
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn iridium(III)-based irreversible protein–protein interaction inhibitor of BRD4 as a potent anticancer agent†
Hai-Jing
Zhong‡
a,
Lihua
Lu‡
b,
Ka-Ho
Leung‡
b,
Catherine C. L.
Wong
c,
Chao
Peng
c,
Siu-Cheong
Yan
d,
Dik-Lung
Ma
 *b,
Zongwei
Cai
*e,
Hui-Min
David Wang
*fg and
Chung-Hang
Leung
*a
*b,
Zongwei
Cai
*e,
Hui-Min
David Wang
*fg and
Chung-Hang
Leung
*a
aState Key Laboratory of Quality Research in Chinese Medicine, Institute of Chinese Medical Sciences, University of Macau, Macao, China. E-mail: duncanleung@umac.mo
bDepartment of Chemistry, Hong Kong Baptist University, Kowloon Tong, Hong Kong, China. E-mail: edmondma@hkbu.edu.hk
cNational Center for Protein Science Shanghai, State Key Laboratory of Cell Biology, Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China
dDepartment of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong, China
ePartner State Key Laboratory of Environmental and Biological Analysis, Department of Chemistry, Hong Kong Baptist University, 224 Waterloo Road, Kowloon Tong, Hong Kong SAR, P. R. China. E-mail: zwcai@hkbu.edu.hk
fDepartment of Fragrance and Cosmetic Science, Kaohsiung Medical University, Kaohsiung 807, Taiwan, Republic of China. E-mail: davidw@kmu.edu.tw
gGraduate Institute of Natural Products, Kaohsiung Medical University, Kaohsiung 807, Taiwan, Republic of China
First published on 30th July 2015
Abstract
Bromodomain-containing protein 4 (BRD4) has recently emerged as an attractive epigenetic target for anticancer therapy. In this study, an iridium(III) complex is reported as the first metal-based, irreversible inhibitor of BRD4. Complex 1a is able to antagonize the BRD4-acetylated histone protein–protein interaction (PPI) in vitro, and to bind BRD4 and down-regulate c-myc oncogenic expression in cellulo. Chromatin immunoprecipitation (ChIP) analysis revealed that 1a could modulate the interaction between BRD4 and chromatin in melanoma cells, particular at the MYC promoter. Finally, the complex showed potent activity against melanoma xenografts in an in vivo mouse model. To our knowledge, this is the first report of a Group 9 metal complex inhibiting the PPI of a member of the bromodomain and extraterminal domain (BET) family. We envision that complex 1a may serve as a useful scaffold for the development of more potent epigenetic agents against cancers such as melanoma.
Introduction
Gene transcription is a dynamic process tightly regulated by chromatin, which is a complex structure comprised of DNA and histone proteins.1 The function of gene regulation is controlled by post-translational modification states of DNA-packing histones in the chromatin complex.2 For example, the N-terminal lysine residues of histone proteins can be acetylated and deacetylated to control gene expression via the interplay of a range of enzymes such as histone acetyltransferase (HAT), histone deacetylase (HDAC) and methyltransferase (MT).1 Hence, these enzymes have become the targets of drug discovery efforts.3,4 However, the reader domains that interrogate post-translational modification states have been less intensively pursued as epigenetic targets.5,6Acetylated histones are recognized by small protein pockets called bromodomains.7 The bromodomain and extraterminal domain (BET) family of bromodomain-containing proteins (BRD2, BRD3, BRD4 and BRDT) are a class of transcriptional regulators containing tandem bromodomains and a carboxyl-terminal recruitment domain.8,9 In particular, BRD4 plays a significant role in cell cycle progression and viability via its effects on growth-related genes at the M/G1 boundary.10,11 Recently, BRD4 has been shown to play an important role in sustaining the proliferation of metastatic melanoma, a mostly incurable disease, thus rendering it as a possible target for epigenetic therapy.12
The selective inhibition of the bromodomain 4 (BRD4)/histone interaction has been demonstrated by several small molecule inhibitors such as (+)-JQ1, which is capable of occupying the ε-N-acetylated lysine residue (Kac) binding site of BRD4 and act as a Kac-competitive inhibitor.13 Subsequent reports have shown that (+)-JQ1 can directly regulate transcription mediated by the c-myc gene and reduce the expression of oncogenic c-myc protein.14,15
The success of the anti-cancer compound cisplatin and its analogues has inspired the investigation of metal-based compounds as therapeutic agents over the past few decades.16–24 While classical metal-based chemotherapeutic agents typically target double-helical DNA, increasing knowledge in molecular biology has uncovered the possibility of specifically targeting therapeutically relevant proteins or enzymes using transition-metal complexes.25–32 Metal-based compounds can offer distinct opportunities in targeting proteins or enzymes compared to organic small molecules due to their interesting structural diversity and electronic properties. Moreover, metal complexes can undergo ligand exchange reactions with biomolecules, and such irreversible inhibitors may show enhanced potency and potentially allow for less frequent and lower dosages in vivo.33 Examples of approved drugs that act via a covalent mechanism include EGFR inhibitors Neratinib (Pfizer), Afatinib/BIBW-2992 (Boehringer Ingelheim) and PF-00299804 (Pfizer), and anti-HCV agents Telaprevir (Vertex Pharmaceuticals and Johnson & Johnson) and Boceprevir/Victrelis (Merck) (Fig. S1†).33 Neratinib, Afatinib/BIBW-2992 and PF-00299804 target cysteine in EGFR, and Carfilzomib/Kyprolis, a selective proteasome inhibitor, targets threonine, while Telaprevir, used for the treatment of HCV, targets serine. Boceprevir/Victrelis also targets serine of HCV protease, and is used for the treatment of hepatitis caused by HCV.
Metal complexes can adopt a wide range of geometrical shapes defined by the oxidation state of the metal center and the nature of the co-ligands, while organic compounds are mainly restricted to linear, trigonal-planar and tetrahedral geometries. Therefore, metal complexes may be able to sample additional chemical space within the active site of enzymes or proteins. In addition, the steric and electronic properties of metal complexes can be easily tuned without lengthy synthetic protocols due to the modular nature of inorganic synthesis. We and others have previously demonstrated that certain Ir(III),34–36 Rh(III)37,38 and Ru(II)39–42 complexes can be developed as inhibitors of enzymes or protein–protein interactions (PPI). Continuing in our quest to explore the therapeutic applications of Group 9 complexes, we report herein the first metal-based epigenetic inhibitor of BRD4 and of any BET protein in general.
 | ||
| Fig. 1 Chemical structures of the cyclometallated Ir(III) and Rh(III) complexes (racemates) used for preliminary screening. Only one enantiomer is shown for clarity; complexes 1 and 17 are OTf salts, and others are PF6 salts. | ||
Results and discussion
Screening of Group 9 metal complexes as BRD4 inhibitors
The Ir(III)/Rh(III) metal complexes 1–27 were synthesized as racemates according to literature methods (Fig. 1).25,43 As lysine-acetylated histone peptides are known substrates for bromodomain-containing proteins, we initially screened the complexes at a concentration of 100 μM for their ability to modulate the protein–protein interaction between tetra-acetylated lysine histone 4 peptide (H4AcK4) and BRD4 using a time-resolved-fluorescent resonance energy transfer (TR-FRET) assay (Cayman Chemical, Ann Arbor, MI, USA). Among the 27 compounds tested, complex 1 emerged as a top candidate, with slightly lower activity compared to the positive control compound (+)-JQ-1 (Fig. S1 and S2†). We analyzed the structure of complexes 1–27 to identify favorable substructures for the development of the next round of complexes. We observed that complex 1 possessed two 2-phenylpyridine C^N ligands and two acetonitrile ligands, whereas the other complexes tested generally possessed bidentate N^N donor ligands. Complex 5, which possessed the dipyrido[3,2-a:2′,3′-c]phenazine (dppz) N^N ligand, showed the weakest potency out of the complexes tested, suggesting that this ligand detracted from biological activity (Fig. 2).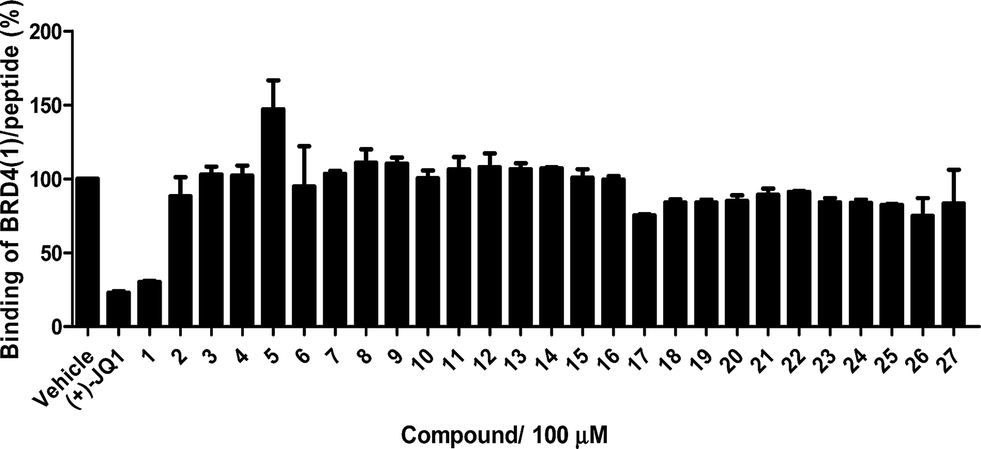 | ||
| Fig. 2 Displacement of a tetra-acetylated H4 peptide from BRD4 by a selection of Ir(III)/Rh(III) complexes at 100 μM in a TR-FRET assay. Error bars represent the standard deviations of the results from three independent experiments. | ||
Synthesis and structure-based optimization of novel BRD4 inhibitors
Based on the structure of 1, a focused library of 10 cyclometalated rhodium(III) and iridium(III) complexes containing different C^N or N-donor ligands (1a–1j) (Fig. 3a) was designed and synthesized. The spectroscopic data of the complexes are presented in Table S1.† This library was enriched in acetonitrile or acetonitrile-based N-donor ligands that were identified in the first round of screening to be favorable substructures for biological potency. In the second round of screening, the iridium(III) complex 1a showed the greatest inhibition of BRD4-H4AcK4 binding and was comparably potent to (+)-JQ1 (Fig. 3b). Notably, complex 1a, which contains two 2-phenyl-6-methylpyridine C^N ligands and two acetonitrile ligands, was more potent than the parent complex 1. Based on these results, preliminary structure-activity relationships (SAR) could be deduced. Varying the N-donor ligands from acetonitrile to other nitrile-based ligands (such as in 1i) did not result in improved activity against BRD4, as complex 1i was one of the least active compounds in the TR-FRET assay. Additionally, substituting the 2-phenyl-6-methylpyridine (as in 1a) or 2-phenylpyridine (as in 1) C^N co-ligands with more extended aromatic systems such as 2-phenylquinoline (as in 1d) or 1-phenylisoquinoline (as in 1f) also decreased the potency of the complex. Moreover, the presence of fluorine substituents on the C^N ligands appeared to be highly detrimental for activity, as complex 1b showed the weakest activity out of this series. Finally, replacing the iridium(III) center of complex 1 with rhodium(III) (as in congener 1h) resulted in greatly decreased activity against BRD4. Taken together, these results suggest that the binding between 1a and BRD4 is highly sensitive to the steric and/or electronic properties of the metal complexes.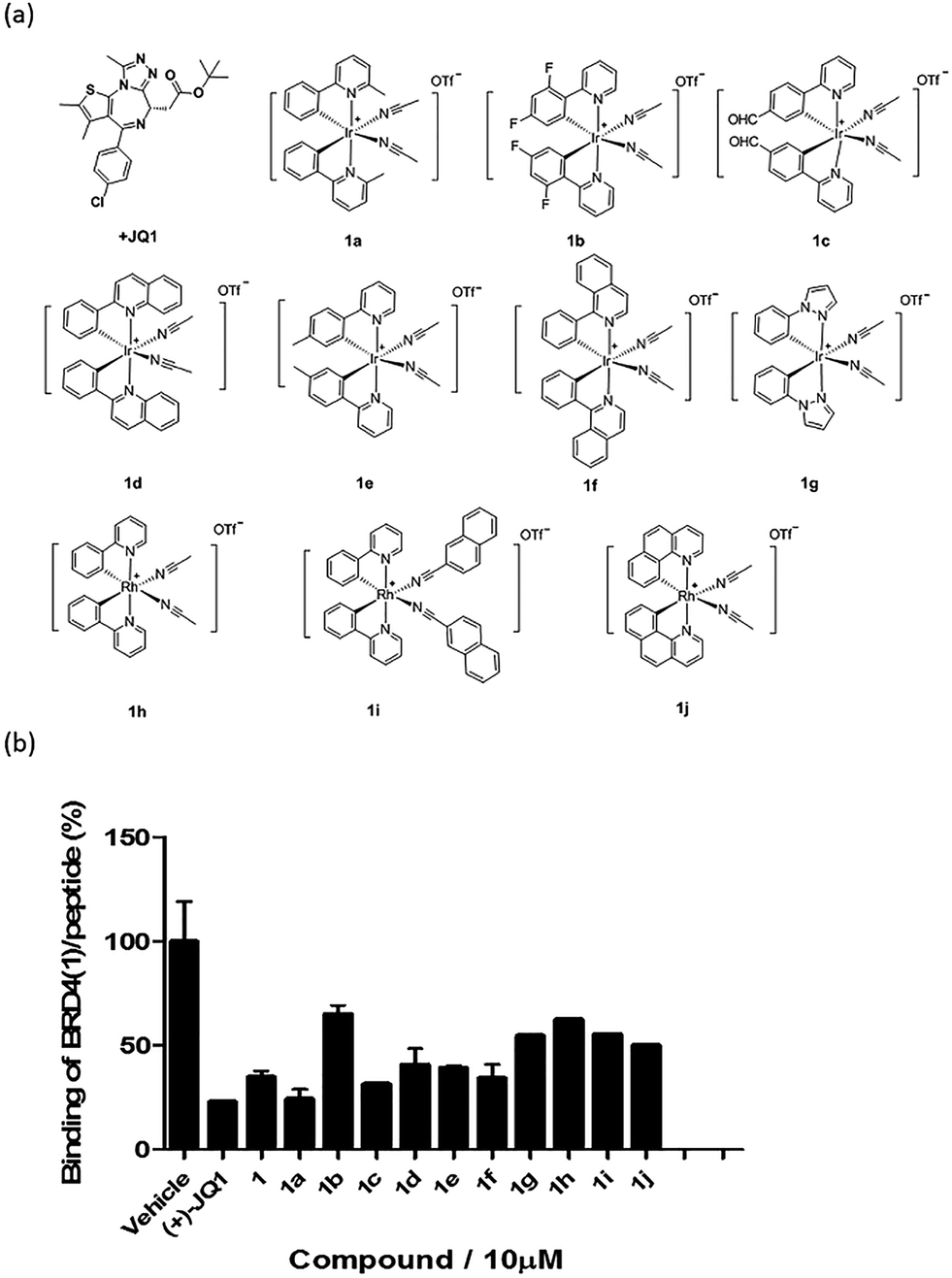 | ||
| Fig. 3 (a) Chemical structures of the cyclometallated Ir(III) and Rh(III) complexes (racemates) used for structure-activity analysis (SAR). (b) Displacement of a tetra-acetylated H4 peptide from BRD4 by complex 1 and analogues 1a–1j at 10 μM in a TR-FRET assay. Error bars represent the standard deviations of the results from three independent experiments. | ||
As complex 1 has been previously reported to bind covalently to histidine and generate a luminescence signal,25 we were interested to investigate whether complex 1a would also show a luminescent response to histidine or various other natural amino acids. In the presence of histidine, 1a exhibits an intense emission enhancement at Imax = 497 nm, whereas no significant changes in its emission were observed upon the addition of other natural amino acids (Fig. S2†). Competition experiments were carried out by incubating 1a with histidine and 10 equivalents of another natural amino acid. No significant difference was observed between the luminescence intensity detected in the competition experiments compared to that in the presence of histidine alone (data not shown). We further analyzed the binding of 1a to histidine by electrospray ionization mass spectrometry. After incubation of 1a (m/z 529.1) with histidine for 30 min at 20 °C, a new peak at m/z 684.2 was observed (Fig. S3b†). This peak corresponds to the covalent attachment of one histidine molecule (m/z 155) to 1a. No mass change was recorded for 1a upon incubation with other natural amino acids under same reaction conditions (Fig. S3c†). This data demonstrates that 1a selectively and covalently binds to histidine, leading to a luminescence response.
To assess the selectivity of complex 1a, we tested its activity against two unrelated proteins, caspase-6 and STAT3, which contain 12 and 13 histidine residues, respectively. The results showed that 1a exhibited no significant effect on caspase-6 activity, and only slightly inhibited STAT3 DNA-binding activity in vitro (Fig. S4†). This data therefore demonstrates that 1a does not bind equally well to all histidine-containing proteins, and suggests that there exists further criteria that determine the activity of 1a against BRD4.
BRD4 inhibitor antagonizes the BRD4-acetylated histone PPI in vitro
To further verify the BRD4 inhibitory activity of iridium(III) complex 1a, the complex was subjected to a dose–response experiment in the TR-FRET assay. The results showed that 1a inhibited the peptide-binding activity of BRD4 with an IC50 value (dose required to inhibit 50% TR-FRET ratio) of 0.07 μM (Fig. 4a). The inhibitory activity of complex 1a against the interaction between BRD4 and H4AcK4 was further confirmed using an AlphaScreen assay (Fig. S5†).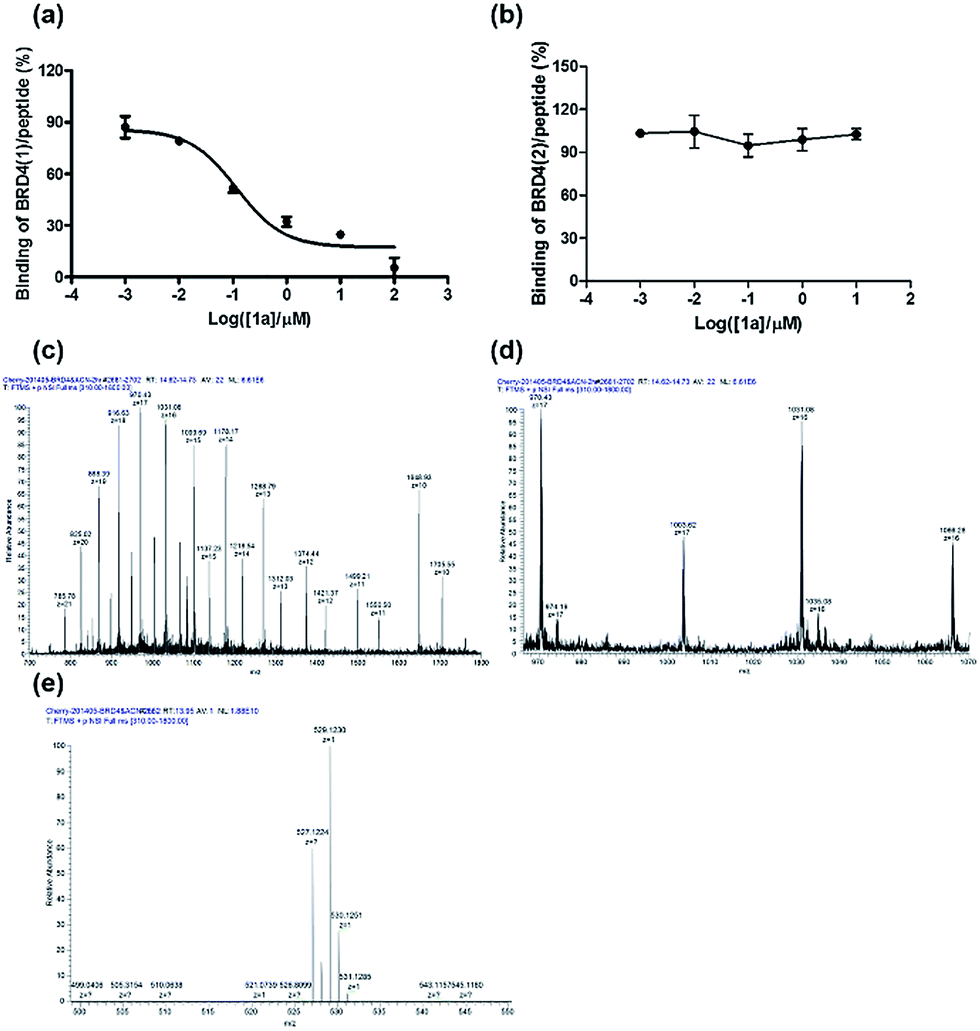 | ||
| Fig. 4 Ability of 1a to displace H4AcK4 peptide from (a) BRD4(1) and (b) BRD4(2) in a time-resolved-fluorescent resonance electron transfer (TR-FRET) assay. Binding of H4AcK4 to BRD4(1) was strongly inhibited by 1a, with half-maximum inhibitory concentration (IC50) value of 0.07 μM. Error bars represent the standard deviations of the results from three independent experiments. LC-MS/MS analysis of (c) BRD4(1), (d) BRD4(1) with 1a and (e) 1a only. BRD4(1) and 1a complex were buffered in 10 mM Tris–HCl, pH = 7.5, 500 mM NaCl and incubated at 25 °C for 2 h. The sample was analyzed by positive ion mass spectra. | ||
BRD4 contains two conserved N-terminal bromodomains BRD4(1) and BRD4(2). The activity of complex 1a against the binding of BRD4(2) to H4AcK4 was also investigated using the TR-FRET assay (Fig. 4b). The results revealed that 1a exhibited no significant inhibition of the interaction between BRD4(2) and H4AcK4. Therefore, complex 1a selectively inhibits the BRD4(1) domain. This result was further corroborated by a fluorescence polarization assay (Fig. S6†).
The interaction between complex 1a with the BRD4 proteins was also monitored by luminescence spectroscopy, since complex 1a exhibits a high luminescence response in the presence of BRD4(1) and BRD4(2) (Fig. S7†). A time-course experiment revealed that the luminescence signal of 1a reached steady-state within 5 and 8 min upon the addition of BRD4(1) or BRD4(2) at 25 °C, respectively (Fig. S8†). These data suggest that complex 1a may react slightly faster with the BRD4(1) protein.
LC-MS/MS further demonstrated the binding of 1a to BRD4(1). Mass spectrometer data was pooled and analyzed for the BRD4(1)-1a complex of 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 036.5 Da or intact BRD4(1) corresponding to 16472.3 Da (Fig. 4c and d). As shown in Fig. 4e, MS/MS fragmentation of the singly-charged ion (m/z 529.1) matched the molecular weight of 1a with cleavage of two ACN ligands. After 2 h of incubation with 1a, a complex was observed corresponding to BRD4(1) binding to C24H20N2Ir (529.1) with an additional buffer adduct NH4+OH− (35.0). The MS data therefore suggests that 1a loses two ACN ligands upon binding to BRD4(1).
036.5 Da or intact BRD4(1) corresponding to 16472.3 Da (Fig. 4c and d). As shown in Fig. 4e, MS/MS fragmentation of the singly-charged ion (m/z 529.1) matched the molecular weight of 1a with cleavage of two ACN ligands. After 2 h of incubation with 1a, a complex was observed corresponding to BRD4(1) binding to C24H20N2Ir (529.1) with an additional buffer adduct NH4+OH− (35.0). The MS data therefore suggests that 1a loses two ACN ligands upon binding to BRD4(1).
To further investigate the mechanism of action of 1a, we incubated the complex in DMSO solution. The results showed that 1a exchanges its acetonitrile ligands for DMSO ligands from the solution (Fig. S9†). This is similar to previous complexes,44 as well as NAMI-A and KP1019. Moreover, since DMSO ligands are also labile, this should not affect the ability of 1a to bind covalently to the protein target, as is the case for the previously described complexes. This makes the mechanism of 1a likely to be similar to that of NAMI-A/KP1019,45–49 which also interact covalently with their biomolecular targets via ligand exchange.50,51 Furthermore, after ligand exchange with DMSO, the complex was stable for at least 24 h in DMSO solution and in plasma under our test conditions, as revealed by the absence of significant changes in the absorbance.
BRD4 inhibition suppresses MYC and Bcl-2 in cellulo and represses cancer cell growth
Given by the promising activity of complex 1a at antagonizing the BRD4-H4AcK4 interaction in vitro, the complex was further examined for its biological activity in cells. We first performed a chromatin immunoprecipitation (ChIP) assay to investigate whether 1a can modulate the binding of BRD4 to chromatin in human malignant melanoma A375 and human caucasian metastatic melanoma A2058 cell lines (Fig. 5). ChIP analysis at the MYC promoter showed that 10 nM of complex 1a decreased the recruitment of BRD4 after 6 h. A similar pattern was observed at Bcl-2 and CDK6 loci, but not at the housekeeping B2M gene. These results suggest that 1a is able to modulate the interaction between chromatin and BRD4 in A375 and A2058 cells, particularly at the MYC promoter.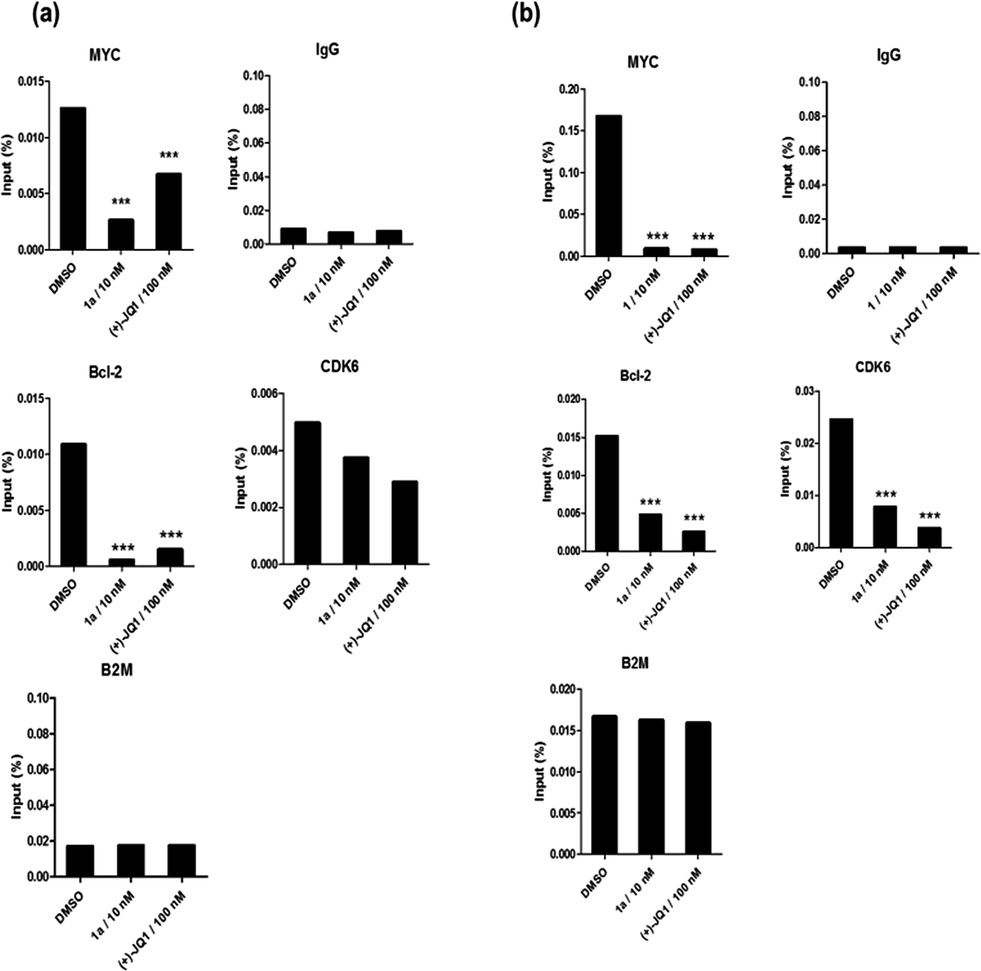 | ||
| Fig. 5 Chromatin immunoprecipitation (ChIP) analysis showed that 1a selectively decreased the binding of BRD4 to MYC, Bcl-2 and CDK6, but not housekeeping genes (B2M) in (a) A375 and (b) A2058 cells. Bar graphs represented the mean enrichment relative to input and error bars reflect standard deviation of results derived from biological triplicate experiments. Significantly different at ***p < 0.01. Error bars represent the standard deviations of the results from three independent experiments. | ||
Furthermore, the impact of complex 1a on c-myc and Bcl-2 expression in A375 and A2058 cells was investigated. Immunoblotting analysis of lysates from treated cells revealed that the expression of c-myc and Bcl-2 proteins was reduced by 1a in a dose-dependent manner (Fig. 6 and Fig. S10†). These observations are also consistent with the result of the ChIP assay described above, which showed that complex 1 could disrupt the binding of BRD4 to the MYC and Bcl-2 promoters. These results therefore suggest that 1a may act as a transcriptional modulator of c-myc and Bcl-2 expression.
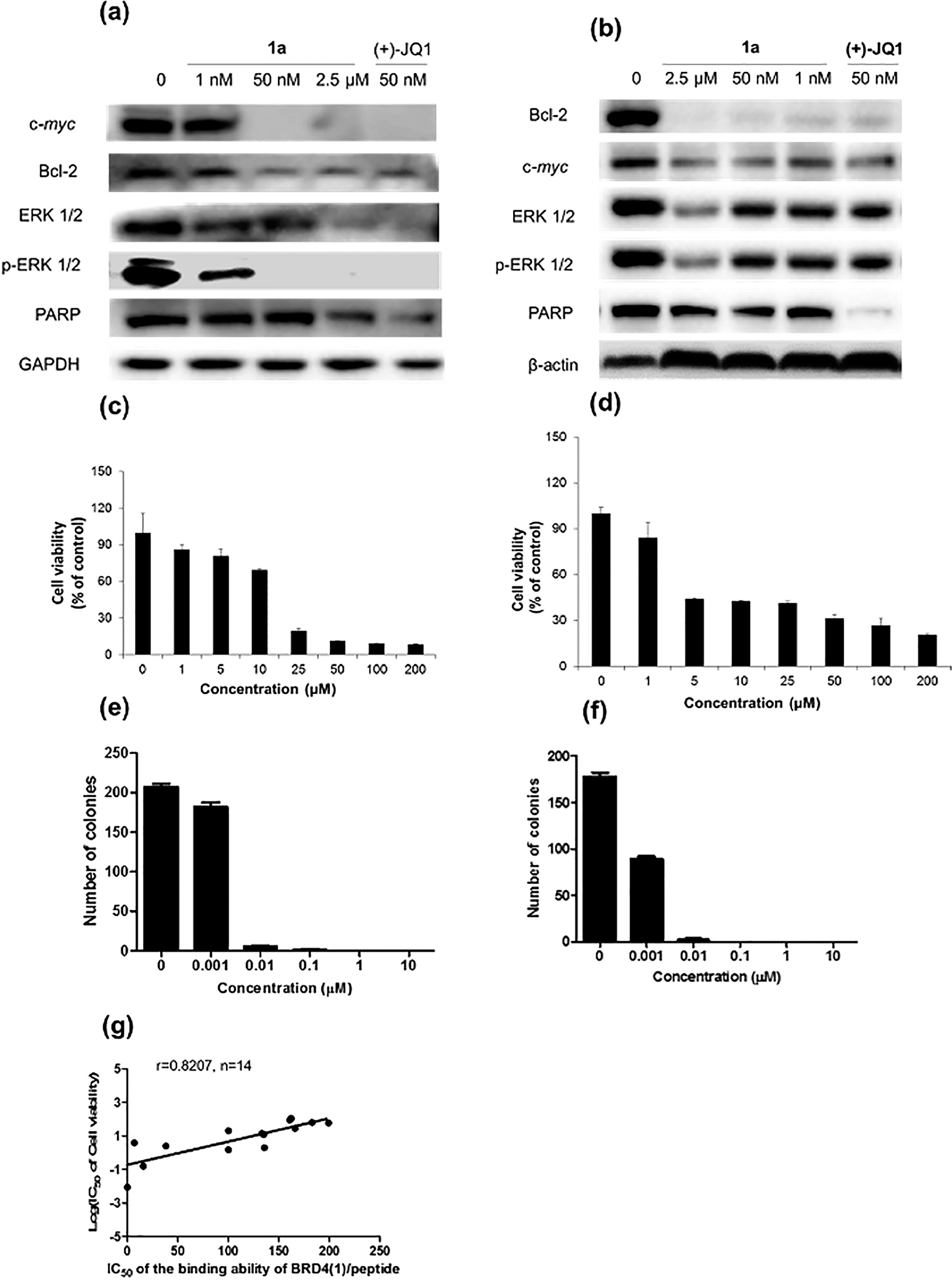 | ||
| Fig. 6 Immunoblotting analysis of the effect of 1a and (+)-JQ1 treatment in (a) A375 and (b) A2058 cells. Densitometry analysis revealed that 1a inhibited c-myc, Bcl-2, ERK 1/2, p-ERK 1/2 and PARP expression. Dose response analysis of cell viability of complex 1a against (c) A375 cells and (d) A2058 cells. Error bars represent the standard deviations of the results from three independent experiments. Normalized proliferation curves in the colony formation assay for (e) A375 and (f) A2058 cells treated with vehicle or 1a (0.001–10 μM) measured by crystal violet staining. Error bars represent the standard deviations of the results from three independent experiments. (g) The relationship between the IC50 of the binding ability of BRD4(1)/peptide and the log of IC50 of A375 cell viability, and a trend of positive correlation was observed (r = 0.8207, n = 14). | ||
c-myc and Bcl-2 have been intensely studied as anti-cancer targets due to their roles in cell cycle progression, cellular transformation and apoptosis.52,53 Therefore, we were interested to investigate whether or not complex 1a could exhibit anti-proliferative effects against cancer cells. In in vitro evaluation, complex 1a exhibited potent cytotoxicity against the A375 (IC50 = 12.5 μM) and A2058 (IC50 = 3 μM) cell lines (Fig. 6c and d). The anti-proliferative activity of complex 1a towards A375 and A2058 cells was further determined using the colony formation assay. The results showed that 1a was cytotoxic against A375 and A2058 melanoma cells with estimated IC50 values (dose required to inhibit 50% cellular growth after 24 h exposure to 1a) of 5 nM and 1 nM, respectively (Fig. 6e and f). We reason that the cytotoxicity imparted by 1a could be attributed, at least in part, to the suppression of c-myc and Bcl-2 transcription via BRD4 inhibition. Additionally, ERK 1/2, p-ERK 1/2 and PARP expression were also down-regulated in A375 and A2058 cells after treatment with 1a (Fig. 6a and b). Aberrant ERK 1/2 activation is implicated in numerous tumors, while PARP promotes cell survival due to its role in DNA repair. Therefore, the inhibition of ERK 1/2 and/or PARP activity by complex 1a may represent an alternative mechanism by which the complex exerts anti-proliferative activity.
BRD4 inhibitor displays anti-proliferative activity in an animal model of melanoma
To further explore the relationship between BRD4 inhibition and cytotoxic activity, we also tested the metal complexes 1, 1a–1j from the second round of screening and 9–11 with chemical structures similar to 1a from the first round of screening for their in vitro anticancer activity against A375 cells. Plotting the antiproliferative IC50 values of the complexes against the IC50 values for the inhibition of BRD4(1)-H4AcK4 binding revealed a positive relationship (r = 0.8207, n = 14) (Fig. 6g), suggesting that the anticancer activity of the metal complexes may be mediated by their inhibition of BRD4 binding activity.To further investigate the antitumor activity exerted by BRD4 inhibition, we explored the biological activity of 1a in a mouse xenograft tumor model. Six to seven week-old male CB.17 SCID mice were injected subcutaneously with human malignant melanoma A375 cells, and after the establishment of palpable tumors, were administrated with 1a (100 mg kg−1) or vehicle (13% DMSO in normal saline) intraperitoneally (i.p.) once daily for 16 days. Encouragingly, the treated tumors were ca. 40% smaller than the control tumors over the course of the treatment, with a significant difference in estimated tumor volume being observed after 16 days (Fig. 7a–c). Tumor weight measurements after sacrifice confirmed a reduction in tumor growth in mice administrated with 1a (Fig. 7d). We also observed that the treated mice exhibited no signs of weight loss over the course of the experiment (Fig. 7e). Taken together, these results indicate that complex 1a significantly inhibited the growth of melanoma tissue in an in vivo xenograft model, without causing overt toxicity to the mice.
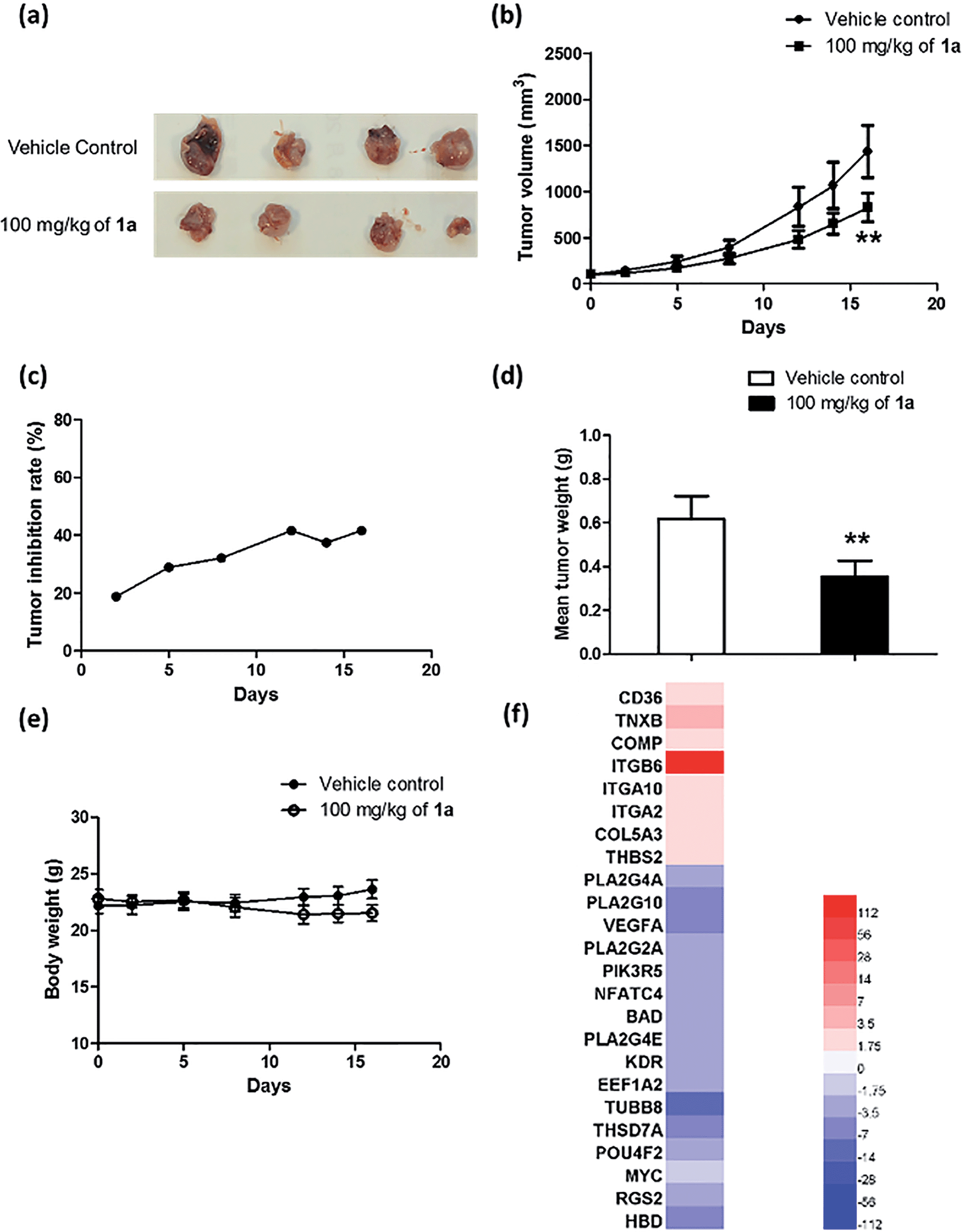 | ||
| Fig. 7 Anti-proliferative activity of 1a in an in vivo xenograft model of melanoma. (a) Photographs of dissected tumors from the control (vehicle) and treatment (1a, 100 mg kg−1). (b) Average A375 tumour volume in the control group and treatment group (1a, 100 mg kg−1). Each group contained six mice and results are reported as the values of the mean ± SEM. (c) Tumour inhibition of A375 xenografts in the treatment group (1a, 100 mg kg−1) expressed as percentage reduction in tumor volume compared to the control group. The results were analyzed using the Student's t-test. Significantly different at 0.01 < **p < 0.05. (d) Average tumor weight of the vehicle control group versus the treatment group (1a, 100 mg kg−1). (e) Average body weight of the two groups. Each group contained six mice, and results were reported as the values of the mean ± SEM. The results were analyzed using the Student's t-test. Significantly different at 0.01 < **p < 0.05. (f) Heat map of regulated genes of the ECM pathway and VEGF signaling pathway following treatment with 1a. The color scale in the inset represents the log-fold change of expression compared with untreated control. | ||
Microarray analysis was performed on the excised tumor tissues to identify signaling pathways that were up-regulated or down-regulated by complex 1a. The results showed that treatment with 1a resulted in MYC down-regulation as well as a significant decrease in expression of the c-myc target gene set in the tumor tissues. Moreover, complex 1a up-regulated genes in the extracellular matrix (ECM) pathway while down-regulating genes in the VEGF signaling pathway (Table S2† and Fig. 7f). Differential ECM gene expression is an important marker of metastatic activity, and BRD4 modulates the expression of many ECM genes that are dysregulated in tumors.54 Moreover, increased MYC activity can also suppress ECM gene expression.55–57 Hence, the up-regulation of the ECM pathway in tumor tissues could potentially be attributed to the inhibition of BRD4-directed transcription by 1ain vivo. Furthermore, MYC promotes angiogenesis through the up-regulation of VEGF.58,59 Therefore, the down-regulation of genes in the VEGF signaling pathway could also be attributed to the effects of 1a on BRD4-mediated transcription in vivo.
Conclusions
In conclusion, an iridium(III) complex 1a was found to be a potential modulator of the epigenetic reader protein BRD4. Complex 1a inhibited the PPI between BRD4 and an acetylated histone peptide as revealed by multiple biochemical assays, including FRET, AlphaScreen and FP assays. Although mass spectrometry data suggested that 1a binds to histidine residues with the loss of ACN ligands, 1a was found not to significantly interact with other histidine-containing proteins such as caspase-6 and STAT3. Additionally, complex 1a displaced BRD4 from chromatin and hence inhibited c-myc expression in melanoma cells through blocking the binding of BRD4 to the c-myc promoter. Cytotoxicity and colony formation experiments suggested 1a is capable of anti-proliferative activity in melanoma cells, possibly through down-regulation of c-myc protein expression. Finally, complex 1a significantly repressed A375 melanoma xenograft growth in an in vivo mouse model without causing visible toxicity to the mice. Preliminary structure–activity analysis indicated that the nature of the metal ion and the C^N and N^N co-ligands were important for the biological activity of 1a. To our knowledge, complex 1a represents the first metal-based inhibitor of BRD4 and of any BET bromodomain-containing protein in general. We envision that complex 1a may serve as a useful scaffold for the development of more potent epigenetic agents against cancers such as melanoma. Additionally, the irreversible and selective nature of BRD4 inhibition by 1a may allow the complex to be used at lower dosages in potential in vivo applications.Experimental
Materials and cell lines
All the chemicals were purchased from Sigma-Aldrich and were used as received. BRD4 bromodomain 1 TR-FRET assay kit and BRD4 bromodomain 2 TR-FRET assay kitware purchased from Cayman Chemical (Ann Arbor, MI, USA). Fetal bovine serum (FBS) and Dulbecco's modified Eagle's medium (DMEM) were obtained from Gibco BRL (Gaithersburg, MD, USA). All other reagents and chemicals were obtained from commercial sources.General experimental
Mass spectrometry was performed at the Mass Spectroscopy Unit at the Department of Chemistry, Hong Kong Baptist University, Hong Kong (China). Deuterated solvents for NMR purposes were obtained from Armar and used as received.1H and 13C NMR were recorded on a Bruker Avance 400 spectrometer operating at 400 MHz (1H) and 100 MHz (13C). 1H and 13C chemical shifts were referenced internally to solvent shift (acetonitrile-d3: 1H, δ 1.94, 13C δ 118.7; acetone-d6: 1H δ 2.05, 13C δ 29.7). Chemical shifts (δ) are quoted in ppm, the downfield direction being defined as positive. Uncertainties in chemical shifts are typically ±0.01 ppm for 1H and ±0.05 for 13C. Coupling constants are typically ±0.1 Hz for 1H–1H and ±0.5 Hz for 1H–13C couplings. The following abbreviations are used for convenience in reporting the multiplicity of NMR resonances: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet. All NMR data was acquired and processed using standard Bruker software (Topspin).Cell cultures
The human melanoma A375, A2058 cell lines were purchased from the Bioresource Collection and Research Center (Hsinchu, Taiwan, ROC) and the American Type Culture Collection (Manassas, VA, USA).Time-resolved-fluorescence resonance energy transfer (TR-FRET) assay
The disruption of BRD4/tetra-acetylated lysine histone 4 peptide (H4AcK4) binding by the tested complexes was evaluated according to the manufacturer's instructions (Cayman Chemical, Carlsbad, CA). Briefly, the tested compounds, BRD4 bromodomain 1 or BRD4 bromodomain 2 europium chelate and BRD4 bromodomain 1 or BRD4 bromodomain 2 ligand/APC acceptor mixture were dissolved in 1 × TR-FRET buffer provided by the manufacturer and stored in −80 °C before use. To each test well, 10 μL of the diluted BRD4 bromodomain 1 europium chelate was added. Indicated concentrations of the tested compounds and (+)-JQ1 were then added to the well and incubated for 15 min at room temperature and dark condition to allow pre-equilibration of the compounds with the BRD4 bromodomain 1 or BRD4 bromodomain 2 europium chelate. Subsequently, 5 μL of the BRD4 bromodomain 1 or BRD4 bromodomain 2 ligand/APC acceptor mixture was added to each tested well. The microtitre plate was sealed and incubated at room temperature in the dark for 1 h. The experimental results were qualified using a spectrophotometer by measuring the dual emissions at 620 nm and 670 nm using an excitation at 340 nm. Data analysis is performed using the TR-FRET ratio (670 nm emission/620 nm emission).Mass spectrometry analysis
1 μg BRD4(1) and 25 μM complex 1a were incubated in reaction buffer (10 mM Tris–HCl, pH = 7.5, 500 mM NaCl) at 25 °C for 2 h. Positive ion mass spectra were acquired as accurate mass centroid data using a Thermo Q Exactive with a nano spray source, connected to a thermo Easy-nLC 1000 HPLC system. Survey full-scan MS spectra (from m/z 310–1800) were acquired in the Orbitrap analyzer with resolution r = 70000 at m/z 400. The elemental composition was calculated using Xcalibur 2.1 for the [M + H]+.
For amino acid mass spectra analysis, histidine were incubated with complex 1a in Tris–HCl buffer (10 mM Tris–HCl, pH = 7.5, 500 mM NaCl) 30 min, then were directly injected into an ESI time-of-flight mass spectrometer at a flow rate of 5 μL min−1. The capillary voltage was set at −4500 V, and the dry N2 gas flow was 4.0 L min−1 at 180 °C. Data were analyzed by the software Bruker Daltonics Data Analysis.
Chromatin immunoprecipitation
Approximately 1 × 108 A375 and A2058 cells were treated with 500 nM complex 1a or DMSO for 24 h and cross-linked with 1× crosslink solution (1.1% formaldehyde, 5 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.3, 10 mM NaCl, 100 μM ethylenediaminetetraacetic acid (EDTA), 50 μM ethylene glycol tetraacetic acid (EGTA)) followed by two washes using phosphate buffered saline (PBS). Cells were scraped and frozen in liquid nitrogen. ChIP-PCR analysis was done following a published protocol.60Immunoblotting
Cells were harvested to obtain whole-cell extracts by the addition of one volume of 250 mM Tris–HCl (pH 6.8), 20% glycerol, 2% sodium dodecyl sulfate (SDS), 5% 2-mercaptoethanol, and 0.2% bromophenol blue to cells in one volume of PBS followed by boiling for 5 min. Samples were resolved on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS/PAGE) gels and transferred to polyvinylidene fluoride (PVDF) membranes. Blots were probed with antibodies to c-myc, Bcl-2, ERK 1/2, p-ERK 1/2, PARP, GAPDH and beta-actin (cell signaling). After incubation with secondary antibodies (Santa Cruz Biotechnology), blots were developed with ECL reagent (ThermoFisher).Animal materials
In this study, the use of animals complied with the Guiding Principles in the Care and Use of Animals of the American Physiology Society and was approved by the Animal Care and Use Committee at the National Kaohsiung Medical University. Six to seven weeks old male CB.17 SCID mice were purchased from BioLascoTaiwan Co., LTD and quarantined for one week. During the experiment period, 5 mice were housed in one cage. All animals were hosted in the Da-Hu animal facility in a 12 h light/12 h dark cycle at 19–25 °C. Animals had free access to rodent pellet food and water ad libitum. The experimental protocol of animal study was reviewed and approved by the Institutional Animal Care and Use Committee, DCB.Xenograft tumor assay
The human A375 melanoma cells used for implantation were harvested during log phase growth and re-suspended in phosphate buffered saline to a concentration containing 1 × 107 cells per mL. Each six to seven weeks' old male CB.17 SCID mice was injected subcutaneously (s.c.) in the flank with 1.5 × 107 cells in 0.15 mL of a 50% matrigel solution (BD Biosciences, MA, USA). When the average tumor volume had reached 100 mm3, the mice were randomly divided into 2 groups and were administrated with 1a (50 or 100 mg kg−1) or vehicle (13% DMSO in normal saline) intraperitoneally (i.p.) once daily for 16 days. The diameters of xenograft tumors were measured with Digimatic caliper (Series no. 500, Mitutoyo Corp., Japan) and the tumor volume (in mm3) was calculated using the formula:| Tumor volume = (w2 × l)/2 |
Tumors were measured twice per week using calipers. The percentage of tumor growth inhibition (TGI) was calculated using the following formula:
| %TGI = [1 – (T/C)] × 100% |
Animals were weighed twice weekly until the completion of the study. The body weight change was calculated as the percentage increase in body weight compared to the initial body weight.
Microarray analysis
Xenografts in the mouse models were removed 16 days after the administration of A375 cells. Total RNA from tumours from three independent vehicle and treatment mice were prepared using TRIzol reagent (Invitrogen). Gene expression profiles were examined using Mouse One Array DNA Microarray chips (Phalanx Biotech Group, Inc., Hsinchu, Taiwan). The criterion for affected genes was a significant difference in tumor RNA levels between the treatment and vehicle groups (p < 0.05), with at least a 2-fold difference. Gene ontology analysis was performed using PANTHER.Statistical analysis
Data were analyzed using the Prism software version 6 (Graph Pad software Inc., San Diego, CA, USA). The Student's t-test was used for comparisons between the two groups. The correlation between BRD4 inhibition and cytotoxic activity was calculated using the Pearson's correlation coefficient equation. Quantitative data are reported as mean ± SEM from at least three independent experiments. Differences were considered statistically significant at p < 0.05. All statistical tests were two-sided.All synthetic methods, characterization and biological assays details are given in the ESI.†
Acknowledgements
This work is supported by Hong Kong Baptist University (FRG2/14-15/004), Centre for Cancer and Inflammation Research, School of Chinese Medicine (CCIR-SCM, HKBU), the Health and Medical Research Fund (HMRF/13121482 and HMRF/14130522), the Research Grants Council (HKBU/201811, HKBU/204612, and HKBU/201913), the French National Research Agency/Research Grants Council Joint Research Scheme (A-HKBU201/12 - Oligoswitch), Interdisciplinary Research Matching Scheme (RC-IRMS/14-15/06), the Science and Technology Development Fund, Macao SAR (103/2012/A3 and 098/2014/A2), the University of Macau (MYRG091(Y3-L2)-ICMS12-LCH, MYRG2015-00137-ICMS-QRCM and MRG023/LCH/2013/ICMS) and the National Science Council under the Grant nos. NSC102-2622-E-037-002, NSC102-2221-E-037-005, NSC102-2622-E-011-001-CC2 and MOST103-2221-E-037-004. We thank the project of Center for Stem Cell Research, Kaohsiung Medical University, Kaohsiung, Taiwan, KMU-TP103G00 and KMU-TP103G02-05. The authors would like to thank Dr. Hiu Yee KWAN, Hong Kong Baptist University for providing technical help.Notes and references
- B. Li, M. Carey and J. L. Workman, Cell, 2007, 128, 707–719 CrossRef CAS PubMed.
- G. E. Zentner and S. Henikoff, Nat. Struct. Mol. Biol., 2013, 20, 259–266 CAS.
- A. J. Ruthenburg, H. Li, D. J. Patel and C. D. Allis, Nat. Rev. Mol. Cell Biol., 2007, 8, 983–994 CrossRef CAS PubMed.
- C. H. Arrowsmith, C. Bountra, P. V. Fish, K. Lee and M. Schapira, Nat. Rev. Drug Discovery, 2012, 11, 384–400 CrossRef CAS PubMed.
- S. D. Taverna, H. Li, A. J. Ruthenburg, C. D. Allis and D. J. Patel, Nat. Struct. Mol. Biol., 2007, 14, 1025–1040 CAS.
- C.-w. Chung, A. W. Dean, J. M. Woolven and P. Bamborough, J. Med. Chem., 2012, 55, 576–586 CrossRef CAS PubMed.
- L. Zeng and M.-M. Zhou, FEBS Lett., 2002, 513, 124–128 CrossRef CAS.
- B. Florence and D. V. Faller, Front. Biosci., 2001, 6, d1008–d1018 CrossRef CAS PubMed.
- C.-w. Chung and D. F. Tough, Drug Discovery Today: Ther. Strategies, 2012, 9, e111–e120 CrossRef PubMed.
- Z. Yang, N. He and Q. Zhou, Mol. Cell. Biol., 2008, 28, 967–976 CrossRef CAS PubMed.
- A. Dey, A. Nishiyama, T. Karpova, J. McNally and K. Ozato, Mol. Biol. Cell, 2009, 20, 4899–4909 CrossRef CAS PubMed.
- M. F. Segura, B. Fontanals-Cirera, A. Gaziel-Sovran, M. V. Guijarro, D. Hanniford, G. Zhang, P. Gonzalez-Gomez, M. Morante, L. Jubierre, W. Zhang, F. Darvishian, M. Ohlmeyer, I. Osman, M. M. Zhou and E. Hernando, Cancer Res., 2013, 73, 6264–6276 CrossRef CAS PubMed.
- P. Filippakopoulos, J. Qi, S. Picaud, Y. Shen, W. B. Smith, O. Fedorov, E. M. Morse, T. Keates, T. T. Hickman, I. Felletar, M. Philpott, S. Munro, M. R. McKeown, Y. Wang, A. L. Christie, N. West, M. J. Cameron, B. Schwartz, T. D. Heightman, N. la Thangue, C. A. French, O. Wiest, A. L. Kung, S. Knapp and J. E. Bradner, Nature, 2010, 468, 1067–1073 CrossRef CAS PubMed.
- J. E. Delmore, G. C. Issa, M. E. Lemieux, P. B. Rahl, J. Shi, H. M. Jacobs, E. Kastritis, T. Gilpatrick, R. M. Paranal, J. Qi, M. Chesi, A. C. Schinzel, M. R. McKeown, T. P. Heffernan, C. R. Vakoc, P. L. Bergsagel, I. M. Ghobrial, P. G. Richardson, R. A. Young, W. C. Hahn, K. C. Anderson, A. L. Kung, J. E. Bradner and C. S. Mitsiades, Cell, 2011, 146, 904–917 CrossRef CAS PubMed.
- J. A. Mertz, A. R. Conery, B. M. Bryant, P. Sandy, S. Balasubramanian, D. A. Mele, L. Bergeron and R. J. Sims, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16669–16674 CrossRef CAS PubMed.
- C. G. Hartinger and P. J. Dyson, Chem. Soc. Rev., 2009, 38, 391–401 RSC.
- A. V. Klein and T. W. Hambley, Chem. Rev., 2009, 109, 4911–4920 CrossRef CAS PubMed.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed.
- A. L. Noffke, A. Habtemariam, A. M. Pizarro and P. J. Sadler, Chem. Commun., 2012, 48, 5219–5246 RSC.
- A. Meyer, C. P. Bagowski, M. Kokoschka, M. Stefanopoulou, H. Alborzinia, S. Can, D. H. Vlecken, W. S. Sheldrick, S. Wolfl and I. Ott, Angew. Chem., Int. Ed., 2012, 51, 8895–8899 CrossRef CAS PubMed.
- A. de Almeida, B. L. Oliveira, J. D. G. Correia, G. Soveral and A. Casini, Coord. Chem. Rev., 2013, 257, 2689–2704 CrossRef CAS PubMed.
- C. G. Hartinger, M. Groessl, S. M. Meier, A. Casini and P. J. Dyson, Chem. Soc. Rev., 2013, 42, 6186–6199 RSC.
- H. Huang, P. Zhang, B. Yu, Y. Chen, J. Wang, L. Ji and H. Chao, J. Med. Chem., 2014, 57, 8971–8983 CrossRef CAS PubMed.
- A. Leonidova, V. Pierroz, L. A. Adams, N. Barlow, S. Ferrari, B. Graham and G. Gasser, ACS Med. Chem. Lett., 2014, 5, 809–814 CrossRef CAS PubMed.
- D. L. Ma, W. L. Wong, W. H. Chung, F. Y. Chan, P. K. So, T. S. Lai, Z. Y. Zhou, Y. C. Leung and K. Y. Wong, Angew. Chem., Int. Ed., 2008, 47, 3735–3739 CrossRef CAS PubMed.
- E. Meggers, Chem. Commun., 2009, 1001–1010 RSC.
- X. Meng, M. L. Leyva, M. Jenny, I. Gross, S. Benosman, B. Fricker, S. Harlepp, P. Hébraud, A. Boos, P. Wlosik, P. Bischoff, C. Sirlin, M. Pfeffer, J.-P. Loeffler and C. Gaiddon, Cancer Res., 2009, 69, 5458–5466 CrossRef CAS PubMed.
- C. M. Che and F. M. Siu, Curr. Opin. Chem. Biol., 2010, 14, 255–261 CrossRef CAS PubMed.
- K. J. Kilpin and P. J. Dyson, Chem. Sci., 2013, 4, 1410–1419 RSC.
- D. L. Ma, H. Z. He, K. H. Leung, D. S. H. Chan and C. H. Leung, Angew. Chem., Int. Ed., 2013, 52, 7666–7682 CrossRef CAS PubMed.
- D. L. Ma, L. J. Liu, K. H. Leung, Y. T. Chen, H. J. Zhong, D. S. H. Chan, H. M. D. Wang and C. H. Leung, Angew. Chem., Int. Ed., 2014, 53, 9178–9182 CrossRef CAS PubMed.
- C. H. Leung, L. J. Liu, L. Lu, B. He, D. W. J. Kwong, C. Y. Wong and D. L. Ma, Chem. Commun., 2015, 51, 3973–3976 RSC.
- J. Singh, R. C. Petter, T. A. Baillie and A. Whitty, Nat. Rev. Drug Discovery, 2011, 10, 307–317 CrossRef CAS PubMed.
- L. Feng, Y. Geisselbrecht, S. Blanck, A. Wilbuer, G. E. Atilla-Gokcumen, P. Filippakopoulos, K. Kräling, M. A. Celik, K. Harms, J. Maksimoska, R. Marmorstein, G. Frenking, S. Knapp, L.-O. Essen and E. Meggers, J. Am. Chem. Soc., 2011, 133, 5976–5986 CrossRef CAS PubMed.
- S. Blanck, Y. Geisselbrecht, K. Kraling, S. Middel, T. Mietke, K. Harms, L.-O. Essen and E. Meggers, Dalton Trans., 2012, 41, 9337–9348 RSC.
- C. H. Leung, H. J. Zhong, H. Yang, Z. Cheng, D. S. H. Chan, V. P. Y. Ma, R. Abagyan, C. Y. Wong and D. L. Ma, Angew. Chem., Int. Ed., 2012, 51, 9010–9014 CrossRef CAS PubMed.
- S. Dieckmann, R. Riedel, K. Harms and E. Meggers, Eur. J. Inorg. Chem., 2012, 813–821 CrossRef CAS PubMed.
- S. Mollin, S. Blanck, K. Harms and E. Meggers, Inorg. Chim. Acta, 2012, 393, 261–268 CrossRef CAS PubMed.
- R. Anand, J. Maksimoska, N. Pagano, E. Y. Wong, P. A. Gimotty, S. L. Diamond, E. Meggers and R. Marmorstein, J. Med. Chem., 2009, 52, 1602–1611 CrossRef CAS PubMed.
- G. E. Atilla-Gokcumen, L. Costanzo and E. Meggers, JBIC, J. Biol. Inorg. Chem., 2011, 16, 45–50 CrossRef CAS PubMed.
- S. Blanck, T. Cruchter, A. Vultur, R. Riedel, K. Harms, M. Herlyn and E. Meggers, Organometallics, 2011, 30, 4598–4606 CrossRef CAS PubMed.
- C. Streu, L. Feng, P. J. Carroll, J. Maksimoska, R. Marmorstein and E. Meggers, Inorg. Chim. Acta, 2011, 377, 34–41 CrossRef CAS PubMed.
- M. S. Lowry, W. R. Hudson, R. A. Pascal and S. Bernhard, J. Am. Chem. Soc., 2004, 126, 14129–14135 CrossRef CAS PubMed.
- C. Li, M. Yu, Y. Sun, Y. Wu, C. Huang and F. Li, J. Am. Chem. Soc., 2011, 133, 11231–11239 CrossRef CAS PubMed.
- F. Piccioli, S. Sabatini, L. Messori, P. Orioli, G. H. Ch and B. K. Keppler, J. Inorg. Biochem., 2004, 98, 1135–1142 CrossRef CAS PubMed.
- A. R. Timerbaev, L. S. Foteeva, A. V. Rudnev, J. K. Abramski, K. Polec-Pawlak, C. G. Hartinger, M. Jarosz and B. K. Keppler, Electrophoresis, 2007, 28, 2235–2240 CrossRef CAS PubMed.
- O. Domotor, C. G. Hartinger, A. K. Bytzek, T. Kiss, B. K. Keppler and E. A. Enyedy, JBIC, J. Biol. Inorg. Chem., 2013, 18, 9–17 CrossRef PubMed.
- R. Trondl, P. Heffeter, C. R. Kowol, M. A. Jakupec, W. Berger and B. K. Keppler, Chem. Sci., 2014, 5, 2925–2932 RSC.
- M. I. Webb and C. J. Walsby, Dalton Trans., 2015 10.1039/c5dt02021b.
- F. Kratz, M. Hartmann, B. Keppler and L. Messori, J. Biol. Chem., 1994, 269, 2581–2588 CAS.
- L. Messori, P. Orioli, D. Vullo, E. Alessio and E. Iengo, Eur. J. Biochem., 2000, 267, 1206–1213 CrossRef CAS.
- M. A. Stoff-Khalili, P. Dall and D. T. Curiel, Cancer Gene Ther., 2006, 13, 633–647 CrossRef CAS PubMed.
- K. J. Falkenberg and R. W. Johnstone, Nat. Rev. Drug Discovery, 2014, 13, 673–691 CrossRef CAS PubMed.
- N. P. S. Crawford, J. Alsarraj, L. Lukes, R. C. Walker, J. S. Officewala, H. H. Yang, M. P. Lee, K. Ozato and K. W. Hunter, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6380–6385 CrossRef CAS PubMed.
- H. A. Coller, C. Grandori, P. Tamayo, T. Colbert, E. S. Lander, R. N. Eisenman and T. R. Golub, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3260–3265 CrossRef CAS.
- J. D. Watson, S. K. Oster, M. Shago, F. Khosravi and L. Z. Penn, J. Biol. Chem., 2002, 277, 36921–36930 CrossRef CAS PubMed.
- E. R. Lawlor, L. Soucek, L. Brown-Swigart, K. Shchors, C. U. Bialucha and G. I. Evan, Cancer Res., 2006, 66, 4591–4601 CrossRef CAS PubMed.
- R. Chen, J. H. Yik, Q. J. Lew and S. H. Chao, BioMed Res. Int., 2014, 2014, 232870 Search PubMed.
- S. W. J. Ember, J. Y. Zhu, S. H. Olesen, M. P. Martin, A. Becker, N. Berndt, G. I. Georg and E. Schönbrunn, ACS Chem. Biol., 2014, 9, 1160–1171 CrossRef CAS PubMed.
- P. B. Rahl, C. Y. Lin, A. C. Seila, R. A. Flynn, S. McCuine, C. B. Burge, P. A. Sharp and R. A. Young, Cell, 2010, 141, 432–445 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic methods, characterization and biological assays details. See DOI: 10.1039/c5sc02321a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |