
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diffusion of macromolecules in self-assembled cellulose/hemicellulose hydrogels
Patricia
Lopez-Sanchez
*a,
Erich
Schuster
bc,
Dongjie
Wang
a,
Michael J.
Gidley
a and
Anna
Strom
cd
aARC Centre of Excellence in Plant Cell Walls, Centre for Nutrition and Food Sciences, Queensland Alliance for Agriculture and Food Innovation, The University of Queensland, Brisbane, 4072, Australia. E-mail: p.lopezsanhcez@uq.edu au
bDepartment of Structure and Material Design, The Swedish Institute for Food and Biotechnology, SIK, Gothenburg, Sweden
cSuMo Biomaterials, Vinn Excellence Center, Chalmers University of Technology, Gothenburg, Sweden
dApplied Chemistry, Department of Chemical and Biological Engineering, Chalmers University of Technology, Gothenburg, Sweden
First published on 15th April 2015
Abstract
Cellulose hydrogels are extensively applied in many biotechnological fields and are also used as models for plant cell walls. We synthesised model cellulosic hydrogels containing hemicelluloses, as a biomimetic of plant cell walls, in order to study the role of hemicelluloses on their mass transport properties. Microbial cellulose is able to self-assemble into composites when hemicelluloses, such as xyloglucan and arabinoxylan, are present in the incubation media, leading to hydrogels with different nano and microstructures. We investigated the diffusivities of a series of fluorescently labelled dextrans, of different molecular weight, and proteins, including a plant pectin methyl esterase (PME), using fluorescence recovery after photobleaching (FRAP). The presence of xyloglucan, known to be able to crosslink cellulose fibres, confirmed by scanning electron microscopy (SEM) and 13C NMR, reduced mobility of macromolecules of molecular weight higher than 10 kDa, reflected in lower diffusion coefficients. Furthermore PME diffusion was reduced in composites containing xyloglucan, despite the lack of a particular binding motif in PME for this polysaccharide, suggesting possible non-specific interactions between PME and this hemicellulose. In contrast, hydrogels containing arabinoxylan coating cellulose fibres showed enhanced diffusivity of the molecules studied. The different diffusivities were related to the architectural features found in the composites as a function of polysaccharide composition. Our results show the effect of model hemicelluloses in the mass transport properties of cellulose networks in highly hydrated environments relevant to understanding the role of hemicelluloses in the permeability of plant cell walls and aiding design of plant based materials with tailored properties.
Introduction
The architecture of the plant cell wall is directly related to its porosity and the transport of water and molecules in the apoplast, the space outside of the cell membrane. Despite being of crucial relevance to understand many biological and industrial processes, little is known about the complex structural organisation and spatial distribution of plant cell wall polysaccharides and their involvement in controlling the porosity and mass transport properties of the cell wall.1 Although the plant cell wall is permeable to water and low molecular weight compounds, it has limited permeability for larger molecules e.g. enzymes and proteins involved in many bioprocesses such as intercellular communication, growth and biomass conversions.The plant cell wall of higher plants is proposed to be a double network of interacting but separated networks of cellulose/hemicelluloses embedded in a pectin network, with generally minor amounts of structural proteins such as extensins.2 Due to the complexity of the cell wall, the role of individual polysaccharides in controlling porosity and permeability is still not well understood, partly due to the complexity of studying these properties in planta. Cellulose composites produced by the bacterium Gluconacetobacter xylinus can be used as a simplified model of the plant cell wall while complexity is added by the incorporation of different hemicelluloses and pectin.3–5
In the primary walls of dicots (and non-grass monocots) pectin, a complex biopolymer composed of different polysaccharides such as homogalacturonan (HG), rhamnogalacturonan I (RG-I) and substituted galacturonans like rhamnogalacturonan II (RG-II), is believed to determine wall porosity creating the network with the smallest pores. Indeed it has been shown that after using pectinase larger molecules could be transported, something that was not observed after the use of cellulase and proteinase; suggesting that pectin controlled the porosity of the wall.6 Homogalacturonan in the wall can be crosslinked with calcium creating a porous network, therefore parameters such as pH and calcium concentration could be used to control the wall porosity by modifying the properties of the pectin network.7 Furthermore the sugar side chains of branched RG-I, mainly arabinan and galactan, have been proposed to play a role in controlling wall porosity.8
The role of hemicelluloses in the permeability of the cell wall has been less investigated and only recently due to the interest from biofuel production to access cellulose in secondary thickened walls e.g. characteristic of wood. Enzymatic degradation of plant cell walls is the most energy efficient route to exploit plant biomass for energy or feed purposes.9 Plant cell walls are however, recalcitrant to degradation by enzymes due to the intermolecular forces between polysaccharide components, such as hemicelluloses and pectin. As for pectin, the presence of hemicelluloses is known to affect the porosity of the cell wall of crops as removal of the hemicelluloses increased the pore size.10 Two major kinds of hemicelluloses are xyloglucans and xylans. Xyloglucan is composed of a cellulose-like backbone of β-(1-4)-linked-D-glucose branched by α-D-xylose molecules which can be further substituted.11 Xyloglucan in plants is found partially covering the cellulose microfibrils, entrapped within some cellulose microfibrils and a minor but structurally highly relevant fraction includes the parts of xyloglucans that crosslink cellulose microfibrils.12 These crosslinks maintain the spaces between cellulose microfibrils and are modulated by xyloglucan endo-transglycosylases and expansins.13,14 An example of xylans is arabinoxylan which has been identified in most cereal endosperms. Arabinoxylan is a linear polymer of xylose molecules substituted at O-3 and/or O-2 by arabinose residues, furthermore phenolics such as ferulic acid have been found esterifying O-5 of occasional arabinose residues. Due to different extractabilities of arabinoxylan fractions, they are claimed to interact in different ways in the plant cell wall: a water extractable weakly bound fraction, held together by physical interactions and an alkali extractable tightly bound fraction, which potentially is connected to other wall polysaccharides by ester-bond phenolic groups.
The diffusivity of molecules in the cell wall is influenced by the cell wall architecture, molecule–molecule interactions and molecule–wall interactions which are different at different structural length scales. Several methods are available to determine diffusion rates.15 The porosity and molecule diffusion in the plant cell wall have been studied using ultrastructural methods such as electron microscopy on isolated cell walls,16 bulk exclusion techniques17 on whole cells and functional assays such as tracking molecules on whole cells under close to physiological conditions.18 However due to the differences intrinsic to the methods used and the heterogeneity of plant materials a wide range or pore sizes, which can be related to cell wall permeability, have been measured. Average pore sizes of 3.5–5.6 nm have been determined using bulk exclusion methods, whereas functional assays suggest sizes of 4.5–9.2 nm. In general a continuous range of pore sizes have been measured, abundant 4–5 nm pores which contribute to bulk uptake or exclusions and less frequent 6–9 nm pores that allow larger molecules to penetrate more slowly.19
Fluorescence recovery after photobleaching (FRAP) in combination with confocal laser scanning microscopy (CLSM) can be used to study molecular self-diffusion through heterogeneous materials. FRAP offers the possibility to determine the diffusion rate locally and monitor the surrounding structure simultaneously. In FRAP, the diffusion rate measurements are based on creating a concentration gradient of fluorescent molecules. This is performed by deactivating the fluorescence (photobleaching) in a region of interest (ROI) by exciting it using a high intensity laser beam. The subsequent diffusion of the photo bleached molecules outside of the ROI and their replacement with adjacent unbleached fluorochromes leads to a recovery of the fluorescence intensity. FRAP is most useful for studying diffusion in the range of 0.1 to 100 μm2 s−1 on a micrometer scale.20,21 FRAP has in the past been used to study the binding reversibility of cellulases to bacterial microcrystalline cellulose fibrils and mats22,23 as well as to study the mobility of labelled xylanases along the xylan surface.24 Using FRAP on soybean root cultured cells with fluorescently labelled dextrans and proteins of graded size, a range of diameters for putative trans-wall channels was determined to be 6.6–8.6 nm.6 FRAP has also been used to study diffusion in pectin gels25 and in feruloylated arabinoxylan gels mixed with cellulose nanocrystals,26 which served as plant cell wall models.
The determination of solute diffusion and molecular interactions is essential when investigating diffusants with binding affinities and in biophysics,27,28 since protein–protein interactions regulate cellular processes. With an appropriate mathematical model, one can then analyze the fluorescence recovery and extract quantitative information on the molecular dynamics. By considering a model that contains an interaction term, it is possible to simultaneously estimate the pseudo-on binding rate, the off binding rate, and the diffusion coefficient via FRAP.27–30
In this work we studied the role of hemicelluloses on the mass transport properties of cellulosic hydrogels as a biomimetic of plant cell walls. Fluorescence recovery after photobleaching was used in combination with confocal laser scanning microscopy to study molecular diffusion in cellulose hydrogels (>95% water) and cellulose composite hydrogels containing xyloglucan or arabinoxylan, selected as model hemicelluloses with different binding abilities to cellulose. A series of fluorescence labelled dextrans and proteins of different molecular weights were used as models representative of a range of plant molecules with different sizes. We also included a fluorescently labelled plant methyl esterase (PME), selected for its lack of specificity to the hydrogel's components. Differences in diffusion coefficients were attributed to microstructural changes introduced by the hemicelluloses, characterised by SEM and 13C NMR. Our results revealed different effects of hemicellulose in cellulose hydrogels and give insights into the potential contribution of different polysaccharides to the permeability of the plant cell wall and man-made cellulose-based composites.
Experimental
Materials
Fluorescein isothiocyanate labelled dextran (FITC-dextran) of three different molecular weights (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (FD 10), 70000 (FD 70), and 500000 (FD 500) g mol−1) were purchased from Invitrogen Molecular Probes, Eugene, OR. FITC labelled bovine serum albumin (FITC-BSA), orange pectin methyl esterase (P5400 – 1KU with 154 units per mg solid or 597 units per mg protein), fluorescein 5(6)-isothiocyanate (FITC, F7256), dimethyl sulfoxide (DMSO) and MES hydrate were purchased from Sigma Aldrich, Steinheim, Germany. Dialysis membranes (Float-A-lyzer G2) with a Mw cut off size of 0.5–1 kDa were obtained from SpectrumLabs, US.
000 (FD 10), 70000 (FD 70), and 500000 (FD 500) g mol−1) were purchased from Invitrogen Molecular Probes, Eugene, OR. FITC labelled bovine serum albumin (FITC-BSA), orange pectin methyl esterase (P5400 – 1KU with 154 units per mg solid or 597 units per mg protein), fluorescein 5(6)-isothiocyanate (FITC, F7256), dimethyl sulfoxide (DMSO) and MES hydrate were purchased from Sigma Aldrich, Steinheim, Germany. Dialysis membranes (Float-A-lyzer G2) with a Mw cut off size of 0.5–1 kDa were obtained from SpectrumLabs, US.
Arabinoxylan extracted from wheat of a Mw of 370000 g mol−1 and xyloglucan extracted from tamarind seed of a Mw of 225000 g mol−1 (both molecular weights are given by the supplier) were purchased from Megazyme International Ltd, Ireland.
The Hestrin and Schramm medium used for incubation of the bacterial strain consisted of 1.15 g l−1 citric acid (Ajax Finechem, Thermo Fisher Scientific, Australia), 2.7 g l−1 Na2HPO4 (Ajax Finechem, Thermo Fisher Scientific, Australia), 5 g l−1 peptone (Oxoid Ltd, Basingstoke, Hampshire, England), 5 g l−1 yeast extract (Becton, Dickinson and Company, Sparks, USA) and 2% (w/v) glucose (Sigma-Aldrich). The pH was adjusted to pH 5 with 10 M HCl.
Preparation of cellulose and cellulose/hemicellulose hydrogels
Xyloglucan and arabinoxylan solutions at a concentration of 1% w/v were prepared by dissolving the polysaccharides in deionised water overnight at room temperature.The bacterial strain Gluconacetobacter xylinus (ATCC 53524 American Type Culture Collection, Manassas, VA, USA) was used to produce cellulose (C), cellulose–xyloglucan (CXG) and cellulose–arabinoxylan (CAX) hydrogels based on the method described by Chanliaud and co-workers3 and Mikkelsen and co-workers31 with minor modifications. Hydrogels were cultivated in the Hestrin and Schramm medium under static conditions at 30 °C. The cellulose–xyloglucan hydrogels were produced by mixing the 1% xyloglucan solution with double concentrated Hestrin and Schramm medium (1:1) before inoculation, leading to a final xyloglucan concentration of 0.5%. A similar preparation method was used for the cellulose–arabinoxylan hydrogels. The samples were harvested from the medium with forceps after 72 hours and washed 6 times with ice-cold deionised water under agitation on an orbital platform shaker (KS 260 IKA-Werke, Staufen, Germany) at 150 rpm to dislodge the bacteria and remove excess medium.
All samples were disks with a diameter of approximately 40 mm, corresponding to the diameter of the containers in which they were cultivated, and variable thickness of ca. 3 mm for C (cellulose), 2.2 mm for CAX (cellulose–arabinoxylan) and 0.3 mm for CXG (cellulose–xyloglucan). Samples were stored in 0.02% NaN3 solution to avoid contamination and microbiological growth at 4 °C until further analysis.
Methods
Concentration of cellulose hydrogels by compression
A mechanical tester machine, Instron 5565 A, was used to compress and concentrate the hydrogels containing cellulose only (C). The samples were placed in the centre of the Instron platform and the crosshead was lowered at a speed of 0.1 mm s−1 until a final thickness of 1 ± 0.1 mm was obtained, a second set of samples was further compressed at 0.001 mm s−1 until a final thickness of 0.5 ± 0.1 mm was reached.Composites composition and microstructural characterisation
000, *25000, *50000. Image analysis was performed using Image J software.34
Preparation of fluorescent probes
Orange pectin methyl esterase was labelled with fluorescein 5(6)-isothiocyanate with some modification of the method described by Videcoq and co-workers.25 The enzyme was dissolved at a concentration of 1% (w/w) in 10 mM MES buffer at pH 7. FITC was dissolved in a mixture composed of DMSO and water in a volumetric ratio of 2:1 to give a final FITC concentration of 0.015 mg ml−1 DMSO and water. PME solution was added to the FITC–DMSO and water solution to yield a final molar ratio between FITC and PME of 5 according to| nFITC/nPME = 5. |
The solution was stirred for 5 hours at 4 °C. The mixture was then dialysed against milliQ water to remove excess FITC for three days, followed by dialysis against MES buffer (10 mM) for one day. The dialysis tube used had a Mw cut off of 0.5–1 kDa (Float-A-lyzer G2).
The FITC-dextran probes were incorporated into the hydrogels by the addition of 200 ppm of each probe to the solution in which the hydrogels were kept. Similarly composites were mixed with 500 ppm of FITC-BSA and FITC-PME. The containers were covered with aluminium foil and left overnight at 5 °C in order to give enough time for the probes to be homogeneously distributed in the gels.
CLSM-FRAP protocol
The CLSM system used consists of a Leica SP2 AOBS (Heidelberg, Germany) utilizing a 20×, 0.5 NA water objective, with the following settings: 256 × 256 pixels, zoom factor 4 (with a zoom-in during bleaching), and 800 Hz, yielding a pixel size of 0.73 μm and an image acquisition rate of two images per second. The FRAP images were stored as 12-bit TIFF-images. The 488 nm line of an argon laser was used to excite the fluorescent probes. The beam expander was set to 1, which lowered the effective NA to ∼0.35 and yielded slightly better bleaching and a more cylindrical bleaching profile. The bleached areas will be called ROI in this study and were 30 μm large discs (nominal radius rn ∼ 15 μm) at 100 μm into the sample. The measurement routine consisted of 20 prebleach images. To obtain an initial bleaching depth of ∼30% of the prebleach intensity in the ROI, one to four bleach images were taken depending on the sample. For every recovery, at least 50 frames were recorded. The FRAP data were normalized by the prebleach fluorescence intensity.The respective diffusion probes were dissolved in deionised water to yield 200/500 ppm solutions. The free diffusion coefficients D0 of the probes in the absence of cellulosic hydrogels were determined at ambient temperature, 7 μl of the probe solutions were placed into secure-seal spacer grids between two cover glass slides, and the FRAP measurements were carried out on such locked samples.
As described above, to prepare the cellulosic hydrogels for FRAP measurements the samples were soaked in the respective probe solutions overnight. An approx. 2 cm × 2 cm sized sample was cut, the surface that was in direct contact with the liquid medium during cellulose synthesis was absorbed on a cover glass slid, then loaded on the microscope stage and FRAP measurements carried out in the upright mode of the microscope at ambient temperature. At least 6 FRAP measurements were performed on different spatial coordinates per sample. To test the reproducibility every sample was remade at least once. All of the recorded recoveries were quick enough to yield Gaussian intensity distributions in the initial recovery images within the bleached area/ROI. Therefore the FRAP model called “most likelihood estimation for FRAP data with a Gaussian starting profile”35 is valid for evaluation of the data. A script provided by Jonasson et al.35 was utilized to analyze the data within this framework in Matlab, Mathworks, U.S.A.
To additionally analyze FRAP data for binding interactions, a quantitative approach to analyze binding-diffusion kinetics by confocal FRAP was developed by Kang and co-workers,29 and a data analysis was carried out as described in ref. 30.
Results and discussion
Chemical and microstructural characterisation of cellulose/hemicellulose hydrogels
Chemical analysis of the composites by GC-MS confirmed an average incorporation of 40% of xyloglucan and 41.2% arabinoxylan in the cellulose hydrogels. The average polysaccharide content in the hydrogels was 1.3% w/w for C, 1.5% w/w for CAX and 2.5% w/w in CXG. The cellulose concentration was 1.3% w/w for C, 0.9% w/w for CAX and 1.5% w/w for CXG. Furthermore the presence of xyloglucan decreased the cellulose crystalline content from 87 to 64% (with the percentage of Iβ allomorph increasing), arabinoxylan did not change the ratio crystalline:amorphous compared to cellulose only samples, in agreement with previously reported data on similar materials.5
A fraction of xyloglucan immobilised in the presence of cellulose was detected by 13C CP/MAS NMR with a peak at 99.5 ppm due to the C1 of xylose (other xyloglucan C-1 signals are coincident with the main cellulose C1 signal), and a mobile fraction was shown by a 13C SP/MAS spectrum attributable to xyloglucan and not cellulose.36 This behaviour is consistent with the crosslinks which could be visualised under SEM as thin strands between cellulose fibres, although the higher density of these composites made it difficult to identify different structural attributes (Fig. 1b). In the cellulose–arabinoxylan hydrogels, aggregates of different sizes were observed deposited on the surface of the cellulose fibres (Fig. 1c). These structures are attributed to aggregates of arabinoxylan.37 Arabinoxylan was still present after extensive washing of the samples suggesting that arabinoxylan was interacting directly with the cellulose fibres. The 13C SP/MAS of arabinoxylan composites revealed 2 peaks in the C1 region typical of arabinoxylan: xylose at 99.5 ppm and arabinose at 104.2 ppm, but only cellulose signals were observed in the CP/MAS spectrum indicating that arabinoxylan is present in the sample but it is not immobilised on the cellulose scaffold. These features of the hydrogels have been previously reported.3,5,37 In the absence of hemicelluloses, bacterial cellulose appeared as a mat of entangled long random oriented cellulose fibres with an average diameter of 75 ± 17 nm estimated from image analysis (Fig. 1a). Similar cellulose networks to the ones reported here after washing have been shown for unwashed pellicles,38 confirming that the speed used in the rotational shaker is not enough to disturbed the tough cellulose–hemicellulose networks. It should also be mentioned that the microstructure of the cellulose hydrogels remains unchanged at a compression speed of 0.1 mm s−1 compared to uncompressed samples whereas at 0.001 mm s−1 the cellulose fibres aggregate resulting in a densification of the structure.39
 | ||
| Fig. 1 Scanning electron micrographs of top and cross sections of cellulose only (a) and (d), cellulose–xyloglucan (b) and (e) and cellulose–arabinoxylan (c) and (f) composites. The magnification bar represents 1 μm in the case of top images (a–c) and 100 μm for the cross sections (d–f). The arrows indicate arabinoxylan aggregates. | ||
Cross section images of the hydrogels revealed a layer by layer structure in which the layers were connected by fibres of different lengths, giving rise to a broad range of pore sizes. This microstructure is the result of the way bacteria produce cellulose under these experimental conditions;40 interestingly the average distance between the layers varied depending on the hydrogels composition. While the cellulose-only hydrogels had an average distance of 7.7 ± 0.9 μm (analysis of 11 images at different magnifications), the distance was increased to 10.7 ± 2 μm when arabinoxylan was present and reduced to 2.8 ± 0.7 μm in the presence of xyloglucan. Although these overall numbers should be treated with caution since they could be influenced by sample preparation for SEM, the trends were clear with distances CAX > C > CXG.
FRAP measurements of probes in solution
The free diffusion coefficients D0 of the probes in the absence of cellulosic hydrogels were determined at ambient temperature. This data yields hydrodynamic radii (rH) – calculated using the Stokes–Einstein relation – and is displayed in Table 1. Additionally, the diffusion rate of the probes in solution is used later to calculate the normalized diffusivity D/D0, which indicates the degree of physical hindrance a probe within a hydrogel (diffusion rate D) encounters.| r H [nm] | D 0 [μm2 s−1] | |
|---|---|---|
| FITC dextran 10 kDa | 2.9 ± 0.3 | 82.8 ± 7.8 |
| FITC dextran 70 kDa | 8.0 ± 0.5 | 30.0 ± 1.8 |
| FITC dextran 500 kDa | 13.5 ± 1.1 | 17.8 ± 1.4 |
| FITC albumin | 4.7 ± 0.4 | 51.1 ± 4.0 |
| FITC PME | 1.2 ± 0.2 | 200 ± 35 |
To identify possible interactions of the probes with the hydrogel components, which might be responsible for their hindrance, 13C NMR was carried out on composites which were soaked in 500000 g mol−1 FITC-dextran solutions. 13C SP/MAS and 13C CP/MAS NMR spectra of cellulose–xyloglucan and cellulose–arabinoxylan soaked in FITC-dextran solutions were similar to those previously reported for these systems in the absence of dextran.5 Dextran is present in very low concentrations in the hydrogels compared to cellulose and xyloglucan or arabinoxylan, therefore it was not possible to detect dextran in the 13C NMR. Literature spectra show only one peak for dextran between 60 and 70 and a C-1 signal at 100.5 ppm, which may be contributing to the larger than expected xylose C-1 signal (Fig. 2).
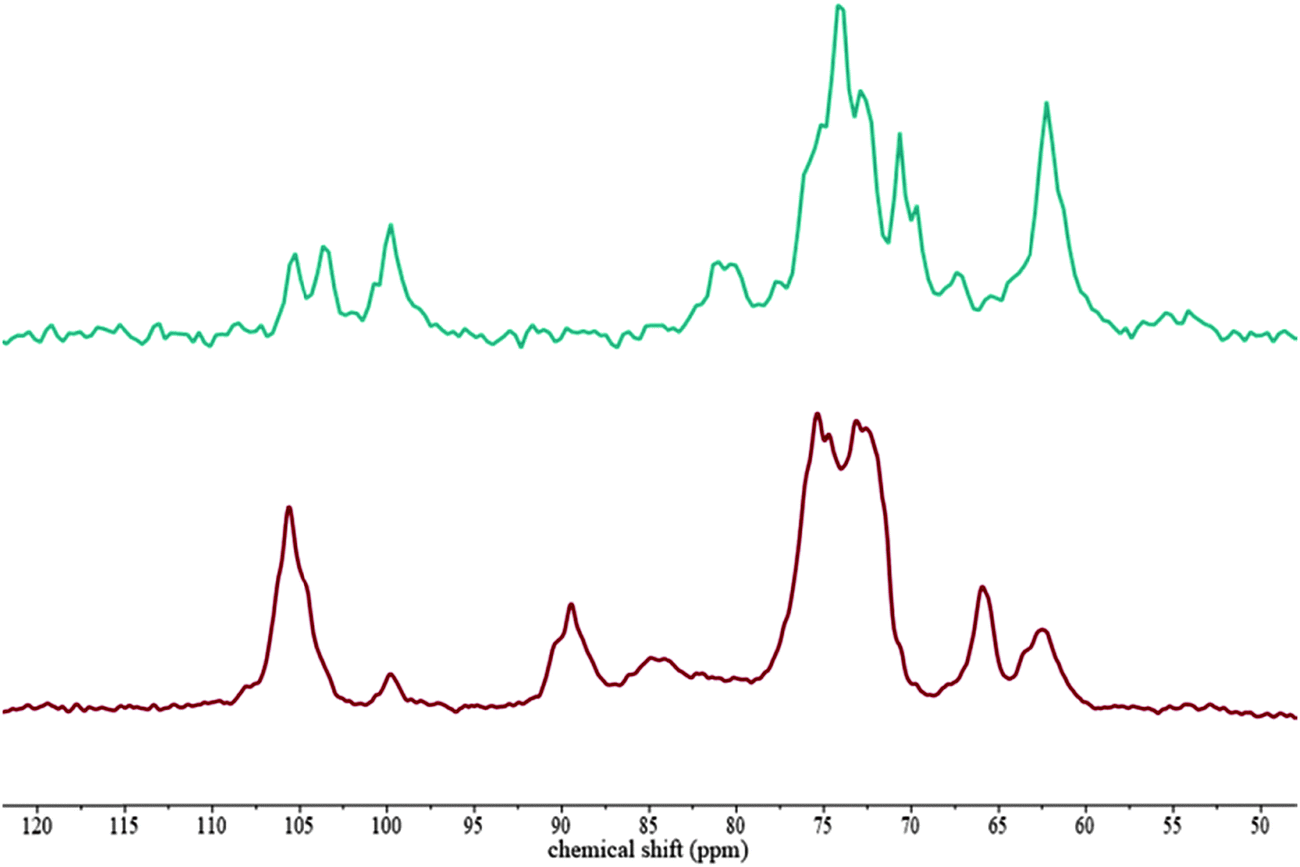 | ||
| Fig. 2
13C SP/MAS and 13C CP/MAS NMR spectra of cellulose–xyloglucan hydrogels soaked in a 500000 g mol−1 FITC-dextran solution. 13C SP/MAS (top) 13C CP/MAS (bottom). | ||
Probe diffusion in cellulose only hydrogels
The diffusion of probes in cellulose-only hydrogels was studied as a function of cellulose concentration. Samples were compressed to different thicknesses; during compression water is released radially from the hydrogels increasing the cellulose concentration. The cellulose concentration of compressed samples can be estimated using the wet and dry weight and adjusting for the volume of water loss.39 Uncompressed cellulose samples had a thickness of 3 mm and a concentration of 1.3% w/w cellulose. Samples compressed at 0.1 mm s−1 to a final thickness of 1 ± 0.1 mm had a cellulose concentration of 3.9%. It has been earlier reported39 that the microstructure, in terms of fibre diameter and pore size, of cellulose hydrogels compressed at rates of 0.1 mm s−1 was very similar to that of uncompressed samples, however lower compression rates induced cellulose fibre aggregation and increased the apparent pore size of the hydrogels. To further investigate the effect of these structural changes on macromolecules diffusion, a second set of samples were compressed at 0.001 mm s−1 to a thickness of 0.5 ± 0.1 mm, the final concentration of these samples was 7.8% w/w cellulose.The diffusion of 10000 g mol−1 FITC-dextran in these different cellulose hydrogels was very similar with D/D0 close to 1, indicating that the probe moved freely in the structure. The D/D0 of 70000 g mol−1 and 500000 g mol−1 dextran probes, was however slowed down in the uncompressed and 1 mm hydrogels compared to the 0.5 mm. The cellulose content of these samples increases from 1.3 to 7.8% upon compression and the result of less hindered diffusion in the sample with higher cellulose content may appear counter intuitive. However the cellulose fibres aggregate in the sample with the higher amount of cellulose,39 thus potentially increasing the pore size of these hydrogels and hence cause less obstruction for the diffusion probes. Alternatively the dynamic movements of the fibres might have been reduced after aggregation and therefore reduced a barrier to diffusion beyond static pore size effects. As would be expected, the diffusion of the probes are increasingly hindered by the cellulose network as their molecular weight increases and thus their radius of hydration estimated to be 2.9 nm for the 10000 g mol−1, 8 nm for 70000 g mol−1 and 13.5 nm for the 500000 g mol−1 dextran respectively (Fig. 3).
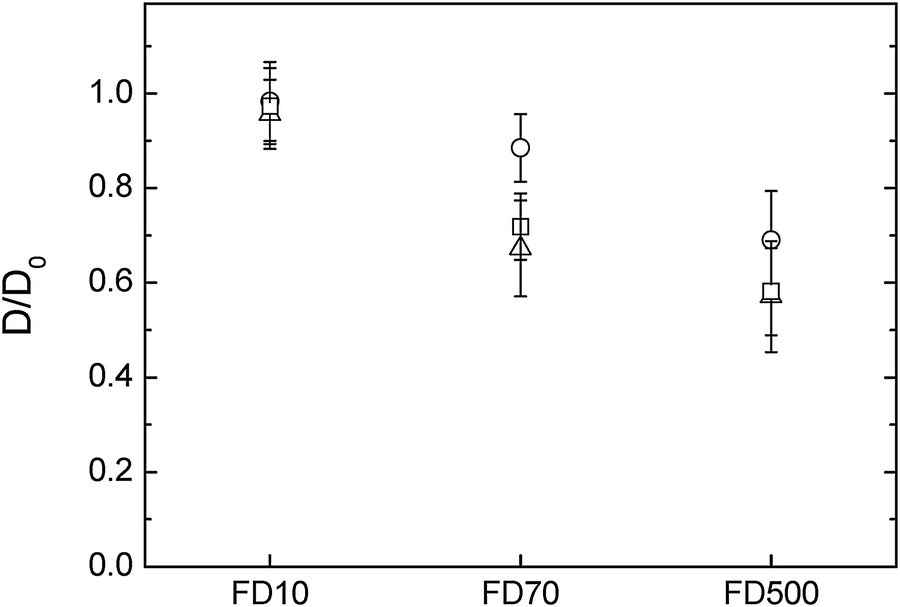 | ||
| Fig. 3 Diffusion of FTIC-dextran molecules in three different cellulosic networks containing (△) 1.3% cellulose (uncompressed ∼3 mm), (□) 3.9% (compressed quickly to ∼1 mm) and (○) 7.8% (compressed slowly to ∼0.5 mm). | ||
Probe diffusion in cellulose/hemicellulose hydrogels
The diffusion in hydrogels containing both cellulose and hemicelluloses was compared with the cellulose-only hydrogels (Fig. 4). As previously described, increasing the molecular weight and thus the radius of hydration of the dextran probes reduced their diffusivity in the pure cellulose hydrogels. The presence of arabinoxylan appears to increase the diffusivity of the dextran probes from the one observed in the cellulose-only hydrogels, especially for the 70000 g mol−1 FTIC-dextran. However, the presence of xyloglucan within the cellulose hydrogel reduced the diffusion of the 70000 g mol−1 and the 500000 g mol−1 dextran considerably more compared to cellulose only. These results suggest in the case of cellulose–xyloglucan that the pore size was reduced compared to cellulose-only hydrogels. The observations made for the hydrogels where arabinoxylan was incorporated suggest either an increased pore structure, reduced dynamics of the network (not likely) or surface energy. The total polysaccharide content increased in the order cellulose < cellulose–arabinoxylan < cellulose–xyloglucan, however the cellulose content was slightly lower in the hydrogels containing arabinoxylan and higher in the hydrogels containing xyloglucan. The effect of the hemicelluloses in the overall cellulose content of the hydrogels has an impact on microstructural effects such as pore size distribution. Indeed, scanning electron micrographs of top and cross sections indicated a denser network in the presence of xyloglucan compared to cellulose only hydrogels furthermore, the distance between the observed fibre layers in the structure was significantly reduced. This is expected for a molecule acting as a cross linker between cellulose fibres which would bring cellulose fibres closer together and lead to increased density of the system. On the other hand the presence of a molecule only interacting at the fibre surface, not crosslinking, as is the case of arabinoxylan, led to a microstructure similar to cellulose only. The coating of arabinoxylan on the cellulose fibre may instead render the arabinoxylan-containing network less hydrophilic as the contact angle between water and washed arabinoxylan is (higher (67–74°)41 than cellulose (40°) and xyloglucan (20°)).42 A change to a less hydrophilic network increases the mobility of fluorescently labelled probes due to repulsion between the probe and the network, similar to observations on systems in which electrostatic repulsion between the probe and the matrix increased the mobility compared to a non-charged reference.43 In principle, the same trend would be expected for the 500000 Da probe, however the repulsion related to probe–network interaction may here be overruled by the physical constraints of the network itself.
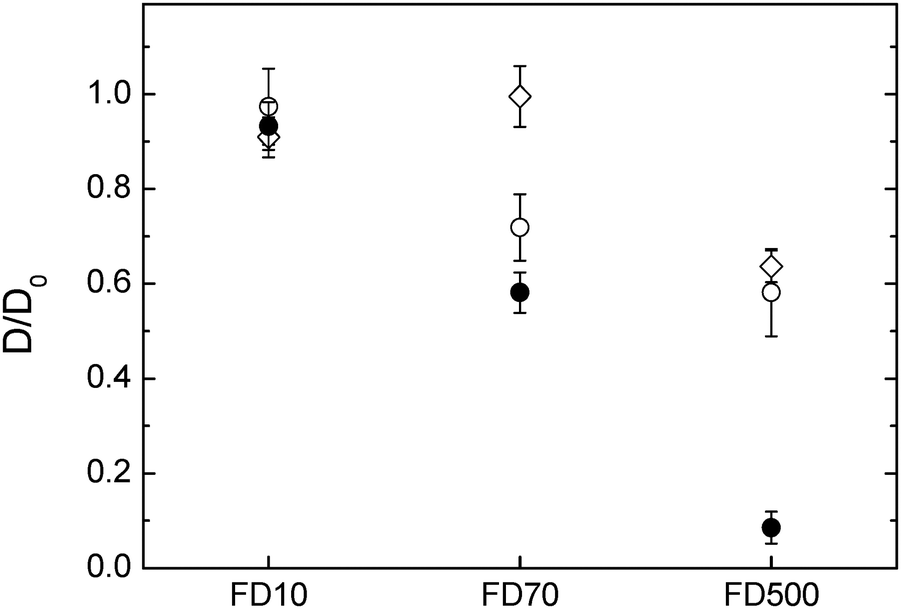 | ||
| Fig. 4 Diffusion of FTIC-dextran molecules in three different hydrogels containing (○) cellulose only (1.8% compressed to 1 mm) (●) cellulose–xyloglucan and (⋄) cellulose–arabinoxylan. | ||
The diffusivity of the two charged probes, albumin and PME, differed depending on the network composition (Fig. 5). It is worthwhile to mention that both albumin and PME can be approximately compared to the 10000 g mol−1 dextran in size as their radius of hydration are close to 4.7 and 1.2 nm respectively where the 10000 g mol−1 dextran is of 2.9 nm in rH. The diffusion of the albumin is less hindered in cellulose–arabinoxylan composite followed by the pure cellulose and nearly immobile in the composite sample containing xyloglucan. The hindrance of the albumin in the different composites (except for the cellulose–arabinoxylan) cannot only be explained by the pore size of the respective networks as the size of the albumin is similar to the size of the 10000 g mol−1 dextran, which is less hindered. An anomalous diffusion i.e. slower diffusion than expected of bovine serum albumin as used in this study was observed also in arabinoxylan gels, prepared as model system for the secondary plant cell wall. In this study the authors concluded that the interaction observed most probably was related to some interaction between the albumin and the gel network itself26 while other studies have observed hindrance of albumin in other polysaccharide solutions.44,45 It is shown in this study that the albumin appears to interact even stronger with the cellulose and cellulose–xyloglucan compared to the cellulose–arabinoxylan network.
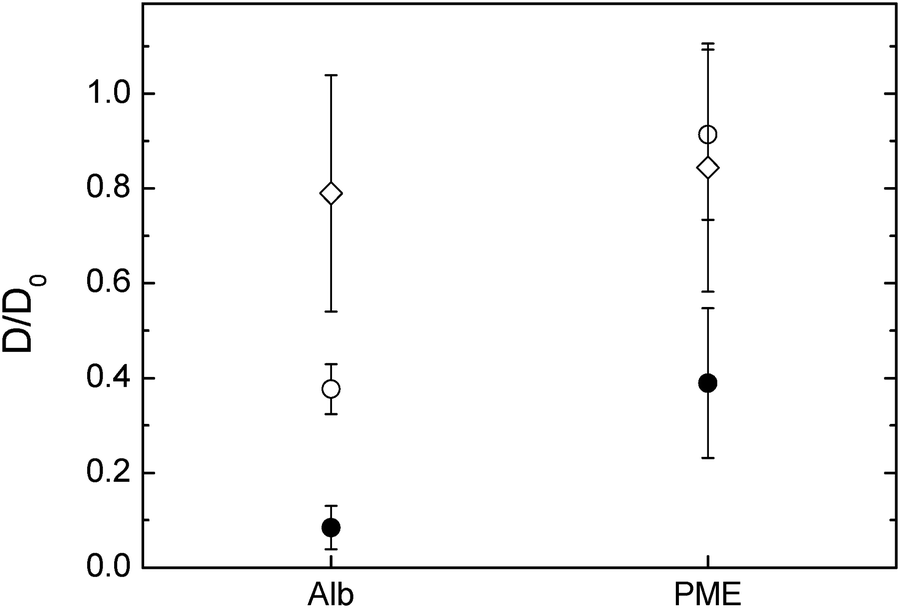 | ||
| Fig. 5 FITC-albumin and FITC-PME diffusion in (○) cellulose only hydrogels (1.8% cellulose) compressed to 1 mm, (●) cellulose–xyloglucan and (⋄) cellulose–arabinoxylan hydrogels. | ||
In the case of PME, its diffusivity in pure cellulose and cellulose arabinoxylan were similar. Furthermore, it was similar to the dextran of 10 kDa i.e., only slightly reduced by the network. This was expected as PME has a rH of ∼1 nm, thus too small to be hindered by the network studied here. Surprisingly, the diffusivity of the PME was largely reduced in the cellulose–xyloglucan gel, more so than 70 kDa dextran with a rH of ∼8 nm. The hindrance of the PME in the cellulose–xyloglucan sample can only be explained by additional interactions between the probe and the polysaccharide matrix rather than hindrance related to pore size. In order to test the behaviour of PME in the presence of xyloglucan and elaborate if there are any interactions which could permanently or temporarily bind the PME, additional experiments on a 1% w/w xyloglucan solution were carried out. FRAP measurements were carried out on FITC-PME in the xyloglucan solution. The recovery curve was analysed in the framework of FRAP and binding, and showed that in a 1% w/w xyloglucan solution PME's mobility is hindered around 20% (D/D0 = 0.79 ± 0.07). Binding with pseudo-on binding rate constant kon* = 0.5 ± 0.4 s−1 and off binding rate constant one magnitude higher ranging koff = 20 ± 10 s−1 indicates that transient interactions on a time-scale of 30 ms–10 s are occurring and a fraction of ∼5% of the PME are in average bound to the xyloglucan.
Our results indicate that PME interacts with xyloglucan in the composites: pectin methyl esterase is an enzyme which de-esterifies methylgalacturonic acid esters in pectins, therefore no interaction was expected with these composites which contain only cellulose and hemicelluloses. It is known that cell wall polysaccharides interact with their specific enzymes by carbohydrate binding sites outside of the active site area. These binding sites can be found on carbohydrate binding modules (CBMs) which are independent domains or they can be present on the surface of enzymes on catalytic domains or other intimately associated domains known as surface binding sites (SBSs).46 Furthermore CBMs have been shown to improve the action of catalytic modules on polysaccharides in plant cell walls through the recognition of non-substrate polysaccharides.1 This function was proved in a pectate lyase, whose degrading pectic homogalacturonan action was increased by cellulose-directed CBMs but not by xylan-directed CBMs. Furthermore the activity of hemicellulosic enzymes such as arabinofuranosidase, which removes side chains from arabinoxylan in xylan-rich and cellulose-poor wheat grain endosperm cell walls, was enhanced by a xylan-binding CBM. Examples in secondary cell walls have also been shown; xylanase degradation of xylan was potentiated by both xylan and cellulose-directed CBMs.1 We propose that PME can potentially have CBM's which might aid the action of this enzyme during cell wall growth and development by interacting with non-substrate polysaccharides such as xyloglucan. The primary plant cell wall is a highly concentrated environment of polysaccharides where pectins and xyloglucans are in close contact, therefore the possibility of enzymes using non substrates to improve their action seems reasonable. Based on the diffusion results of PME in xyloglucan solutions this interaction cannot be only steric but of physical/adhesive nature. Further work is required to characterise this interaction between PME and xyloglucan.
Conclusions
Composition of cellulose-based hydrogels (cellulose, cellulose–arabinoxylan, cellulose–xyloglucan) influence the diffusion of FITC labelled dextran at rH > 4 nm and Mw >10 kDa and protein probes even at rH as low as 1 nm. Cellulose–xyloglucan hydrogels reduce the mobility of all probes to a larger extent than cellulose and cellulose–arabinoxylan. The reduced mobility of the probes in the cellulose–xyloglucan hydrogel can in the case of dextran be explained by change in microstructure. The diffusion of fluorescently labelled PME was slightly reduced in the cellulose and the cellulose–arabinoxylan gel but greatly reduced in the cellulose–xyloglucan hydrogel. An interaction between PME and xyloglucan has to our knowledge not been reported previously. Our results indicate the possibility of such an interaction, an observation which merits further investigation. Using proteins as model probes for diverse enzymes does not give adequate information on its own as it ignores specific interactions as shown by the fact that the mobility of e.g. PME was not reduced in the presence of cellulose and arabinoxylan while albumin mobility was reduced in all networks.Acknowledgements
Cherie T. Beahan is gratefully acknowledged for performing monosaccharide analysis on the composites. Bernadine Flanagan is acknowledged for performing 13C NMR experiments. The financial support from VINN EXcellence SuMo Biomaterials (Supramolecular Biomaterials – Structure dynamics and properties) to AS and ES as well as VINNMER to AS are acknowledged. This work was carried out with the financial support of a UQ Start-up research grant to PLS.References
- C. Herve, A. Rogowski, A. W. Blake, S. E. Marcus, H. J. Gilbert and J. P. Knox, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 15293–15298 CrossRef CAS PubMed.
- Y. B. Park and D. J. Cosgrove, Plant Physiol., 2012, 158, 1933–1943 CrossRef CAS PubMed.
- E. Chanliaud, K. M. Burrows, G. Jeronimidis and M. J. Gidley, Planta, 2002, 215, 989–996 CrossRef CAS PubMed.
- S. E. C. Whitney, M. G. E. Gothard, J. T. Mitchell and M. J. Gidley, Plant Physiol., 1999, 121, 657–663 CrossRef CAS PubMed.
- P. Lopez-Sanchez, J. Cersosimo, D. Wang, B. Flanagan, J. R. Stokes and M. J. Gidley, PLoS One, 2015, 10, e0122132 Search PubMed.
- O. Baronepel, P. K. Gharyal and M. Schindler, Planta, 1988, 175, 389–395 CrossRef CAS PubMed.
- M. C. Jarvis, Plant, Cell Environ., 1984, 7, 153–164 CAS.
- T. J. Foster, S. Ablett, M. C. McCann and M. J. Gidley, Biopolymers, 1996, 39, 51–66 CrossRef CAS.
- M. Pauly and K. Keegstra, Plant J., 2008, 54, 559–568 CrossRef CAS PubMed.
- F. Adani, G. Papa, A. Schievano, G. Cardinale, G. D'Imporzano and F. Tambone, Environ. Sci. Technol., 2011, 45, 1107–1113 CrossRef CAS PubMed.
- A. Ebringerova, Z. Hromadkova and T. Heinze, Adv. Polym. Sci., 2005, 186, 1–67 CrossRef CAS.
- M. Pauly, P. Albersheim, A. Darvill and W. S. York, Plant J., 1999, 20, 629–639 CrossRef CAS PubMed.
- M. C. McCann, B. Wells and K. Roberts, J. Cell Sci., 1990, 96, 323–334 Search PubMed.
- T. Fujino, Y. Sone, Y. Mitsuishi and T. Itoh, Plant Cell Physiol., 2000, 41, 486–494 CrossRef CAS PubMed.
- B. A. Westrin, A. Axelsson and G. Zacchi, J. Controlled Release, 1994, 30, 189–199 CrossRef CAS.
- M. C. McCann, B. Wells and K. Roberts, J. Cell Sci., 1990, 96, 323–334 Search PubMed.
- M. Tepfer and I. E. P. Taylor, Science, 1981, 213, 761–763 CAS.
- A. B. StephenandM. Readin Modern methods of plant analysis. Plant cell analysis, ed. H. F. L. a. J. F. Jackson, Springer-Verlag, Berlin, Heilderberg, 1996, vol. 17 Search PubMed.
- M. A. Horn, P. F. Heinstein and P. S. Low, Plant Physiol., 1992, 98, 673–679 CrossRef CAS PubMed.
- H. Deschout, J. Hagman, S. Fransson, J. Jonasson, M. Rudemo, N. Loren and K. Braeckmans, Opt. Express, 2010, 18, 22886–22905 CrossRef PubMed.
- E. Schuster, J. Eckardt, A. M. Hermansson, A. Larsson, N. Loren, A. Altskar and A. Strom, Soft Matter, 2014, 10, 357–366 RSC.
- J. M. Moran-Mirabal, J. C. Bolewski and L. P. Walker, Biophys. Chem., 2011, 155, 20–28 CrossRef CAS PubMed.
- J. M. Moran-Mirabal, Cellulose, 2013, 20, 2291–2309 CrossRef CAS.
- S. Cuyvers, J. Hendrix, E. Dornez, Y. Engelborghs, J. A. Delcour and C. M. Courtin, J. Phys. Chem. B, 2011, 115, 4810–4817 CrossRef CAS PubMed.
- P. Videcoq, K. Steenkeste, E. Bonnin and C. Garnier, Soft Matter, 2013, 9, 5110–5118 RSC.
- G. Paes and B. Chabbert, Biomacromolecules, 2012, 13, 206–214 CrossRef CAS PubMed.
- B. L. Sprague, R. L. Pego, D. A. Stavreva and J. G. McNally, Biophys. J., 2004, 86, 3473–3495 CrossRef CAS PubMed.
- F. Mueller, D. Mazza, T. J. Stasevich and J. G. McNally, Curr. Opin. Cell Biol., 2010, 22, 403–411 CrossRef CAS PubMed.
- M. C. Kang, C. A. Day, E. DiBenedetto and A. K. Kenworthy, Biophys. J., 2010, 99, 2737–2747 CrossRef CAS PubMed.
- E. Schuster, A. M. Hermansson, C. Ohgren, M. Rudemo and N. Loren, Biophys. J., 2014, 106, 253–262 CrossRef CAS PubMed.
- D. Mikkelsen and M. J. Gidley, in Plant Cell Wall: Methods and Protocols, ed. Z. A. Popper, 2011, vol. 715, pp. 197–208 Search PubMed.
- F. A. Pettolino, C. Walsh, G. B. Fincher and A. Bacic, Nat. Protoc., 2012, 7, 1590–1607 CrossRef CAS PubMed.
- B. A. McKenna, D. Mikkelsen, J. B. Wehr, M. J. Gidley and N. W. Menzies, Cellulose, 2009, 16, 1047–1055 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- J. K. Jonasson, N. Loren, P. Olofsson, M. Nyden and M. Rudemo, J. Microsc., 2008, 232, 260–269 CrossRef CAS PubMed.
- M. J. Gidley, P. J. Lillford, D. W. Rowlands, P. Lang, M. Dentini, V. Crescenzi, M. Edwards, C. Fanutti and J. S. G. Reid, Carbohydr. Res., 1991, 214, 299–314 CrossRef CAS PubMed.
- D. Mikkelsen, M. J. Gidley and B. A. Williams, J. Agric. Food Chem., 2011, 59, 4025–4032 CrossRef CAS PubMed.
- D. Lin, P. Lopez-Sanchez and M. J. Gidley, Carbohydr. Polym., 2015, 126, 108–121 CrossRef CAS PubMed.
- P. Lopez-Sanchez, M. Rincon, D. Wang, S. Brulhart, J. R. Stokes and M. J. Gidley, Biomacromolecules, 2014, 15, 2274–2284 CrossRef CAS PubMed.
- M. Y. Iguchi, S. Yamanaka and A. Budhiono, J. Mater. Sci., 2000, 35, 9 CrossRef.
- I. Egues, A. M. Stepan, A. Eceiza, G. Toriz, P. Gatenholm and J. Labidi, Carbohydr. Polym., 2014, 102, 12–20 CrossRef CAS PubMed.
- G. Raj, E. Balnois, M. A. Helias, C. Baley and Y. Grohens, J. Mater. Sci., 2012, 47, 2175–2181 CrossRef CAS.
- N. Fatin-Rouge, A. Milon, J. Buffle, R. R. Goulet and A. Tessier, J. Phys. Chem. B, 2003, 107, 12126–12137 CrossRef CAS.
- T. C. Laurent and H. Persson, Biochim. Biophys. Acta, 1964, 83, 141–147 CAS.
- S. C. De Smedt, A. Lauwers, J. Demeester, Y. Engelborghs, G. Demey and M. Du, Macromolecules, 1994, 27, 141–146 CrossRef CAS.
- D. Cockburn, C. Wilkens, C. Ruzanski, S. Andersen, J. W. Nielsen, A. M. Smith, R. A. Field, M. Willemoes, M. Abou Hachem and B. Svensson, Biologia, 2014, 69, 705–712 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |