
Can mid-infrared biomedical spectroscopy of cells, fluids and tissue aid improvements in cancer survival? A patient paradigm
Caryn
Hughes
*ab and
Matthew J.
Baker
*b
aSchool of Chemical Engineering & Analytical Sciences, Faculty of Engineering & Physical Science, University of Manchester, Brunswick Street, Manchester, M13 9PL, UK. E-mail: caryn.hughes@manchester.ac.uk; Web: http://www.twitter.com/Dr_Caryn_Hughes
bWestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, Technology and Innovation Centre, 99 George Street, Glasgow, G1 1RD, UK. E-mail: matthew.baker@strath.ac.uk; Web: http://www.twitter.com/ChemistryBaker
First published on 19th October 2015
Abstract
This review will take a fresh approach from the patient perspective; offering insight into the applications of mid-infrared biomedical spectroscopy in a scenario whereby the patient presents with non-specific symptoms and via an extensive diagnostic process multiple lesions are discovered but no clear sign of the primary tumour; a condition known as cancer of unknown primary (CUP). With very limited options to diagnose the cancer origin, treatment options are likely to be ineffective and prognosis is consequentially very poor. CUP has not yet been targeted by infrared biospectroscopy, however, this timely, concise dissemination will focus on a series of research highlights and breakthroughs from the field for the management of a variety of cancer-related diseases – many examples of which have occurred within this year alone. The case for integration of mid-infrared (MIR) technology into clinical practice will be demonstrated largely via diagnostic, but also therapeutic and prognostic avenues by means of including cytological, bio-fluid and tissue analysis. The review is structured around CUP but is relevant for all cancer diagnoses. Infrared spectroscopy is fast developing a reputation as a valid and powerful tool for the detection and diagnosis of cancer using a variety of sample formats. The technology will produce data and tools that are designed to complement routine clinical practice; enhancing the ability of the clinician to make a reliable and non-subjective decision and enabling decreased levels of mortality and morbidity and gains in patient quality of life.
 Caryn Hughes | Caryn Hughes obtained a Master of Chemistry MChem with Industrial Experience in 2007, followed by an Analytical Science PhD entitled ‘Development of FTIR for Drug Response Analysis’ at the University of Manchester in 2011. Caryn has since worked as an interdisciplinary Postdoctoral Research Associate in groups including the Genito Urinary Cancer Research Group (led Prof. Noel Clarke) at the Cancer Research UK Manchester Institute, the Analytical and Medical Vibrational Spectroscopy Group at the Manchester Institute of Biotechnology (led Prof. Peter Gardner), the Instrumentation and Analytical Science group (led Dr Philip Martin) at the University of Manchester and the Bioanalytical Science Research Group (led by Dr Matthew Baker) at the University of Strathclyde. The focus of her research is biomedical infrared spectroscopy, working with biofluids, cells and tissue sample formats, for diagnostic and investigative disease state analysis. |
 Matthew J. Baker | Matt Baker obtained his PhD from the Department of Chemical Engineering and Analytical Science, University of Manchester based in the Manchester Interdisciplinary Biocentre. Following this he was awarded an EPSRC Fellowship conducting research at the Robert Koch Institute and Harvard Medical School. He was then a Project Manager, Senior Scientist and Research Fellow for Dstl and Senior Lecturer at UCLan. He is currently a Senior Lecturer in the Department of Pure and Applied Chemistry, University of Strathclyde based in the Technology Innovation Centre. His research aims are to understand the composition and behavior of molecules within complex matrices related to real-world detection challenges. |
Introduction
Imagine the scenario of an individual who presents with non-specific symptoms including a loss of appetite, unexplained weight loss, constantly lethargy and breathlessness. All the symptoms can be caused by conditions other than cancer, but the GP makes a referral to hospital for tests and for specialist advice. Following exhaustive analysis of medical history, full blood count, urinalysis, stool occult, imaging including chest X-ray computed topography of the thorax, abdomen and pelvis and well as mammography, magnetic resonance imaging and position emission topography, there was evidence of metastatic growths throughout the body, but a failure in detection of the primary tumour.1 In this situation, the patient is diagnosed with a cancer of unknown primary (CUP) following an extensive diagnostic pathway.CUP ranks among the 10 most common malignancies in developed societies, accounting for 3–5% of incidence in both genders.1 The clinical presentation of CUP is extremely heterogeneous. Patients are known to present with a single sight of involvement such as the liver, bone, lung or lymph nodes. However, over half of patients present with multiple sites of involvement.2 Cancer Research UK reported data of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 470 cases of CUP in 2010 broken down into four classifications in order of incidence rate 1 Malignant neoplasm without specification of site, 48%; 2 Secondary malignant neoplasm of respiratory and digestive organs, 32%; 3 Secondary malignant neoplasm of other sites, 12%; 4 Secondary and unspecified malignant neoplasm of lymph nodes, 9%. 1 relates to cases where there has been insufficient information to determine the site of primary origin, whereas (2–4) are cases where 1 is true but secondary malignancies have been located. (For full information on CUP breakdown see http://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/cancer-of-unknown-primary). Interestingly, incidence rates differed when broken down across UK region (the most common class in Wales and Northern Ireland was 3).
470 cases of CUP in 2010 broken down into four classifications in order of incidence rate 1 Malignant neoplasm without specification of site, 48%; 2 Secondary malignant neoplasm of respiratory and digestive organs, 32%; 3 Secondary malignant neoplasm of other sites, 12%; 4 Secondary and unspecified malignant neoplasm of lymph nodes, 9%. 1 relates to cases where there has been insufficient information to determine the site of primary origin, whereas (2–4) are cases where 1 is true but secondary malignancies have been located. (For full information on CUP breakdown see http://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/cancer-of-unknown-primary). Interestingly, incidence rates differed when broken down across UK region (the most common class in Wales and Northern Ireland was 3).
Arguably CUP present the most challenging of diagnostic situations and patient survival rates are extremely poor. It is of great importance to determine the cell lineage and likely primary sites of the cancer as tumours are amenable to distinct therapies. A personalised medicine approach may also be key to identifying favourable or treatable subsets of patients.
In order to improve cancer survival rates in general, we must adapt new and effective technologies to complement existing methods for faster and more reliable detection or chemotherapeutic analysis. Mid-infrared (IR) spectroscopy captures the energies of fundamental vibrational modes of biological molecules that generally lie within the range of 400–4000 cm−1, a spectral region corresponding to mid-infrared (mid-IR) electromagnetic radiation. The method involves a non-destructive and label-free process to probe the composition of the chemical environment of the sample, resulting in a unique fingerprint of global biochemistry.
CUP is yet to be targeted by the clinical mid-infrared spectroscopy community however pioneering research has been performed in diagnosis and prognosis of primary and secondary cancers. For a case where there is no clear answer as to where the site of primary malignant origin may have occurred, by analysing the biomolecular fingerprint for cases of known secondary sites, IR spectroscopy could potentially offer indicative spectral biomarkers for diagnostic or prognostic outcomes. CUP does ultimately present as challenging target for the future of the field; in order to offer a glimpse of the potentials for improving the ominous outlook of CUP using mid-IR biomedical spectroscopy, breakthrough milestones and advances will be highlighted out of the extensive research carried out in the field on multiple cancer targets.
Diagnostics: strategies for early detection
It has been reported that cancer patient outcomes in the UK are considerably lower than in other western countries.3,4 Numerous delays can occur to the patients primary and secondary care; however, delays in symptomatic cancer diagnosis play a major role leading to more advanced stages of disease at diagnosis. In 2010 the National Patient Safety Agency (NPSA) published a review entitled ‘Delayed diagnosis of cancer: Thematic review’ presenting findings where patient safety incidents relating to actual or potential delayed cancer diagnosis. The types of patient safety incident were classified as 53% diagnostics (41% pathology, 12% radiology).5 There is therefore, a considerable requirement to make interventions in order to minimise delays and lead to earlier-stage diagnosis.MIR biomedical diagnostics offers numerous platforms in which cells, fluids or tissue can be probed, label-free, in order to achieve a potentially rapid and clinically relevant result. In the case of CUP, early detection is not possible; there are no current screening programmes. Analysis of blood provides an ideal starting point for rapid and minimally invasive sample collection. A patient with CUP may undergo a variety of different blood tests for tumour markers to suggest a certain type of cancer, including prostate-specific antigen (PSA) (prostate), human chorionic gonadotrophin (HCG) (germ cell), CA19-9 (pancreas/bile ducts) and CA125 (ovarian). It is well known, however, that many of these markers are not completely reliable; PSA for is example is notorious for generating false positive results6 and although CA125 is the gold standard tumour marker in ovarian cancer however, different studies have reported contradictory results.7
Attenuated Total Reflection-Fourier Transform Infra Red (ATR-FTIR) spectroscopy is gaining increasing acceptance as a reliable diagnostic tool for detecting cancer using blood based samples and there have been a number of breakthrough examples. Gajjar et al.8 have previously demonstrated such an application to distinguish ovarian and endometrial cancer against non-cancer controls with classification accuracies of up to 96.7% and 81.7% respectively. More recently Lima et al.9 have extended this research to exploitation of IR spectral biomarkers derived from blood plasma or serum analysis to evaluate ovarian cancer stage and type. Remarkably, in a case study for stage I vs. stage II–IV (plasma samples), they reported accuracies of 100% sensitivity and specificity based on the use of 33 spectral features.
The use of ATR-FTIR is certainly a tangible accessory for rapid point of care diagnostics; its operation is simple and requires minimal sample preparation, especially for biofluids.10 Hands et al. have recently reported an FTIR-ATR based diagnostic test for brain cancer that is achievable within 4 minutes.11 They reported that, when used in conjunction with a complementary Bioplex immunoassay, taking 4 hours to complete, a dual result can be made available within a total of 5 hours from sample collection. Furthermore, during a separate study, they were also able distinguish between gliomas (high-grade and low-grade) from non-cancer with an average sensitivity of 93.75% and specificity of 96.53% from blood serum samples of 97 patients.12
In addition to point of need applications, FTIR spectroscopy can also be utilised for high-throughput (HT) diagnostic screening. Ollesch et al.13 have demonstrated such methodology for blood samples of urinary bladder cancer (UBC) patients where robotically prepared thin dried film for measurements in transmission mode. They were able to achieve a sensitivity of 93 ± 10% and a specificity of 46 ± 18% for bladder cancer (low specificity was most likely attributed to the unbalanced and small number of control samples).
Investigations of specific cell types are also important for new diagnostic pathways. For example, peripheral blood mononuclear cells (PBMCs) present a new route for cancer detection based on the immune system response to the presence of tumour, rather than on observation of the tumour cells themselves.14 Zelig et al. have recently evaluated the feasibility of this approach for early detection of breast cancer. The results provide evidence that the PBMCs of patients with breast cancer are biochemically distinct from the PBMCs of healthy subjects, including patients with benign tumours, with no significant differences in PBMC spectra between patients with benign tumours and healthy subjects.
Although there are no specific examples of MIR spectroscopy applied to patients with non-specific symptoms and CUP patient samples, much can be learned, however, from the recent breakthroughs for other cancer targets. During diagnosis is it essential to determine specific cell lineage, such as lymphoma, sarcoma or melanoma in order to make a more informed therapeutic decision. The issue with CUP is that the tumours are often poorly differentiated. This is also an issue for as certain primary tumour sub-variants and sometimes it is often not possible to tissue architecture or cell morphology alone. Fortunately, however, there are have been many MIR-based in vitro studies that have been performed on sub-variants of cancers that have shown it is possible to discriminate between cell types using the spectral bio-fingerprint;15,16 despite situations where the overall cell morphology is not necessarily distinctly unique. Previous studies have inferred that spectral abnormalities have been observed not only detectable in malignant cervical scrapings but also in those with the premalignant condition cervical dysplasia.17 Townsend et al. have recently conducted an IR study on cervical cytological samples (Fig. 1) concluding that MIR spectroscopy indeed can distinguish truly normal cells and cells with normal morphology from the vicinity of abnormalities. During this study, despite, in some cases cells being pathologically diagnosed as healthy squamous, their spectra suggested otherwise, displaying early spectral markers of disease18
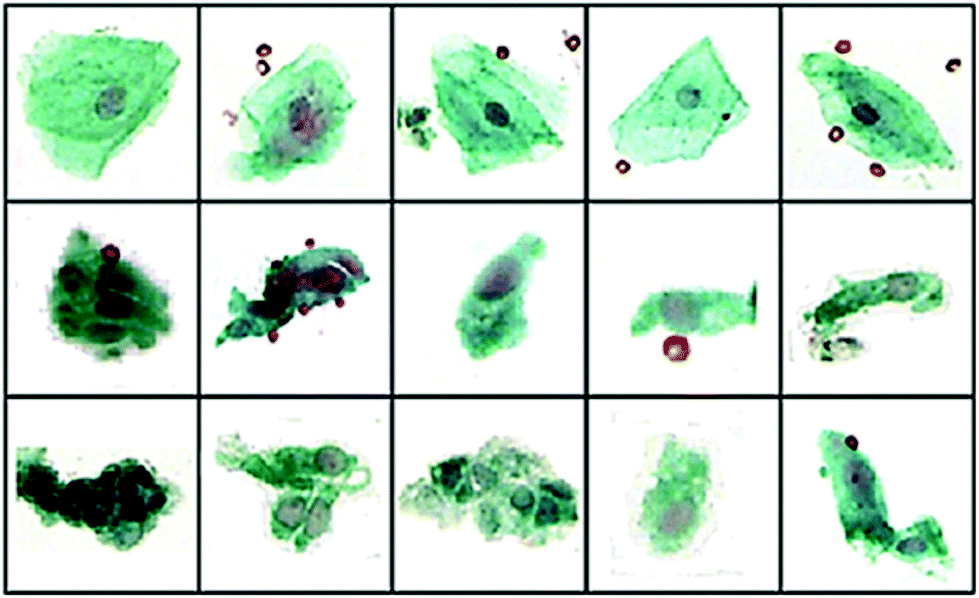 | ||
| Fig. 1 40× visual images of annotated cells some different pathological groups: (top) squamous SQ), (middle) Barrett's Esophageal (BE) and (bottom) dysplasia (DYS). Using an artificial neural network classification of IR spectral data sensitivities and specificities of 95.5% and 94.7% (BE vs. SQ), 88.7% and 91.4% (BE vs. DYS) and 93.4% and 90.0% (SQ vs. DYS) were reported. Adapted with permission from [ref. Townsend et al.18]. | ||
A pathological diagnosis using a tissue biopsy will typically involve the use immunohistochemical/immunofluorescent stains and observation of tissue architecture; a laboriously manual method used for over 150 years, relying on human decision-making.19 Tissue can be imaged with mid-infrared spectrometers coupled to a microscope with a focal plane array detector, creating a stain-free, global molecular fingerprint within a spatially-resolved hyperspectral map. This can be performed on both formalin fixed paraffin embedded (FFPE) tissue (either dewaxed20 or still embedded in paraffin wax21) or snap-frozen tissue. Typically analysis of tissue using this method involves identification of tissue structures (such as epithelial, stroma, blood, bone) before diagnosis in terms healthy, benign, malignant and/or extent of malignancy.22
Bird et al.23 have previously shown that for lung cancer, MIR biomedical spectroscopy can distinguish major classes (adenocarcinoma, squamous cell carcinoma, small cell carcinoma and bronchioloalveolar carcinoma). Specifically, this can be achieved with high sensitivity, specificity and accuracy for small cell carcinoma vs. non-small cell carcinoma (91.2%, 98.0% and 94.6%) but low sensitivity, specificity and accuracy for adenocarcinoma vs. bronchioloalveolar carcinoma (88.8%, 47.2% and 68.0%). Similarly, Großerueschkamp et al. recently presented (Fig. 2) the ability to discriminate between small cell, hamartochondroma, squamous cell, carcinoid, pleura mesothelioma, adenocarcinoma, thymoma and inflammation/necrosis but were able to perform adenocarcinoma subtyping (acinar, lepidic, papillary and micropapillary)24 as Akalin et al.25 have also reported similar findings and as aforementioned, the differentiation of subtypes is especially important for prognosis and therapeutic decision.
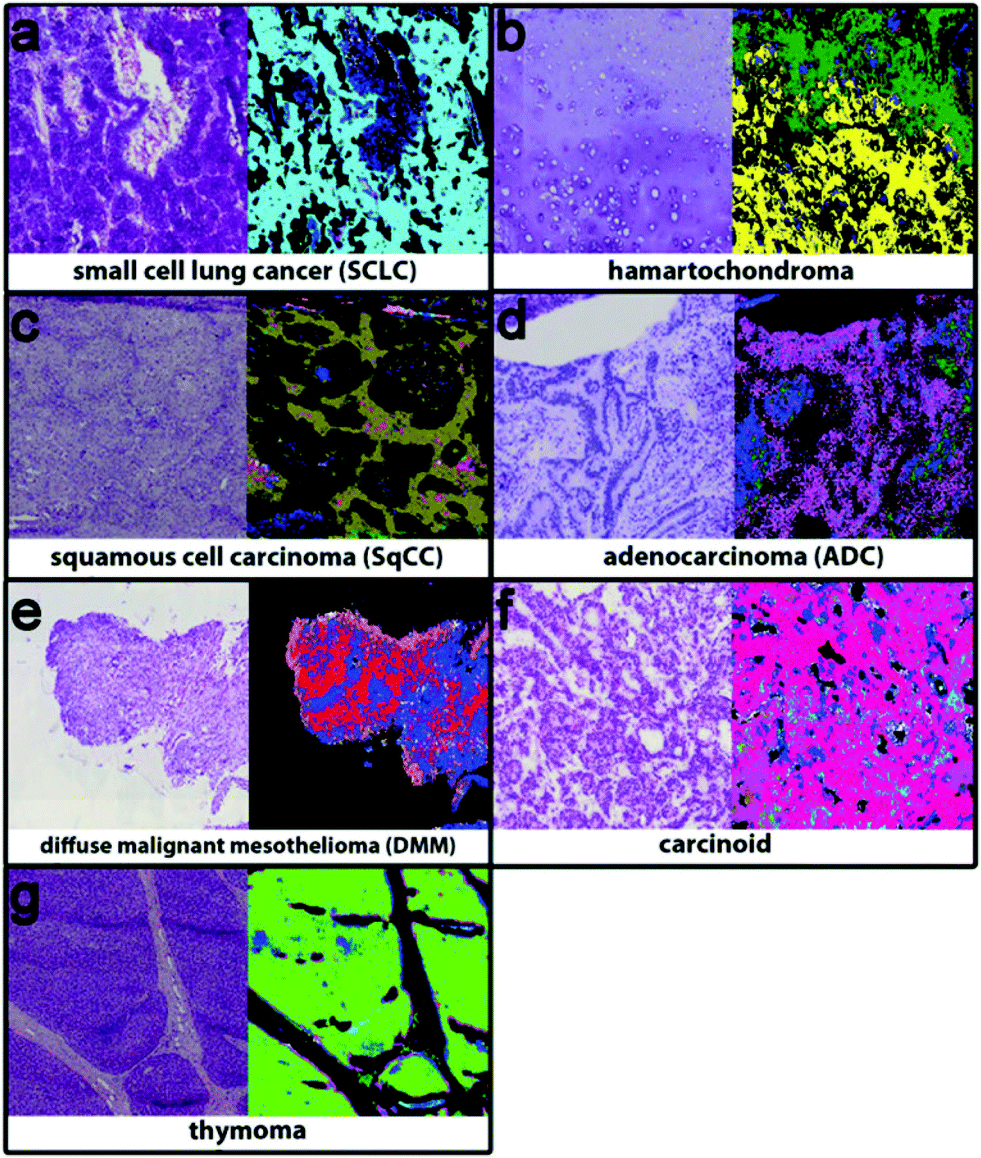 | ||
| Fig. 2 Using IR imaging and random forest classifiers to discriminate between (a) small cell cancer (cyan), (b) hamartochondroma (yellow), (c) squamous cell carcinoma (olive), (d) carcinoid (magenta), (e) pleura mesothelioma (red), (f) adenocarcinoma (pink), and (g) thymoma (light green), and inflammation/necrosis (blue and dark green). Adapted with permission from [ref. Großerueschkamp et al.24]. | ||
As a further example, transitional cell carcinoma (TCC), accounting for 85–90% of all epithelial tumours of the bladder and urinary collecting system, is heterogeneous with a pronounced ability for divergent differentiation and has many sub-variant phenotypes as a consequence. Tissue structure discrimination IR-Imaging has also been successfully applied to TCC and its sub-variants such as clear cell, which is difficult to identify in poorly differentiated carcinoma of the upper tract.26
Until recently, one of the limiting factors in IR imaging has been the limited pixel resolution (typically ∼5.5 μm). Although higher magnification optics have previously been available, the use of non-customised objectives, have generally resulted in extremely poor SNR making them unsuitable for routine applications. The most advanced focal plane array setup with a synchrotron-source in terms of pixel resolution was developed at the IRENI beamline (Synchrotron Radiation (SR) Center, WI, USA) which combined 12 separate SRIR beam to enable diffraction-limited imaging using a pixel size of ∼0.5 μm, allowing for subcellular chemical imaging.27–29 Now, however, high definition (HD) IR imaging is more routinely possible on a bench-top FTIR spectrometer, achieving a pixel resolution of ∼1.1 μm using a standard thermal source.30,31 For some clinical applications, a pixel resolution of ∼5.5 μm would be appropriate for cytological screening when isolating single cell spectra from cultured cells as demonstrated by Mazur et al.32 and Filik et al.33 However, for instances where there may be a clinical requirement to isolate the IR signature of single cells condensed within a tissue cross-section where cells therefore appear smaller in magnitude, increasing the effective pixel size is of use to more accurately resolve the cells spatially and to reduce ‘pixel mixing’ effects of neighbouring tissue structures.
The availability of benchtop instrument HD FT-IR imaging is a very recent technological advancement and is an exciting progressive step in terms of targeting and differentiating cancer-related tissue sub-structure. As an example target, lymph nodes are particularly important as immune response initiators they are routinely used in diagnostic and prognostic evaluations for cancer and chronic inflammation or infection. In the case of the CUP patient, up to 10% of all neck lymph node metastases present without a known primary site and the optimal treatment strategy for these patients is still undefined.34 Evaluation of lymph nodes is often involved in efforts to attempt the determination of the primary site. Lymph nodes are highly heterogeneous, however, due to a constant stream of lymphocytes, macrophages, and antigen cells ever present in the fibrovascular tissue. Excitingly, Leslie et al.23 been able to demonstrate that cells of eight distinct classes (naïve and memory B cells, T cells, erythrocytes, connective tissue, fibrovascular network, smooth muscle, and light and dark zone activated B cells) currently identifiable only through immunohistochemical stains and manual recognition via optical microscopy, can be distinguished through a single IR spectroscopic image; this was achievable for the first time through the benefits of HD-IR imaging. Furthermore, they were able to achieve this in healthy, reactive, and malignant lymph node biopsies using a random forest classifier.
The advent of commercially available quantum cascade laser (QCL) IR microscopes will only further enhance the diagnostic tool status of mid-infrared imaging. With this technology, the conventional thermal source in a benchtop IR system has been exchanged with high-intensity tuneable lasers with a customisable frequency range (Fig. 3). Coupled with a room-temperature detector, high definition IR images can be collected allowing the operator to scan faster (in discrete frequency mode) and for longer (without the time-limit worry associated with the eventual warming of typical FTIR spectrometer detectors cooled at cryogenic temperatures).35–38
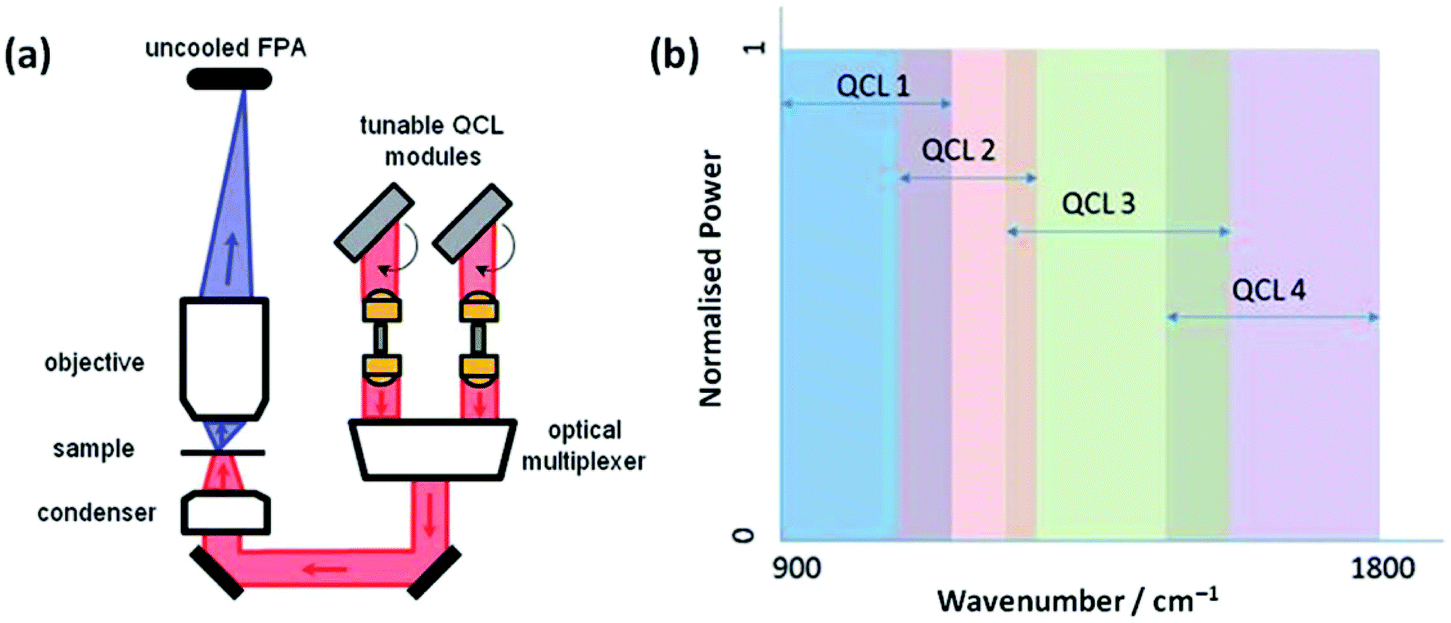 | ||
| Fig. 3 (a) Schematic diagram of the QCL-based Spero spectral imaging microscope shown for transmission mode. The system also operates in reflection mode. (b) Plot illustrating the tunable ranges of the 4 individual QCLs providing continuous coverage of the 900–1800 cm−1 wavenumber range. Adapted with permission from [ref. Bassan et al.36]. | ||
Therapeutic: personalised treatment
Optimal therapeutic management of patients with CUP remains controversial as there is a lack of randomized studies comparing different treatment options and as a result, treatment is mainly based on non-randomized evidence and institutional policy. Recommendations include surgery alone, radiotherapy with or without concomitant chemotherapy. However, the morbidity and quality of life cost of these combined treatments may be high. No randomized or prospective studies are currently available to specifically support these approaches and such a study may be difficult to undertake, as this disease is rare.39If a patient was able to benefit from personalised oncology, a resolved indicative signature of chemosensitivity could have the power to guide treatment decisions. The genomics revolution has great potential and a genomic signature that can predict a patient outcome is revolutionary, but it is still early days and there have been very few assays that have come out of the genomics revolution in the last 12 years.40 In addition to genetic mutation analysis, cellular chemosensitive assays grown from primary patient cells could also be used as a treatment-guiding tool.
There have been many in vitro based infrared studies of chemotherapeutic action on cancer cell lines which has involved both single point measurements and FPA imaging of chemically fixed and dried cells.41–46 As a clinical example, Zelig et al. have used FTIR spectroscopic markers of leukaemia in peripheral blood mononuclear cells (PBMCs) for initial pre-screening and follow-up of individual patient response to chemotherapy.47 They were able to follow progression during chemotherapy largely based on lipid IR absorptions, which have been demonstrated as a spectral marker of apoptosis.48,49 They were unable to predict the treatment outcomes based on the PBMC spectra obtained in pre-treatment days, however, it was shown that FTIR spectroscopy could provide additional information on chemotherapeutic response of an individual patient basis; an application that may be vital to identify unexpected complications in real time as soon as they may appear during treatment.
In a similar fashion to cytotoxic assays used in drug discovery, there is a lack of appropriate in vitro models that have the capability to predict the chemotherapeutic response for each patient. In vitro models often fail to predicting in vivo efficacy of specific chemotherapeutic agents and are thus starting to be replaced by multicellular tumour spheroid (MTS) models that more realistically reflect the behaviour of cells in tumour tissues.50 In terms of IR research in this area, Smolina et al. recently reported direct comparison of cell phenotypes using a triple-negative breast cancer cell line, MDA-MB-231 grown in either standard two-dimensional (2D) or 3-dimentional (3D) laminin-rich extracellular matrix (lrECM) culture (Fig. 4).51 In a comparison with spectra of carcinoma cells from clinical patient samples, principal component analysis (PCA) indicated a separation between the 2D versus the 3D and clinical scores across the first principal component. While cell lines will never have phenotypes absolutely identical to those found in tissues, it does show promise that, as an early deduction, chemosensitivity may possibly be inferred in cells harvested from clinical biopsies of a suspected phenotype via correlation with features in 3D cultured cell lines of known phenotype.
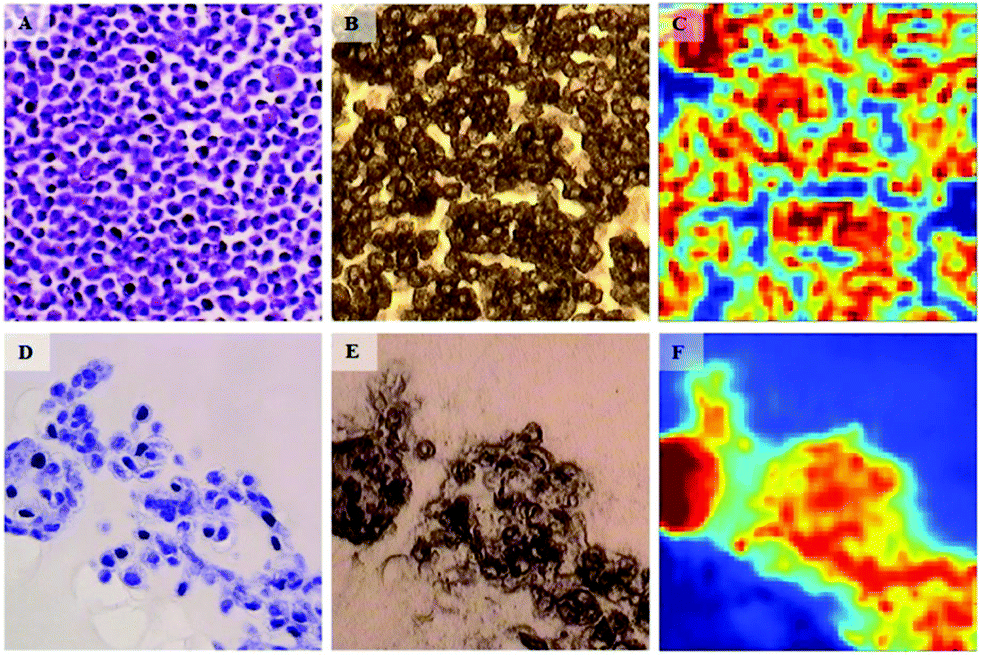 | ||
| Fig. 4 Bright-field microscopy (A, B, D, E) and IR (C, F) images of adjacent 4 μm-thick sections of MDA-MB-231 breast cancer cells grown in 2D (A–C) and 3D (D–F) cultures. For 2D culture, image A shows the H&A stained section. The adjacent, unstained section is presented in image B. The IR raw image (absorbance at 1654 cm−1) of the same region is reported in image C. For the 3D culture, image D shows the H&A stained section. The adjacent, unstained section is presented in image E, and the IR raw image (absorbance at 1654 cm−1) of the same region is reported in F. One covers an area of 180 × 180 μm2. Adapted with permission from [ref. Smolina et al.51]. | ||
Ruppen et al.52 have recently reported on the design of a microfluidic device that is able to form single spheroids of uniform size from either cell line or a limited number of human primary cells thus pinpointing an exciting direction of personalized oncology and live cell analysis would be a great advantage. Single cell IR measurements in the aqueous environment have initially benefitted by the use of ultrabright synchrotron radiation sources due to the fact that transmission measurements in an aqueous environment are constrained by the strong IR absorption of water which consequentially decreases light throughput.53,54 Chan and Kazarian, however, have recently pioneered IR imaging of live cancer cells in the natural aqueous environment by ATR-FTIR using a regular benchtop thermal source and were able to monitor fine chemical and physical changes in situ with very fast acquisition time, good spatial resolution and high molecular specificity.55 Furthermore, Marcsisin et al. have demonstrated that with even without ATR or a synchrotron source, the use of live cell FTIR imaging can produce interpretable spectral differences from live cells treated with chemotherapeutic drugs with optimised instrumentation and data processing methodology.56 The ability to collect data on a commercially available system with no required modification increases the potential to integrate the technology more easily into the clinical setting. The data processing methodology means that a larger quantity of cellular data can be acquired at a faster rate without compromising on spectral quality, therefore allowing results to be more statistically meaningful.
Prognosis: risk of progression/recurrence
Metastatic spread is generally considered the final step of tumour progression, however this is not necessarily the case for CUP. There are at least two different hypotheses which attempt to explain CUP biology; CUP are a heterogeneous group of site-specific tumours which share the properties of the small primary from which they derive, or conversely, CUP are a distinct biological entity that involve specific genetic and phenotypic alterations. At present, there are no known and validated molecular features to clearly distinguish these cancers, making them again an ideal target for assessment by MIR spectroscopy.57There is MIR spectroscopic evidence to suggest tumour progression to the metastatic state involves structural modifications in DNA that are markedly different from the modifications associated with the formation of the primary tumour.58 Cancer cells acquire cell-autonomous capacities to undergo limitless proliferation and as a result are thought to require the support of the tumour microenvironment (TME) (containing carcinoma-associated fibroblasts, leukocytes, bone marrow-derived cells, blood and lymphatic vascular endothelial cells) in order to progress.59 In a controlled study, Ukkonen et al. recently identified the influence of cancer cells in the TME using MIR imaging of an in vitro myoma organotypic model with invading melanoma and oral tongue carcinoma cell lines. They determined that amide and collagen triplet absorption regions could serve as spectral markers for cancer-induced modifications in the TME.60
These preliminary results are also supported by an independent MIR imaging study applied to histopathological specimens of breast cancer of different tumour histological grades.61 MIR spectral changes were observed when examining the extracellular matrix close to and far from carcinoma, in particular in the relative intensities of the collagen bands at 1640 and 1630 cm−1 illustrated a change in collagen spectral features while moving away from the tumour (Fig. 5). These results support the notion that the 1700–1600 cm−1 spectral range could be used as spectral markers to identify cancer-induced modifications in the TME.
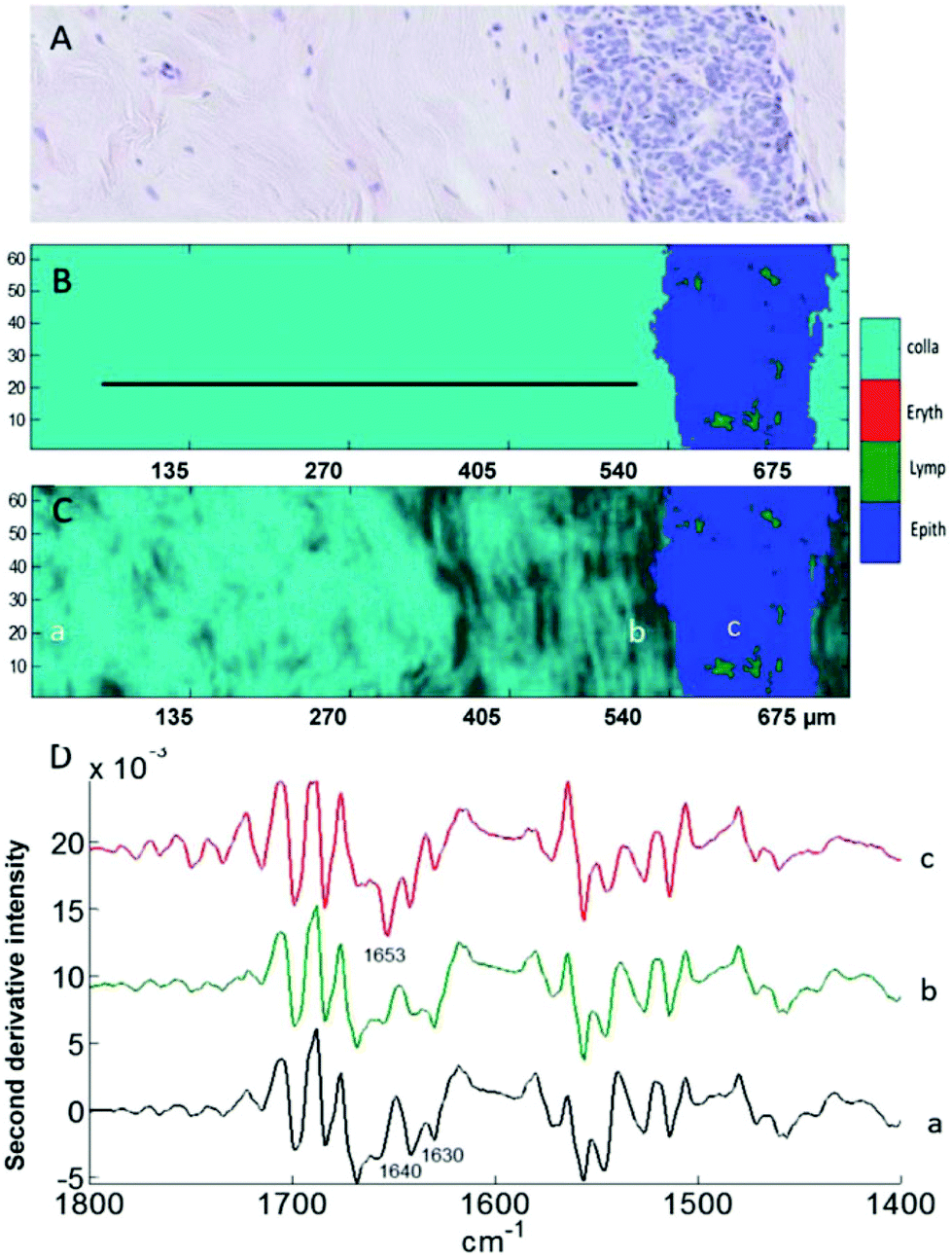 | ||
| Fig. 5 Analysis of a breast tissue sample. (A) H&E stained section of breast tissue showing the extracellular matrix on the left and a mammary duct invaded by grade 2 carcinoma cells on the right. (B and C) Images generated by discriminant partial least squares. The ratio of intensities at 1630/1640 cm−1 is used in (C) for sub-classification of collagen (light to dark). (D) Mean second derivative spectra taken from the three areas a, b, and c indicated in (C) corresponding to far-from-tumour collagen (a), close-to-tumour collagen (b) and to the cells present inside the tumour region (c). Adapted with permission from [ref. Kumar et al.61]. | ||
There will be some cancers that can be managed via waitful watching or active surveillance. Active surveillance is suitable when the cancer is considered to be in the early stages and localised. It usually involves regular hospital tests, such as biopsies and MRI scans (waitful watching requires less frequent tests). There are a number of reasons why it may be the case not to intervene with aggressive chemotherapy of radical surgery. As a primarily age-related disease it is sometimes wise not take drastic measures in attempt to neutralise the risk due to the change of considerably affecting quality of life; in the case of prostate cancer, for example, there is an element of overtreatment in lower risk patient cohorts.62 Certainly, in terms of prognostic markers, much more research still needs to be done. The integration of gene expression, immunohistochemistry, magnetic resonance imaging and tissue morphology has met with limited success and as yet there is no widespread clinical acceptance. While much progress has been made in diagnostic applications of MIR biospectroscopy, there are very few reports of prognostic capability; naturally as it is such a challenging area. In an exciting breakthrough reported this year, Kwak et al.63 have shown that a tissue-based IR spectral pathology approach outperforms Kattan nomogram and CAPRA-score for predicting risk of recurrence in mid-grade dominant cases of prostate cancer. They also determined that an IR score has a truly independent predictive power; a combined score (IR + Kattan + CAPRA-S) was not able to significantly improve upon the use of IR score alone.
Knowledge of the underlying factors of tumour recurrence is also not fully understood. Over the last few decades, a number of hypotheses have been proposed including clonal selection and angiogenic dormancy. More recently the cancer stem cell (CSC) hypothesis64 has sparked controversial option. CSCs are believed to persist in tumours as distinct populations that are fundamentally associated with drug resistance, tumour recurrence, and metastasis. Assessment of CSCs via standard techniques in molecular biology often require large quantities of cells (often to the power of 105/6), however, yet IR spectroscopy offers a unique opportunity to study and characterise CSCs on the single cell level which may help to increased understanding of the cancer pathology.65,66 Moreover, the migration ability of CSCs have also been proposed to explain the non-existence of the primary cancer in CUP25 and in for many cancers CSC-targeted therapeutic strategies for therapeutic treatment are being assessed.67 As discussed in the previous section, there would also be an opportunity for MIR spectroscopy to serve as an investigative tool for therapeutic research.
Conclusions
Many milestones and improvements have been made not only in the technology but also in fundamental understanding of the MIR techniques. As demonstrated, in countless examples of research across cell fluid and tissue investigations, the ability to distinguish between non-cancer and cancer states using label-free infrared spectral biomarkers is undeniable. The number of oncological research applications will inevitably rise as a result and hopefully the field will become increasingly internationally recognised and accepted.For the CUP and for non-symptomatic patients, outlook is currently very poor in terms of diagnostic, therapeutic and prognostic options. It is evident, as demonstrated by the breadth and success of oncological research in this review, that MIR spectroscopy is on course to be a major authoritative tool in oncology. Fig. 6 portrays a schematic of the potential use of MIR throughout the patient's clinical journey. As a positive prospect, the limitations of the MIR biomedical spectroscopy could be tested by playing a significant role in CUP research; alongside the advent of modern molecular techniques such as DNA, gene expression profiling and proteomics.
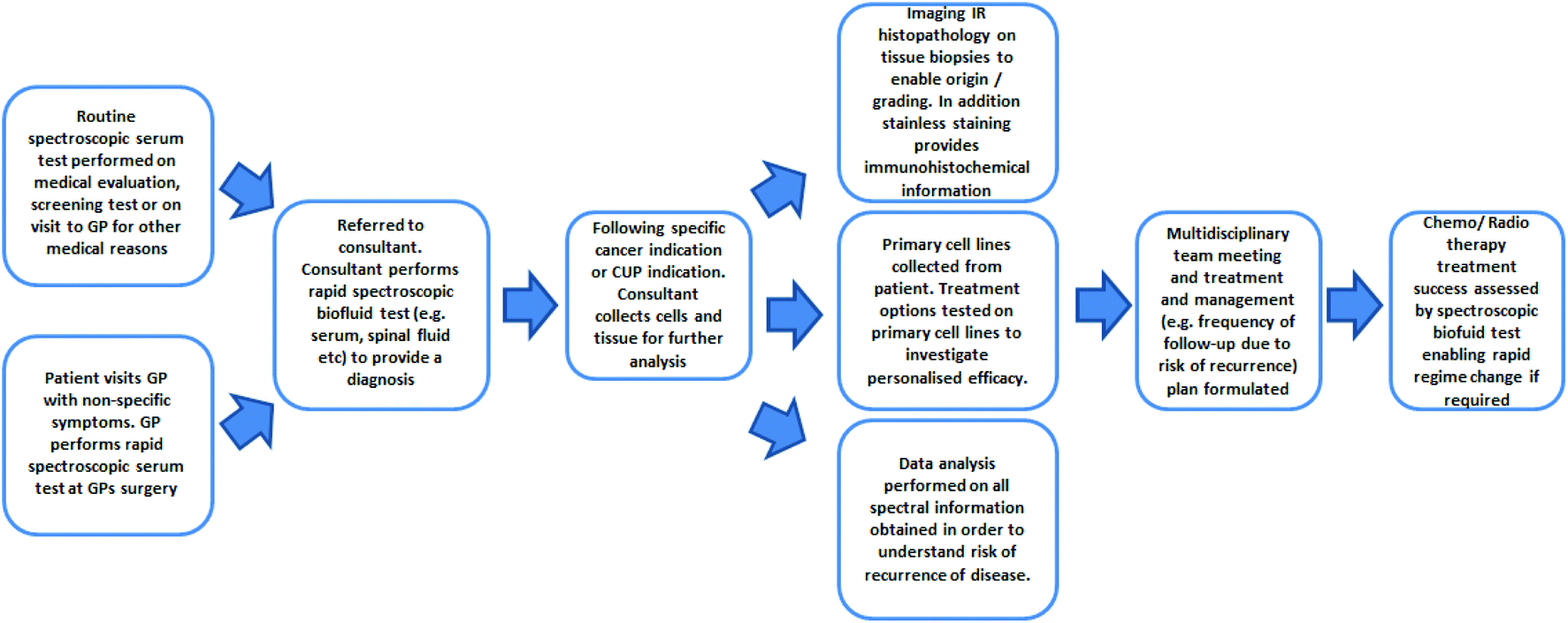 | ||
| Fig. 6 Schematic of the potential use of FTIR in the patient's clinical journey (adapted and updated from [ref. Dorling and Baker69]). | ||
MIR biomedical spectroscopy has recently gained significant recognition as part of the creation of the government-funded Clinical Infrared and Raman Spectroscopy for Medical Diagnosis Network68 which is a step forward in gaining clinical acceptance. In order to realise the goal of translating biomedical spectroscopy benefits to address unmet needs in oncology, however, great efforts must be made on behalf of academic, industrial and clinical partners in order to secure its place in the clinical environment. As demonstrated, proof of the concept has been demonstrated in many small but controlled studies and clinically significant results are required to truly capture the interest of healthcare practitioners. Reaching this next plateau, therefore, requires the pooled community to consider sample size and power in clinical trials. Finally, as commercialisation is key to translating the science to the clinic, industrial partners will then be more likely to enter these untapped markets in the clinical and life sciences, making FTIR spectroscopy-based healthcare products and services a reality.
Acknowledgements
MJB would like to thank EPSRC, Dstl, Rosemere Cancer Foundation, Brain Tumour North West, and the Sydney Driscoll Neuroscience Foundation for funding. This review is dedicated to the memory of Stephen Tattum, who passed away due to cancer of unknown primary origin.References
- N. Pavlidis and G. Pentheroudakis, Ann. Oncol., 2010, 21, vii303–vii307 CrossRef PubMed.
- E. Briasoulis and N. Pavlidis, Oncologist, 1997, 2, 142–152 Search PubMed.
- M. P. Coleman, D. Forman, H. Bryant, J. Butler, B. Rachet, C. Maringe, U. Nur, E. Tracey, M. Coory, J. Hatcher, C. E. McGahan, D. Turner, L. Marrett, M. L. Gjerstorff, T. B. Johannesen, J. Adolfsson, M. Lambe, G. Lawrence, D. Meechan, E. J. Morris, R. Middleton, J. Steward and M. A. Richards, Lancet, 2011, 377, 127–138 CrossRef CAS.
- R. D. Neal, Br. J. Cancer, 2009, 101, S9–S12 CrossRef PubMed.
- E. Minghella, M. Lakhani, C. Hughes and B. Thomas, Delayed diagnosis of cancer: Thematic Review, Ref. 0968, 2010, NHS National Patient Safety Agency, 1–46.
- H. B. Carter, BJU Int., 2013, 112, 543–547 CrossRef PubMed.
- D. Gupta and C. Lis, J. Ovarian Res., 2009, 2, 13 CrossRef PubMed.
- K. Gajjar, J. Trevisan, G. Owens, P. J. Keating, N. J. Wood, H. F. Stringfellow, P. L. Martin-Hirsch and F. L. Martin, Analyst, 2013, 138, 3917–3926 RSC.
- K. M. G. Lima, K. B. Gajjar, P. L. Martin-Hirsch and F. L. Martin, Biotechnol. Prog., 2015, 31, 832–839 CrossRef CAS PubMed.
- F. Bonnier, F. Petitjean, M. J. Baker and H. J. Byrne, J. Biophotonics, 2014, 7, 167–179 CrossRef CAS PubMed.
- J. Hands, P. Abel, K. Ashton, T. Dawson, C. Davis, R. Lea, A. McIntosh and M. Baker, Anal. Bioanal. Chem., 2013, 405, 7347–7355 CrossRef CAS PubMed.
- J. R. Hands, K. M. Dorling, P. Abel, K. M. Ashton, A. Brodbelt, C. Davis, T. Dawson, M. D. Jenkinson, R. W. Lea, C. Walker and M. J. Baker, J. Biophotonics, 2014, 7, 189–199 CrossRef CAS PubMed.
- J. Ollesch, S. L. Drees, H. M. Heise, T. Behrens, T. Bruning and K. Gerwert, Analyst, 2013, 138, 4092–4102 RSC.
- U. Zelig, E. Barlev, O. Bar, I. Gross, F. Flomen, S. Mordechai, J. Kapelushnik, I. Nathan, H. Kashtan, N. Wasserberg and O. Madhala-Givon, BMC Cancer, 2015, 15, 408 CrossRef PubMed.
- T. J. Harvey, E. Gazi, A. Henderson, R. D. Snook, N. W. Clarke, M. Brown and P. Gardner, Analyst, 2009, 134, 1083–1091 RSC.
- T. J. Harvey, A. Henderson, E. Gazi, N. W. Clarke, M. Brown, E. C. Faria, R. D. Snook and P. Gardner, Analyst, 2007, 132, 292–295 RSC.
- M. A. Cohenford and B. Rigas, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 15327–15332 CrossRef CAS.
- D. Townsend, M. Miljkovic, B. Bird, K. Lenau, O. Old, M. Almond, C. Kendall, G. Lloyd, N. Shepherd, H. Barr, N. Stone and M. Diem, Analyst, 2015, 140, 2215–2223 RSC.
- N. Pavlidis and K. Fizazi, Crit. Rev. Oncol. Hematol., 2005, 54, 243–250 CrossRef PubMed.
- C. Hughes, L. Gaunt, M. Brown, N. W. Clarke and P. Gardner, Anal. Methods, 2014, 6, 1028–1035 RSC.
- P. Bassan, A. Sachdeva, J. H. Shanks, M. D. Brown, N. W. Clarke and P. Gardner, Progress in Biomedical Optics and Imaging - Proceedings of SPIE, San Diego, CA, 2014 Search PubMed.
- M. J. Baker, J. Trevisan, P. Bassan, R. Bhargava, H. J. Butler, K. M. Dorling, P. R. Fielden, S. W. Fogarty, N. J. Fullwood, K. A. Heys, C. Hughes, P. Lasch, P. L. Martin-Hirsch, B. Obinaju, G. D. Sockalingum, J. Sulé-Suso, R. J. Strong, M. J. Walsh, B. R. Wood, P. Gardner and F. L. Martin, Nat. Protocols, 2014, 9, 1771–1791 CAS.
- B. Bird, M. S. Miljkovic, S. Remiszewski, A. Akalin, M. Kon and M. Diem, Lab. Invest., 2012, 92, 1358–1373 CrossRef PubMed.
- F. Großerueschkamp, A. Kallenbach-Thieltges, T. Behrens, T. Brüning, M. Altmayer, G. Stamatis, D. Theegarten and K. Gerwert, Analyst, 2015, 140, 2114–2120 RSC.
- A. Akalin, X. Mu, M. A. Kon, A. Ergin, S. H. Remiszewski, C. M. Thompson, D. J. Raz and M. Diem, Lab. Invest., 2015, 95, 406–421 CrossRef PubMed.
- C. Hughes, J. Iqbal-Wahid, M. Brown, J. H. Shanks, A. Eustace, H. Denley, P. J. Hoskin, C. West, N. W. Clarke and P. Gardner, J. Biophotonics, 2013, 6, 73–87 CrossRef CAS PubMed.
- E. C. Mattson, M. Unger, B. Manandhar, Z. Alavi and C. J. Hirschmugl, J. Phys.: Conf. Ser., 2013, 425 Search PubMed.
- M. J. Nasse, M. J. Walsh, E. C. Mattson, R. Reininger, A. Kajdacsy-Balla, V. MacIas, R. Bhargava and C. J. Hirschmugl, Nat. Methods, 2011, 8, 413–416 CrossRef CAS PubMed.
- M. J. Walsh, M. J. Nasse, F. N. Pounder, V. Macias, A. Kajdacsy-Balla, C. Hirschmugl and R. Bhargava, AIP Conference Proceedings, Banff, AB, 2010 Search PubMed.
- C. Hughes, A. Henderson, M. Kansiz, K. M. Dorling, M. Jimenez-Hernandez, M. D. Brown, N. W. Clarke and P. Gardner, Analyst, 2015, 140, 2080–2085 RSC.
- C. R. Findlay, R. Wiens, M. Rak, J. Sedlmair, C. J. Hirschmugl, J. Morrison, C. J. Mundy, M. Kansiz and K. M. Gough, Analyst, 2015, 140, 2493–2503 RSC.
- A. I. Mazur, E. J. Marcsisin, B. Bird, M. Miljković and M. Diem, Anal. Chem., 2012, 84, 8265–8271 CrossRef CAS PubMed.
- J. Filik, A. V. Rutter, J. Sule-Suso and G. Cinque, Analyst, 2012, 137, 5723–5729 RSC.
- H. Christiansen, R. Hermann, A. Martin, M. Nitsche, H. Schmidberger and O. Pradier, Strahlenther. Onkol., 2005, 181, 355–362 CrossRef PubMed.
- L. S. Leslie, T. P. Wrobel, D. Mayerich, S. Bindra, R. Emmadi and R. Bhargava, PLoS One, 2015, 10, e0127238 Search PubMed.
- P. Bassan, M. J. Weida, J. Rowlette and P. Gardner, Analyst, 2014, 139, 3856–3859 RSC.
- C. Kristament, M. Brandstetter, A. Schwaighofer, M. R. Alcaraz and B. Lendl, CLEO: Science and Innovations, CLEO-SI 2015, 2015.
- K. Yeh, S. Kenkel, J. N. Liu and R. Bhargava, Anal. Chem., 2014, 87, 485–493 CrossRef PubMed.
- S. M. Zhuang, X.-F. Wu, J.-J. Li and G.-H. Zhang, Mol. Clin. Oncol., 2014, 2, 917–922 Search PubMed.
- D. Hayes, H. Markus, R. Leslie and E. Topol, BMC Med., 2014, 12, 37 CrossRef PubMed.
- G. Bellisola, G. Cinque, M. Vezzalini, E. Moratti, G. Silvestri, S. Redaelli, C. G. Passerini, K. Wehbe and C. Sorio, Analyst, 2013, 138, 3934–3945 RSC.
- G. Bellisola, M. Della Peruta, M. Vezzalini, E. Moratti, L. Vaccari, G. Birarda, M. Piccinini, G. Cinque and C. Sorio, Analyst, 2010, 135, 3077–3086 RSC.
- A. Derenne, M. Verdonck and E. Goormaghtigh, Analyst, 2012, 137, 3255–3264 RSC.
- F. Draux, P. Jeannesson, C. Gobinet, J. Sule-Suso, J. Pijanka, C. Sandt, P. Dumas, M. Manfait and G. D. Sockalingum, Anal. Bioanal. Chem., 2009, 395, 2293–2301 CrossRef CAS PubMed.
- C. Hughes, M. D. Brown, N. W. Clarke, K. R. Flower and P. Gardner, Analyst, 2012, 137, 4720–4726 RSC.
- J. Sulé-Suso, D. Skingsley, G. D. Sockalingum, A. Kohler, G. Kegelaer, M. Manfait and A. J. El Haj, Vib. Spectrosc., 2005, 38, 179–184 CrossRef.
- U. Zelig, S. Mordechai, G. Shubinsky, R. K. Sahu, M. Huleihel, E. Leibovitz, I. Nathan and J. Kapelushnik, Biochim. Biophys. Acta, Gen. Subj., 2011, 1810, 827–835 CrossRef CAS PubMed.
- S. Gaudenzi, D. Pozzi, P. Toro, I. Silvestri, S. Morrone and A. Congiu Castellano, Spectroscopy, 2004, 18, 415–422 CrossRef CAS.
- M. Jimenez-Hernandez, M. D. Brown, C. Hughes, N. W. Clarke and P. Gardner, Analyst, 2015, 140, 4453–4464 RSC.
- L. E. Jamieson, D. J. Harrison and C. J. Campbell, Analyst, 2015, 140, 3910–3920 RSC.
- M. Smolina and E. Goormaghtigh, Analyst, 2015, 140, 2336–2343 RSC.
- J. Ruppen, F. D. Wildhaber, C. Strub, S. R. R. Hall, R. A. Schmid, T. Geiser and O. T. Guenat, Lab Chip, 2015, 15, 3076–3085 RSC.
- M. J. Tobin, L. Puskar, R. L. Barber, E. C. Harvey, P. Heraud, B. R. Wood, K. R. Bambery, C. T. Dillon and K. L. Munro, Vib. Spectrosc., 2010, 53, 34–38 CrossRef CAS.
- L. Quaroni, T. Zlateva, B. Sarafimov, H. W. Kreuzer, K. Wehbe, E. L. Hegg and G. Cinque, Biophys. Chem., 2014, 189, 40–48 CrossRef CAS PubMed.
- K. L. A. Chan and S. G. Kazarian, Analyst, 2013, 138, 4040–4047 RSC.
- E. J. Marcsisin, C. M. Uttero, A. I. Mazur, M. Miljković, B. Bird and M. Diem, Analyst, 2012, 137, 2958–2964 RSC.
- G. Stella, R. Senetta, A. Cassenti, M. Ronco and P. Cassoni, J. Transl. Med., 2012, 10, 12 CrossRef PubMed.
- C. Petibois and G. Déléris, Trends Biotechnol., 2006, 24, 455–462 CrossRef CAS PubMed.
- G. Lorusso and C. Ruegg, Histochem. Cell Biol., 2008, 130, 1091–1103 CrossRef CAS PubMed.
- H. Ukkonen, S. Kumar, J. Mikkonen, T. Salo, S. P. Singh, A. P. Koistinen, E. Goormaghtigh and A. M. Kullaa, Vib. Spectrosc., 2015, 79, 24–30 CrossRef CAS.
- S. Kumar, C. Desmedt, D. Larsimont, C. Sotiriou and E. Goormaghtigh, Analyst, 2013, 138, 4058–4065 RSC.
- M. B. Amin, D. W. Lin, J. L. Gore, J. R. Srigley, H. Samaratunga, L. Egevad, M. Rubin, J. Nacey, H. B. Carter, L. Klotz, H. Sandler, A. L. Zietman, S. Holden, R. Montironi, P. A. Humphrey, A. J. Evans, J. I. Epstein, B. Delahunt, J. K. McKenney, D. Berney, T. M. Wheeler, A. M. Chinnaiyan, L. True, B. Knudsen and M. E. H. Hammond, Arch. Pathol. Lab. Med., 2014, 138, 1387–1405 CrossRef PubMed.
- J. T. Kwak, A. Kajdacsy-Balla, V. Macias, M. Walsh, S. Sinha and R. Bhargava, Sci. Rep., 2015, 5 Search PubMed.
- F. Islam, B. Qiao, R. A. Smith, V. Gopalan and A. K. Y. Lam, Exp. Mol. Pathol., 2015, 98, 184–191 CrossRef CAS PubMed.
- C. Hughes, M. Liew, A. Sachdeva, P. Bassan, P. Dumas, C. A. Hart, M. D. Brown, N. W. Clarke and P. Gardner, Analyst, 2010, 135, 3133–3141 RSC.
- O. Uckermann, R. Galli, M. Anger, C. Herold-Mende, E. Koch, G. Schackert, G. Steiner and M. Kirsch, Int. J. Radiat. Biol., 2014, 90, 710–717 CrossRef CAS PubMed.
- K. Chen, Y.-h. Huang and J.-l. Chen, Acta Pharmacol. Sin., 2013, 34, 732–740 CrossRef CAS PubMed.
- P. Gardner and M. J. Baker, Clinical Infrared and Raman Spectroscopy Network (CLIRSPEC), Engineering and Physical Sciences Research Council and -. (EP L012952 1).
- K. M. Dorling and M. J. Baker, Trends Biotechnol., 2013, 31, 327–328 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |