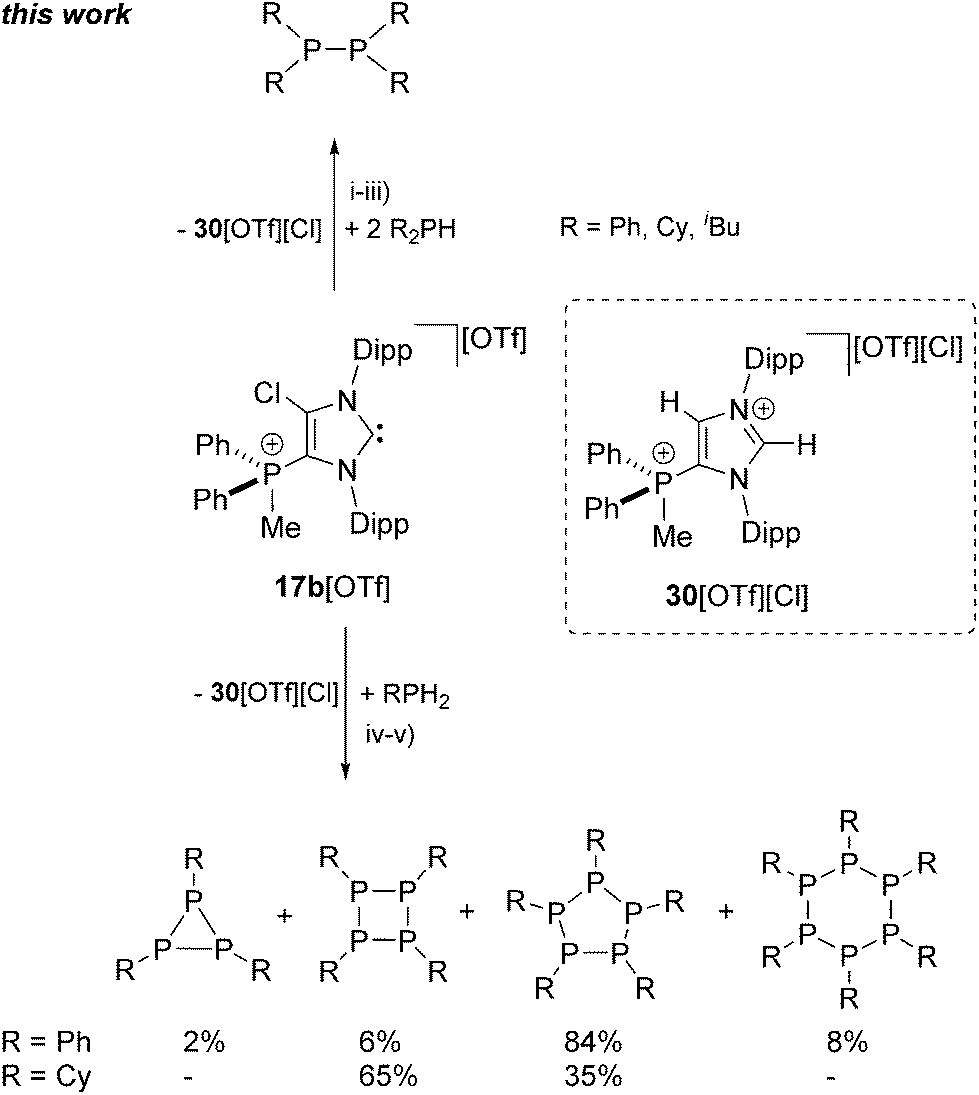DOI:
10.1039/C6DT01871H
(Paper)
Dalton Trans., 2016,
45, 11384-11396
Cationic 5-phosphonio-substituted N-heterocyclic carbenes†
Received
11th May 2016
, Accepted 3rd June 2016
First published on 23rd June 2016
Abstract
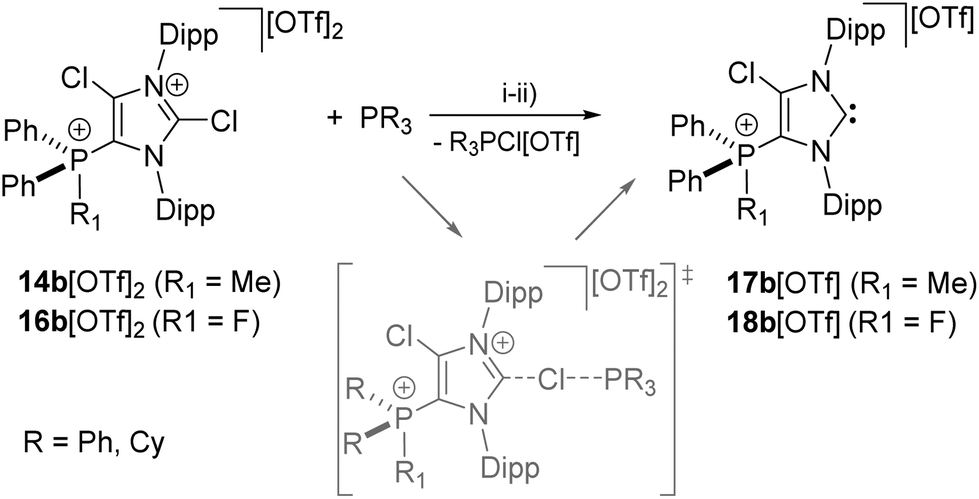
2-Phosphanyl-substituted imidazolium salts 2-PR2(4,5-Cl-Im)[OTf] (9a,b[OTf]) (4,5-Cl-Im = 4,5-dichloro-1,3-bis(2,6-di-isopropylphenyl)-imidazolium) (a: R = Cy, b: R = Ph) are prepared from the reaction of R2PCl (R = Cy, Ph) with NHC 8 (4,5-dichloro-1,3-bis(2,6-di-isopropylphenyl)-imidazolin-2-ylidene) in the presence of Me3SiOTf. 5-Phospanyl-substituted imidazolium salts 5-PR2(2,4-Cl-Im)[OTf] (10a,b[OTf]) are obtained in quantitative yield when a slight excess of the NHC 8 is used. 5-Phosphonio-substituted imidazolium salts 5-PR2Me(2,4-Cl-Im)[OTf]2 (14a,b[OTf]2) and 5-PR2F(2,4-Cl-Im)[OTf]2 (16a,b[OTf]2) result from methylation reaction or oxidation of 10a,b[OTf] with XeF2 and subsequent fluoride abstraction. According to our quantum chemical studies the Cl1 atom at the 2-position at the imidazolium ring of dication 14b2+ carries a slightly positive charge and is therefore accessible for nucleophilic attack. Accordingly, the reaction of 14a,b[OTf]2 and 16a,b[OTf]2 with R3P (R = Cy, Ph) affords cationic 5-phosphonio-substituted NHCs 5-PR2Me(4-Cl-NHC)[OTf] (17a,b[OTf]) and 5-PR2F(4-Cl-NHC)[OTf] (18a,b[OTf]) via a SN2(Cl)-type reaction. A series of transition metal complexes such as [AuCl(5-PPh2Me(4-Cl-NHC))][OTf] (19[OTf]), [CuBr(5-PPh2Me(4-Cl-NHC))][OTf] (20[OTf]), [AuCl(5-PPh2F(4-Cl-NHC))[OTf] (21[OTf]) and [RhCl(cod)(5-PPh2Me(4-Cl-NHC))][OTf] (23[OTf]) are prepared to prove the coordination abilities of carbenes 17b[OTf] and 18b[OTf]. The isolation of a rare example of a tricationic bis-carbene silver complex [Ag(5-PPh2Me(4-Cl-NHC))2][OTf]3 (22[OTf]3) is achieved by reacting 14b[OTf] with Cy3P in the presence of AgOTf. NHC 17b[OTf] represents a very effective dehydrocoupling reagent for secondary (R2PH, R = Ph, Cy, iBu) and primary (RPH2, R = Ph, Cy) phosphanes to give diphosphanes of type R4P2 (R = Ph, Cy, iBu) and oligophosphanes R4P4, R5P5 (R = Ph, Cy), respectively. Methylation of 17b+ and subsequent deprotonation reaction with LDA affords the cationic NHO (N-heterocyclic olefin) 35+ of which the gold complex 36+ is readily accessible via the reaction with AuCl(tht).
Introduction
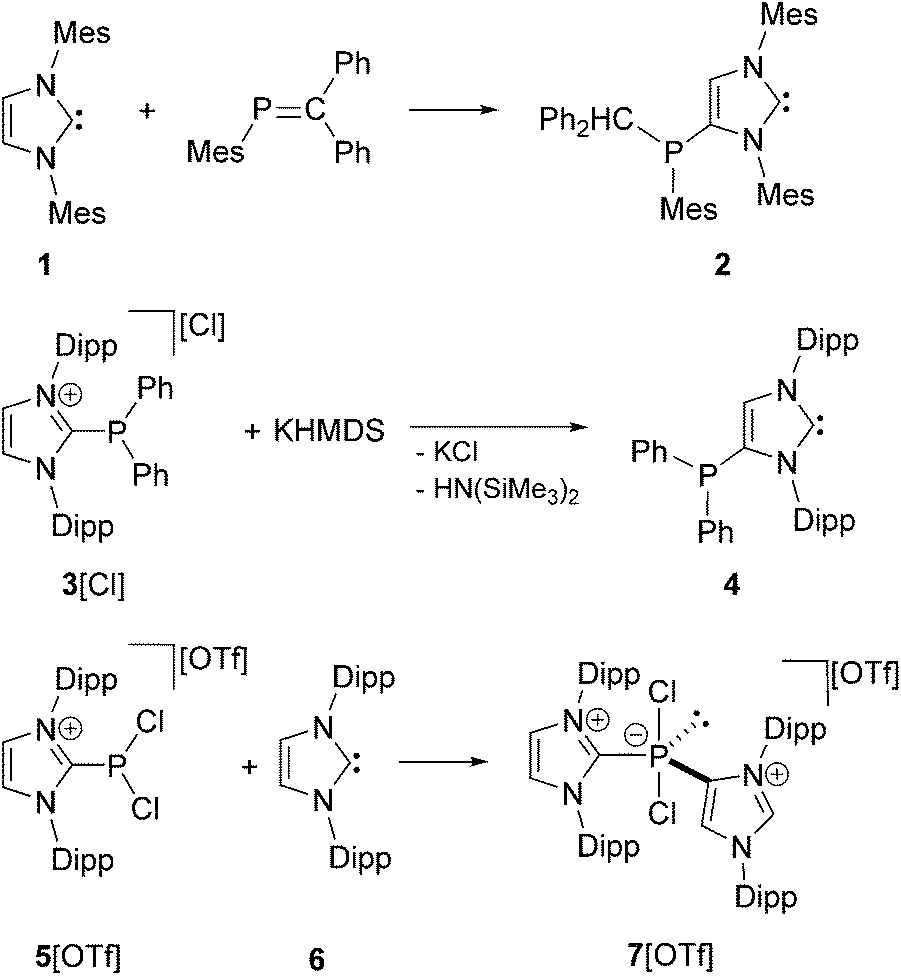
The application of N-heterocyclic carbenes (NHCs) in phosphorus chemistry has led to some remarkable discoveries in recent years and is a growing field with considerable impact. The most general feature of NHCs is their tendency to react at the 2-position with electrophilic PIII compounds, leading to the corresponding 2-phosphanyl-substituted imidazolium salts or to donor–acceptor complexes of the NHC and the respective P-centered moiety.1,2 In contrast, only a few examples are known where the 4/5-position of NHCs can be selectively addressed to yield 4/5-phosphanyl-substituted NHCs or imidazolium salts. Reactions are reported involving inter alia phosphaalkenes3 and chlorophosphanes.4,5 In 2009 Gates and co-workers observed the unusual reaction of 1,3-di(mesityl)imidazolin-2-ylidene 1 with phosphaalkene MesP![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CPh2 to afford the first example of a neutral 4-phosphanyl-substituted NHC 2 (Scheme 1, top).3 Shortly afterwards, Bertrand and co-workers observed the rearrangement reaction of 2-phosphanyl-substituted imidazolium salt 3[Cl] to the 4-substituted derivative 4 when treated with a base (e.g. KHMDS; Scheme 1, middle).4 The neutral 4-phosphanyl-substituted NHCs 2 and 4 were used as ligands in transition metal chemistry to generate mono metallic,6 homo-6,7 and hetero-7 bimetallic complexes. In 2013 our group reported on the reaction of P-centered imidazolium salt 5[OTf] with NHC 6 leading to the cationic phosphoranide derivative 7[OTf] where the second imidazoliumyl-substituent is bonded via the 4-position to the P atom (Scheme 1, bottom). The proposed mechanism includes the formation of an abnormal carbene8 followed by an intermolecular rearrangement, which we believe is related to Bertrand`s mechanism for the formation of 4.4
CPh2 to afford the first example of a neutral 4-phosphanyl-substituted NHC 2 (Scheme 1, top).3 Shortly afterwards, Bertrand and co-workers observed the rearrangement reaction of 2-phosphanyl-substituted imidazolium salt 3[Cl] to the 4-substituted derivative 4 when treated with a base (e.g. KHMDS; Scheme 1, middle).4 The neutral 4-phosphanyl-substituted NHCs 2 and 4 were used as ligands in transition metal chemistry to generate mono metallic,6 homo-6,7 and hetero-7 bimetallic complexes. In 2013 our group reported on the reaction of P-centered imidazolium salt 5[OTf] with NHC 6 leading to the cationic phosphoranide derivative 7[OTf] where the second imidazoliumyl-substituent is bonded via the 4-position to the P atom (Scheme 1, bottom). The proposed mechanism includes the formation of an abnormal carbene8 followed by an intermolecular rearrangement, which we believe is related to Bertrand`s mechanism for the formation of 4.4
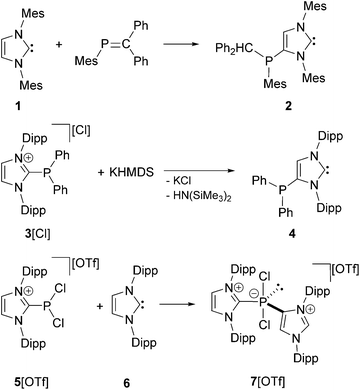 |
| | Scheme 1 Preparation of 2, 4, and 7[OTf] (Mes = 1,3,5-trimethylphenyl, Dipp = 2,6-diisopropylphenyl, Ph = phenyl, HMDS = bis(trimethylsilyl)amide); for all examples, only one representative Lewis structure is depicted. | |
Only very few examples of 4-, 4,5-bisphosphoryl-9 and 4,5-bis-phosphanyl-10 substituted imidazolium salts and 4-phosphanido-substituted NHC11 are reported in the literature so far. The aforementioned compounds exhibit either one or two Lewis basic functionalities. However, so far there is no example of a cationic NHC featuring a phosphonium functionality at the 4/5-position, which should have a significant influence on the reactivity of the carbene.
Results and discussion
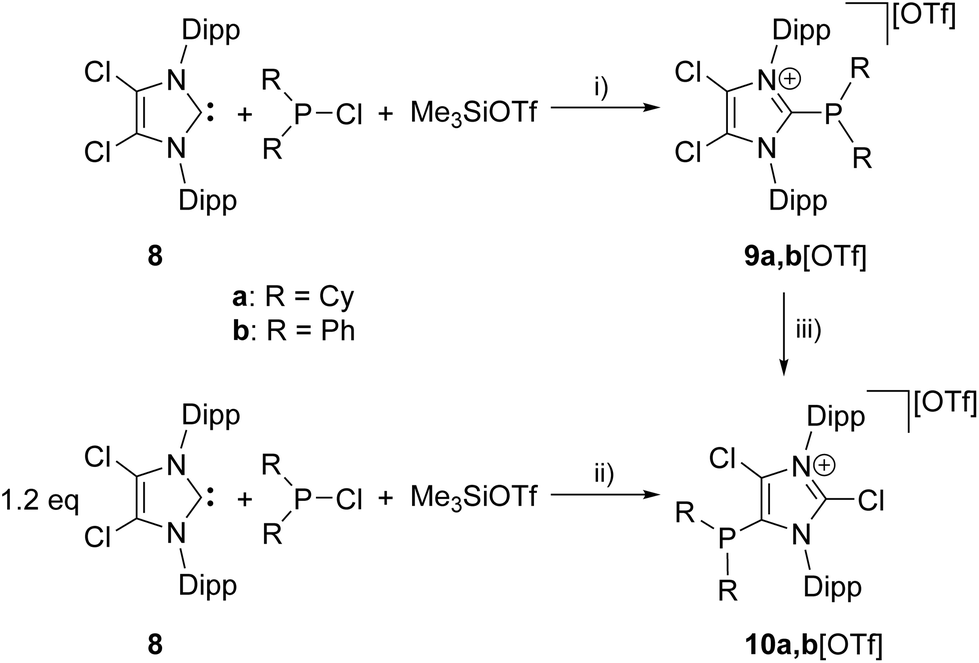
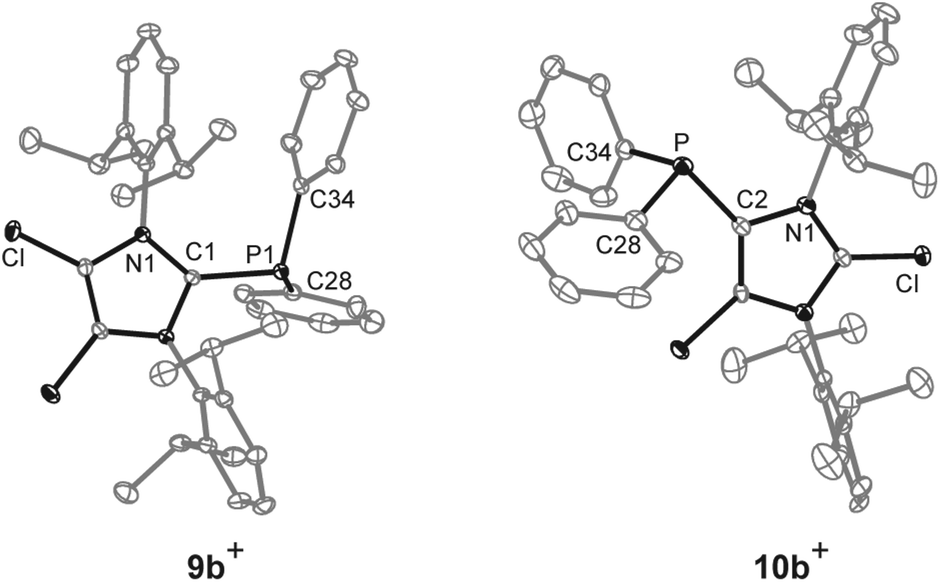
The reaction of chlorophosphanes R2PCl (R = Cy, Ph) with Me3SiOTf and NHC 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 in a precise 1:1:1 reaction gives the corresponding 2-phosphanyl-substituted imidazolium salts 9a,b[OTf] in excellent yield (9a[OTf]: 91%; 9b[OTf]: 99%; Scheme 2). The 31P NMR spectra of the purified, colorless solids show a singlet resonance in each case (9a+: δ(P) = 12.7 ppm; 9b+: δ(P) = −5.1 ppm), which is significantly upfield shifted compared to the respective chlorophosphanes (Cy2PCl: δ(P) = 127.1 ppm; Ph2PCl: δ(P) = 81.9 ppm)13 due to the presence of the imidazolium substituents. Suitable single crystals for X-ray analysis are obtained by slow diffusion of n-hexane into a saturated CH2Cl2 solution of 9b[OTf] (Fig. 1). The P–C bond lengths (P–C1 1.8437(9) Å, P–C28 1.8205(10) Å, P–C34 1.8249(10) Å) are in the expected range of a P–C single bond (1.83 Å) involving a tri-coordinate phosphorus atom.14
12 in a precise 1:1:1 reaction gives the corresponding 2-phosphanyl-substituted imidazolium salts 9a,b[OTf] in excellent yield (9a[OTf]: 91%; 9b[OTf]: 99%; Scheme 2). The 31P NMR spectra of the purified, colorless solids show a singlet resonance in each case (9a+: δ(P) = 12.7 ppm; 9b+: δ(P) = −5.1 ppm), which is significantly upfield shifted compared to the respective chlorophosphanes (Cy2PCl: δ(P) = 127.1 ppm; Ph2PCl: δ(P) = 81.9 ppm)13 due to the presence of the imidazolium substituents. Suitable single crystals for X-ray analysis are obtained by slow diffusion of n-hexane into a saturated CH2Cl2 solution of 9b[OTf] (Fig. 1). The P–C bond lengths (P–C1 1.8437(9) Å, P–C28 1.8205(10) Å, P–C34 1.8249(10) Å) are in the expected range of a P–C single bond (1.83 Å) involving a tri-coordinate phosphorus atom.14
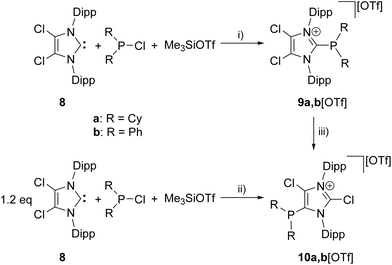 |
| | Scheme 2 Preparation of 9a,b[OTf], (i): C6H5F, r.t., 6 h, 9a[OTf] 91%; 9b[OTf] 99%; preparation of 10a,b[OTf]; (ii): o-C6H4F2, r.t. 14 h, 10a[OTf] 92%; 10b[OTf] 97%; conversion of 9a,b[OTf] to 10a,b[OTf], (iii) 0.3 eq. 8, o-C6H4F2, r.t. 30 h, quantitative. | |
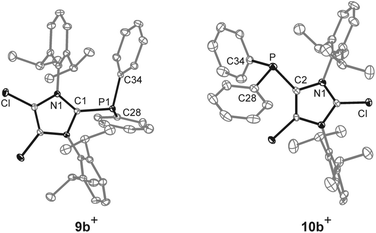 |
| | Fig. 1 Molecular structures of cations 9b+ and 10b+ of the respective triflate salts; hydrogen atoms, solvate molecules and anions are omitted for clarity and thermal ellipsoids are displayed at 50% probability; selected bond lengths (Å) and angles (°) for 9b+: P–C1 1.8437(9), P–C28 1.8205(10), P–C34 1.8249(10); C1–P–C28 107.15(4), C1–P–C34 100.59(4), C28–P–C34 101.71(4) and 10b+: P–C2 1.831(4), P–C28 1.827(4), P–C34 1.830(4); C2–P–C28 100.22(16), C2–P–C34 103.41(16), C28–P–C34 103.52(17). | |
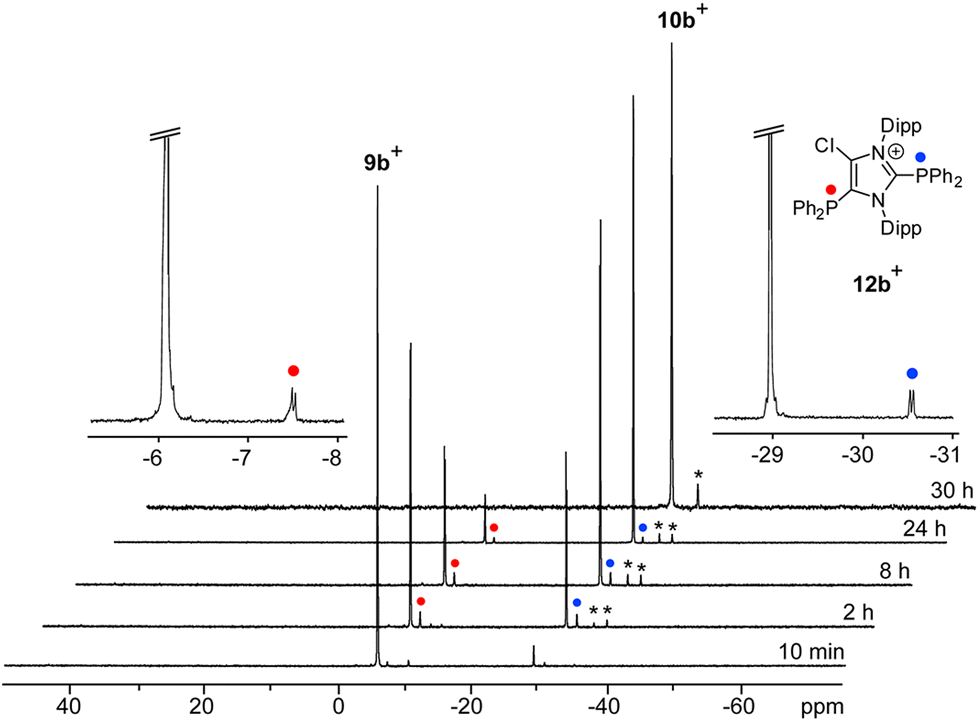
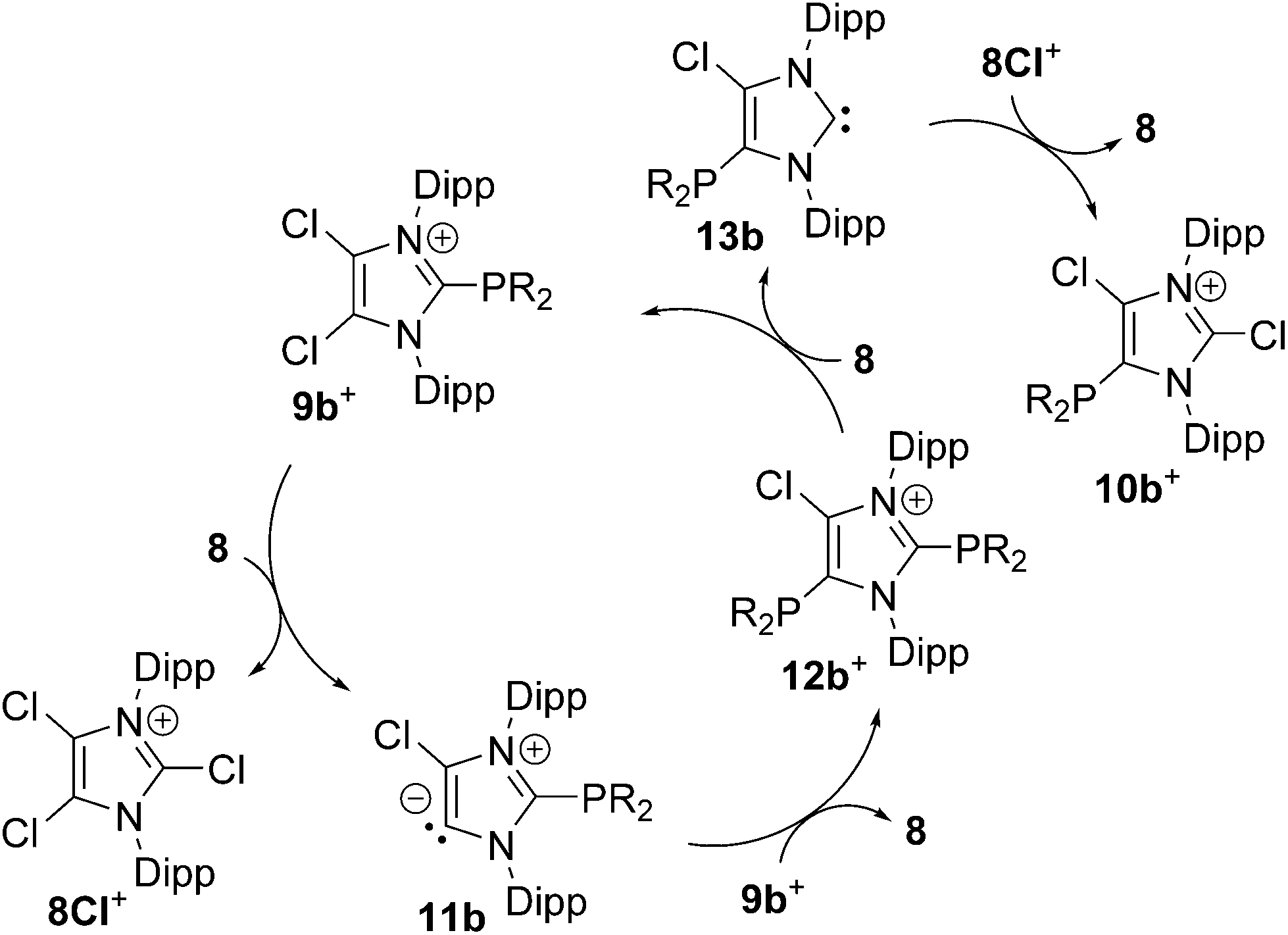
Increasing the amount of NHC 8 to 1.2 equivalents in the reaction leads to the quantitative formation of 5-phosphanyl-substituted imidazolium salts 10a,b[OTf] (10a[OTf]: 92%; 10b[OTf]: 97%). The molecular structure of cation 10b+ is depicted in Fig. 1. The P–C bond lengths in this cation (P–C2 1.831(4) Å, P–C28 1.827(4) Å, P–C34 1.830(4) Å) are in the same range as those observed for the structural isomer 9b+ and compound 4 (P–C2 1.8124(12) Å, P–CPh1 1.8318(14) Å, P–CPh2 1.8388(13) Å). Compared to 9a,b[OTf], a pronounced high field shift is observed in the 31P NMR spectra for the 5-substituted derivatives 10a,b[OTf] (10a+: δ(P) = −15.2 ppm; 10b+: δ(P) = −28.9 ppm). This can be explained by the stronger σ-donor abilities of the carbon atom in the 5-position compared to the 2-position,15–18 and the weaker electron withdrawing effect of only one N atom in β-position to the P atom. The 1H NMR spectra of 10a,b[OTf] show the expected additional sets of resonances for the isopropyl-groups of the Dipp-substituents due to a lower symmetry compared to 9a,b[OTf] (see Fig. S2.1†). Compounds 9a,b[OTf] are cleanly converted to the 5-substituted derivatives 10a,b[OTf] within 30 h when adding 0.3 equivalents of NHC 8 to the dissolved material in C6H4F2. The time dependent 31P NMR spectra also confirm our suggestion that 9a,b+ is first formed and slowly converted into 10a,b+ during the reaction (Fig. 2). After 2 h, significant amounts of 9b+ are consumed along with the formation of the structural isomer 10b+ (9b+: δ(P) = −5.1 ppm and 10b+: δ(P) = −28.9 ppm). The presence of two doublet resonances at δ(P) = −7.5 ppm and 30.7 ppm, (marked with a red and blue dot) with low intensity and a coupling constant of 4JPP = 7 Hz indicates the formation of intermediately formed cation 12b+ (Fig. 2). We thus propose for the rearrangement reaction for the first step a chlorenium ion abstraction from cation 9b+ by NHC 8 to give chloroimidazolium salt 8Cl+ and mesoionic NHC 11b (Scheme 3). The latter reacts with 9b+via phosphanyl-abstraction to NHC 8 and cation 12b+ whose formation is observed in the time dependent 31P NMR spectra (Fig. 2). NHC 8 abstracts a PR2-moiety from cation 12b+ to liberate cation 9b+ and intermediately formed NHC 13b. The catalytic cycle is closed by the chlorenium ion abstraction from 8Cl+ by 13b to yield 5-phosphanyl-substituted imidazolium cation 10b+ and catalyst NHC 8. This catalytic cycle also explains the formation of cation 10a+ and is related to a mechanism we proposed recently elsewhere.5
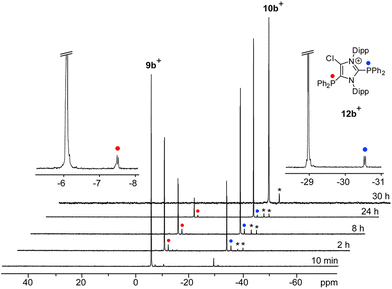 |
| | Fig. 2 Time-dependent 31P NMR spectra for the reaction of 9b[OTf] with 0.3 eq. of NHC 8 (o-C6H4F2, C6D6-capillary, 300 K). The intermediately formed cation 12b+ is indicated by red and blue dots, not identified intermediates by asterisks. | |
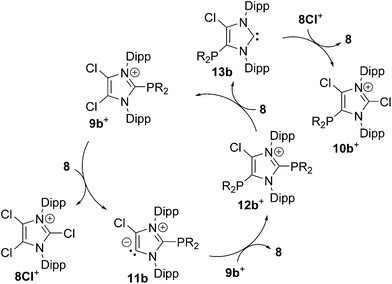 |
| | Scheme 3 Proposed mechanism for the formation of cations 10b+; anions are omitted for clarity. | |
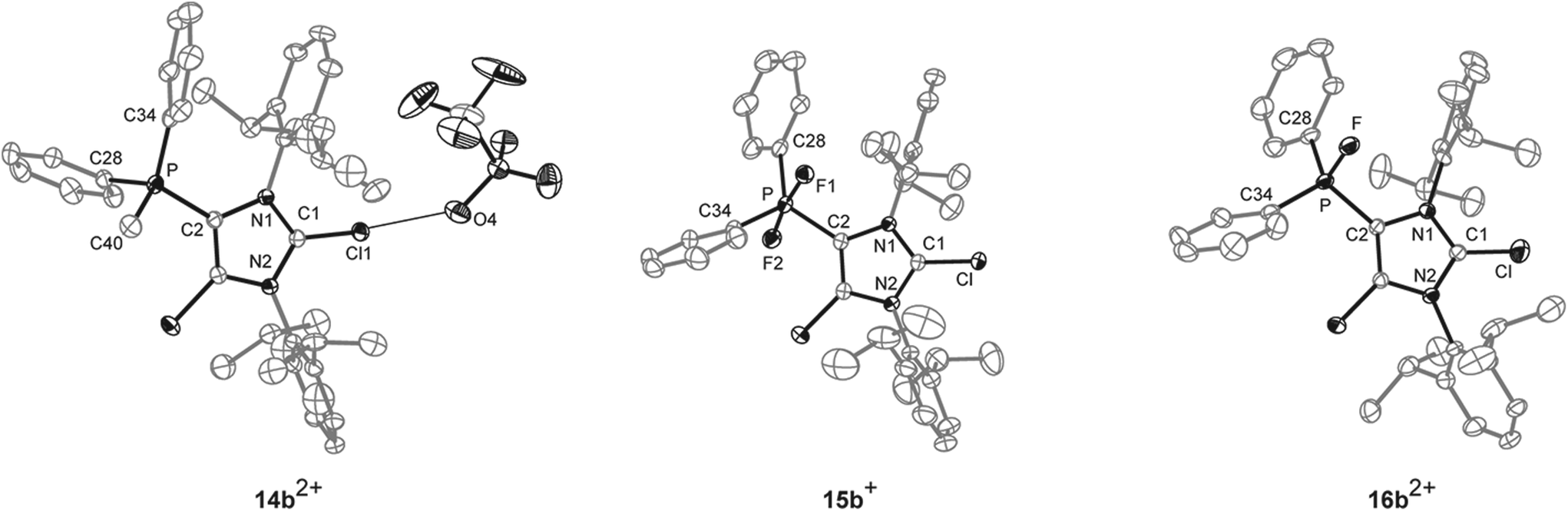
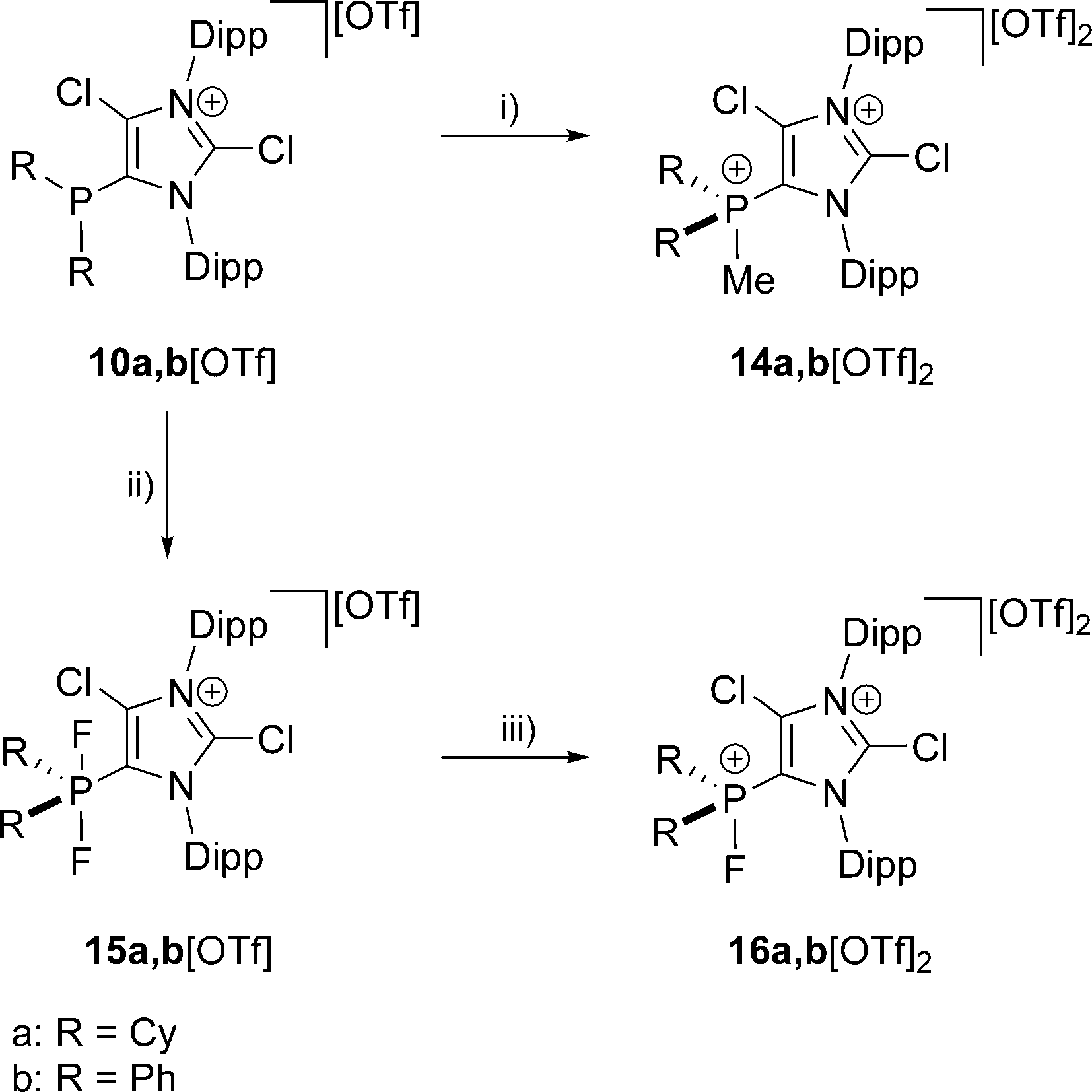
The reaction of 10a,b[OTf] with methyltriflate (MeOTf) in CH2Cl2 leads to the quantitative formation of the corresponding 5-phosphonium ions 14a,b2+ as triflate salts (isolated yield 14a[OTf]2: 68%; 14b[OTf]2: 79%; Scheme 4). This is indicated by a pronounced downfield shifted quartet resonance (14a2+: δ(P) = 38.8 ppm, 2JPH = 12 Hz; 14b2+: δ(P) = 16.9 ppm, 2JPH = 14 Hz) in the respective 31P NMR spectrum. Single crystals of 14b+ suitable for X-ray crystallography are obtained by slow diffusion of n-hexane into a saturated CH2Cl2 solution of 14b[OTf]2 (Fig. 3). The molecular structure of cation 14b2+ shows the expected tetra-coordinate bonding environment at the P atom and slightly shortened P–C bond distances (P–C2 1.810(2) Å, P–C28 1.782(2) Å, P–C34 1.787(2) Å, P–C40 1.790(2) Å) compared to cation 10b+. Selected geometrical parameters are given in Table 1.
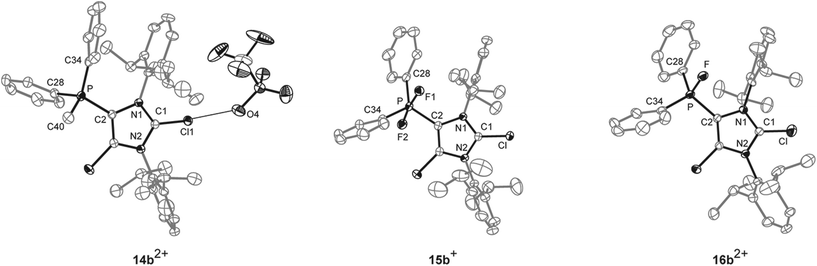 |
| | Fig. 3 Molecular structures of 14b2+, 15b+, and 16b2+ of the respective triflate salts; hydrogen atoms, solvent molecules and anions are omitted for clarity and thermal ellipsoids are displayed at 50% probability. Selected geometrical parameters are given in Tables 1 and 2. | |
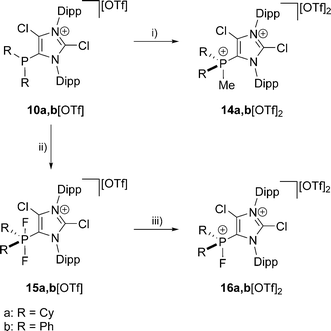 |
| | Scheme 4 Preparation of: 14a,b[OTf]2, (i): + MeOTf, C6H5F, r.t., 6 h, 14a[OTf]2: 68%; 14b[OTf]2: 79%; 15a,b[OTf], (ii): +XeF2, CH2Cl2, r.t. 2 h, −Xe, 15a[OTf]: 81%; 15b[OTf]: 89%; 16a,b[OTf]2, (iii): +Me3SiOTf, o-C6H4F2, r.t. 5 h, −Me3SiF, 16a[OTf]2: 72%; 16b[OTf]2: 71%. | |
Table 1 Selected geometrical parameters of crystallographically characterized cations 14b2+, 17b+, 19+, 20+, 223+ and 23+
| |
14b
2+
|
17b
+
|
19
+
|
20
+
|
22
3+c |
23
+
|
|
M = transition metal.
X = halogen atom.
Average bond lengths and angles are given.
|
| P–C2 in Å |
1.810(2) |
1.779(2) |
1.794(3) |
1.788(2) |
1.792 |
1.785(2) |
| P–C28 in Å |
1.782(2) |
1.785(5) |
1.780(3) |
1.783(2) |
1.787 |
1.795(2) |
| P–C34 in Å |
1.787(2) |
1.790(2) |
1.772(3) |
1.782(2) |
1.80 |
1.792(2) |
| P–C40 in Å |
1.790(2) |
1.785(2) |
1.782(3) |
1.787(3) |
1.790 |
1.786(2) |
| C1–N1 in Å |
1.335(2) |
1.375(3) |
1.350(4) |
1.346(3) |
1.359 |
1.360(3) |
| C1–N2 in Å |
1.337(2) |
1.362(3) |
1.354(4) |
1.365(3) |
1.358 |
1.380(3) |
| C1–Ma in Å |
— |
— |
1.981(3) (M = Au) |
1.884(2) (M = Cu) |
2.139 (M = Ag) |
2.039(2) (M = Rh) |
| N1–C1–N2 in (°) |
110.42(14) |
102.40(16) |
106.2(3) |
104.81(19) |
104.35 |
104.78(19) |
| C1–M–Xb in (°) |
— |
— |
176.75(10) (X = Cl) |
174.88(7) (X = Br) |
178.27(15) (X = C) |
86.85(6) (X = Cl) |
| Cl1⋯Otriflate in Å |
2.813(2) |
— |
— |
— |
— |
— |
One of the oxygen atoms of one triflate anion shows a close contact to the Cl1 atom that is well within the sum of the van der Waals radii (O4⋯Cl1 2.813(2) Å; rA(O) + rA(Cl) = 3.21 Å).19 Also the almost linear C1–Cl1–O4 angle of 170.879(6)° is indicative for a strongly directional rather than purely electrostatic interaction, namely a halogen bonding.20 This effect might be attributed to a slightly increased electrophilic character of the Cl1 atom caused by the high group electronegativity of dication 14b2+ (vide infra).20–22
The quantitative oxidation of 10a,b[OTf] via the slow addition of a solution of XeF2 in CH2Cl2 affords the difluorophosphoranes 15a,b[OTf] (isolated yield >80%; Scheme 4). Analytically pure compounds are obtained via precipitation with n-hexane. For both compounds the 31P NMR spectra show the expected triplet resonance with a typical 1JPF coupling due to the presence of two chemically equivalent fluorine atoms (15a+: δ(P) = −28.6 ppm, 1JPF = 722 Hz; 15b+: δ(P) = –64.9 ppm, 1JPF = 715 Hz). Single crystals suitable for structure investigation are obtained by slow diffusion of n-hexane into a saturated CH2Cl2 solution of 15b[OTf]. Cation 15b+ shows a trigonal bipyramidal bonding environment at the P atom with the F atoms being in the axial positions displaying an almost linear F–P–F angle of 172.91(10)° and typical P–F bond lengths (P–F1 1.6523(19) Å, P–F2 1.6582(19) Å; Fig. 3).
In contrast to a related difluorophosphorane,23 which requires a very strong fluoride abstracting reagent such as Et3Si[B(C6F5)4]·2(C7H8), the reaction of 15a,b[OTf] with Me3SiOTf in CH2Cl2 quantitatively gives the fluorophosphonium salts 16a,b[OTf]. Analytically pure compounds are obtained after the addition of n-hexane (isolated yield >70%). The 31P NMR spectra of the isolated compounds display a pronounced downfield shifted doublet resonance (16a2+: δ(P) = 109.6 ppm, 1JPF = 988 Hz; 16b2+: δ(P) = 70.5 ppm, 1JPF = 995 Hz), coinciding with the electron deficiency at the P atom as observed for similar Lewis acidic fluorophosphonium derivatives.23,24 Accordingly, the much larger 1JPF coupling constant in 16a,b2+ compared to 15a,b+ results from a decreased electron density at the P atom.25 The formation of dication 16b2+ was also confirmed by X-ray crystallography (Fig. 3). The P–F bond (1.5612(19) Å) is substantially shortened compared to the P–F bond lengths in 15b+ (vide supra), which is in agreement with the change of hybridization from sp2 to sp3 at the P atom. The same trend is observed for the P–C bonds to the phenyl substituents in 16b2+ (P–C28 1.778(3) Å, P–C34 1.769(3) Å) compared to 15b+ (P–C28 1.808(3) Å, P–C34 1.804(3) Å). Similarly, the P–C2 bond length in 16b2+ (P–C2 1.782(3) Å) is significantly shortened compared to that in 15b+ (P–C2 1.826(3) Å). Selected geometrical parameters are given in Table 2.
Table 2 Selected geometrical parameters of crystallographically characterized cations 15b+, 16b2+ and 21+
| |
15b
+
|
16b
2+
|
21
+
|
| P–C2 in Å |
1.826(3) |
1.782(3) |
1.774(3) |
| P–C28 in Å |
1.808(3) |
1.778(3) |
1.754(3) |
| P–C34 in Å |
1.804(3) |
1.769(3) |
1.748(3) |
| P–F1 in Å |
1.6523(19) |
1.561(2) |
1.5422(17) |
| P–F2 in Å |
1.6582(19) |
— |
— |
| C1–N1 in Å |
1.332(4) |
1.320(3) |
1.350(3) |
| C1–N2 in Å |
1.334(4) |
1.335(3) |
1.366(3) |
| C1–Au in Å |
— |
— |
1.973(2) |
| N1–C1–N2 in (°) |
109.5(2) |
110.0(2) |
105.2(2) |
| C1–Au–Cl in (°) |
— |
— |
177.74(8) |
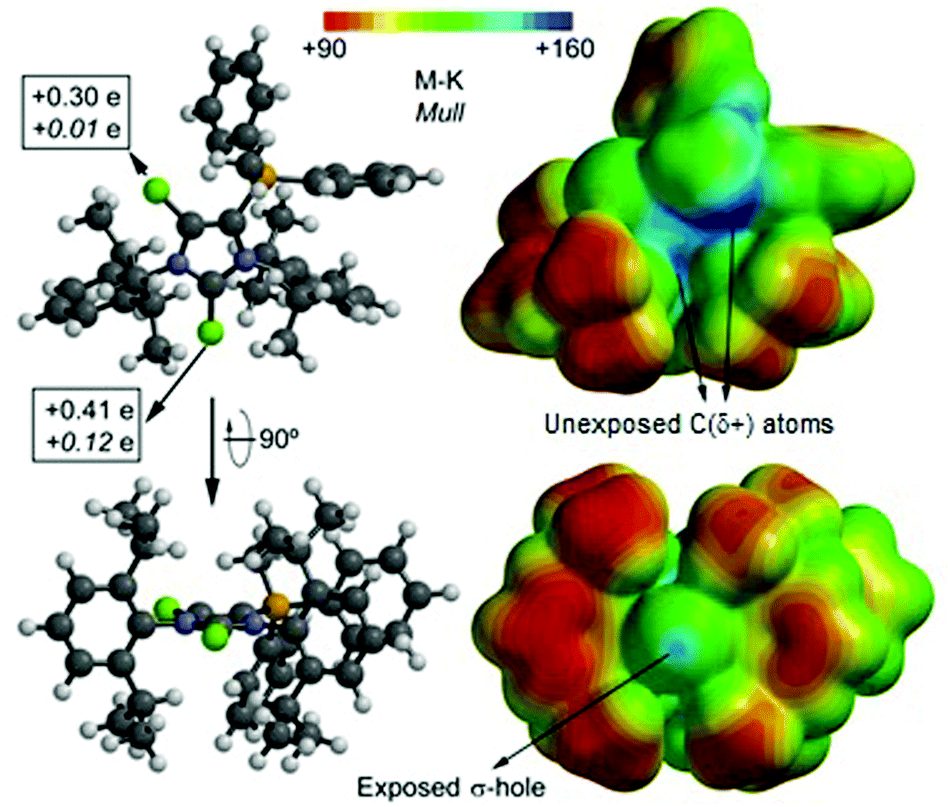
In order to shed light on the reactivity of dications 14a,b2+ and 16a,b2+ we analyzed the Merz–Kollman (M–K) and the Mulliken (Mull) charge distribution of 14b2+ which are shown in Fig. 4 (left). Moreover, the Molecular Electrostatic Potential (MEP) distribution of 14b2+ was calculated and plotted onto the van der Waals surface were regions of high charge are blue and regions with lower charge red (Fig. 4, right). For this study the geometries and energies were calculated applying the density functional theory (DFT) model BP8626 with the latest correction of dispersion (D3),27 together with the def2-TZVP basis set.28,29 Both methods confirm a significant positive charge of the Cl atoms (Cl1: +0.41e (M–K), +0.12e (Mull); Cl2: +0.30e (M–K), +0.01e (Mull)) which are bonded to the imidazolium ring, whereas the Cl1 atom, bonded to the 2-position, is more positive. The MEP surface illustrates that the imidazolium ring has the highest charge, however, it is not accessible for a nucleophilic attack due to steric effects. In sharp contrast, the Cl1 atom is very exposed and presents a σ-hole – a well defined positively charged region on the extension of the C1–Cl1 bond.20,30 Therefore, the most reactive site towards voluminous nucleophiles in dication 14b2+ is indeed the Cl1 atom bonded at the 2-position.
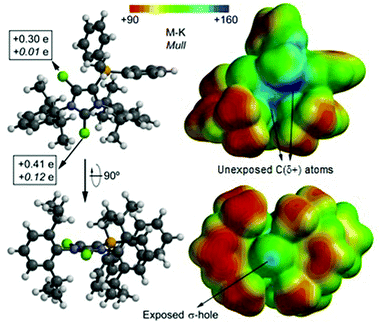 |
| | Fig. 4 Two orientations of cation 14b2+ calculated at BP86-D3/def2-TZVP level of theory; Merz–Kollman (M–K) and the Mulliken (Mull, italics) charge distribution (left); Molecular Electrostatic Potential (MEP) distribution plotted onto the van der Waals surface (right). | |
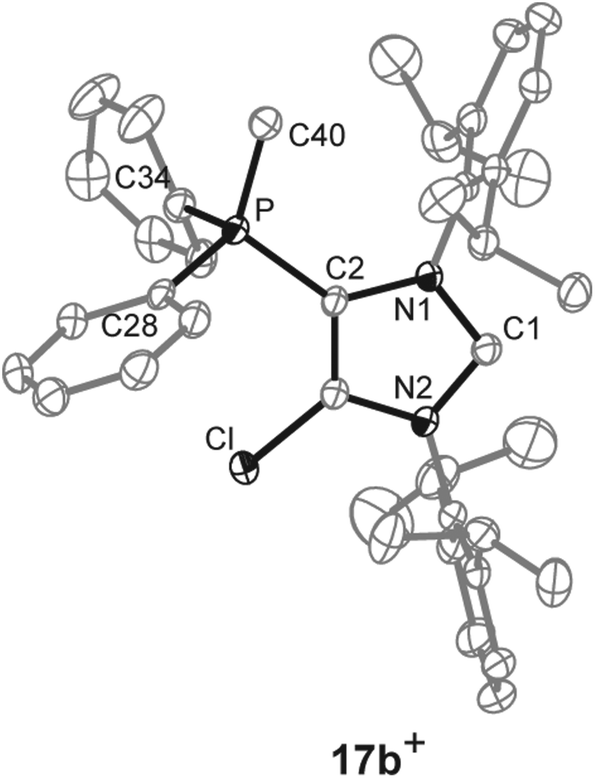
As for dications 14a,b2+ and 16a,b2+ we aimed at the synthesis of the corresponding cationic 5-phosphonio-substituted NHCs and proceeded to react dications 14a,b2+ and 16a,b2+ with Cy3P or PPh3, respectively (Scheme 5). The reaction of 14a,b[OTf]2 with Cy3P in C6H5F gives quantitatively Cy3PCl[OTf], of which the cation Cy3PCl+ was unambiguously identified by its 31P NMR chemical shift of δ(P) = 104.2 ppm (ref. 31) in the reaction mixture. The 31P NMR spectra of the reaction mixtures also show a second resonance which can be assigned to the cationic NHCs 17a,b+. The isolation of 17a[OTf] was hampered due to a similar solubility of 17a[OTf] and Cy3PCl[OTf]. Analytically pure 17b[OTf] was isolated as colorless powder in good yield (75%) via precipitation and washing with benzene. For cation 17b+ a slightly high field shifted singlet resonance in the 31P NMR spectrum (δ(P) = 10.6 ppm) compared to 14b2+ (δ(P) = 16.9 ppm) is observed. The 13C NMR spectrum displays a resonance at δ(C) = 230.2 ppm confirming the formation of an NHC.16,32 This signal, which is downfield shifted compared to NHC 8 (δ(C) = 219.4 ppm, in C6D6),12 splits into a doublet due to the coupling to the P atom in 5-position (3JCP = 6 Hz). As compound 17b[OTf] can be handled in solvents such as CH2Cl2 and CH3CN without decomposition the scope of application is significantly broadened. Single crystals suitable for X-ray crystallography are obtained by slow diffusion of n-hexane into a saturated C6H5F solution of 17b[OTf] (Fig. 5). Major differences in this cation compared to dication 14b2+ are the elongation of the C1–N1/N2 bond lengths (17b+: C1–N1 1.375(3) Å, C1–N2 1.362(3) Å vs.14b2+: C1–N1 1.335(2) Å, C1–N2 1.337(2) Å)) and the reduced bond angle N1–C1–N2 (17b+: 102.40(16)° vs.14b2+: N1–C1–N2 110.42(14)°). These effects are explained by a diminished π-delocalization16,32 in the N1–C1–N2 moiety and an increased p-orbital character of the C1–N1/N2 bonds. As a result of the diminished π-delocalization within the imidazole ring the P–C2 bond length in 17b+ (1.779(2) Å) is significantly shortened compared to that in 14b+ (P–C2 1.810(2) Å). Selected geometrical parameters are summarized in Table 1.
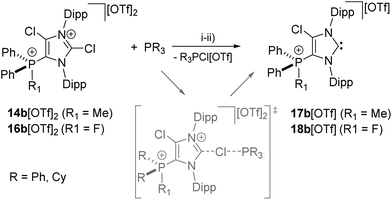 |
| | Scheme 5 Preparation of 17b[OTf], (i): +Cy3P, C6H5F, r.t., 2 h, −Cy3PCl[OTf], 75%; preparation of 18b[OTf], (ii): +Ph3P, C6H5F, r.t., 2 h, −Ph3PCl[OTf], 55%. Proposed transition state of a SN2(Cl) type reaction (grey). | |
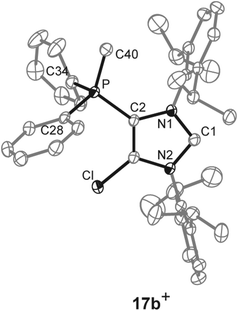 |
| | Fig. 5 Molecular structure of 17b+ of the triflate salt; hydrogen atom and anions are omitted for clarity and thermal ellipsoids are displayed at 50% probability. Selected geometrical parameters are given in Table 1. | |
Similarly, the reaction of 16a,b[OTf]2 with Ph3P leads to the quantitative formation of chlorophosphonium salt Ph3PCl[OTf] (δ(P) = 65.5 ppm)31 and NHCs 18a,b[OTf]. The isolation of NHC 18a[OTf] was hampered due to similar solubility of 18a[OTf] and PPh3Cl[OTf]. 18b[OTf] shows a slightly downfield shifted doublet resonance at δ(P) = 76.8 ppm (1JPF = 996 Hz) in the 31P NMR spectrum compared to the starting material 15[OTf]2 (δ(P) = 70.5 ppm). The confirmation of the carbene was given by the 13C{1H} NMR spectrum showing a resonance at δ(C) = 230.8 ppm,16,32 which splits into a pseudo triplet (3JCP = 5 Hz, 4JCF = 5 Hz) due to the coupling to the phosphorus atom and the fluorine atom, respectively. Mechanistically, the formation of NHCs 17a,b+ and 18a,b+ proceeds via a SN2(Cl) type reaction33 which is initiated by the nucleophilic attack of R3P (R = Ph, Cy) on the σ-hole of the Cl1 atom bonded to the C2-position. This suggestion is supported by a detailed computational study considering a SN2(Cl) type reaction. The calculations were performed using the high level ab initio method RI-MP234 with the def2-TZVP28 basis set to obtain reliable reaction barriers (Fig. 6, left).29 In initial calculations a minimalistic model was used considering PH3 as nucleophile which reacts with 14b2+ where the Dipp-substituents are replaced by H atoms. The reaction is initiated by the formation of a hypercoordinate complex I in which a P⋯Cl halogen bond20 with a comparatively short bond distance of 3.151 Å is observed. This intermediate was found to be 9.2 kcal mol−1 lower in energy than the corresponding starting materials and is well organized for the subsequent SN2(Cl) type reaction33 as it presents an ideal directionality for the nucleophilic attack. The transition state TS is only 9.4 kcal mol−1 higher in energy than the intermediate I, thus confirming that the formation of the carbene is energetically feasible. The resulting adduct P is 6 kcal mol−1 lower in energy than the intermediate I and in total 15.2 kcal mol−1 more stable than the starting materials. These findings were then transferred to the complete model with dicationic 14b2+ and PPh3 as nucleophile using the density functional theory methods BP86-D3/def2-TZVP26–29 (Fig. 6, right). The hypercoordinate complex I′ with a P⋯Cl bond distance of 2.702 Å and the product P′ were computed. The obtained energy difference between I′ and P′ with a value of −9 kcal mol−1 is in good agreement with the high level ab initio MP2/def2-TZVP calculation of the minimalistic model.
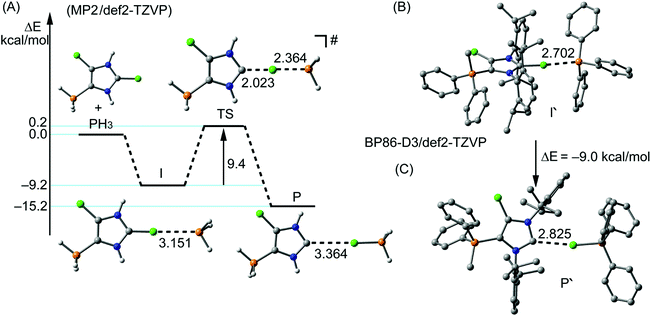 |
| | Fig. 6 MP2/def2-TZVP reaction coordinate, energies and geometries of the SN2(Cl) type reaction (left, A); DFT optimized geometries of the intermediate I and products P of the experimental system (right, B and C; distances are given in Å). | |
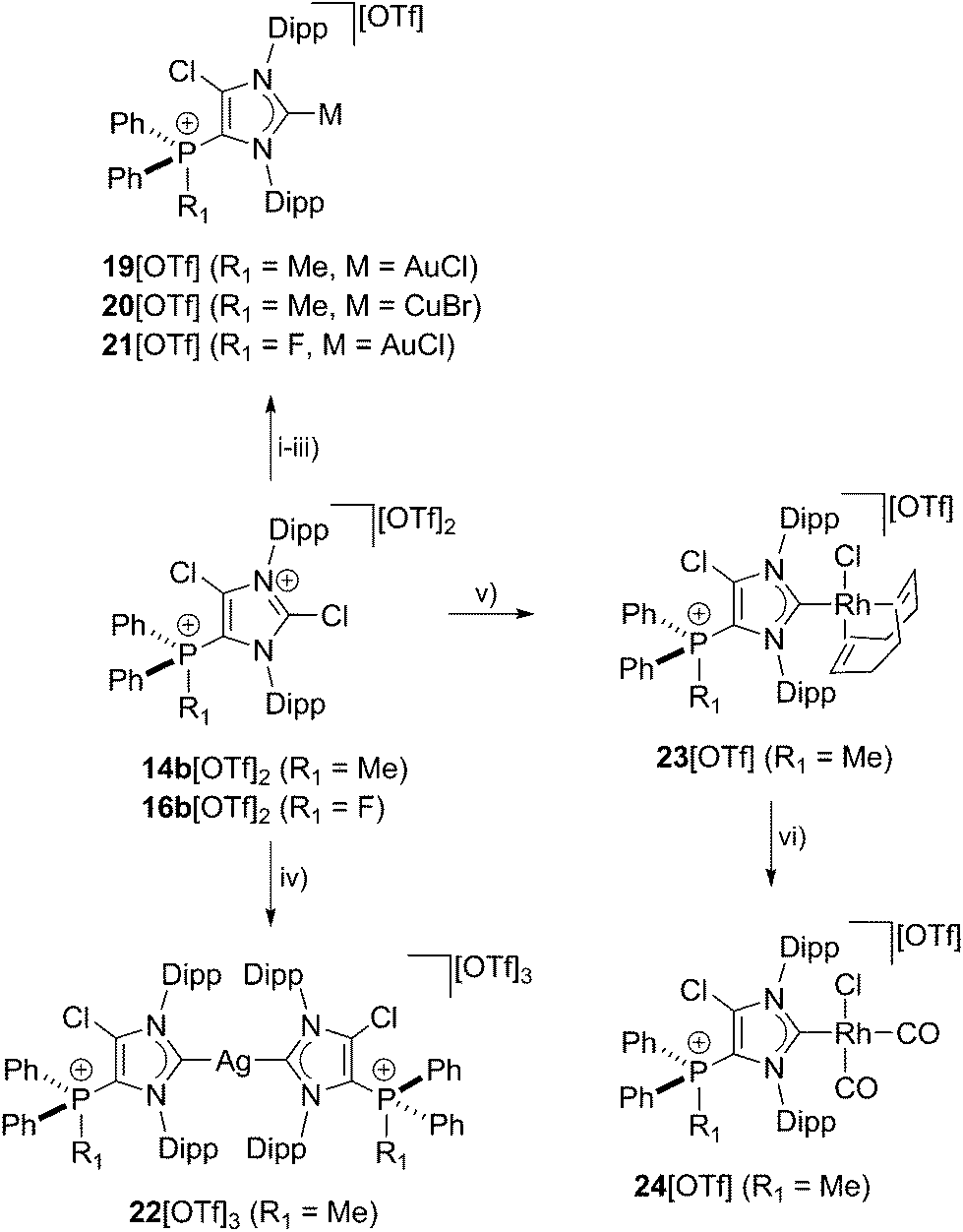
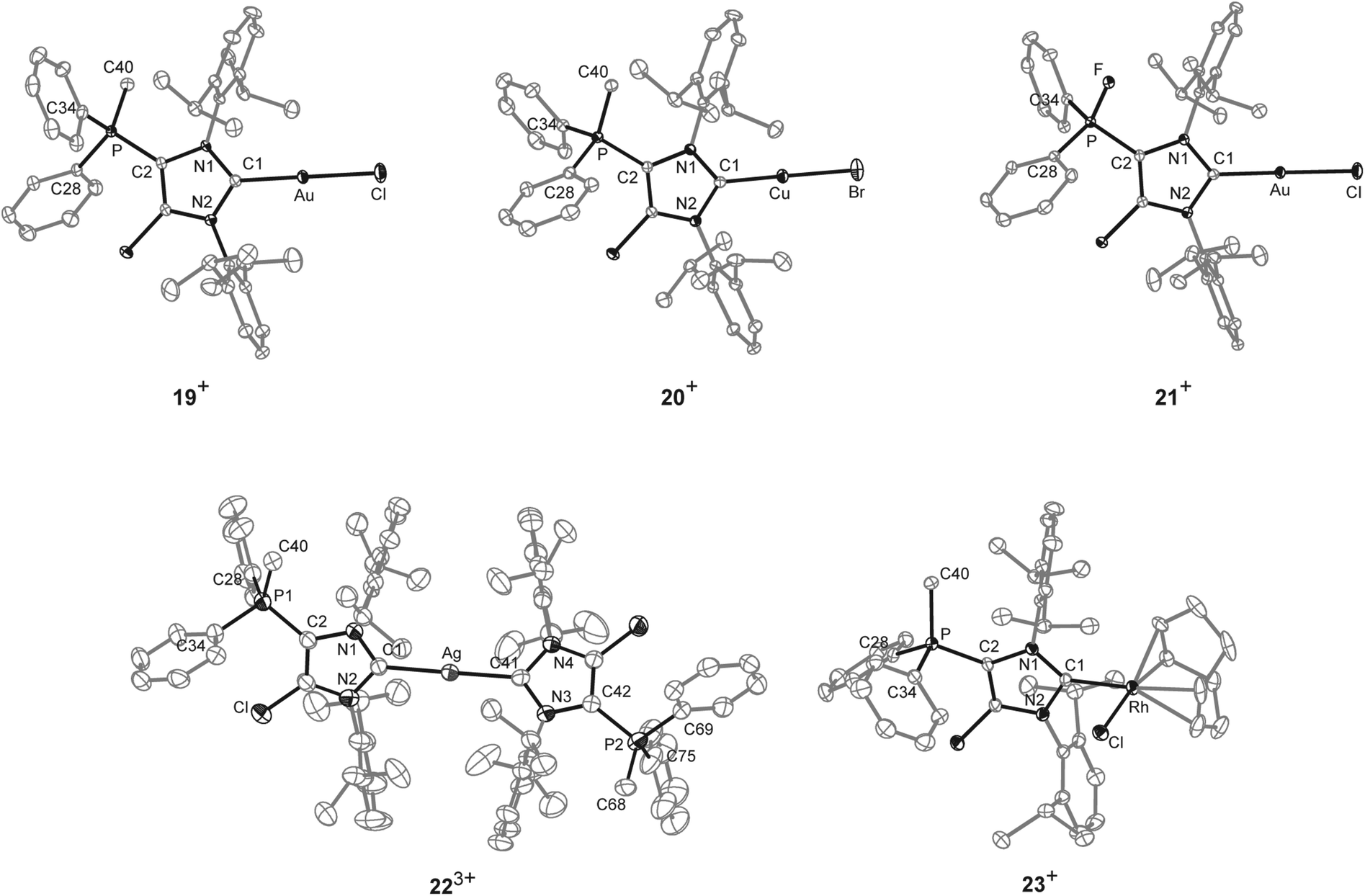
As NHCs are extensively used in coordination chemistry of transition metals, the formation of a series of complexes was investigated. The isolation of transition metal complexes 19[OTf], 20[OTf], 21[OTf] and 23[OTf] succeeds via the in situ reaction of compounds 14b[OTf]2 or 16b[OTf]2 with R3P (R = Cy, for 19[OTf], 20[OTf], and 23[OTf]; R = Ph, for 21[OTf]) in the presence of one equivalent of the corresponding transition metal salts at ambient temperature (Scheme 6; AuCl(tht) for 19[OTf] and 21[OTf], CuBr(tht) for 20[OTf], [RhCl(cod)]2 for 23[OTf]) in THF. The formation of the complexes is indicated by the 13C{1H} NMR spectra of the respective compounds showing resonances at δ(C) = 183.0 ppm for 19[OTf], δ(C) = 186.5 ppm for 21[OTf], δ(C) = 189.51 ppm for 20[OTf] and δ(C) = 206.03 ppm for 23[OTf].16 The reaction of 14b[OTf]2 with Cy3P and one eq. AgOTf in THF leads to the formation of 22[OTf]3 which is a rare example of a tricationic bis-carbene silver complex.35 The investigation of the NMR spectra of isolated 22[OTf]3 show only one set of resonances (e.g. δ(C) = 186.5 ppm, d, 1JCAg107 = 202 Hz, 1JCAg109 = 234 Hz; δ(P) = 14.1 ppm), indicating a symmetric arrangement of the two ligands around the Ag atom. Single crystals suitable for X-ray crystallography of the triflate salt of 223+ are obtained by slow diffusion of Et2O into a saturated CH3CN solution (Fig. 7). The Ag atom is in an almost linear arrangement between the two carbenes (C1–Ag–C41 178.27(15)°) but the NHC ligands are slightly twisted as illustrated by the torsion angle of N1–C1–C41–N3 34.380°. Molecular structures of the transition metal complexes 19+, 20+, 21+, 223+ and 23+ are depicted in Fig. 7 and selected geometrical parameters are given in Tables 1 or 2. As expected, the average N1–C1–N2 bond angles of cationic NHC complexes (N1–C1–N2: range of 103.2(3)°–106.2(3)°) are wider than the corresponding bond angle in free cationic NHC 17b+ (N1–C1–N2 102.40(16)°).16,35 Accordingly, the N1–C1/N2–C1 bond lengths in complexes 19+, 20+, 21+, 223+, 23+ (N1–C1/N2–C1: range of 1.346(3) Å–1.366(3) Å) are slightly shortened compared to those in 17b+ (av. N1–C1/N2–C1 1.369 Å), which is also in line with the coordination to transition metals.16 The C1–metal bond lengths in cationic complexes 19+, 20+, 21+, 223+, and 23+ are comparable to those observed in other neutral NHC metal complexes.35,36
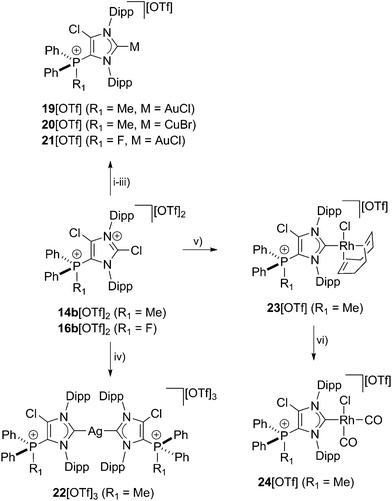 |
| | Scheme 6 Preparation of: 19[OTf], (i): +Cy3P, +AuCl(tht), THF, r.t., 2 h, −[Cy3PCl][OTf], −tht, 88%; 20[OTf], (ii): +Cy3P, +CuBr(tht), THF, r.t., 2 h, −[Cy3PCl][OTf], −tht, 93%; 21[OTf], (iii): +Ph3P, +AuCl(tht), THF, 2 h, −[Ph3PCl][OTf], −tht, 87%; 22[OTf]3, (iv): +Cy3P, +AgOTf, THF, r.t., 4 h, −[Cy3PCl][OTf], 82%; 23[OTf], (v): +Cy3P, +RhCl(cod)2, THF, r.t., [Cy3PCl][OTf], −cod, 88%; 24[OTf], (vi): +2CO, CH2Cl2, r.t., 1.5 h, −cod, 86%. | |
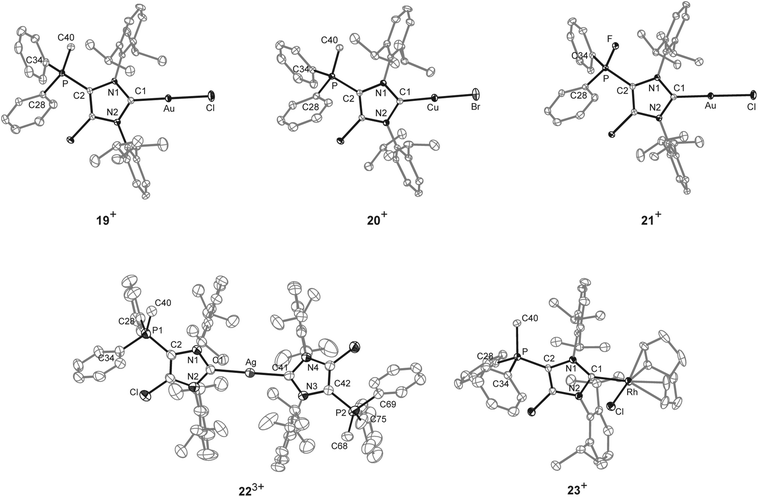 |
| | Fig. 7 Molecular structures of 19+, 20+, 21+, 223+ and 23+ of the respective triflate salts; hydrogen atoms, solvent molecules and anions are omitted for clarity and thermal ellipsoids are displayed at 50% probability. Selected geometrical parameters are given in Tables 1 and 2. | |
In order to elucidate the donor/acceptor properties of cationic NHC 17b[OTf], we synthesized the cis-chlorodicarbonylrhodium complex 24[OTf] via the reaction of carbon monoxide with 23[OTf] (Scheme 6)29 and subsequently investigated the IR stretching frequencies for the carbonyl ligands of the formed carbonyl complex (see Fig. S2.10†). The average CO stretching frequency (νav(CO) = 2040.3 cm−1, TEP = 2051 cm −1)18 indicate a weak donor ability of cationic NHC 17b[OTf]. According to a recent work by Ganter and co-workers, the 1JCH coupling constant in imidazolium salts correlates with the σ-donor strength of the corresponding carbene.37 It was found that poor σ-donors reveal high coupling constants for the C–H bond, which can be explained by a higher s-orbital character of the corresponding C–H bond38 (compare [ImMes-H]+:391JCH = 206 Hz and [4,5-Cl-ImDipp-H]+: 1JCH = 229 Hz).37 For imidazolium salt 17bH2+ (17b+ protonated at C1), we observed a 1JCH coupling constant of 233 Hz (see Fig. S2.2†)29 rendering cationic NHC 17b+ a weak σ-donor. We thus conclude that our cationic NHCs are very electron deficient (weak σ-donor), which is promising for interesting follow up chemistry (vide infra).
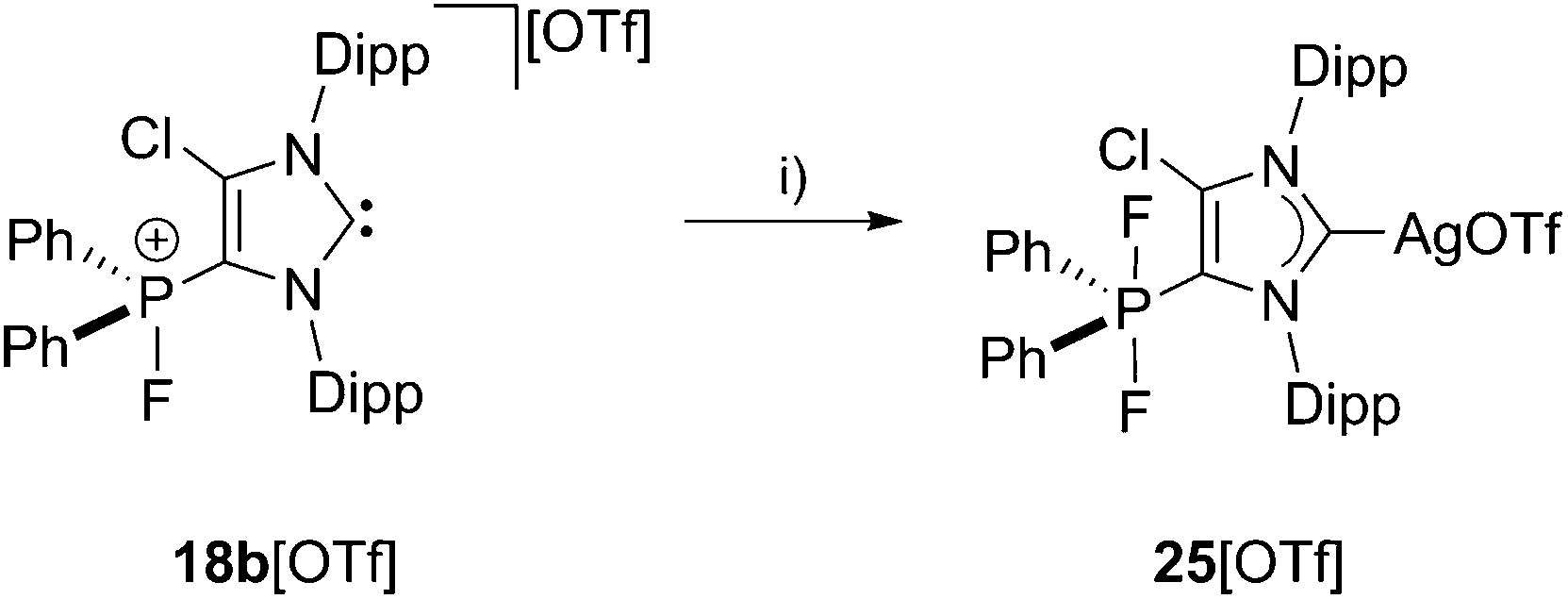
The ambiphilic nature of 18b+ is displayed by the reaction of 18b[OTf] with silver fluoride (AgF) in a 1 to 1 stoichiometry at ambient temperature in C6H5F yielding complex 25[OTf] (Scheme 7). The 31P NMR spectrum of 25[OTf] shows a triplet resonance at δ(P) = −65.3 ppm (1JPF = 701 Hz) indicating a clean formation of cation 25+ (see Fig. S2.3†). Additionally, the 13C{1H} NMR spectrum shows two doublets at δ(C) = 184.3 ppm (1JCAg109 = 232 Hz, 1JCAg107 = 200 Hz; see Fig. S2.4†), indicative for the coordination of carbene to the silver atom.35
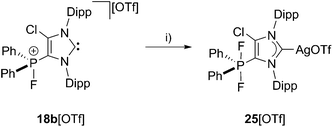 |
| | Scheme 7 Reaction of 18b[OTf] with AgF: (i) C6H5F, r.t., 10 h.29 | |
25[OTf] is extremely sensitive and decomposes readily in solution which prevents its isolation. In 2010 Bertrand and co-workers reported on the activation of the primary phosphane PhPH2 using NHC 2639 and isolated the insertion product of the oxidative addition 27 in high yield (Scheme 8I).39 Only recently, Radius and co-workers reported on the dehydrogenative coupling of primary and secondary phosphanes utilizing the more electron deficient NHC 28 (Scheme 8II).40 For the 1 to 2 reaction of 28 with Ph2PH an oxidative addition of the phosphane into the carbene moiety followed by a reductive elimination of the corresponding diphosphane and the 2,3-dihydro-1H-imidazole 29 was assumed.40
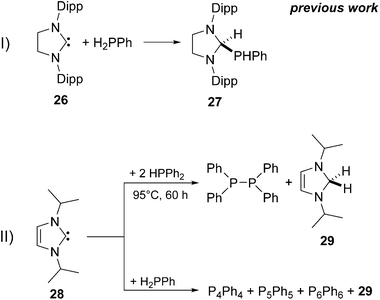 |
| | Scheme 8 Reaction of 26 with H2PPh (I) and dehydrocoupling reactions of HPPh2 or H2PPh with 28 (II). | |
Similarly, the reaction of prim. and sec. phosphanes with the electron deficient cationic NHC 17b+ was performed. The reaction of two equivalents Ph2PH with 17b+ in 1,2-dichloroethane (DCE) at ambient temperature affords the quantitative formation of diphosphane Ph4P2 after 12 h (Scheme 9, Fig. S2.5†). The 31P NMR spectrum of the reaction mixture shows a singlet resonance at δ(P) = −16.3 ppm which can be attributed to Ph4P240 and a singlet at δ(P) = 12.1 ppm, corresponding to the byproduct which is formed during the reaction. To our surprise, in depth NMR spectroscopic studies reveal dication 302+ as byproduct of this reaction and not the corresponding 2,3-dihydro-1H-imidazole. It was reported that aryl substituents at the phosphane are beneficial for the NHC mediated40,41 or transition metal catalysed42 dehydrocoupling reaction, since electron rich alkyl phosphanes gave either no, or a non-selective conversion.
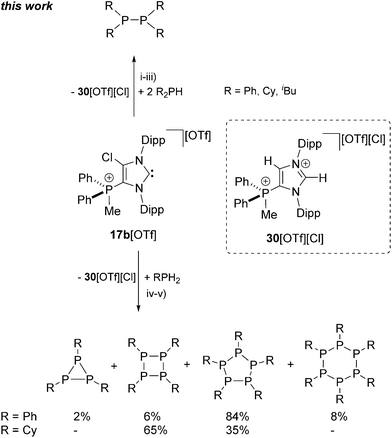 |
| | Scheme 9 P–P coupling reactions of R2PH (R = Ph, Cy, iBu; top) and PhPH2 (bottom) using 17b[OTf] as hydrogen acceptor; (i) +2 eq. Ph2PH, DCE, r.t., 12 h; (ii) +2 eq. Cy2PH, CH3CN, r.t., 12 h; (iii) +2 eq. iBu2PH, CH3CN, r.t., 10 h; (iv) +PhPH2, r.t., 10 h, product distribution estimated by integration of the 31P NMR spectrum of the reaction mixture; (v) +CyPH2, r.t., 24 h, product distribution estimated by integration of the 31P NMR spectrum of the isolated solid. | |
When alkyl phosphanes R2PH (R = Cy, iBu) are reacted with 17b[OTf] in a 2 to 1 stoichiometry in CH3CN at ambient temperature, a clean formation of the corresponding diphosphane R4P2 (R = Cy, iBu) is observed after 10 h reaction time. The reaction of 2 eq. Cy2PH with 17b[OTf] in CH3CN leads to the formation of a colorless precipitate. The 31P NMR spectrum of the isolated precipitate reveals a singlet resonance at δ(P) = −21.3 ppm illustrating a clean and quantitative formation of diphosphane Cy4P2 (see Fig. S2.6†). The 31P NMR spectrum for the reaction of 2 eq. iBu2PH with 17b[OTf] shows a singlet at δ(P) = 13.5 ppm for 302+ and a singlet resonance at δ(P) = −52.4 ppm which is attributed to iBu4P2. Small amounts of unreacted iBu2PH give rise to a doublet resonance at δ(P) = −83.7 ppm due to a slight excess of the inserted phosphane in the reaction (see Fig. S2.7†). No conversion is observed when 2 eq. of the more sterically encumbered tBu2PH is reacted with 17b[OTf], leading to the assumption that the dehydrogenative coupling reaction maybe limited by the steric demand but not necessarily by electronic effects of the corresponding substituents on the phosphane.
The equimolar reaction of cationic NHC 17b+ with primary phosphane PhPH2 in 1,2-dichloroethane at ambient temperature led to the formation of cyclo-phosphanes Ph3P3, Ph4P4, Ph5P5 and Ph6P6 (>90%) along with small amounts of unidentified side products and imidazolium dication 302+. The proton decoupled 31P NMR spectrum of the reaction mixture revealed an approximate product distribution of the cyclo-phosphanes of which Ph5P5 (ca. 84%) is found to be the main product (see Fig. S2.8†). Reacting the alkyl phosphane CyPH2 with 17b[OTf] in a 1:1 stoichiometry in CH3CN afforded the quantitative precipitation of Cy4P4 and Cy5P5 after 24 h. The 31P NMR spectrum of the isolated solid shows two singlet resonances which are assigned to Cy4P4 (δ(P) = −68.8 ppm,43 65%) and Cy5P5 (δ(P) = 7.7 ppm,44 35%; see Fig. S2.9†), illustrating that NHC 17b+ is also suitable for the dehydrocoupling of prim. phosphanes to cyclo-phosphanes.
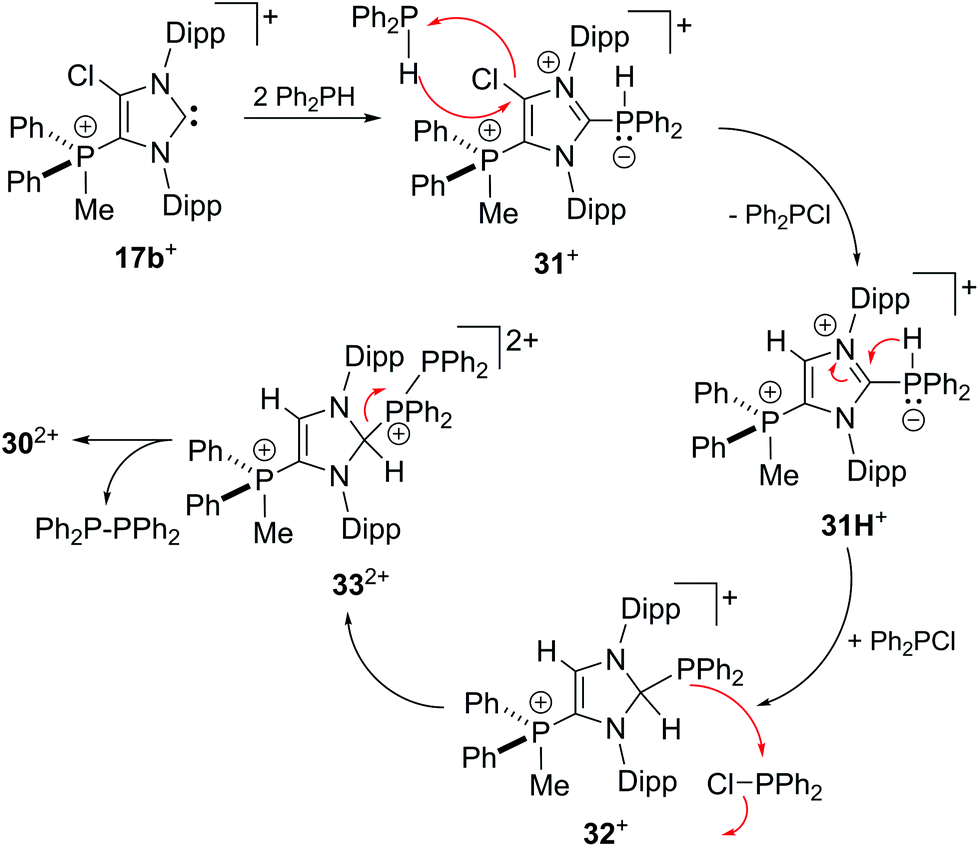
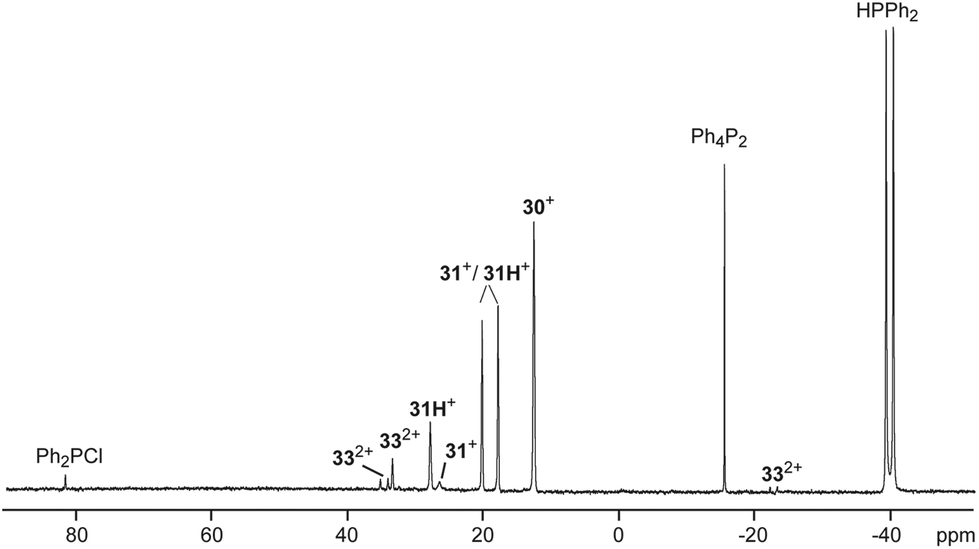
To support a possible reaction mechanism (Scheme 10), 5 eq. of Ph2PH and 17b[OTf] were mixed together in DCE and the reaction mixture was investigated by means of 31P NMR spectroscopy (Fig. 8). We assume that the first step of this reaction involves the nucleophilic attack of cation 17b+ towards Ph2PH to give the hyper-coordinate intermediate 31+. Related phosporanides were recently reported.1g,5 This intermediate reacts with a second eq. of Ph2PH to 31H+, accompanied by the formation of Ph2PCl (δ(P) = 81.8 ppm).13 Cation 31H+ shows a doublet resonance due to proton coupling at δ(P) = 18.9 ppm (1JPH = 477 Hz) and a singlet at δ(P) = 27.6 ppm for the tetra-coordinate phosphorus atom. Small amounts of cation 31+ are present in the spectrum as indicated by the observation of a singlet of low intensity at δ(P) = 26.4 ppm. The corresponding doublet for the penta-coordinate P-atom coincides with that of 31H+. Intermediate 31H+ rearranges to 2,3-dihydro-1H-imidazole 32+ in accordance to the work by Bertrand, Röschenthaler and Radius.1g,39,40 This cation readily reacts with the liberated Ph2PCl to dication 332+ which is indicated by the observation of two doublets at δ(P) = −23.4 ppm and δ(P) = 34.6 (1JPP = –227 Hz) and a singlet resonance at δ(P) = 33.3 ppm for the phosphonium moiety in the backbone. In the last step, cation 332+ liberates Ph4P2 accompanied by the formation of dication 302+.
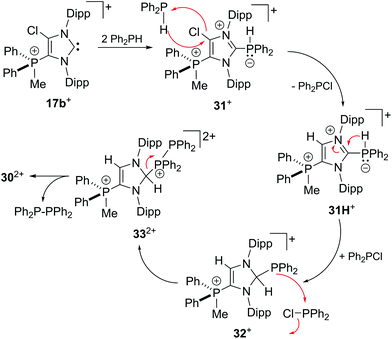 |
| | Scheme 10 Possible reaction mechanism for the P–P coupling reaction of 2 eq. R2PH with 17b+ to R2P–PR2. | |
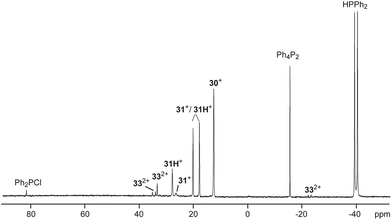 |
| | Fig. 8
31P NMR spectrum for the reaction of 17b[OTf] with 5 eq. Ph2PH after 8 h (1,2-dichloroethane, C6D6-capillary, 300 K). Resonances are assigned to possible intermediates for the mechanism of the P–P coupling reaction. | |
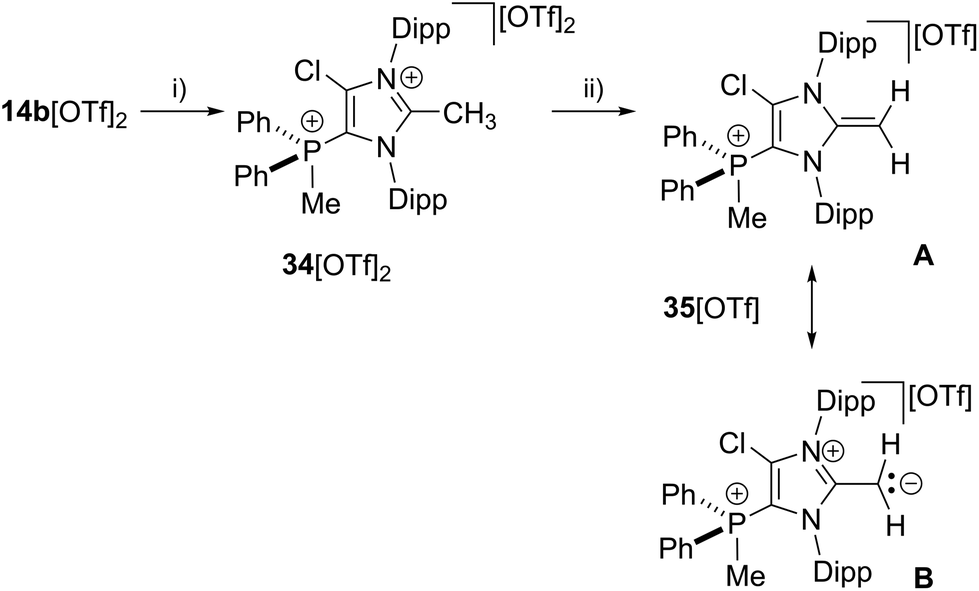
Further reactivity studies of 17b[OTf] were directed towards the synthesis of a cationic N-heterocyclic olefin (NHO).45,46 Therefore we methylated in a first step the in situ formed cation 17b+ obtained from 14b[OTf]2 and Cy3P with one equivalent of MeOTf to give dication 342+ (Scheme 11). Dication 342+ was isolated as triflate salt (isolated yield 91%) and investigated by means of multinuclear NMR spectroscopy and X-ray analysis (Fig. 9). The 1H NMR spectrum of dissolved 34[OTf]2 in CD2Cl2 reveals a doublet resonance at δ(H) = 2.91 ppm (2JHP = 13.8 Hz) for the CH3-group which is bonded to the phosphorus atom whereas the CH3-group at the 2-position of the imidazolium ring gives a singlet resonance at δ(H) = 2.38 ppm. The 31P NMR spectrum shows a quartet resonance at δ(P) = 15.6 ppm which is slightly upfield shifted compared to 14b2+ (δ(P) = 16.9 ppm). Derivative 34[OTf]2 is readily deprotonated in a subsequent step by lithium diisopropylamide (LDA) in THF to give 35[OTf] as yellow powder in excellent yield (93%). The 31P NMR spectrum shows an upfield shifted quartet resonance at δ(P) = 9.6 ppm compared to dication 342+ (δ(P) = 15.6 ppm).
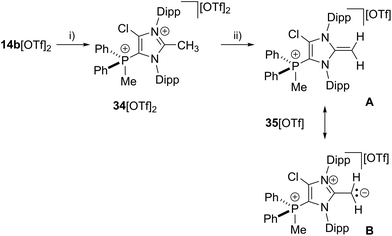 |
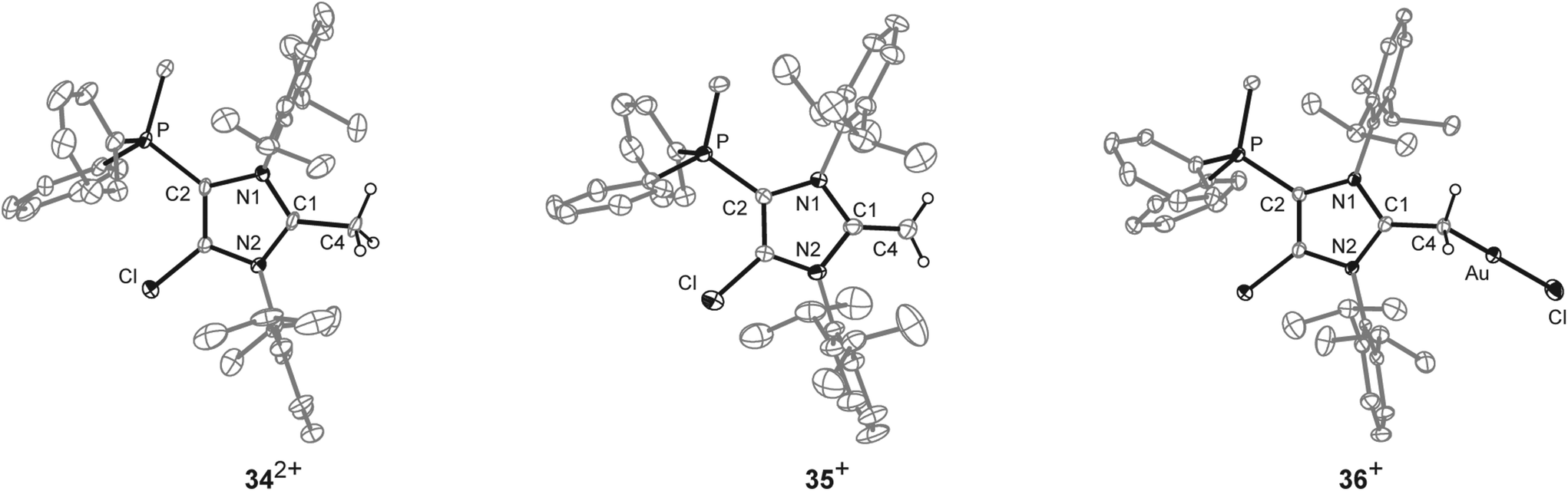
| | Scheme 11 Preparation of 34[OTf]2, (i) +Cy3P, MeOTf, C6H5F, r.t., 14 h, −Cy3PCl[OTf], 91%; and 35[OTf], (ii) +LDA THF, r.t., 30 min, −Li[OTf], −HDA (diisopropylamine), 93%; two representative resonance structures (A, B) of 35a[OTf] are displayed. | |
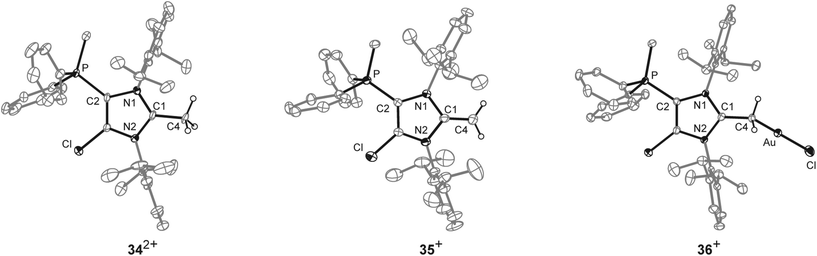 |
| | Fig. 9 Molecular structures of 342+, 35+, and 36+, of the respective triflate salts; hydrogen atoms, solvent molecules and anions are omitted for clarity and thermal ellipsoids are displayed at 50% probability; selected bond lengths (Å) and angles (°) for 342+: C1–C4 1.499(5), C1–N1 1.347(5), C1–N2 1.332(5); N1–C1–N2 109.141(9); 35+: C1–C4 1.334(4), C1–N1 1.404(3), C1–N2 1.339(3); N1–C1–N2 104.6(2); for 36+: C1–C4 1.453(4), C1–N1 1.344(4), C1–N2 1.356(4), C28–Au 2.062(3), Au–Cl 2.3080(8); N1–C1–N2 107.5(3), C1–C4–Au 116.2(2), C4–Au–Cl 174.90(9). | |
The doublet resonance for the CH3-group bound to the P atom is shifted to higher field (δ(H) = 1.83 ppm; 2JHP = 13.6 Hz) compared to the corresponding CH3-group in 342+ (δ(H) = 2.91 ppm; 2JHP = 13.8 Hz). The CH2-group at the 2-position of the imidazole ring gives rise to a doublet resonance at δ(H) = 2.54 ppm and a doublet of doublet resonance at δ(H) = 2.61 ppm due to the hindered rotation around the C1–CH2 bond. Both signals exhibit a geminal coupling constant of 2JHH = 3.7 Hz and the latter resonance shows an additional coupling to the P atom (5JHP = 1.6 Hz) which is explained by the spatial arrangement of this proton (see Fig. S2.11†). The C atom of the CH2-group resonates at δ(P) = 55.1 ppm which is in the typical range for an olefinic C atom. Single crystals suitable for X-ray crystallography are obtained by slow diffusion of Et2O into a saturated solution of 35[OTf] in CH3CN at −35 °C (Fig. 9). Compared to the molecular structure of 342+ the major differences are the elongated C1–N1/N2 bond distances (C1–N1 1.404(3) Å, C1–N2 1.399(3) Å in 35+ and C1–N1 1.347(5) Å, C1–N2 1.332(5) Å) in 342+) and the reduced bond angle between N1–C1–N2 (104.6(2)° in 35+ and N1–C1–N2 109.141(9)° in 342+) and are explained by a diminished degree of π-delocalization between the N1–C1–N2 moiety and an increased p-orbital character of the C1–N1/N2 bonds. The C1–C4 bond length of 1.334(4) Å is well within the range of a typical olefinic CC double bond (1.34 Å).47 Cation 35+ is regarded as cationic N-heterocyclic olefin (NHO) of which the main resonance structures (A, B) are depicted in Scheme 11, indicating the highly polarized nature of the exocyclic CC double bond. As already discussed by others, NHOs are of great interests as strong two-electron donors due to their considerable nucleophilic character.45,46
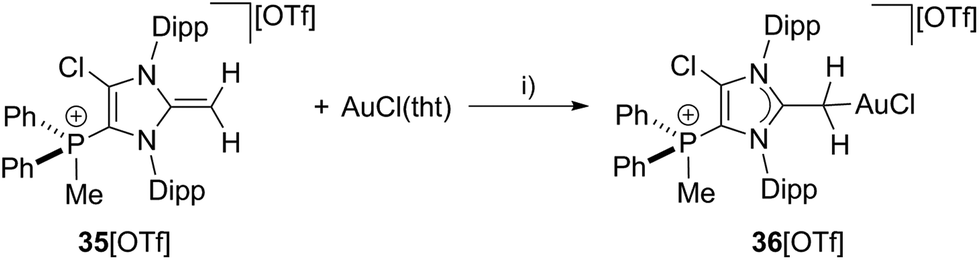
To proof the donor properties of cationic NHO 35+, the triflate salt was reacted with one equivalent AuCl(tht) in THF at ambient temperature to afford the NHO gold complex 36[OTf] in good yield (71%, Scheme 12). The 31P NMR spectrum of 36[OTf] reveals a slightly downfield shifted resonance at δ(P) = 9.6 ppm compared to the free ligand 35+ (δ(P) = 14.3 ppm). The 1H NMR spectrum of 36+ shows a singlet resonance for the CH2-group at δ(H) = 2.36 ppm, thus the two protons of the CH2 moiety become magnetically equal upon coordination to the transition metal due to the free rotation around the C1–CH2 bond (see Fig. S2.11†). The coordination of the gold chloride fragment to the CH2-group causes a pronounced high field shift in the 13C NMR spectrum (δ(C) = 8.5 ppm) compared to cation 35+ (δ(C) = 55.1 ppm). Single crystals suitable for X-ray crystallography are obtained by slow diffusion of Et2O into a saturated solution of 36[OTf] in CH3CN at −35 °C (Fig. 9). The molecular structure of cation 36+ displays a tetra-coordinate bonding environment at the C4 atom with a C1–C4–Au bond angle of 116.2(2)° and an elongated C1–C4 bond length of 1.453(4) Å compared to that in 35+ (C1–C4 1.399(3) Å). This elongation is a result of the reduced π-bond character of the C1–C4 bond caused by the coordination of the gold chloride fragment.
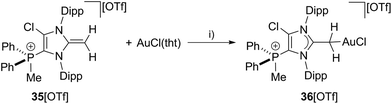 |
| | Scheme 12 Preparation of NHO gold complex 36[OTf]; I) THF, r.t., 3 h, −tht, 78%. | |
Conclusions
In this contribution, we present a facile and high yielding syntheses of 2-phosphanyl-(9a,b[OTf]) and 5-phosphanyl-(10a,b[OTf]) substituted imidazolium salts from the reaction of the corresponding chlorophosphanes and NHC 8. Methylation or oxidation with XeF2 and subsequent fluoride abstraction of these salts affords 5-phosphonio substituted imidazolium salts 14a,b[OTf]2 and 16a,b[OTf]2 in good to very good yields. Quantum chemical calculations revealed the Cl1 atom (σ-hole) the most reactive position towards bulky nucleophiles. Chlorenium abstraction is achieved by the addition of R3P (R = Ph, Cy) in a SN2(Cl) type reaction to yield the first cationic 5-phosphonio-substituted NHCs 17a,b+ and 18a,b+ as triflate salts. These salts are a new class of electron deficient cationic NHCs which conveniently form transition metal complexes with corresponding metals salts (AuCl(tht) for 19[OTf], and 21[OTf], CuBr(tht) for 20[OTf], [RhCl(cod)]2 for 23[OTf]) and AgOTf for 22[OTf]3). As shown for derivative 17b[OTf], the methylated derivatives can be handled also in solvents such as CH2Cl2 and CH3CN without decomposition which significantly broadens the scope of application. 17b[OTf] was used in dehydrocoupling reactions of prim. and sec. aryl and even alkyl phosphanes. The preparation of a highly nucleophilic N-heterocyclic olefin (NHO, 35[OTf]) proceeds via a two step synthesis involving the reaction of in situ formed 17b[OTf] with MeOTf and subsequent deprotonation with LDA. Cation 35+ acts as a two-electron donor as confirmed by the isolation of the AuCl complex 36[OTf].
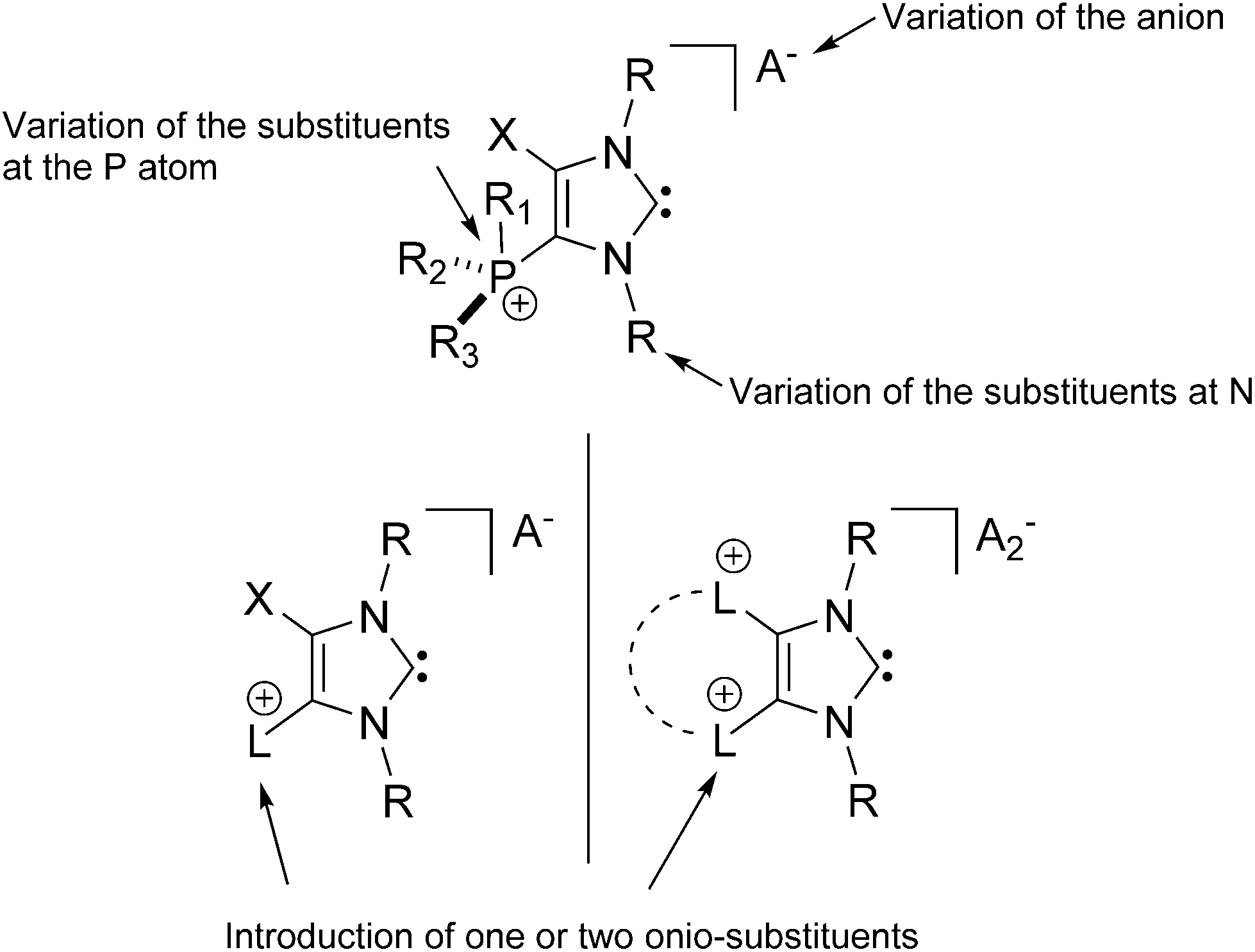
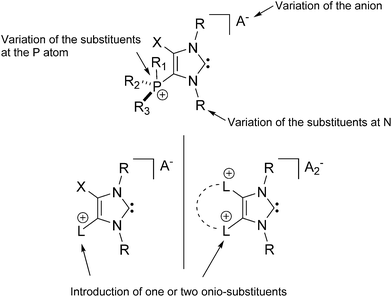
The isolation of the first NHC salts does impact the broad field of NHC chemistry since it allows the design of new charged systems (Chart 1). Thus, the opportunity of including a variety of different onio-substituents (phosphanes, amines, pyridines48etc.)49 allows for the introduction of additional cationic charges in 4/5-position. This should have a tremendous influence on the reactivity of the resulting carbenes which was already shown by a in situ formed pyridinio-substituted NHC derivative.48 The introduction of a stereogenic center is feasible by the variation of the substituents at the phosphonium center, the substituents of the N atoms in the heterocycle, as well as the choice of counter-ions which makes the resulting systems interesting candidates for stereoselective transition metal or organo catalysis.
 |
| | Chart 1 Possible variations for a new class of NHC systems. | |
Acknowledgements
This work was supported by the Fonds der Chemischen Industrie (FCI, Kekulé scholarship for F. H.), the German Science Foundation (DFG, grant number WE 4621/2-1) and the ERC (ERC starting grant SynPhos: 307616). A. B. and A. F. thank DGICYT of Spain (projects CTQ2014-57393-C2-1-P and CONSOLIDER INGENIO CSD2010-00065, FEDER funds) for funding. We also thank Philipp Lange and Stephen Schulz for experimental assistance.
References
- Selected examples of P-centered NHC adducts and imidazoliumyl salts:
(a) N. Kuhn, J. Fahl, D. Bläser and R. Boese, Z. Anorg. Allg. Chem., 1999, 625, 729 CrossRef CAS;
(b) A. J. Arduengo III, J. C. Calabrese, A. H. Cowley, H. V. R. Dias, J. R. Goerlich, W. J. Marshall and B. Riegel, Inorg. Chem., 1997, 36, 2151 CrossRef;
(c) A. J. Arduengo III, C. J. Carmalt, J. A. C. Clyburne, A. H. Cowley and R. Pyati, Chem. Commun., 1997, 981 RSC;
(d) C. A. Dyker, A. Decken, B. D. Ellis and C. L. B. Macdonald, Chem. Commun., 2005, 1965 Search PubMed;
(e) Y. Wang, Y. Xie, P. Wei, R. B. King, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2008, 130, 14970 CrossRef CAS PubMed;
(f) Y. Wang, Y. Xie, M. Y. Abraham, R. J. Gilliard Jr., P. Wei, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, Organometallics, 2010, 29, 4778 CrossRef CAS;
(g) T. Böttcher, B. S. Bassil, L. Zhechkov, T. Heine and G.-V. Röschenthaler, Chem. Sci., 2013, 4, 77 RSC;
(h) A. Doddi, D. Bockfeld, T. Bannenberg, P. G. Jones and M. Tamm, Angew. Chem., Int. Ed., 2014, 53, 13568 (
Angew. Chem.
, 2014
, 126
, 13786
) CrossRef CAS PubMed;
(i) K.-O. Feldmann, F. D. Henne and J. J. Weigand, J. Am. Chem. Soc., 2010, 132, 16321 CrossRef PubMed;
(j) F. D. Henne, E.-M. Schnöckelborg, K.-O. Feldmann, J. Grunenberg, R. Wolf and J. J. Weigand, Organometallics, 2013, 32, 6674 CrossRef CAS;
(k) M. H. Holthausen, S. K. Surmiak, P. Jerabek, G. Frenking and J. J. Weigand, Angew. Chem., Int. Ed., 2013, 52, 11078 (
Angew. Chem.
, 2013
, 125
, 11284
) CrossRef CAS PubMed;
(l) F. D. Henne, A. T. Dickschat, F. Hennersdorf, K.-O. Feldmann and J. J. Weigand, Inorg. Chem., 2015, 54, 6849 CrossRef CAS PubMed.
- For reviews see also:
(a) S. Gaillard and J.-L. Renaud, Dalton Trans., 2013, 42, 7255 RSC;
(b) Y. Canac, C. Maaliki, I. Abdellah and R. Chauvin, New J. Chem., 2012, 36, 17 RSC.
-
(a) J. I. Bates, P. Kennepohl and D. P. Gates, Angew. Chem., Int. Ed., 2009, 48, 9844 (
Angew. Chem.
, 2009
, 121
, 10028
) CrossRef CAS PubMed;
(b) P. K. Majhi, K. C. F. Chow, T. H. H. Hsieh, E. G. Bowes, G. Schnakenburg, P. Kennepohl, R. Streubel and D. P. Gates, Chem. Commun., 2016, 52, 998 RSC.
- D. Mendoza-Espinosa, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2010, 132, 7264 CrossRef CAS PubMed.
- K. Schwedtmann, M. H. Holthausen, K.-O. Feldmann and J. J. Weigand, Angew. Chem., Int. Ed., 2013, 52, 14204 (
Angew. Chem.
, 2013
, 125
, 14454
) CrossRef CAS PubMed.
- J. I. Bates and D. P. Gates, Organometallics, 2012, 31, 4529 CrossRef CAS.
- D. Mendoza-Espinosa, B. Donnadieu and G. Bertrand, Chem. – Asian J., 2011, 6, 1099 CrossRef CAS PubMed.
- E. Aldeco-Perez, A. J. Rosenthal, B. Donnadieu, P. Parameswaran, G. Frenking and G. Bertrand, Science, 2009, 326, 556 CrossRef CAS PubMed.
- K. Majhi, S. Sauerbrey, G. Schnakenburg, A. J. Arduengo III and R. Streubel, Inorg. Chem., 2012, 51, 10408 CrossRef PubMed.
-
(a) J. Ruiz and A. F. Mesa, Chem. – Eur. J., 2012, 18, 4485 CrossRef CAS PubMed;
(b) J. Ruiz, A. F. Mesa and D. Sol, Organometallics, 2015, 34, 5129 CrossRef CAS.
- P. K. Majhi, G. Schnakenburg, Z. Kelemen, L. Nyulaszi, D. P. Gates and R. Streubel, Angew. Chem., Int. Ed., 2013, 52, 10080 (
Angew. Chem.
, 2013
, 125
, 10264
) CrossRef CAS PubMed.
- A. J. Arduengo III, F. Davidson, H. V. R. Dias, J. R. Goerlich, D. Khasnis, W. J. Marshall and T. K. Prakasha, J. Am. Chem. Soc., 1997, 119, 12742 CrossRef.
- NMR spectra of the purified chlorophosphanes were measured independently.
-
H. Goldwhite, Introduction to phosphorus chemistry, Cambridge University Press, Cambridge, 1981 Search PubMed.
- O. Back, M. Henry-Ellinger, C. D. Martin, D. Martin and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 2939 (
Angew. Chem.
, 2013
, 125
, 3011
) CrossRef CAS PubMed.
- For reviews see:
(a) T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 6940 (
Angew. Chem.
, 2010
, 122
, 7094
) CrossRef PubMed;
(b) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122 (
Angew. Chem.
, 2008
, 120
, 3166
) CrossRef CAS PubMed.
- J. B. Waters and J. M. Goicoechea, Coord. Chem. Rev., 2015, 293–294, 80 CrossRef CAS.
- The Tolman Electronic Paramter (TEP) has been employed in transition metal complexes to evaluate the donor abilities of normal and mesoionic NHCs:
(a) R. A. Kely III, H. Clavier, S. Guidice, N. M. Scott, E. D. Stevens, J. Bordner, I. Samardjiev, C. D. Hoff, L. Cavallo and S. P. Nolan, Organometallics, 2008, 27, 202 CrossRef;
(b) D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723 RSC.
- H. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef.
- P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114 (
Angew. Chem.
, 2008
, 120
, 6206
) CrossRef CAS PubMed , and references reported therein.
-
(a) N. Inamoto and S. Masuda, Chem. Lett., 1982, 11, 1003 CrossRef;
(b) F. De Proft, W. Langenaeker and P. Gerlings, J. Phys. Chem., 1993, 97, 1826 CrossRef CAS.
- M. Alcarazo, Chem. – Eur. J., 2014, 20, 7868 CrossRef CAS PubMed.
- M. Holthausen, M. Mehta and D. W. Stephan, Angew. Chem., Int. Ed., 2014, 53, 6538 (
Angew. Chem.
, 2014
, 126
, 6656
) CrossRef CAS PubMed.
-
(a) M. Pérez, L. J. Hounjet, C. B. Caputo, R. Dobrovetsky and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 18308 CrossRef PubMed;
(b) C. B. Caputo, L. J. Hounjet, R. Dobrovetsky and D. W. Stephan, Science, 2013, 341, 1374 CrossRef CAS PubMed;
(c) L. J. Hounjet, C. B. Caputo and D. W. Stephan, Dalton Trans., 2013, 42, 2629 RSC;
(d) M. Holthausen, J. M. Bayne, I. Mallov, R. Dobrovetsky and D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 7298 CrossRef CAS PubMed.
-
(a)
O. Kühl, Phosphorus-31-NMR Spectroscopy, Springer, Berlin, 2008 Search PubMed;
(b)
D. G. Gorenstein, Phosphorus-31 NMR Principles and Applications, Academic Press. Inc., Cambridge, 1984 Search PubMed.
-
(a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS;
(b) J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 33, 8822 CrossRef;
(c) J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 34, 7406 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- For further details see ESI.†.
- A. Bauzá, J. T. Mooibkoek and A. Frontera, ChemPhysChem, 2015, 16, 2496 CrossRef PubMed , and references reported therein.
- For comparison of the chemical shift, [Cy3PCl]+ and [Ph3PCl]+ were independently synthesized according to a literature procedure: J. K. Ruff, Inorg. Chem., 1963, 2, 813 CrossRef CAS.
- A. J. Arduengo III, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 363 CrossRef.
- R. Weiss and S. Engel, Angew. Chem., Int. Ed., 1992, 31, 216 (
Angew. Chem.
, 1992
, 104
, 239
) CrossRef.
- F. Weigend, M. Häser, H. Patzelt and R. Ahlrich, Chem. Phys. Lett., 1998, 294, 143 CrossRef CAS.
-
(a) P. de Fremont, N. M. Scott, E. D. Stevens and S. P. Nolan, Organometallics, 2005, 24, 2411 CrossRef CAS;
(b) P. de Frémont, N. Marion and S. P. Nolan, J. Organomet. Chem., 2009, 694, 551 CrossRef.
- P. de Frémont, N. M. Scott, E. D. Stevens, T. Ramnial, O. C. Lightbody, C. L. B. Macdonald, J. A. C. Clyburne, C. D. Abernethy and S. P. Nolan, Organometallics, 2005, 24, 6301 CrossRef.
- K. Verlinden, H. Buhl, W. Frank and C. Ganter, Eur. J. Inorg. Chem., 2015, 2416 CrossRef CAS.
- H. A. Bent, Chem. Rev., 1961, 61, 275 CrossRef CAS.
- G. D. Frey, J. D. Masuda, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 9444 (
Angew. Chem.
, 2010
, 122
, 9634
) CrossRef CAS PubMed.
- H. Schneider, D. Schmidt and U. Radius, Chem. Commun., 2015, 51, 10138 RSC.
- S. Molitor, J. Becker and V. H. Gessner, J. Am. Chem. Soc., 2014, 136, 15517 CrossRef CAS PubMed.
- Selected examples of transition metal catalyzed dehydrogenative coupling of phosphanes:
(a) V. P. W. Böhm and M. Brookhart, Angew. Chem., Int. Ed., 2001, 40, 4694 (
Angew. Chem.
, 2001
, 113
, 4832
) CrossRef;
(b) A. K. King, A. Buchard, M. F. Mahon and R. L. Webster, Chem. – Eur. J., 2015, 21, 15960 CrossRef CAS PubMed.
- A. Henderson Jr., M. Epstein and F. S. Seichter, J. Am. Chem. Soc., 1963, 85, 2462 CrossRef.
- J. D. Masuda, A. J. Hoskin, T. W. Graham, C. Beddie, M. C. Fermin, N. Etkin and D. W. Stephan, Chem. – Eur. J., 2006, 12, 8696 CrossRef CAS PubMed.
-
(a) N. Kuhn, H. Bohnen, J. Kreutzberg, D. Bläser and R. Boese, J. Chem. Soc., Chem. Commun., 1993, 1136 RSC;
(b) S. M. I. Al-Rafia, A. C. Malcolm, S. K. Liew, M. J. Ferguson, R. McDonald and E. Rivard, Chem. Commun., 2011, 47, 6987 RSC;
(c) A. C. Malcolm, K. J. Sabourin, R. McDonald, M. J. Ferguson and E. Rivard, Inorg. Chem., 2012, 51, 12905 CrossRef CAS PubMed;
(d) S. M. I. Al-Rafia, M. R. Momeni, M. J. Ferguson, R. McDonald, A. Brown and E. Rivard, Angew. Chem., Int. Ed., 2013, 52, 6390 (
Angew. Chem.
, 2013
, 125
, 6518
) CrossRef CAS PubMed;
(e) Y. Wang, M. Y. Abraham, R. J. Gilliard Jr., D. R. Sexton, P. Wei and G. H. Robinson, Organometallics, 2013, 32, 6639 CrossRef CAS;
(f) R. S. Ghadwal, S. O. Reichmann, F. Engelhardt, D. M. Andrada and G. Frenking, Chem. Commun., 2013, 49, 9440 RSC;
(g) S. M. I. Al-Rafia, M. R. Momeni, M. J. Ferguson, R. McDonald, A. Brown and E. Rivard, Organometallics, 2013, 32, 6201 CrossRef;
(h) S. Kronig, P. G. Jones and M. Tamm, Eur. J. Inorg. Chem., 2013, 2301 CrossRef CAS;
(i) A. Fürstner, M. Alcarazo, R. Goddard and C. W. Lehmann, Angew. Chem., Int. Ed., 2008, 120, 3254 (
Angew. Chem.
, 2008
, 120
, 3254
) CrossRef.
- For a review of NHOs see: E. Rivard, Dalton Trans., 2014, 43, 8577 RSC ; and references reported therein.
-
W. M. Haynes, D. R. Lide and T. J. Bruno, Handbook of Chemistry and Physics, CRC Press, New York, 96th edn, 2015 Search PubMed.
- H. Buhl and C. Ganter, Chem. Commun., 2013, 49, 5417 RSC.
- For transition metal-substituted derivatives see:
(a) B. Hildebrandt, W. Frank and C. Ganter, Organometallics, 2011, 30, 3483 CrossRef CAS;
(b) B. Hildebrandt, S. Raub, W. Frank and C. Ganter, J. Organomet. Chem., 2012, 717, 83 CrossRef CAS; B. Hildebrandt, S. Raub, W. Frank and C. Ganter, Chem. – Eur. J., 2012, 18, 6670 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2016 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b,
Robert
Weiss
c and
Jan J.
Weigand
*a
b,
Robert
Weiss
c and
Jan J.
Weigand
*a