt all exposure routes, through (for example) animal, child, and adult fecal management; dedicated safe water treatment and storage; hand hygiene; and limiting contacts with infected household members. Notably, child feces was described as “the most important contaminant in the household environment with the highest risk of exposure to young infants” in a 2004 review of infant and young child feces management.142 When a person inside the home is infected with Cryptosporidium spp. or norovirus, others are also at an increased risk (between 2- to 26-times), so limiting household contacts is also important.143–145
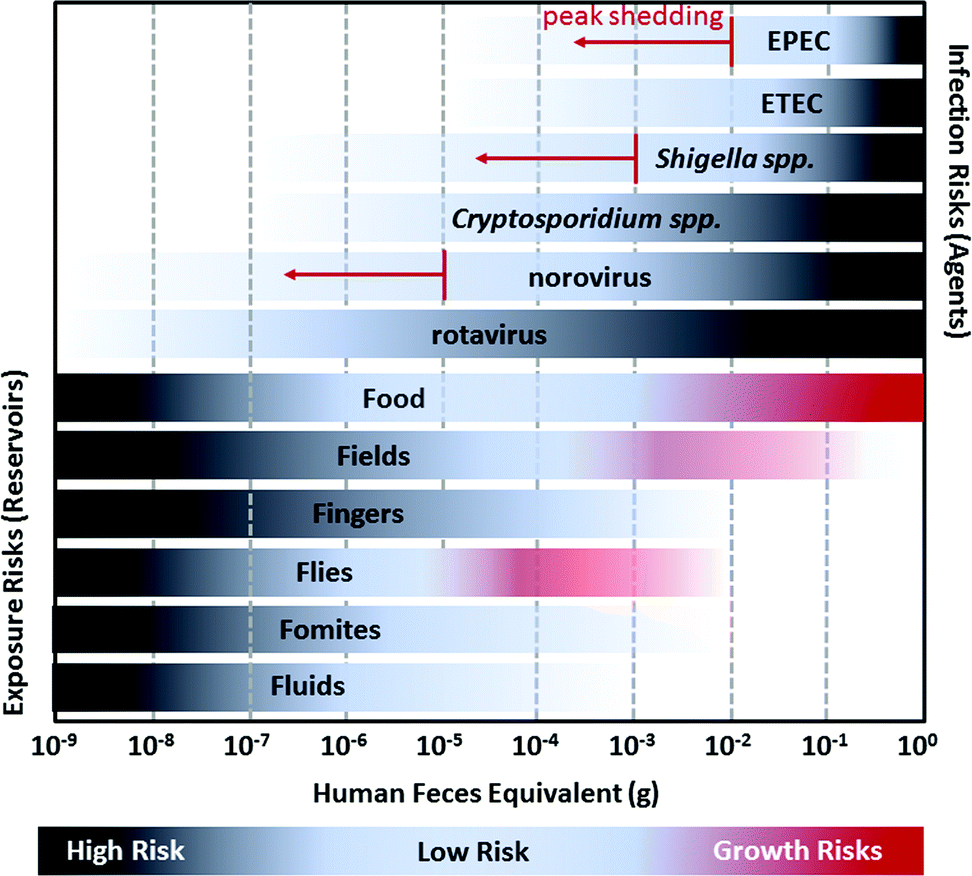 | ||
| Fig. 3 A visualization of the relationship between the estimated amount of human feces equivalents required to initiate infection for diarrheal pathogens (EPEC, ETEC, Shigella spp., Cryptosporidium spp., norovirus, and rotavirus) and the approximate amount of fecal contamination observed in environmental reservoirs (Food, Fields, Fingers, Flies, Fomites, and Fluids). The human feces equivalents for infection risks are based on the ratio of the 50% human infectious dose (HID50) to the number of pathogens shed in feces. For example, the HID50 for Shigella spp. is approximately 1000 cells and Shigella spp. is typically shed in feces at concentrations of 104 to 106 cells per g, so infection risks are highest for people exposed to more than ∼10−3 g feces of an infected person. However, Shigella spp. may reach as many as 1010 cells per g during peak shedding so there is a non-negligible risk of infection for exposure to more than 10−7 g feces. Darker shading for pathogens refers to increased risk of infection; when exposed to a higher feces equivalent there is a greater likelihood of infection. Risks from peak shedding are indicated by red arrows. Fecal contamination in environmental reservoirs is estimated from the ratio of E. coli contamination on surfaces as reported in previous studies to E. coli concentration of feces (conservatively estimated as 105 colony forming units per g). Darker shading for environmental reservoirs refers to increased likelihood of contamination: it is more likely that a reservoir will have 10−9 g feces than 10−8 g. For Food, Fields, and Flies, the possibility of, bacterial pathogen (EPEC, ETEC, and Shigella spp.) growth can further increase risks; this is depicted by red shading.12,13 | ||
Unfortunately, preventing rotavirus infection is nearly impossible. The high infectivity combined with a high rate of shedding require near complete avoidance of the infected person (Fig. 3). It is therefore unsurprising that nearly everyone is infected by the age of five, including children in industrialized countries.60,146 Efforts to reduce rotavirus-related child morbidity and mortality should be focused on vaccination, nutrition, safe and plentiful water, and health care including oral rehydration therapy.60 The goal should be to delay infection until after a child is 1 year old when likelihood of hospitalization and risk of mortality are reduced.60
Primary data on etiology of diarrheal disease is expensive and technically challenging in many settings. Expenses increase when accounting for geographic and seasonal changes in pathogens.9,147–149 In the absence of primary data, secondary sources may provide guidance for intervention design. Examples of secondary sources include data obtained from local clinics and hospitals or from prior studies (e.g., GEMS and MAL-ED) from similar (e.g., socioeconomic status, climate, geography) sites. Development of low cost clinical diagnostic tools, epidemiological studies to identify risk factors for specific causes of diarrhea, and infectious disease modeling14,150 may provide additional data on etiology when other sources are not available. It is important to note that imperfect information on important causes of diarrhea is likely more useful to intervention design than no information. Researchers should also consider monitoring child diarrheal disease infections as indicators of child health as opposed to diarrheal disease generally.151
Interventions also need to consider prevailing water, sanitation, and hygiene infrastructure and practices. Previous research has shown intervention effectiveness varies by location and by water, sanitation, and hygiene conditions.11,152 Information on water, sanitation, and hygiene is generally available at the country-level (i.e., Demographic Health Surveys, Multiple Indicator Cluster Surveys), but more site-specific data can be obtained through deployment of additional surveys, such as the WHO's Core Questions on Drinking Water and Sanitation for Household Surveys.153 Given the diversity of diarrheal disease agents and the mounting evidence regarding the importance of multiple environmental reservoirs in transmission, observed variation in intervention efficacy should not be surprising.11,15–20 Greater reductions in diarrheal disease in LMICs can be achieved when the design of interventions is informed by site-specific conditions – including etiology of disease and existing water, sanitation, and hygiene infrastructure and practices.
Acknowledgements
Thanks to Sara J. Marks, Ana K. Pitol, and anonymous reviewers for providing suggestions to improve the manuscript. This work was funded by the Swiss National Science Foundation (SNSF) and Eawag.References
- C. L. F. Walker, I. Rudan, L. Liu, H. Nair, E. Theodoratou, Z. A. Bhutta, K. L. O'Brien, H. Campbell and R. E. Black, Lancet, 381, 1405–1416 CrossRef.
- L. Liu, S. Oza, D. Hogan, J. Perin, I. Rudan, J. E. Lawn, S. Cousens, C. Mathers and R. E. Black, Lancet, 385, 430–440 CrossRef.
- M. Chan and A. Lake, Lancet, 381, 1436–1437 CrossRef.
- M. Kosek, C. Bern and R. L. Guerrant, Bull. W. H. O., 2003, 81, 197–204 Search PubMed.
- C. L. F. Walker, J. Perin, M. J. Aryee, C. Boschi-Pinto and R. E. Black, BMC Public Health, 2012, 12, 220 CrossRef PubMed.
- Z. A. Bhutta, J. K. Das, N. Walker, A. Rizvi, H. Campbell, I. Rudan and R. E. Black, Lancet, 2013, 381, 1417–1429 CrossRef.
- D. Cutler and G. Miller, Demography, 2005, 42, 1–22 CrossRef PubMed.
- A. Prüss, D. Kay, L. Fewtrell and J. Bartram, Environ. Health Perspect., 2002, 110, 537–542 CrossRef.
- K. L. Kotloff, J. P. Nataro, W. C. Blackwelder, D. Nasrin, T. H. Farag, S. Panchalingam, Y. Wu, S. O. Sow, D. Sur and R. F. Breiman, Lancet, 2013, 382, 209–222 CrossRef.
- The MAL-ED Network Investigators, Clin. Infect. Dis., 2014, 59, S193–S206 CrossRef PubMed.
- J. N. S. Eisenberg, J. C. Scott and T. Porco, Am. J. Public Health, 2007, 97, 846–852 CrossRef PubMed.
- K. Kawata, Am. J. Clin. Nutr., 1978, 31, 2114–2123 CAS.
- E. G. Wagner and J. N. Lanoix, Excreta Disposal for Rural Areas and Small Communities, 1958 Search PubMed.
- J. E. Goldstick, J. Trostle and J. N. S. Eisenberg, Am. J. Epidemiol., 2014, 179, 1247–1254 CrossRef PubMed.
- L. Fewtrell, R. B. Kaufmann, D. Kay, W. Enanoria, L. Haller and J. M. Colford Jr, Lancet Infect. Dis., 2005, 5, 42–52 CrossRef PubMed.
- T. Clasen, W.-P. Schmidt, T. Rabie, I. Roberts and S. Cairncross, BMJ, 2007, 334, 782 CrossRef PubMed.
- B. F. Arnold and J. M. Colford, Am. J. Trop. Med. Hyg., 2007, 76, 354–364 Search PubMed.
- M. C. Freeman, M. E. Stocks, O. Cumming, A. Jeandron, J. Higgins, J. Wolf, A. Prüss-Ustün, S. Bonjour, P. R. Hunter and L. Fewtrell, Trop. Med. Int. Health, 2014, 19, 906–916 CrossRef PubMed.
- J. A. Fuller, J. A. Westphal, B. Kenney and J. N. S. Eisenberg, Trop. Med. Int. Health, 2015, 20, 284–292 CrossRef PubMed.
- T. Clasen, S. Boisson, P. Routray, B. Torondel, M. Bell, O. Cumming, J. Ensink, M. Freeman, M. Jenkins and M. Odagiri, Lancet Global Health, 2014, 2, e645–e653 CrossRef PubMed.
- C. F. Lanata, C. L. Fischer-Walker, A. C. Olascoaga, C. X. Torres, M. J. Aryee and R. E. Black, PLoS One, 2013, 8, e72788 CAS.
- R. E. Black, C. G. Victora, S. P. Walker, Z. A. Bhutta, P. Christian, M. De Onis, M. Ezzati, S. Grantham-McGregor, J. Katz and R. Martorell, Lancet, 2013, 382, 427–451 CrossRef.
- J. H. Humphrey, Lancet, 2009, 374, 1032–1035 CrossRef.
- M. N. N. Mbuya and J. H. Humphrey, Matern. Child Nutr., 2016, 12, 106–120 CrossRef PubMed.
- B. F. Arnold, C. Null, S. P. Luby, L. Unicomb, C. P. Stewart, K. G. Dewey, T. Ahmed, S. Ashraf, G. Christensen, T. Clasen, H. N. Dentz, L. C. H. Fernald, R. Haque, A. E. Hubbard, P. Kariger, E. Leontsini, A. Lin, S. M. Njenga, A. J. Pickering, P. K. Ram, F. Tofail, P. J. Winch and J. M. Colford, BMJ Open, 2013, 3, e003476 CrossRef PubMed.
- M. C. M. Mattioli, J. Davis and A. B. Boehm, Environ. Sci. Technol., 2015, 49, 1912–1920 CrossRef CAS PubMed.
- J. Ottoson and T. A. Stenström, Water Res., 2003, 37, 645–655 CrossRef CAS PubMed.
- D. D. Mara and J. Oragui, Bull. W. H. O., 1985, 63, 773 CAS.
- R. Wright, J. Hyg., 1982, 89, 69–78 CrossRef CAS PubMed.
- O. Lukjancenko, T. M. Wassenaar and D. W. Ussery, Microb. Ecol., 2010, 60, 708–720 CrossRef CAS PubMed.
- A. von Mentzer, T. R. Connor, L. H. Wieler, T. Semmler, A. Iguchi, N. R. Thomson, D. A. Rasko, E. Joffre, J. Corander, D. Pickard, G. Wiklund, A.-M. Svennerholm, A. Sjoling and G. Dougan, Nat. Genet., 2014, 46, 1321–1326 CrossRef CAS PubMed.
- M. A. Croxen, R. J. Law, R. Scholz, K. M. Keeney, M. Wlodarska and B. B. Finlay, Clin. Microbiol. Rev., 2013, 26, 822–880 CrossRef CAS PubMed.
- D. J. Ingle, M. Tauschek, D. J. Edwards, D. M. Hocking, D. J. Pickard, K. I. Azzopardi, T. Amarasena, V. Bennett-Wood, J. S. Pearson, B. Tamboura, M. Antonio, J. B. Ochieng, J. Oundo, I. Mandomando, S. Qureshi, T. Ramamurthy, A. Hossain, K. L. Kotloff, J. P. Nataro, G. Dougan, M. M. Levine, R. M. Robins-Browne and K. E. Holt, Nat. Rev. Microbiol., 2016, 1, 15010 CrossRef.
- J. S. Yoder, S. Cesario, V. Plotkin, X. Ma, K. K. Shannon and M. S. Dworkin, Clin. Infect. Dis., 2006, 42, 1513–1517 CrossRef PubMed.
- M. J. Darsley, S. Chakraborty, B. DeNearing, D. A. Sack, A. Feller, C. Buchwaldt, A. L. Bourgeois, R. Walker and C. D. Harro, Clin. Vaccine Immunol., 2012, 19, 1921–1931 CrossRef CAS PubMed.
- D. J. Freedman, C. O. Tacket, A. Delehanty, D. R. Maneval, J. Nataro and J. H. Crabb, J. Infect. Dis., 1998, 177, 662–667 CAS.
- C. Harro, S. Chakraborty, A. Feller, B. DeNearing, A. Cage, M. Ram, A. Lundgren, A.-M. Svennerholm, A. L. Bourgeois and R. I. Walker, Clin. Vaccine Immunol., 2011, 18, 1719–1727 CrossRef CAS PubMed.
- T. J. Ochoa and C. A. Contreras, Curr. Opin. Infect. Dis., 2011, 24, 478–483 CrossRef PubMed.
- T. V. Nguyen, P. Le Van, C. Le Huy, K. N. Gia and A. Weintraub, J. Clin. Microbiol., 2005, 43, 755–760 CrossRef CAS PubMed.
- S. Panchalingam, M. Antonio, A. Hossain, I. Mandomando, B. Ochieng, J. Oundo, T. Ramamurthy, B. Tamboura, A. K. M. Zaidi, W. Petri, E. Houpt, P. Murray, V. Prado, R. Vidal, D. Steele, N. Strockbine, P. Sansonetti, R. I. Glass, R. M. Robins-Browne, M. Tauschek, A.-M. Svennerholm, K. Kotloff, M. M. Levine and J. P. Nataro, Clin. Infect. Dis., 2012, 55, S294–S302 CrossRef CAS PubMed.
- T. H. Hazen, M. S. Donnenberg, S. Panchalingam, M. Antonio, A. Hossain, I. Mandomando, J. B. Ochieng, T. Ramamurthy, B. Tamboura, S. Qureshi, F. Quadri, A. Zaidi, K. L. Kotloff, M. M. Levine, E. M. Barry, J. B. Kaper, D. A. Rasko and J. P. Nataro, Nat. Rev. Microbiol., 2016, 1, 15014 CrossRef.
- F. Barletta, T. J. Ochoa, E. Mercado, J. Ruiz, L. Ecker, G. Lopez, M. Mispireta, A. I. Gil, C. F. Lanata and T. G. Cleary, Clin. Infect. Dis., 2011, 53, 1223–1229 CrossRef CAS PubMed.
- J. E. Afset, L. Bevanger, P. Romundstad and K. Bergh, J. Med. Microbiol., 2004, 53, 1137–1144 CrossRef PubMed.
- N. N. Rang, S. T. Louise, T. Marija and M. R.-B. Roy, Emerg. Infect. Dis., 2006, 12, 597 CrossRef PubMed.
- F. Yang, J. Yang, X. Zhang, L. Chen, Y. Jiang, Y. Yan, X. Tang, J. Wang, Z. Xiong and J. Dong, Nucleic Acids Res., 2005, 33, 6445–6458 CrossRef PubMed.
- R. Lan and P. R. Reeves, Microbes Infect., 2002, 4, 1125–1132 CrossRef CAS PubMed.
- S. Livio, N. A. Strockbine, S. Panchalingam, S. M. Tennant, E. M. Barry, M. E. Marohn, M. Antonio, A. Hossain, I. Mandomando, J. B. Ochieng, J. O. Oundo, S. Qureshi, T. Ramamurthy, B. Tamboura, R. A. Adegbola, M. J. Hossain, D. Saha, S. Sen, A. S. G. Faruque, P. L. Alonso, R. F. Breiman, A. K. M. Zaidi, D. Sur, S. O. Sow, L. Y. Berkeley, C. E. O'Reilly, E. D. Mintz, K. Biswas, D. Cohen, T. H. Farag, D. Nasrin, Y. Wu, W. C. Blackwelder, K. L. Kotloff, J. P. Nataro and M. M. Levine, Clin. Infect. Dis., 2014, 59, 933–941 CrossRef PubMed.
- K. L. Kotloff, J. P. Winickoff, B. Ivanoff, J. D. Clemens, D. L. Swerdlow, P. J. Sansonetti, G. Adak and M. Levine, Bull. W. H. O., 1999, 77, 651–666 CAS.
- M. M. Levine, H. L. DuPont, S. B. Formal, R. B. Hornick, A. Takeuchi, E. J. Gangarosa, M. J. Snyder and J. P. Libonati, J. Infect. Dis., 1973, 127, 261–270 CrossRef CAS PubMed.
- A. V. Hardy and S. P. Halbert, Public Health Rep., 1896-1970, 1948, 790–792 Search PubMed.
- W. Mokhtari, S. Nsaibia, A. Gharbi and M. Aouni, Mol. Cell. Probes, 2013, 27, 53–59 CrossRef CAS PubMed.
- E. A. Groisman, Principles of Bacterial Pathogenesis, Academic Press, 2001 Search PubMed.
- L. Robertson, A. Campbell and H. Smith, Appl. Environ. Microbiol., 1992, 58, 3494–3500 CAS.
- D. Korich, J. Mead, M. Madore, N. Sinclair and C. R. Sterling, Appl. Environ. Microbiol., 1990, 56, 1423–1428 CAS.
- S. H. Elsafi, T. N. Al-Maqati, M. I. Hussein, A. A. Adam, M. M. A. Hassan and E. M. Al Zahrani, Parasitol. Res., 2013, 112, 1641–1646 CrossRef PubMed.
- C. L. Chappell, P. C. Okhuysen, C. R. Sterling, C. Wang, W. Jakubowski and H. L. Dupont, Am. J. Trop. Med. Hyg., 1999, 60, 157–164 CAS.
- R. C. Shepherd, C. L. Reed and G. P. Sinha, J. Clin. Pathol., 1988, 41, 1104–1106 CrossRef CAS PubMed.
- P. C. Okhuysen, C. L. Chappell, J. H. Crabb, C. R. Sterling and H. L. DuPont, J. Infect. Dis., 1999, 180, 1275–1281 CrossRef CAS PubMed.
- P. F. M. Teunis, N. J. D. Nagelkerke and C. N. Haas, Risk Analysis, 19, 1251–1260 CAS.
- U. D. Parashar, E. G. Hummelman, J. S. Bresee, M. A. Miller and R. I. Glass, Emerging Infect. Dis., 2003, 9, 565 CrossRef PubMed.
- World Health Organization, Generic protocol for monitoring impact of rotavirus vaccination on gastroenteritis disease burden and viral strains, World Health Organization, Geneva, WHO/IVB/08.16, 2008, 77 Search PubMed.
- T. Flewett, Br. Med. J., 1983, 287, 568–569 CrossRef CAS PubMed.
- T. M. Fumian, J. P. G. Leite, M. S. Rocha, J. S. R. de Andrade, J. M. Fioretti, R. M. S. de Assis, M. R. S. Assis, A. M. Fialho and M. P. Miagostovich, J. Virol. Methods, 2016, 228, 123–129 CrossRef CAS PubMed.
- I. Mukhopadhya, R. Sarkar, V. K. Menon, S. Babji, A. Paul, P. Rajendran, T. V. Sowmyanarayanan, P. D. Moses, M. Iturriza-Gomara, J. J. Gray and G. Kang, J. Med. Virol., 2013, 85, 1661–1668 CrossRef CAS PubMed.
- R. L. Ward, D. I. Bernstein, E. C. Young, J. R. Sherwood, D. R. Knowlton and G. M. Schiff, J. Infect. Dis., 1986, 154, 871–880 CrossRef CAS PubMed.
- L. Lindesmith, C. Moe, S. Marionneau, N. Ruvoen, X. Jiang, L. Lindblad, P. Stewart, J. LePendu and R. Baric, Nat. Med., 2003, 9, 548–553 CrossRef CAS PubMed.
- B. R. Bank-Wolf, M. König and H.-J. Thiel, Vet. Microbiol., 2010, 140, 204–212 CrossRef PubMed.
- A. M. Hutson, F. Airaud, J. LePendu, M. K. Estes and R. L. Atmar, J. Med. Virol., 2005, 77, 116–120 CrossRef CAS PubMed.
- R. L. Atmar, A. R. Opekun, M. A. Gilger, M. K. Estes, S. E. Crawford, F. H. Neill, S. Ramani, H. Hill, J. Ferreira and D. Y. Graham, J. Infect. Dis., 2014, 209, 1016–1022 CrossRef PubMed.
- M. J. Messner, P. Berger and S. P. Nappier, Risk Analysis, 2014, 34, 1820–1829 CrossRef PubMed.
- P. J. Schmidt, Risk Analysis, 2015, 35, 1364–1383 CrossRef PubMed.
- R. L. Atmar, A. R. Opekun, M. A. Gilger, M. K. Estes, S. E. Crawford, F. H. Neill and D. Y. Graham, Emerging Infect. Dis., 2008, 14, 1553 CrossRef PubMed.
- K. Ozawa, T. Oka, N. Takeda and G. S. Hansman, J. Clin. Microbiol., 2007, 45, 3996–4005 CrossRef CAS PubMed.
- M. Chan, J. Sung, R. Lam, P. Chan, N. Lee, R. Lai and W. K. Leung, Emerging Infect. Dis., 2006, 12, 1278–1280 CrossRef PubMed.
- U.S. EPA, Exposure Factors Handbook 2011 Edition (Final), U.S. Environmental Protection Agency, Washington DC, EPA/600/R-09/052F, 2011, 1436 Search PubMed.
- K. Onda, J. LoBuglio and J. Bartram, Int. J. Environ. Res. Public Health, 2012, 9, 880 CrossRef PubMed.
- S. Rufener, D. usezahl, H.-J. Mosler and R. Weingartner, J. Health Popul. Nutr., 2010, 28, 34–41 Search PubMed.
- A. R. Harris, J. Davis and A. B. Boehm, J. Water Health, 2013, 11, 543–554 CrossRef CAS PubMed.
- D. Ahmed, M. S. Islam, Y. A. Begum, A. Janzon, F. Qadri and Å. Sjöling, J. Appl. Microbiol., 2013, 114, 1223–1229 CrossRef CAS PubMed.
- A. Samie, M. B. Mashao, P. O. Bessong, T. F. NKgau, M. N. B. Momba and C. L. Obi, J. Health Popul. Nutr., 2012, 30, 241–249 CAS.
- N. Taneja, M. Singh, P. Rao, M. Biswal, S. Priya, R. Chander and M. Sharma, J. Commun. Dis., 2011, 43, 193–199 Search PubMed.
- A. S. Ferguson, A. C. Layton, B. J. Mailloux, P. J. Culligan, D. E. Williams, A. E. Smartt, G. S. Sayler, J. Feighery, L. D. McKay, P. S. Knappett, E. Alexandrova, T. Arbit, M. Emch, V. Escamilla, K. M. Ahmed, M. J. Alam, P. K. Streatfield, M. Yunus and A. van Geen, Sci. Total Environ., 2012, 431, 314–322 CrossRef CAS PubMed.
- M. C. Mattioli, A. B. Boehm, J. Davis, A. R. Harris, M. Mrisho and A. J. Pickering, PLoS One, 2014, 9, e84939 Search PubMed.
- P. Kelly, K. S. Baboo, P. Ndubani, M. Nchito, N. P. Okeowo, N. P. Luo, R. A. Feldman and M. J. Farthing, J. Infect. Dis., 1997, 176, 1120–1123 CAS.
- A. E. Abo-Amer, S. M. Soltan el and M. A. Abu-Gharbia, Acta Microbiol. Immunol. Hung., 2008, 55, 311–326 CrossRef CAS PubMed.
- W. H. Organization, Guidelines for Drinking-water Quality: Recommendations, World Health Organization, 2004 Search PubMed.
- A. K. Camper, G. A. McFeters, W. G. Characklis and W. L. Jones, Appl. Environ. Microbiol., 1991, 57, 2233–2239 CAS.
- Å. Lothigius, Å. Sjöling, A. M. Svennerholm and I. Bölin, J. Appl. Microbiol., 2010, 108, 1441–1449 CrossRef PubMed.
- Å. Lothigius, Å. Sjöling, A. M. Svennerholm and I. Bölin, J. Appl. Microbiol., 2010, 108, 1441–1449 CrossRef PubMed.
- J. E. Mellor, J. A. Smith, A. Samie and R. A. Dillingham, J. Environ. Eng., 2013, 139, 1152–1161 CrossRef CAS PubMed.
- B. A. Sackey, P. Mensah, E. Collison and E. Sakyi-Dawson, Int. J. Food Microbiol., 2001, 71, 21–28 CrossRef CAS PubMed.
- A. J. Pickering, T. R. Julian, S. J. Marks, M. C. Mattioli, A. B. Boehm, K. J. Schwab and J. Davis, Environ. Sci. Technol., 2012, 46, 5736–5743 CrossRef CAS PubMed.
- M. A. Islam, T. Ahmed, A. S. G. Faruque, S. Rahman, S. K. Das, D. Ahmed, V. Fattori, R. Clarke, H. P. Endtz and A. Cravioto, Eur. J. Clin. Nutr., 2012, 66, 1242–1246 CrossRef CAS PubMed.
- P. Antwi-Agyei, S. Cairncross, A. Peasey, V. Price, J. Bruce, K. Baker, C. Moe, J. Ampofo, G. Armah and J. Ensink, PLoS One, 2015, 10, e0142346 Search PubMed.
- F. M. Mosupye and A. von Holy, J. Food Prot., 1999, 62, 1278–1284 CAS.
- M. Steele and J. Odumeru, J. Food Prot., 2004, 67, 2839–2849 Search PubMed.
- S. Ram, S. Khurana, S. B. Khurana, D. V. Vadehra, S. Sharma and R. S. Chhina, Indian J. Med. Res., 1996, 103, 253–258 CAS.
- M. S. Islam, M. K. Hasan and S. I. Khan, Appl. Environ. Microbiol., 1993, 59, 652–654 CAS.
- L. D. Fantasia, L. Mestrandrea, J. P. Schrade and J. Yager, Appl. Microbiol., 1975, 29, 179–185 CAS.
- V. E. Letschert and M. A. McNeil, Material world: Forecasting household appliance ownership in a growing global economy, Lawrence Berkeley National Laboratory, Berkeley, LBNL 2403E, 2009, 8 Search PubMed.
- A. J. Pickering, T. R. Julian, S. Mamuya, A. B. Boehm and J. Davis, Trop. Med. Int. Health, 2011, 16, 233–239 CrossRef PubMed.
- P. K. Ram, I. Jahid, A. K. Halder, B. Nygren, M. S. Islam, S. P. Granger, J. W. Molyneaux and S. P. Luby, Am. J. Trop. Med. Hyg., 2011, 84, 510–516 CrossRef PubMed.
- A. V. Hardy and J. Watt, Public Health Rep., 1948, 63, 363–378 CrossRef CAS PubMed.
- M. C. M. Mattioli, A. J. Pickering, R. Gilsdorf, J. Davis and A. B. Boehm, Environ. Sci. Technol., 2012, 47, 355–363 CrossRef PubMed.
- A. J. Pickering, J. Davis, S. P. Walters, H. M. Horak, D. P. Keymer, D. Mushi, B. Strickfaden, J. S. Chynoweth, J. Liu, A. Blum, K. Rogers and A. B. Boehm, Environ. Sci. Technol., 2010, 44, 3267–3272 CrossRef CAS PubMed.
- M. C. M. Mattioli, J. Davis, M. Mrisho and A. B. Boehm, Am. J. Trop. Med. Hyg., 2015, 93, 478–484 CrossRef CAS PubMed.
- S. A. Ansari, S. A. Sattar, V. S. Springthorpe, G. A. Wells and W. Tostowaryk, J. Clin. Microbiol., 1988, 26, 1513–1518 CAS.
- P. Rusin, S. Maxwell and C. Gerba, J. Appl. Microbiol., 2002, 93, 585–592 CrossRef CAS PubMed.
- L. Utsi, S. J. Smith, R. M. Chalmers and S. Padfield, Epidemiol. Infect., 2016, 144, 1000–1009 CrossRef CAS PubMed.
- C. f. D. C. a. P. (CDC), MMWR Morb Mortal Wkly Rep, 2011, 60, 1456.
- Y. C. Lin, E. Hipfl, I. Lederer, F. Allerberger and D. Schmid, Int. J. Infect. Dis., 2015, 37, 25–29 CrossRef PubMed.
- J. D. Greig, E. C. Todd, C. A. Bartleson and B. S. Michaels, J. Food Prot., 2007, 70, 1752–1761 Search PubMed.
- K. Rochon, T. J. Lysyk and L. B. Selinger, J. Med. Entomol., 2004, 41, 1082–1089 CrossRef CAS PubMed.
- S. Collinet-Adler, S. Babji, M. Francis, D. Kattula, P. S. Premkumar, R. Sarkar, V. R. Mohan, H. Ward, G. Kang, V. Balraj and E. N. Naumova, Appl. Environ. Microbiol., 2015, 81, 6053–6058 CrossRef CAS PubMed.
- K. Khalil, G. B. Lindblom, K. Mazhar and B. Kaijser, Epidemiol. Infect., 1994, 113, 435–444 CrossRef CAS PubMed.
- M. Kobayashi, T. Sasaki, N. Saito, K. Tamura, K. Suzuki, H. Watanabe and N. Agui, Am. J. Trop. Med. Hyg., 1999, 61, 625–629 CAS.
- M. J. Alam and L. Zurek, Appl. Environ. Microbiol., 2004, 70, 7578–7580 CrossRef CAS PubMed.
- A. J. De Jesús, A. R. Olsen, J. R. Bryce and R. C. Whiting, Int. J. Food Microbiol., 2004, 93, 259–262 CrossRef PubMed.
- L. Wasala, J. L. Talley, U. Desilva, J. Fletcher and A. Wayadande, Phytopathology, 2013, 103, 373–380 CrossRef CAS PubMed.
- A. N. Schaupp, MPH Thesis, Emory University, 2013.
- F. M. Ngure, J. H. Humphrey, M. N. Mbuya, F. Majo, K. Mutasa, M. Govha, E. Mazarura, B. Chasekwa, A. J. Prendergast and V. Curtis, Am. J. Trop. Med. Hyg., 2013, 89, 709–716 CrossRef PubMed.
- M. Byappanahalli and R. Fujioka, Water Sci. Technol., 1998, 38, 171–174 CrossRef CAS.
- T. R. Julian, L. H. MacDonald, Y. Guo, S. J. Marks, M. Kosek, P. P. Yori, S. R. Pinedo and K. J. Schwab, Am. J. Trop. Med. Hyg., 2013, 5, 869–872 CrossRef PubMed.
- R. G. Sinclair and C. P. Gerba, Lett. Appl. Microbiol., 2010, 52, 144–149 CrossRef PubMed.
- C. E. Stauber, A. Walters, A. M. F. de Aceituno and M. D. Sobsey, Int. J. Environ. Res. Public Health, 2013, 10, 1586–1597 CrossRef PubMed.
- J. Vujcic, P. K. Ram, F. Hussain, L. Unicomb, P. S. Gope, J. Abedin, Z. H. Mahmud, M. Sirajul Islam and S. P. Luby, Trop. Med. Int. Health, 2014, 19, 528–536 CrossRef PubMed.
- V. Keshav, C. A. Kruger, A. Mathee, N. Naicker, A. Swart and T. G. Barnard, J. Water, Sanit. Hyg. Dev., 2015, 5, 351–358 CrossRef.
- B. Torondel, Y. Gyekye-Aboagye, P. Routray, S. Boisson, W. Schimdt and T. Clasen, Trans. R. Soc. Trop. Med. Hyg., 2015, 109, 386–392 CrossRef PubMed.
- T. R. Julian and A. J. Pickering, PLoS One, 2015, 10, e0136158 Search PubMed.
- G. U. Lopez, C. P. Gerba, A. H. Tamimi, M. Kitajima, S. L. Maxwell and J. B. Rose, Appl. Environ. Microbiol., 2013, 79, 5728–5734 CrossRef CAS PubMed.
- T. Julian, J. Leckie and A. Boehm, J. Appl. Microbiol., 2010, 109, 1868–1874 CrossRef CAS PubMed.
- A. Kramer, I. Schwebke and G. Kampf, BMC Infect. Dis., 2006, 6, 130 CrossRef PubMed.
- S. A. Boone and C. P. Gerba, Appl. Environ. Microbiol., 2007, 73, 1687–1696 CrossRef CAS PubMed.
- D. J. Weber and W. A. Rutala, Infect. Control Hosp. Epidemiol., 2001, 22, 306–315 CAS.
- D. L. Taylor, T. M. Kahawita, S. Cairncross and J. H. J. Ensink, PLoS One, 2015, 10, e0135676 Search PubMed.
- S. A. Esrey, R. G. Feachem and J. M. Hughes, Bull. W. H. O., 1985, 63, 757 CAS.
- K. S. Enger, K. L. Nelson, J. B. Rose and J. N. S. Eisenberg, Water Res., 2013, 47, 1181–1190 CrossRef CAS PubMed.
- M. S. Islam, Z. H. Mahmud, P. S. Gope, R. U. Zaman, Z. Hossain, M. S. Islam, D. Mondal, M. A. Sharker, K. Islam, H. Jahan, A. Bhuiya, H. P. Endtz, A. Cravioto, V. Curtis, O. Toure and S. Cairncross, Trop. Med. Int. Health, 2013, 18, 250–258 Search PubMed.
- D. Cohen, M. Green, C. Block, R. Slepon, R. Ambar, S. S. Wasserman and M. M. Levine, Lancet, 1991, 337, 993–997 CrossRef CAS.
- M. U. Khan, Trans. R. Soc. Trop. Med. Hyg., 1982, 76, 164–168 CrossRef CAS PubMed.
- M. D. Cattaneo, S. Galiani, P. J. Gertler, S. Martinez and R. Titiunik, Am. Econ. J., 2009, 75–105 Search PubMed.
- A. Gil, C. Lanata, E. Kleinau and M. Penny, Children's Feces Disposal Practices in Developing Countries and Interventions to Prevent Diarrheal Diseases, U.S. Agency for International Development 2004 Search PubMed.
- P. A. Gastañaduy, Y. Vicuña, F. Salazar, N. Broncano, N. Gregoricus, J. Vinjé, M. Chico, U. D. Parashar, P. J. Cooper and B. Lopman, The Pediatric infectious disease journal, 2015, 34, 1031 CrossRef PubMed.
- B. Lopman, Y. Vicuña, F. Salazar, N. Broncano, M. D. Esona, C. Sandoval, N. Gregoricus, M. D. Bowen, D. Payne, M. Vaca, M. Chico, U. Parashar and P. J. Cooper, PLoS One, 2013, 8, e67763 CAS.
- J. Perera and G. Lucas, Ceylon Med J., 1990, 35, 11–14 CAS.
- I. o. M. U. C. o. I. a. P. f. N. V. Development, Journal, 1986.
- R. L. Guerrant, L. V. Kirchhoff, D. S. Shields, M. K. Nations, J. Leslie, M. A. de Sousa, J. G. Araujo, L. L. Correia, K. T. Sauer, K. E. McClelland, F. L. Trowbridge and J. M. Hughes, J. Infect. Dis., 1983, 148, 986–997 CrossRef CAS PubMed.
- J. Yu, H. Jing, S. Lai, W. Xu, M. Li, J. Wu, W. Liu, Z. Yuan, Y. Chen, S. Zhao, X. Wang, Z. Zhao, L. Ran, S. Wu, J. D. Klena, L. Feng, F. Li, X. Ye, Y. Qiu, X. Wang, H. Yu, Z. Li and W. Yang, J. Infect., 2015, 71, 19–27 CrossRef PubMed.
- S. Breurec, N. Vanel, P. Bata, L. Chartier, A. Farra, L. Favennec, T. Franck, T. Giles-Vernick, J.-C. Gody, L. B. Luong Nguyen, M. Onambélé, C. Rafaï, R. Razakandrainibe, L. Tondeur, V. Tricou, P. Sansonetti and M. Vray, PLoS Neglected Trop. Dis., 2016, 10, e0004283 Search PubMed.
- A. Lal, T. Ikeda, N. French, M. G. Baker and S. Hales, PLoS One, 2013, 8, e83484 Search PubMed.
- A. J. Pickering and M. L. Alzua, Lancet Global Health, 2016, 4, e160 CrossRef PubMed.
- J. A. Fuller, J. A. Westphal, B. Kenney and J. N. S. Eisenberg, Trop. Med. Int. Health, 2015, 20, 284–292 CrossRef PubMed.
- UNICEF and World Health Organization, Core questions on drinking water and sanitation for household surveys, World Health Organization, Geneva, 2006, 24 Search PubMed.
- The Sanitation Hygiene Infant Nutrition Efficacy (SHINE) Trial Team, Clin. Infect. Dis., 2015, 61, S685–S702 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2016 |