
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Coordination of Zn2+ and Cu2+ to the membrane disrupting fragment of amylin†
M.
Rowińska-Żyrek
Faculty of Chemistry, University of Wroclaw, F. Joliot-Curie 14, 50383 Wroclaw, Poland. E-mail: magdalena.rowinska-zyrek@chem.uni.wroc.pl
First published on 5th April 2016
Abstract
Amylin, a small peptide co-secreted from pancreatic β-cells together with insulin, is one of the hallmarks of type II diabetes. In the course of this disease, it misfolds into small oligomers or into an aggregated β-sheet amyloid fiber. The misfolding mechanism is not yet well understood, but it is clear that metal ions such as zinc and copper play an important role in the process. In this work, the coordination chemistry of Zn2+ and Cu2+ with the membrane-disrupting part of amylin (amylin1–19) is discussed. Cu2+ alters the structure of amylin1–19 only locally, by binding to His18 imidazole and to three preceding amides at the N-terminal side of this residue. Zn2+ binds to the imidazole of His18 and the amine group of Lys1, imposing a kink in the peptide between these residues. This zinc-induced kink might be a partial explanation of the formation of prefibrillar oligomeric aggregates of amylin, which are much more toxic to β-cells than large fibrillar deposits.
Introduction
Nowadays, the impact of metal ions on the structure and function of biomolecules has become undoubtable. Understanding the relationship between metal coordination abilities of certain proteins can help explain the molecular basis and, in turn, various macroscopic symptoms present in the course of illnesses. Metal coordination abilities of amyloid beta, α-synuclein or the prion protein in Alzheimer's, Parkinson's and prion disease could be good examples. These proteins which are the hallmarks of the diseases are all linked by being related to metal-coordinated amyloidogenic peptides.1,2 Such peptides undergo a misfolding process from a random coil; their secondary structure is changed into an α-helical intermediate prior to a conformational change into a β-sheet, which then aggregates to form elongated fibers.3,4 For decades, there has been inconsistency in whether the presence of excess metal ions (such as Cu2+, Zn2+ or Fe(III) and Al2+) triggers or hinders the pathological deposition of amyloid deposits in the brain.5–9In the beginning of the initial debate, the prevailing opinion suggested that the excess of metal ions is the trigger for misfolding, and often, chelation therapy was even suggested.10 The process was later found to be much more complicated, involving not only metal coordination and disruption of metal homeostasis, but also various other factors, such as the interaction of hydrophobic regions or interactions with membranes that are also factors which trigger the formation of the thermodynamically unfavored nucleus, by which fiber formation is triggered.11
There are numerous parallels between amyloidogenic proteins involved in neurodegenerative diseases and a small polypeptide involved in glucose regulation – amylin, also known as the islet amyloid polypeptide (IAPP). Amylin is a 37 residue peptide, which is co-secreted from pancreatic β-cells together with its synergistic partner, insulin, and stored in the secretory granules of the islets of Langerhans in pancreatic β-cells.12,13 Amylin takes part in glucose regulation and appetite suppression, preventing post-prandial spikes in blood glucose levels.14 It is also an amyloidogenic peptide that undergoes misfolding from a monomeric random coil conformation to an aggregated β-sheet amyloid fiber, found in the islet beta cells of over 95% of the patients with type II diabetes (T2D)15,16 – a common, chronic, degenerative metabolic disease characterized by elevated blood glucose levels, abnormal insulin secretion and insulin resistance, which affects over 300 million people worldwide.17 The misfolding and amyloid fibril formation of amylin result in the death of islet beta cells and are thought to be important in the pathogenesis of T2D.18 The aggregation of small amylin oligomers results in oxidative stress and in the disruption of cell membranes.19–21 An increasing amount of evidence shows that most toxic species are not mature amyloid fibrils, but small oligomers, which most likely cause membrane disruption (another parallel to the proteins involved in neurodegeneration).22–24
The native form of amylin is amidated at the C-terminus and has a disulfide bridge between Cys2 and Cys7. The hormone peptide comprises three regions: (i) the membrane disrupting fibrillogenic N-terminus (residues 1–19, the fragment studied in this work), which influences the overall kinetics of fibril formation, (ii) the amyloidogenic region (residues 20–29), and the C-terminal part (residues 30–37), which enhances amyloid formation (Fig. 1).
 | ||
| Fig. 1 A comparison of the native sequence of human and rat amylin. Sequential differences are marked in green. Histidine 18 is the potential metal binding site. | ||
One of the differences between the human and the rat form of the peptide is the H18R substitution (Fig. 1), which results in the change of cytotoxicity on β-cells: the human form can become highly cytotoxic, while the non-aggregating rat amylin has negligible influence on β-cell survival.25,26 It might also be worth to point out that the N-terminal (1–19) region of the human amylin is toxic like the full-length peptide, but the respective sequence of the rat amylin is not (Fig. 1).27,28
It is clear that amylin is very prone to aggregation.29 Why doesn't this process occur in vivo, in the granules where it is stored in very high millimolar concentrations? There are several compounds in the granule that prevent amylin from fibrilising under these conditions: (i) the relatively low pH of the granule (pH = 6.0), (ii) the high concentration of zinc (∼14 mM) and (iii) the high concentration of insulin (∼4 times more than amylin).
The increasing number of diabetic patients causes an elevated amount of attention drawn towards understanding the molecular basis of the disease. As a result of this, numerous recent studies prove that the presence of Zn2+ and Cu2+ can inhibit the aggregation of amylin, although the underlying mechanism is far from being understood.29,30 Zinc is of particular interest, since its concentration in pancreatic β-cells is one of the highest in the body, and since zinc deficiency is quite a common symptom of type II diabetes.31 What is already clear is the fact that the presence of zinc inhibits the membrane disruption ability of amylin and reduces the amount of formed amyloids, but it does not alter the morphology of the fibers. It is interesting that zinc has a dual effect on fibrillogenesis of amylin: it increases the lag-time for fibre formation and increases the rate of addition of amylin to existing fibres at high concentrations, while having the opposite effect at lower concentrations.
Some studies show that also copper(II) levels are significantly elevated in diabetic patients,32,33 which suggests a correlation between copper homeostasis and the molecular basis of type 2 diabetes. A local disruption of copper homeostasis triggers the formation of oxidative species, stimulating the production of H2O2 with amylin34 (another parallel to amyloid beta).35 Several studies correlated the ability of copper(II) to inhibit amylin fibrillation with toxicity,36,37 and confirmed the non-fibrillogenic nature of the copper(II)–amylin aggregates.38
This metal has also been found to mediate the membrane-interactions of the 17–29 fragment of amylin.39
Recently, the 15–22 fragment was studied by using different spectroscopy techniques, showing that Cu2+ anchors to His18 and, which is quite unusual, binds to the subsequent amide groups toward the C-terminus, forming a thermodynamically unfavorable seven-membered chelate ring with an equatorial {3N,O} coordination mode at physiological pH.40 Copper binding to a monomeric IAPP was expected to compete with conformation changes needed to form β-sheet structures, and thus delay fibril formation.
The interactions of Cu2+ with the rat fragments of amylin were also extensively studied, showing the formation of a square planar complex, in which copper(II) is bound to four amide nitrogens.41
Knowing the importance of the impact of copper and zinc on the aggregation and membrane disrupting abilities of amylin, this work tries to understand the interactions between Cu2+ and Zn2+ with the membrane disrupting fragment of human amylin, K![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) NTATATQRLANFLVHS-NH2 (amylin1–19), focusing on the thermodynamics of such complex formation and precisely pointing out metal binding sites. The work provides a detailed understanding of the coordination chemistry of amylin–metal interactions and gives insight into bioinorganic chemistry of type 2 diabetes.
NTATATQRLANFLVHS-NH2 (amylin1–19), focusing on the thermodynamics of such complex formation and precisely pointing out metal binding sites. The work provides a detailed understanding of the coordination chemistry of amylin–metal interactions and gives insight into bioinorganic chemistry of type 2 diabetes.
Experimental
Synthesis
The C-protected disulphide-bridged amylin1–19 (KNTATATQRLANFLVHS-NH2) was purchased from KareBay Biochem (USA) (certified purity: 99.30%) and was used as received. Its purity was checked potentiometrically. Cu(ClO4)2 and Zn(ClO4)2 were extra pure products (Sigma-Aldrich); the concentrations of their stock solutions were determined by ICP-MS. The carbonate-free stock solution of 0.1 mol dm−3 KOH was purchased from Sigma-Aldrich and then potentiometrically standardized with potassium hydrogen phthalate.
Potentiometric measurements
Stability constants for proton, Cu2+ and Zn2+ complexes were calculated from titration curves carried out in the pH range 2–11 at 25 °C and ionic strength 0.1 M (KClO4) using a total volume of 3 cm3. The potentiometric titrations were performed using a Dosimat 665 Metrohm titrator connected to a Metrohm 691 pH-meter and a Metrohm LL Unitrode glass electrode. The thermostabilized glass-cell was equipped with a magnetic stirring system, a microburette delivery tube and an inlet–outlet tube for argon. Solutions were titrated with 0.1 M carbonate-free KOH. The electrodes were daily calibrated for hydrogen ion concentrations by titrating HClO4 with KOH under the same experimental conditions as above. The purities and the exact concentrations of the ligand solutions were determined by the Gran method.42 The ligand concentration was 1 mM, and the Zn2+ and Cu2+-to-ligand ratios were both 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
:1.
HYPERQUAD 2006 and SUPERQUAD programs were used for stability constant calculations.43,44 Standard deviations were computed by HYPERQUAD 2006 and refer to random errors only. The constants for hydrolytic Zn2+ species were used in these calculations.45 The speciation and competition diagrams were computed with the HYSS program.46
Spectroscopy studies
Solutions were of similar concentrations with respect to those used in the potentiometric studies. Absorption spectra were recorded on a Cary 300 Bio spectrophotometer. Circular dichroism (CD) spectra were recorded on a Jasco J 715 spectropolarimeter in the 800–230 nm range.The UV-Vis and CD spectroscopy parameters were calculated from the spectra obtained at the pH values corresponding to the maximum concentration of each particular species, on the basis of potentiometric studies.
Mass spectrometry measurements
High-resolution mass spectra were obtained on a BrukerQ-FTMS spectrometer (Bruker Daltonik, Bremen, Germany), equipped with an Apollo II electrospray ionization source with an ion funnel. The mass spectrometer was operated in positive ion mode. The instrumental parameters were as follows: scan range m/z 300–3000, dry gas – nitrogen, temperature 170 °C, and ion energy 5 eV. The capillary voltage was optimized to the highest S/N ratio and it was 4500 V. The small changes in voltage (±500 V) did not significantly affect the optimized spectra. The samples (metal:ligand in a 1:1, 1:2 and 2:1 stoichiometry, [ligand]tot = 10−4 M) were prepared in a 1:1 acetonitrile–water mixture at pH 5, 7.4 and 9. The variation of the solvent composition down to 5% of acetonitrile did not change the species composition. The sample was infused at a flow rate of 3 μL min−1. The instrument was calibrated externally with a Tunemix™ mixture (Bruker Daltonik, Germany) in quadratic regression mode. Data were processed by using Bruker Compass DataAnalysis 4.0 program. The mass accuracy for the calibration was better than 5 ppm, enabled together with the true isotopic pattern (using SigmaFit) an unambiguous confirmation of the elemental composition of the obtained complex.
NMR measurements
NMR spectra were recorded at 14.1 T on a Bruker Avance III 600 MHz equipped with a Silicon Graphics workstation. The temperatures were controlled with an accuracy of ±0.1 K. Suppression of the residual water signal was achieved by excitation sculpting, using a selective square pulse on water, 2 ms long. All the samples were prepared in a 90% H2O and 10% D2O (99.95% from Merck) mixture. Proton resonance assignment was accomplished by 2D 1H–1H total correlation spectroscopy (TOCSY) and nuclear Overhauser effect spectroscopy (NOESY) experiments were carried out with standard pulse sequences. Spectral processing and analysis were performed using Bruker TOPSPIN 2.1 and Sparky. The samples of complexes were prepared by adding metal ions to an acidic solution of a ligand (pH 3.5), and the pH was then increased to a higher value.Results and discussion
Structural and thermodynamic properties of Zn2+ and Cu2+ complexes with the membrane disrupting part of amylin were studied and compared to each other by using mass spectrometry, potentiometry, and several spectroscopy techniques. Mass spectrometry measurements provided the information on the stoichiometry of the interactions and the combined UV-Vis and CD results allowed us to conclude that the binding mode of copper(II) and the geometry of these species formed in solution and potentiometric titrations were the basis for the determination of precise stability constants and pH-dependent species distribution diagrams for the studied systems. NMR spectra recorded both in the presence and in the absence of metal ions pointed out precise metal binding sites. A combination of all the used methods allowed us to explain coordination geometries and perform thermodynamic analysis.The KNTATATQRLANFLVHS-NH2 peptide (amylin1–19) behaves as an LH3 acid, with the deprotonating groups corresponding to the histidine imidazole, the N-terminal amine group and the lysine side chain group, with pKa values of 6.04, 7.91 and 10.83, respectively. The cysteine groups are bridged with a disulfide bond, as in the wild type form of amylin, and the C-terminal serine is amidated, in order to be a better mimic of the full-length amylin.
Amylin1–19 shows an NMR behavior typical of a disordered peptide. NMR spectra recorded at different pH values and temperatures did not suggest any specific rearrangements of the apo peptide.
Mass spectrometry proves that amylin1–19 forms only mononuclear complexes with both copper and zinc ions. ESI-MS peak assignments were based on the comparison between the precise calculated and experimental m/z values and their isotopic patterns. The prevailing signals correspond to the free ligand (m/z = 1038.6, z = 2+) and to the chloride adducts of equimolar complexes with Cu2+ (m/z = 1088.5, z = 2+) and with Zn2+ (m/z = 1089.1, z = 2+) (Fig. S1†). The signals are most abundant under all the studied pH conditions (pH 5, 7.4 and 9), and also when 1:2, 1:1 and 2:1 metal to ligand ratios are analyzed. Other minor signals correspond to various chloride and sodium adducts of the free ligand and of their metal complexes.
There are major differences in the coordination mode of amylin copper and zinc complexes. The only parallel for both metals is the first anchoring site – the His18 imidazole; the remaining coordinating groups are different for the two metals.
In the case of copper species, the first complex observed at low pH, CuH2L, involves only His18 in binding – this is clearly visible in the NMR spectra (Fig. 2A), whereafter the addition of 0.3 Cu2+ equivalents, only the aromatic imidazole protons are broadened (since copper is a paramagnetic metal ion, a reasonably small amount of this metal causes selective broadening of the residues involved in binding). The coordination mode is further confirmed by a d–d band at 655 nm in UV-Vis spectroscopy, and by the lack of pronounced circular dichroism signals (Table 1, Fig. S2 and S3†). However, the possible co-existence of minor CuHL species cannot be unequivocally excluded.
 | ||
| Fig. 2 Selected regions of 1H–1H TOCSY spectra of 3 mM amylin1–19, T = 298 K, in the absence (black contours) and in the presence (green contours) of metal ions; (A) 0.3 Cu2+ equivalents, pH 4; (B) 0.3 Cu2+ equivalents, pH 5.5; (C) 1 Zn2+ equivalent, pH 7.2. | ||
| Species | logβ |
pKa | UV-Vis | CD | ||
|---|---|---|---|---|---|---|
| λ/nm | ε/M−1 cm−1 | λ/nm | Δε/M−1 cm−1 | |||
| HL | 10.83 | 10.83 (Lys) | ||||
| H2L | 18.74 | 7.91 (N-t) | ||||
| H3L | 24.78 | 6.04 (His) | ||||
| Cu2+ complexes | ||||||
| CuH2L | 24.92 (2) | 655 | 36 | 279 | −0.09 | |
| 317 | −0.06 | |||||
| 690 | −0.11 | |||||
| CuL | 14.19 (2) | 5.39 (amide) | 560 | 123 | 288 | −27.3 |
| 319 | 6.74 | |||||
| 495 | 2.72 | |||||
| 635 | 11.56 | |||||
| CuH−1L | 8.80 (3) | 8.11 (N-t) | 545 | 145 | 292 | −18.90 |
| 316 | 8.22 | |||||
| 503 | 8.06 | |||||
| 595 | −17.75 | |||||
| CuH−2L | 0.69 (4) | 9.74 (Lys) | 540 | 158 | 295 | −9.50 |
| 316 | 17.58 | |||||
| 495 | 6.13 | |||||
| 570 | −18.49 | |||||
| CuH−3L | −9.05 (5) | 535 | 154 | 292 | −11.05 | |
| 316 | 19.45 | |||||
| 560 | −19.21 | |||||
| Zn2+ complexes | ||||||
| ZnH2L | 21.33 (2) | 6.27 (N-t) | ||||
| ZnHL | 15.06 (2) | 8.07 (H2O) | ||||
| ZnL | 6.99 (4) | 8.22 (H2O) | ||||
| ZnH−1L | −1.23 (4) | 9.84 (Lys) | ||||
| ZnH−2L | −11.07 (7) | |||||
Above pH 5, drastic changes can be observed in all spectroscopy methods. The UV-Vis band undergoes a blue shift to 560 nm, and the CD spectra are typical of a {Nim,2N−} coordination (Table 1, Fig. S2 and S3†). Also, selective broadening on NH gamma and NH beta of Val17 observed on the NMR spectra strongly supports the involvement of amide nitrogens in binding (Fig. 2B).
Above pH 6, the CuL complex deprotonates to CuH−1L, with a pKa of 5.39 (Fig. 3A). The strong blue shift and increase in intensity in the CD (from 635 to 595 nm, Table 1 and Fig. S3†) and UV-Vis spectra (from 560 to 545 nm, Table 1 and Fig. S2†) confirm the involvement of a third amide in the coordination, resulting in {Nim, 3N−} binding mode. This mode does not change with the further increase of pH; pKa constants of 8.11 and 9.74 correspond to the deprotonation of the N-terminal amine and of the side chain of lysine, respectively; both the groups do not participate in binding (Table 1).
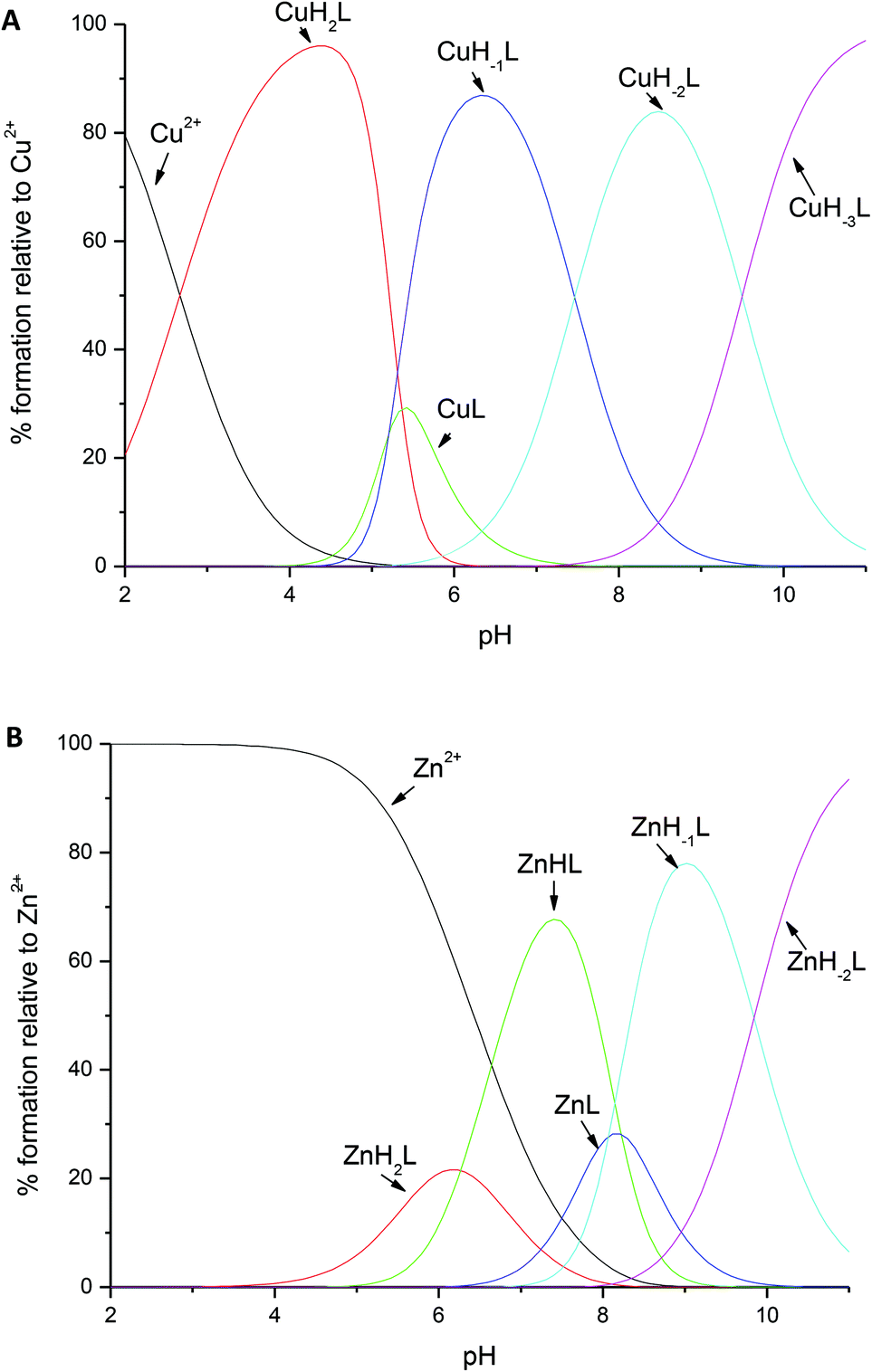 | ||
| Fig. 3 Distribution diagrams for the formation of (A) Cu2+ and (B) Zn2+ complexes with amylin1–19 at 25 °C and I = 0.1 M. [M2+] = 0.5 × 10−3 M; M/L molar ratio = 1:1. | ||
The coordination of Zn2+ to amylin1–19 differs significantly when compared to that of Cu2+; for ZnH2L, the anchoring site is the histidine imidazole, just as it was in the first complex observed for copper at low pH. However, in the species observed at a physiological pH (with a maximum at pH 7.2), ZnHL, the imidazole nitrogen and the N-terminal amino group are the residues which are directly involved in coordination; the vacant zinc binding sites are occupied by water molecules. The pKa value of 6.27 strongly suggests the involvement of this group in binding (the corresponding pKa for the free ligand = 7.91, Table 1). NMR spectroscopy confirms the {Nim,NH2} binding mode. The addition of Zn2+ to amylin1–19 causes selective line broadening of His18 Hα–Hβ correlation and a broadening and shift of the N-terminal Lys1 Hα–Hβ, Hα–Hδ and Hα–Hγ NMR signals (Fig. 2C). The terminal amine protons are not visible in either the metal bound or the apo form of the peptide, most probably due to a fast water exchange.
The ZnHL complex dominates in solutions up to pH 8. At higher pH, two stepwise deprotonations with the corresponding pKa values of 8.07 and 8.22 occur (Fig. 3B and Table 1). Most likely, those two protons arise from two water molecules, present in the two vacant coordination sites not occupied by peptide residues. This scenario is very likely, since Zn2+ is not able to deprotonate amide nitrogens, in contrast to Cu2+. Another scenario could include the carbonyl oxygen of His18 or Lys1 having a stabilization role in the coordination of Zn2+.
The binding mode does not change with the increase of pH; the ZnH−2L complex results from the deprotonation of the unbound lysine residue (with a pKa value of 9.84).
It becomes obvious that His18 is crucial for the coordination of both copper and zinc to amylin1–19. This was expected, since it was already shown in previous studies that the binding of zinc to amylin causes a local disruption of the secondary structure in the vicinity of His-18.47 Biological studies showed that the substitution of this residue to arginine severely reduces the membrane disrupting ability of the 1–19 fragment.48 The situation becomes very similar after the addition of Zn2+ to the full length amylin–membrane disruption also becomes far less pronounced. Both the H to R substitution and zinc binding change the overall charge distribution of the peptides – most likely, such peptides would be more surface associated, since the positive His-18 imidazole would make it impossible for them to penetrate into the hydrophobic core of a lipid bilayer. Also, zinc may create an energetic barrier for the formation of amyloids by promoting the formation of prefibrillar aggregates and thus inhibiting the formation of amyloid fibrils.49,50 It is worth keeping in mind that (just as it was in the case of amyloid β) mature amyloid fibers show relatively little toxicity to β-cells, and most likely the small, prefibrillar aggregates are the toxic species.23
It is tempting to suggest that the zinc–amylin binding mode described in this work might be at least a partial explanation of how these prefibrillar aggregates are formed – the coordination mode which involved the imidazole of His18 and the amine group of Lys1 imposes a kink between those residues (Fig. 4). This special structure might be the thermodynamical basis of prefibrillar aggregate formation. Zinc is bound to His18 and Lys1 at a physiological pH (ZnHL species, in our case, with a maximum abundance at pH 7.2). Literature data show that at a similar pH, the presence of low concentrations of Zn2+ in the incubation solution decreases the rate of amylin amyloid formation, whereas a higher zinc concentration has an opposite effect.48
 | ||
| Fig. 4 Suggested binding modes of Cu2+ and Zn2+ complexes at pH 7.2. Explicit hydrogens are omitted for clarity. | ||
Again, it can be hypothesised that in excess of zinc, more than one Zn2+ ion can be bound to the peptide (e.g. one to His18 and another to the N-terminal amine of Lys1) – in this case, the special kink observed in the case of mononuclear species would no longer be present.
Copper, on the other hand, alters the structure of amylin1–19 only locally, by binding to His18 imidazole and to three preceding amides at the N-terminal side of this residue (Fig. 4). It is quite a common binding mode for Cu2+, and in the case of amylin1–19 (CuH−1L), it already starts to prevail in solution at pH 6. This coordination mode is different from the one reported by Rivillas-Acevedo et al.;40 here copper deprotonated amides from the 15–24 amylin fragment are towards the C-terminus of the peptide. Clearly, the membrane disrupting amylin1–19 region has only one amide that can be deprotonated by Cu(II), once it is bound to His18 and the data cannot be directly compared to that from Rivillas-Acevedo's work. The binding mode of the highly prone to aggregation full length amylin requires more studies, e.g. on the non-aggregating analogue – pramlintide.
Biological studies emphasize the role of copper-dependent generation of H2O2, which was found to directly contribute to the toxicity of human amylin.51,52 Again, a similar scenario was observed in the case of β-amyloid-copper or prion-copper complexes.7,53 Does similar coordination chemistry result in a similar biological outcome? Most probably, numerous biological studies and a detailed kinetical description of the amyloid aggregation in the presence of metal ions are necessary, before the answer to this question becomes something more than a very far-reaching hypothesis about the protective role of copper–amylin complex in vivo.
Conclusions
Misfolding processes of certain proteins are a common hallmark of numerous diseases. Most common examples are those involved in neurodegeneration – α-synuclein, β-amyloid, huntingtin or the prion protein. For almost two decades, we have been experiencing an ongoing debate about the impact of metal ions on the molecular basis and, in turn, on the course of these illnesses. Recently, a similar discussion has been started for amylin, which shows numerous parallels to metal binding amyloid-forming neuro-proteins.This work summarizes the details of Zn2+ and Cu2+ coordination to the membrane-disrupting fragment of amylin and amylin1–19. The anchoring site for Cu2+ is His18, and at physiological pH, the histidine imidazole and three amides are in the first coordination sphere. Such a typical, square-planar copper complex might contribute to the generation of H2O2, which is one of the factors responsible for the toxicity of human amylin. From the point of view of bioinorganic chemistry, the binding mode of Zn2+ to amylin1–19 seems to be much more interesting – at physiological pH, it is bound to His18 imidazole and to the N-terminal amine group of Lys1. This type of binding induces a kink in the peptide backbone, which might be the first step of prefibrillar aggregate formation.
This work gives us very precise details about the coordination of copper and zinc to the membrane disrupting fragment of amylin, but in the end, it brings up even more questions that still need to be answered: does the binding of zinc and copper indeed have an influence on the pathogenesis of amylin? How does it influence the aggregation of the full-length amylin? And, above all – are the metal-bound aggregates the cause, or rather the consequence of T2D?
Acknowledgements
The author would like to express her thankfulness to Ms. Agnieszka Przybyła for her skillful assistance in solution studies. The work was supported by the National Science Centre (nr UMO-2014/13/D/ST5/02868) and by the Ministry of Science and Higher Research (1493/M/WCH/15).References
- R. A. Floyd and D. A. Butterfield, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 3270 CrossRef.
- B. J. Tabner, S. Turnbull, O. El-Agnaf and D. Allsop, Curr. Top. Med. Chem., 2001, 1, 507 CrossRef CAS PubMed.
- G. Yamin, C. B. Glaser, V. N. Uversky and A. L. Fink, J. Biol. Chem., 2003, 278, 27630 CrossRef CAS PubMed.
- C. Talmard, R. L. Yona and P. Faller, J. Biol. Inorg. Chem., 2009, 14, 449 CrossRef CAS PubMed.
- E. Gaggelli, H. Kozłowski, D. Valensin and G. Valensin, Chem. Rev., 2006, 106, 1995 CrossRef CAS PubMed.
- H. Kozłowski, M. Łuczkowski, M. Remelli and D. Valensin, Coord. Chem. Rev., 2012, 256, 2129 CrossRef.
- M. Rowińska-Żyrek, M. Salerno and H. Kozłowski, Coord. Chem. Rev., 2015, 284, 298 CrossRef.
- S. S. Leal, H. M. Botelho and C. M. Gomes, Coord. Chem. Rev., 2012, 256, 2253 CrossRef CAS.
- B. R. Roberts, T. M. Ryan, A. I. Bush, C. L. Masters and J. A. Duce, J. Neurochem., 2012, 1, 149 CrossRef PubMed.
- A. Gaeta and R. C. Hider, Br. J. Pharmacol., 2005, 146, 1041 CrossRef CAS PubMed.
- M. Jucker and L. C. Walker, Ann. Neurol., 2001, 70, 532 CrossRef PubMed.
- S. E. Khan, D. A. D'Alessio, M. W. Schwartz, W. Y. Fujimoto, J. W. Ensinck, G. J. J. Taborsky and D. J. J. Porte, Diabetes, 1990, 5, 634 CrossRef.
- A. Kanatsuka, H. Makino, H. Ohsawa, Y. Tokuyama, T. Yamaguchi, S. Yoshida and M. Adachi, Fed. Eur. Biochem. Soc. J., 1989, 259, 199 CrossRef CAS.
- T. K. Reda, A. Geliebter and F. X. Pi-Sunyer, Obes. Res., 2002, 10, 1087 CrossRef CAS PubMed.
- P. Westermark and E. Wilander, Diabetologia, 1978, 15, 417 CrossRef CAS PubMed.
- J. W. Hoppener and C. J. Lips, Int. J. Biochem. Cell Biol., 2006, 38, 726 CrossRef PubMed.
- J. M. Olefsky, in Endocrinology, ed. L. J. De Groot, Saunders, London, 1989, vol. 2, p. 1369 Search PubMed.
- J. W. Hoppener, B. Ahren and C. J. Lips, N. Engl. J. Med., 2000, 343, 411 CrossRef CAS PubMed.
- K. Ono, M. M. Condron and D. B. Teplow, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14745 CrossRef CAS PubMed.
- J. R. Brender, S. Salamekh and A. Ramamoorthy, Acc. Chem. Res., 2012, 45, 454 CrossRef CAS PubMed.
- S. A. Jayasinghe and R. Langen, Biochemistry, 2005, 44, 12113 CrossRef CAS PubMed.
- J. R. Brender, E. L. Lee, K. Hartman, P. T. Wong, A. Ramamoorthy, D. G. Steel and A. Gafni, Biophys. J., 2011, 100, 685 CrossRef CAS PubMed.
- C. Haass and D. J. Selkoe, Nat. Rev. Mol. Cell Biol., 2007, 8, 101 CrossRef CAS PubMed.
- J. P. Cleary, D. M. Walsh, J. J. Hofmeister, G. M. Shankar, M. A. Kuskowski, D. J. Selkoe and K. H. Ashe, Nat. Neurosci., 2005, 8, 79 CrossRef CAS PubMed.
- G. Pappalardo, D. Milardi, A. Magri, F. Attanasio, G. Impellizzeri, C. La Rosa, D. Grasso and E. Rizzarelli, Chem. – Eur. J., 2007, 13, 10204 CrossRef PubMed.
- R. P. R. Nanga, J. R. Brender, J. Xu, G. Veglia and A. Ramamoorthy, Biochemistry, 2008, 47, 12689 CrossRef CAS PubMed.
- A. N. Roberts, B. Leighton, J. A. Todd, D. Cockburn, P. N. Schofield and R. Suttonet, et al. , Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 9662 CrossRef CAS.
- J. R. Brender, K. Hartman, K. R. Reid, R. T. Kennedy and A. Ramamoorthy, Biochemistry, 2008, 47, 12680 CrossRef CAS PubMed.
- V. Wineman-Fisher, Y. Atsmon-Raz and Y. Miller, Biomacromolecules, 2015, 16, 156 CrossRef CAS PubMed.
- F. Belliaa and G. Grasso, J. Mass Spectrom., 2014, 49, 274 CrossRef PubMed.
- C. G. Taylor, BioMetals, 2005, 18, 305 CrossRef CAS PubMed.
- A. H. Zargar, N. A. Shah, S. R. Masoodi, B. A. Laway, F. A. Dar, A. R. Khan, F. A. Sofi and A. I. Wani, Postgrad. Med. J., 1998, 74, 665 CrossRef CAS PubMed.
- T. Naka, H. Kaneto, N. Katakami, T. Matsuoka, A. Harada, Y. Yamasaki, M. Matsuhisa and I. Shimomura, Endocr. J., 2013, 60, 393 CrossRef CAS PubMed.
- A. Masad, L. Hayes, B. J. Tabner, S. Turnbull, L. J. Cooper, N. J. Fullwood, M. J. German, F. Kametani, O. M. El-Agnaf and D. Allsop, FEBS Lett., 2007, 581, 3489 CrossRef CAS PubMed.
- D. Schubert, C. Behl, R. Lesley, A. Brack, R. Dargusch, Y. Sagara and H. Kimura, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 1989 CrossRef CAS.
- B. Ward, K. Walker and C. Exley, J. Inorg. Biochem., 2008, 102, 371 CrossRef CAS PubMed.
- C. E. E. House, T. Patel, L. Wu and P. E. Fraser, J. Inorg. Biochem., 2010, 104, 1125 CrossRef PubMed.
- A. Sinopoli, A. Magrı, D. Milardi, M. Pappalardo, P. Pucci, A. Flagiello, J. J. Titman, V. G. Nicoletti, G. Caruso, G. Pappalardo and G. Grasso, Metallomics, 2014, 6, 1841 RSC.
- G. Pappalardo, D. Milardi, A. Magri, F. Attanasio, G. Impellizzeri, C. La Rosa, D. Grasso and E. Rizzarelli, Chem. – Eur. J., 2007, 13, 10204 CrossRef PubMed.
- L. Rivillas-Acevedo, C. Sánchez-López, C. Amero and L. Quintanar, Inorg. Chem., 2015, 54, 3788 CrossRef CAS PubMed.
- A. David, C. Kallay, D. Sanna, N. Lihi, I. Sovago and K. Varnagy, Dalton Trans., 2015, 44, 1709 RSC.
- G. Gran, Acta Chem. Scand., 1950, 4, 559 CrossRef CAS.
- P. Gans, A. Sabatini and A. Vacca, J. Chem. Soc., Dalton Trans., 1985, 1195 RSC.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739 CrossRef CAS PubMed.
- L. Pettit and H. K. J. Powell, The IUPAC Stability Constants Database, IUPAC, London, 1992–2002 Search PubMed.
- L. Alderighi, P. Gans, A. Ienco, D. Peters, A. Sabatini and A. Vacca, Coord. Chem. Rev., 1999, 184, 311 CrossRef CAS.
- J. R. Brender, K. Hartman and R. P. R. Nanga, et al. , J. Am. Chem. Soc., 2010, 132, 8973 CrossRef CAS PubMed.
- J. R. Brender, K. Hartman, K. R. Reid, R. T. Kennedy and A. Ramamoorthy, Biochemistry, 2008, 47, 12680 CrossRef CAS PubMed.
- J. R. Brender, J. Krishnamoorthy and G. M. L. Messina, et al. , Chem. Commun., 2013, 49, 3339 RSC.
- S. Salamekh, J. R. Brender and S. J. Hyung, et al. , J. Mol. Biol., 2011, 410, 294 CrossRef CAS PubMed.
- A. Masad, L. Hayes and B. J. Tabner, et al. , FEBS Lett., 2007, 581, 3489 CrossRef CAS PubMed.
- A. Masad, B. J. Tabner, J. Mayes and D. Allsop, Free Radicals Biol. Med., 2011, 51, 869 CrossRef CAS PubMed.
- B. J. Tabner, O. M. A. El-Agnaf and S. Turnbull, et al. , J. Biol. Chem., 2005, 280, 35789 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6dt00628k |
| This journal is © The Royal Society of Chemistry 2016 |