
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterometallic titanium–gold complexes inhibit renal cancer cells in vitro and in vivo†‡
Jacob
Fernández-Gallardo§
 a,
Benelita T.
Elie§
ab,
Tanmoy
Sadhukha
c,
Swayam
Prabha
cd,
Mercedes
Sanaú
e,
Susan A.
Rotenberg
bf,
Joe W.
Ramos
*g and
María
Contel
*abg
a,
Benelita T.
Elie§
ab,
Tanmoy
Sadhukha
c,
Swayam
Prabha
cd,
Mercedes
Sanaú
e,
Susan A.
Rotenberg
bf,
Joe W.
Ramos
*g and
María
Contel
*abg
aDepartment of Chemistry, Brooklyn College and The Graduate Center, The City University of New York, Brooklyn, NY 11210, USA. E-mail: mariacontel@brooklyn.cuny.edu
bBiology PhD Program, The Graduate Center, The City University of New York, 365 Fifth Avenue, New York, NY 10016, USA
cDepartment of Pharmaceutics, College of Pharmacy, University of Minnesota, MN 55455, USA
dCenter for Translational Drug Delivery, University of Minnesota, MN 55455, USA
eDepartamento de Química Inorgánica, Universidad de Valencia, Burjassot, Valencia, 46100, Spain
fDepartment of Chemistry and Biochemistry, Queens College, The City University of New York, Flushing, NY 11367, USA
gCancer Biology Program, University of Hawaii Cancer Center, University of Hawaii at Manoa, Honolulu, HI 96813, USA
First published on 23rd June 2015
Abstract
Following recent work on heterometallic titanocene–gold complexes as potential chemotherapeutics for renal cancer, we report here on the synthesis, characterization and stability studies of new titanocene complexes containing a methyl group and a carboxylate ligand (mba = S–C6H4–COO−) bound to gold(I)-phosphane fragments through a thiolate group [(η-C5H5)2TiMe(μ-mba)Au(PR3)]. The compounds are more stable in physiological media than those previously reported and are highly cytotoxic against human cancer renal cell lines. We describe here preliminary mechanistic data involving studies on the interaction of selected compounds with plasmid (pBR322) DNA used as a model nucleic acid, and with selected protein kinases from a panel of 35 protein kinases having oncological interest. Preliminary mechanistic studies in Caki-1 renal cells indicate that the cytotoxic and anti-migration effects of the most active compound 5 [(η-C5H5)2TiMe(μ-mba)Au(PPh3)] involve inhibition of thioredoxin reductase and loss of expression of protein kinases that drive cell migration (AKT, p90-RSK, and MAPKAPK3). The co-localization of both titanium and gold metals (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) in Caki-1 renal cells was demonstrated for 5 indicating the robustness of the heterometallic compound in vitro. Two compounds were selected for further in vivo studies on mice based on their selectivity in vitro against renal cancer cell lines when compared to non-tumorigenic human kidney cell lines (HEK-293T and RPTC) and the favourable preliminary toxicity profile in C57BL/6 mice. Evaluation of Caki-1 xenografts in NOD.CB17-Prkdc SCID/J mice showed an impressive tumor reduction (67%) after treatment for 28 days (3 mg per kg per every other day) with heterometallic compound 5 as compared with the previously described [(η-C5H5)2Ti{OC(O)-4-C6H4-P(Ph2)AuCl}2] 3 which was non-inhibitory. These findings indicate that structural modifications on the ligand scaffold affect the in vivo efficacy of this class of compounds.
:1 ratio) in Caki-1 renal cells was demonstrated for 5 indicating the robustness of the heterometallic compound in vitro. Two compounds were selected for further in vivo studies on mice based on their selectivity in vitro against renal cancer cell lines when compared to non-tumorigenic human kidney cell lines (HEK-293T and RPTC) and the favourable preliminary toxicity profile in C57BL/6 mice. Evaluation of Caki-1 xenografts in NOD.CB17-Prkdc SCID/J mice showed an impressive tumor reduction (67%) after treatment for 28 days (3 mg per kg per every other day) with heterometallic compound 5 as compared with the previously described [(η-C5H5)2Ti{OC(O)-4-C6H4-P(Ph2)AuCl}2] 3 which was non-inhibitory. These findings indicate that structural modifications on the ligand scaffold affect the in vivo efficacy of this class of compounds.
Introduction
For over three decades, complexes with metals other than platinum have been explored as potential cancer chemotherapeutics1 in order to overcome the drawbacks of cisplatin and some of its derivatives. The challenges posed by these compounds include both acquired or intrinsic resistance, and a limited therapeutic window due to high toxicity leading to undesired side effects.2,3Titanocene dichloride (TDC, [(η-C5H5)2TiCl2]) was the first organometallic complex to enter clinical trials in 1993.4 This compound exhibited considerable antitumor activity in both in vitro and in vivo experimental models even against cisplatin-resistant cells and tumors that are generally difficult to treat.5,6 However, the efficacy of titanocene dichloride in phase II clinical trials in patients with metastatic renal cell carcinoma7 or metastatic breast cancer was too low to be pursued.8 To overcome these problems, different follow-up TDC derivatives have been developed, e.g. Titanocene Y, Titanocene Y* and Titanocene T22 (ref. 9–14) that were shown to be effective as potential anti-cancer agents in vitro and in vivo. These derivatives display a different mode of action than cisplatin since they produce selected significant renal tumor growth inhibition that warrants further pharmacological and clinical investigation.
Gold complexes have emerged as another family of metallodrugs with encouraging pharmacological profiles. Gold compounds have displayed significant effects against tumors resistant to cisplatin.15,16 In addition, it has been demonstrated that for most gold compounds DNA is not the primary target. A variety of alternative biochemical targets such as deacetylases, specific proteases, protein kinases and polymerases have been proposed for some of the gold(III) and gold(I) complexes.17–28
Seeking an enhancement of the anti-cancer properties of these two families of metallodrugs, we and others have worked on the design of titanium–gold heterometallic complexes. The hypothesis is that the incorporation of two different cytotoxic metals in the same molecule may improve their activity as anti-tumor agents due to interaction of the different metals with multiple biological targets (cooperative effect) or by the improved chemicophysical properties of the resulting heterometallic compound (synergism). It has been demonstrated that a number of titanocene–gold complexes are more cytotoxic and selective towards human cancer cell lines than the corresponding monometallic moieties.29,30
Our first synthetic approach was based on titanocene–gold compounds with a cyclopentadienyl-phosphane linker moiety30 (compound 1, Chart 1). Species of this type showed enhanced anti-cancer properties with respect to the monometallic counterparts.29,30 However, due to the well-known hydrolysis of Cp–Ti bonds at pH 7, we hypothesized that these heterometallic compounds could potentially break into monometallic species in physiological media or in vivo before reaching the tumors. Indeed, we found that in some cases in solution the stability of the compounds was low.30
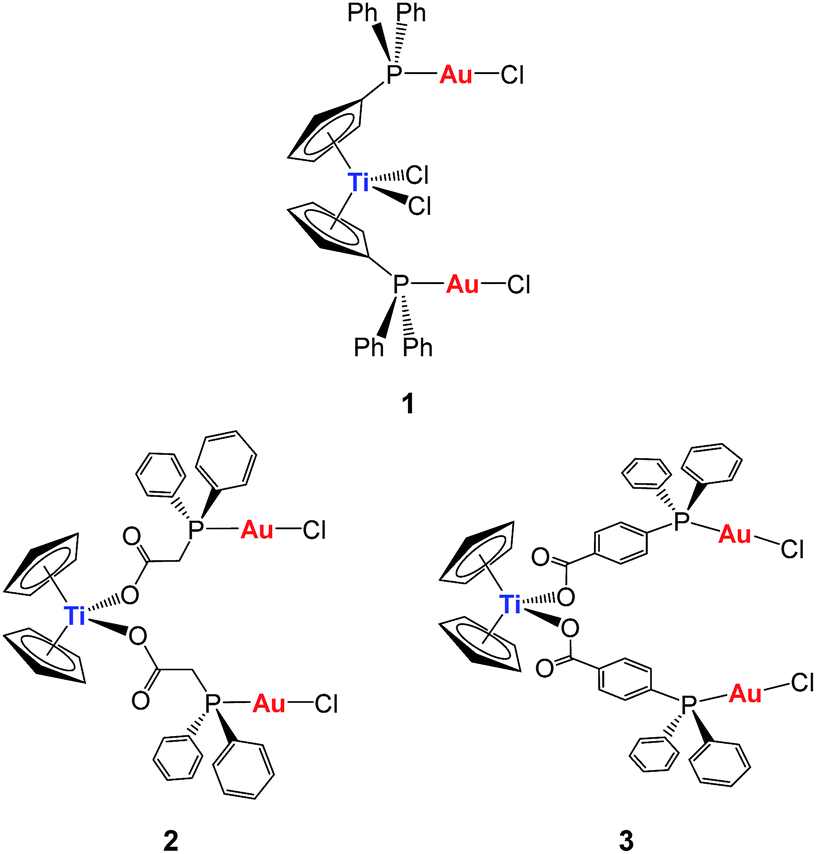 | ||
| Chart 1 Previous heterometallic Ti–Au complexes synthesized in our group.30,31 | ||
In order to increase stability, we incorporated the second metal to a ligand strongly bound to the titanium(IV) center to ensure that heterometallic Ti–M species would remain after Ti–Cp hydrolysis occurs under physiological pH conditions. Since Ti–O bonds are considerably stronger (ΔHf298 = 662(16) kJ mol−1) than Ti–C (ΔHf298 = 439 kJ mol−1) or Ti–Cl (ΔHf298 = 494 kJ mol−1) bonds, we envisioned a carboxylate as the ideal group to bind titanium(IV) centers (compounds 2 and 3, Chart 1).31 This strategy rendered compounds more stable than TDC and 1 in solution, and endowed them with relevant activity and selectivity against human renal cancer cell lines as well as a favourable preliminary toxicity profile in C57BL/6 mice.31
Thiolato-gold(I) phosphane complexes (such as Auranofin) have displayed relevant biological properties which are related in most cases to their inhibitory properties on thioredoxin reductase.32–34 We aimed to increase the cytotoxicity of heterometallic Ti–Au compounds by the incorporation of thiolato ligands coordinated to gold. Bifunctional ligand mba (derived from 4-mercaptobenzoic acid (H2mba) Chart 2) was chosen as an attractive candidate to covalently link both metal centres.
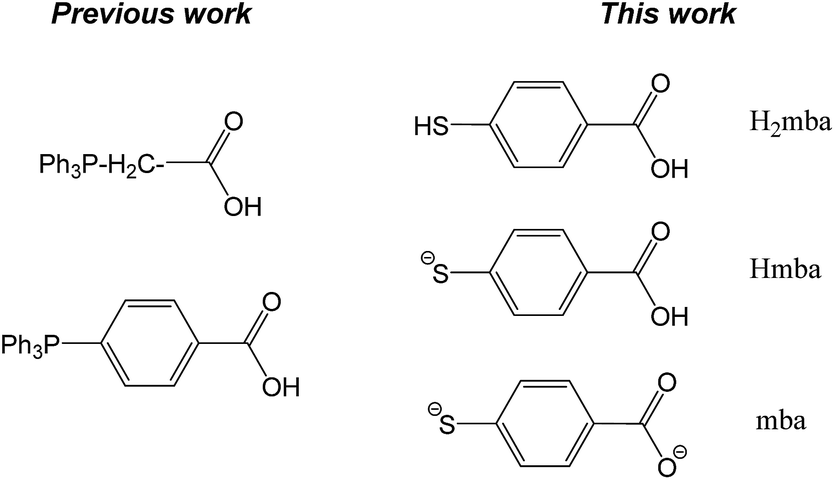 | ||
| Chart 2 Previous and new bifunctional ligands employed by this laboratory. | ||
We report here on the synthesis and characterization of new organometallic Ti–Au compounds with mba of the type [(η-C5H5)2TiMe(μ-mba)Au(PR3)] (R = Ph, 5, R = MPPF, 7). The stability of these compounds in dimethyl sulfoxide (DMSO) and phosphate buffer saline (PBS) was monitored by NMR and UV-vis spectroscopy. We describe their in vitro activity against human renal Caki-1 cancer cell lines, non-tumorigenic human embryonic kidney cell lines HEK-293T, and human renal proximal tubular cells (RPTC). We present studies of the effect of compounds on cell death, interactions with DNA, and inhibitory effects with thioredoxin reductase and certain protein kinases implicated in our earlier study. In addition, we report on the verification of activity in renal cancer cell lines against these targets by 5 and the previously reported [(η-C5H5)2Ti{OC(O)-C6H4-PPh2AuCl}2] (3, Chart 1) and compare the pharmacokinetics and efficacy of compounds 3 and 5 in an in vivo evaluation of Caki-1 xenografts in NOD.CB17-Prkdc SCID/J mice.
Results and discussion
Synthesis and characterization
The synthesis of the new heterometallic TiAu complexes is depicted in Scheme 1. The reaction of one equivalent of the corresponding mononuclear gold(I) complexes, 4 or 6, with one equivalent of [(η-C5H5)2TiMe2] (with concomitant elimination of one equivalent of methane) afforded [Cp2TiMe(μ-mba)Au(PR3)] (R = Ph, 5, R = MPPF, 7) in high yields as air- and moisture-stable orange-coloured solids. Both heterometallic compounds 5 and 7 turned out to be less acidic than titanocene dichloride (see experimental section) and are soluble in DMSO/H2O, DMSO/PBS or DMSO/media (1:99) mixtures at micromolar concentrations, which is relevant for subsequent biological testing.
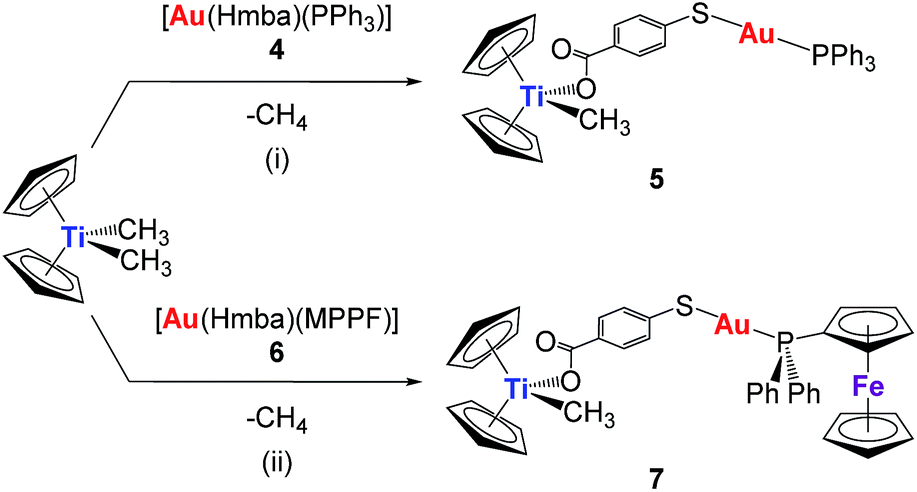 | ||
| Scheme 1 Preparation of heterometallic titanocene–gold complexes [(η-C5H5)2Ti(CH3){OC(O)C6H4SAu(PR2R′)}] (R = R′ = Ph, 5; R = Ph, R′ = ferrocene, 7). (i) THF/toluene 1.5:1, RT, 1 h; (ii) acetonitrile/toluene 3:1, RT, 1 h. | ||
The reaction in Scheme 1 did not yield the expected trinuclear TiAu2 species (like 2 and 3 in Chart 1 with the carboxylato-phosphane linker) or as reported for ZrAu2 species with mba ligands.35 The smaller size of titanium with respect to zirconium may be the reason that the di-substitution reaction did not proceed and bimetallic compounds TiAu were obtained instead. Compounds 5 and 7 are examples of unusually stable methyl-containing titanocene moieties that are stable as solids at air and RT and in CDCl3 solution for at least 5 days. To the best of our knowledge, this is the first air- and moisture-stable Ti–Me derivative.
Although the synthesis of gold compound 4 had been previously reported,36,37 compound 4 as well as new compound 6 were synthesized in a more efficient manner31 following the strategy established by Sordo and co-workers38 for the synthesis of gold thiocarboxylate derivatives via selective deprotonation of the thiol over the carboxylic acid (see Experimental section). Compound 6 was isolated as a pale orange solid in high yield. By layering n-hexane over a solution of compound 6 in dichloromethane, we obtained crystals suitable for X-ray diffraction.
The crystals of compound 6 were determined to be triclinic (space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) with Z = 2 formula units in the unit cell. The individual monomeric units (Fig. 1) form chains which show hydrogen bonding at one end and Au–S groups at the other (Fig. 2). The environment of the gold atoms is close to linear [P–Au–S 177.31(5)] (Fig. 1). A selection of structural parameters is given in Table 1. In the polymeric units (Fig. 2), the (MPPF)Ph2P–Au–S moieties of neighbouring molecules are aligned head to tail to form parallelograms with edges Au–S 2.3128(16), Au–S* 3.374 Å and a diagonal Au–Au* of 4.351 Å.
) with Z = 2 formula units in the unit cell. The individual monomeric units (Fig. 1) form chains which show hydrogen bonding at one end and Au–S groups at the other (Fig. 2). The environment of the gold atoms is close to linear [P–Au–S 177.31(5)] (Fig. 1). A selection of structural parameters is given in Table 1. In the polymeric units (Fig. 2), the (MPPF)Ph2P–Au–S moieties of neighbouring molecules are aligned head to tail to form parallelograms with edges Au–S 2.3128(16), Au–S* 3.374 Å and a diagonal Au–Au* of 4.351 Å.
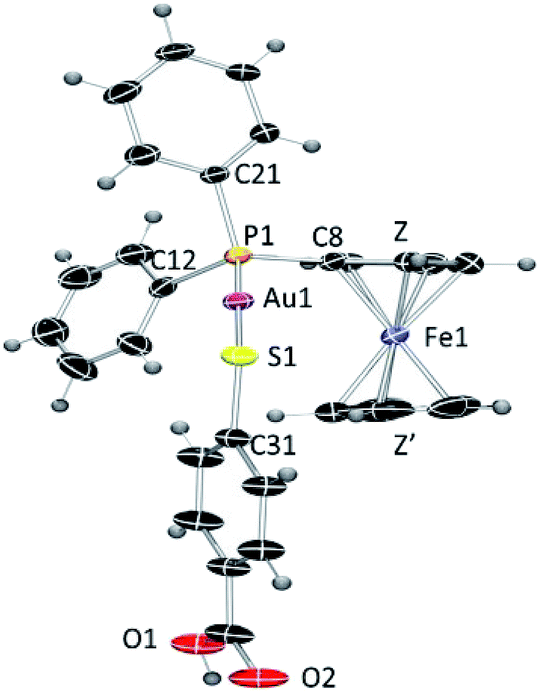 | ||
| Fig. 1 ORTEP view of the molecular structure of 6 showing the labelling scheme. Labelling for hydrogen and some carbon atoms is omitted for clarity. A drawing of the molecular structure containing carbon atoms labelled is provided in the ESI.‡ | ||
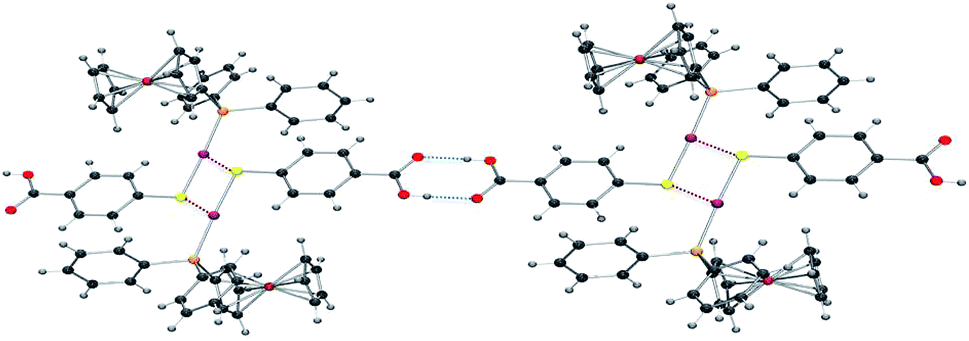 | ||
| Fig. 2 ORTEP view of the polymeric structure of 6 showing hydrogen bonds (blue dotted line) and the Au–S weak interactions (red-brown dotted line). Applied colour code as in Fig. 1. | ||
| Au(1)–P(1) | 2.2647(15) | P(1)–Au(1)–S(1) | 177.31(5) |
| Au(1)–S(1) | 2.3128(16) | P(1)–C(8)–Fe(1) | 128.7(3) |
| P(1)–C(8) | 1.789(6) | Au(1)–S(1)–C(31) | 105.1(2) |
| P(1)–C(11) | 1.823(6) | Z–Fe(1)–Z′ | 175.86 |
| P(1)–C(21) | 1.822(6) | ||
| S(1)–C(31) | 1.770(6) | ||
| Distance Fe–Z (centroid of C5H4) | 1.640 | ||
| Distance Fe–Z′ (centroid of C5H5) | 1.667 |
This polymeric structure was previously described for similar Hmba complexes.37 The main difference herein lies in the intermolecular Au–Au* and AuS* distances: in this case, the intermolecular Au–S* distances are considerably shorter than the Au–Au* distances and very similar to the van der Waals radii of the AuS pair [3.35 Å]. Therefore, since gold–gold distance is larger than the sum of the van der Waals radii for two gold atoms [3.60 Å], the AuS* contact is more likely to be the dominating contributor to the aggregation. No structural anomaly was observed in the hydrogen bonds between the two carboxylic groups.
The structures of complexes 5 and 7 depicted in Scheme 1 have been proposed on the basis of NMR and UV-vis spectroscopy, mass spectrometry, and elemental analysis (see Experimental section). Moreover, IR experiments and DFT calculations were carried out in order to shed light on the coordination mode of the carboxylate groups. The solid state IR spectra of compounds 5 and 7 show that the carboxylate groups are bonded to the titanium centre in a monodentate fashion since the differences found between the symmetric and antisymmetric stretching bands39,40 are 346 cm−1 and 348 cm−1, respectively, i.e. larger than 200 cm−1.
These data are supported by the theoretical values obtained by DFT calculations where the differences between the symmetric and antisymmetric stretching bands39,40 were found to be 328 cm−1 and 330 cm−1 for compounds 5 and 7, respectively, thus indicating the monodentate coordination mode of the carboxylic moieties (Fig. 3). In the ESI‡ section, data on different optimizations are provided. All calculations performed led to the species containing a monodentate carboxylate.
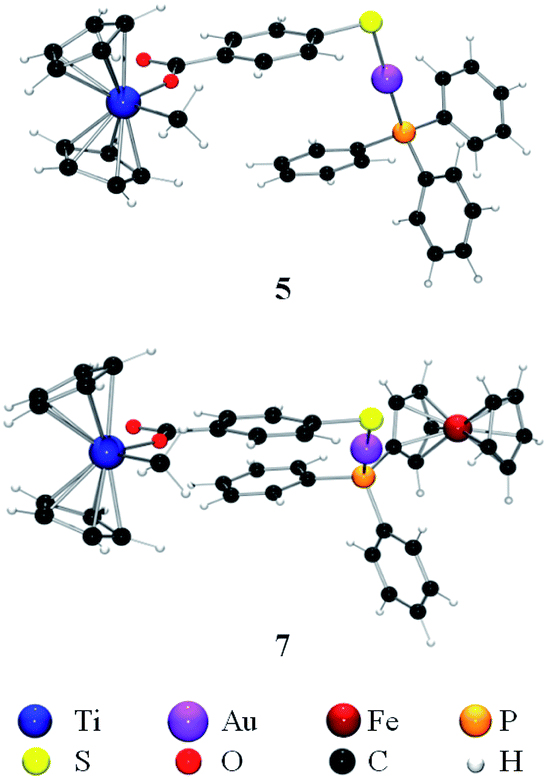 | ||
| Fig. 3 Optimized structures for heterometallic complexes 5 and 7. | ||
The stability of compounds 5 and 7 was evaluated by 31P{1H} and 1H NMR spectroscopy in DMSO-d6, UV-vis spectroscopy in DMSO/PBS solution and by mass spectrometry over time (see ESI‡). NMR experiments were performed in neat DMSO-d6 due to the low solubility of the compounds in mixtures of DMSO-d6/D2O in concentrations larger than 100 micromolar. Titanocene dichloride is known to hydrolyze with a higher rate in DMSO than in water.41 The half-life of 5 and 7 derivatives in DMSO-d6 was found to be 8 and 32 hours respectively, i.e. 16-fold or 64-fold larger than that of 2 and 3 in the same solvent.31 As reported for TDC, compounds 5 and 7 lose the cyclopentadienyl groups in DMSO (most plausibly being replaced by OH groups).31 Surprisingly, the methyl groups of compounds 5 and 7 are hydrolysed at the same rate than the cyclopentadienyl groups. Therefore, the longer half-life found for compounds 5 and 7 in DMSO-d6 could indicate the positive influence of the di-covalent linker (μ-mba) between the titanium and the gold centres.
The UV-vis spectra of compounds 5 and 7 (micromolar concentration) in 1:99 DMSO/PBS solution did not change over time (24 h, see ESI‡) indicating that the hydrolysis is much slower in solutions at physiological pH. Importantly, mass spectrometry also indicated the presence of species containing both titanium and gold in 1% DMSO/PBS solution after 24 h (see ESI‡).
Biological activity
Cytotoxicity studies. The cytotoxicity of the heterometallic complexes [(η-C5H5)2 TiMe(μ-mba)Au(PR3)] (R = Ph, 5, R = MPPF, 7) and monometallic gold(I) complexes [Au(Hmba)PR3] (PR3 = PPh34;31 MPPF 6 in Scheme 1) was assayed by monitoring their ability to inhibit cell growth using the XTT assay (see Experimental section). The cytotoxic activity of the compounds was determined as described in the Experimental section. In this assay, human renal Caki-1 cancer cell lines, non-tumorigenic human embryonic kidney cell lines HEK-293T, and human renal proximal tubular cells (RPTC) were incubated with the indicated compound for 72 hours and compared for sensitivity to cisplatin, titanocene dichloride and Titanocene Y.42 The results are summarized in Table 2; data comparing both 24 and 72 hours can be found in the ESI.‡
| Caki-1 | HEK-293T | RPTC | |
|---|---|---|---|
| [Au(Hmba)(PPh3)] 4 | 2.76 ± 0.35 | 1.11 ± 0.65 | 3.87 ± 0.15 |
| Ti-Au 5 | 0.12 ± 0.003 | 0.49 ± 0.008 | 2.67 ± 0.12 |
| [Au(Hmba)MPPF] 6 | 3.6 ± 0.342 | 3.0 ± 0.07 | 3.78 ± 0.13 |
| Ti–Au 7 | 4.11 ± 0.64 | 3.09 ± 0.003 | 3.76 ± 0.21 |
| Cisplatin | 29 ± 4.11 | 3.27 ± 0.13 | — |
| [(η-C5H5)2TiCl2] | >200 | >200 | — |
| Titanocene Y | 29.42 ± 4.18 | >200 | — |
The heterometallic compounds are considerably more toxic to the renal cancer cell line (Caki-1 cells) than cisplatin, titanocene dichloride and Titanocene Y. In addition, the heterometallic compound 5 is far more toxic in the nanomolar range than the monometallic gold compound 4 on these cells. Importantly, compound 5 is considerably less toxic to the non-tumorigenic human embryonic kidney cell line (HEK-293T) and human renal proximal tubular cells than is 4. Heterometallic compound 7 with phosphine MPPF exhibits cytotoxicity with both Caki-1 and non-tumorigenic cell lines similar to that of parent monometallic compound 6, but is much less selective than 5 after 72 h incubation.
We studied the effect of the combination of monometallic gold compound 4 and titanocene dichloride in renal cancer cell lines at 24 h which in all cases gave IC50 values > 100 μM (data not shown). This fact supports the idea that there is indeed a synergistic effect for the heterometallic complexes in their in vitro activity on renal cancer cell lines.
Robustness of compound 5 and the co-localization of Au/Ti metals in Caki-1 cells. In an attempt to determine the co-localization of Au and Ti metals in the Caki-1 cells, lysates of Caki-1 cells treated with 5 were analysed by Inductively Coupled Plasma-Mass Spectromety (ICP-MS) to determine the amounts of both metals (see Experimental section for details). Lysates of Caki-1 cells receiving no drugs were employed as controls. It was found that these cells have some basal levels of both the elements present (3.5 μg Ti and 0.1 μg Au per mg cell protein). When corrected for the background levels of the two elements in the cell lysates, it was found that the levels of Ti and Au demonstrated a stoichiometric ratio close to unity suggesting that the compound is stable in intracellular environment. Fig. 4 shows that cellular levels of compound 5 increased with an increase in the concentration of drug in the incubation media. Increasing the drug concentration from 1 μM to 5 μM resulted in a four-fold increase in the cellular levels of compound 5. Elemental analysis (ICP-MS) confirmed that the stoichiometric ratio of Au to Ti (0.96 in 1 μM and 0.95 in 5 μM) in the cell lysate is close to unity, thus confirming that the compound is stable when taken up in the cells.
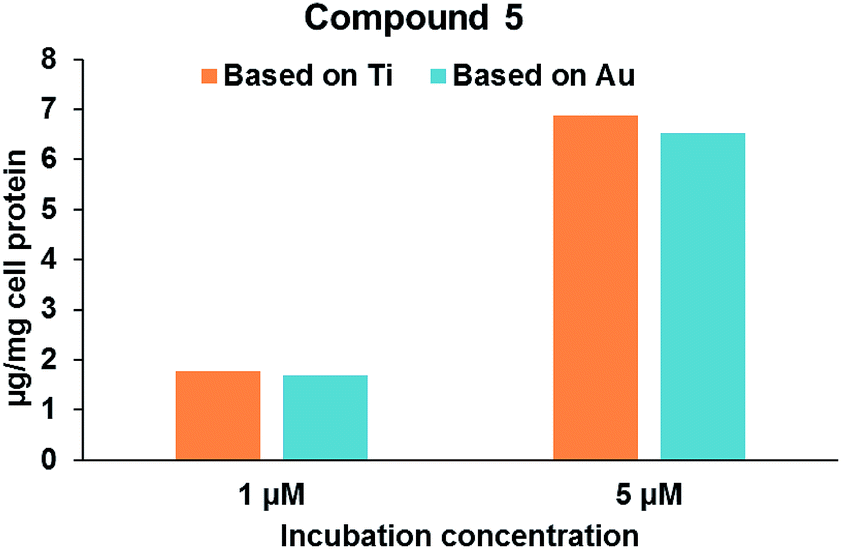 | ||
| Fig. 4 Cellular uptake of compound 5 in Caki-1 cells. The concentration of compound 5 calculated based on Ti and Au content in the cell lysates is similar suggesting that the compound is robust and both the elements are co-localized in the cells. | ||
Cell death assays. The induction of both apoptosis and necrosis are known for gold compounds43 while titanocenes like titanocene C, X and Y are known to induce apoptosis in different cancer cell lines.13 In order to gain some insight into the nature of the cytotoxicity that the most toxic and selective heterometallic complex 5 induces in renal cancer cell lines, we performed cell death assays for apoptosis with this compound dissolved in 1% DMSO (see Experimental section for details) and used either 1% DMSO alone in media (vehicle control) or 10 μM staurosporine (a known inducer of apoptosis) (positive control).31
After incubation for 6 hours with 10 μM compound 5, the appearance of peaks similar to that of staurosporine implicates apoptosis as one basis for the cytotoxic effect of this compound (Fig. 5).
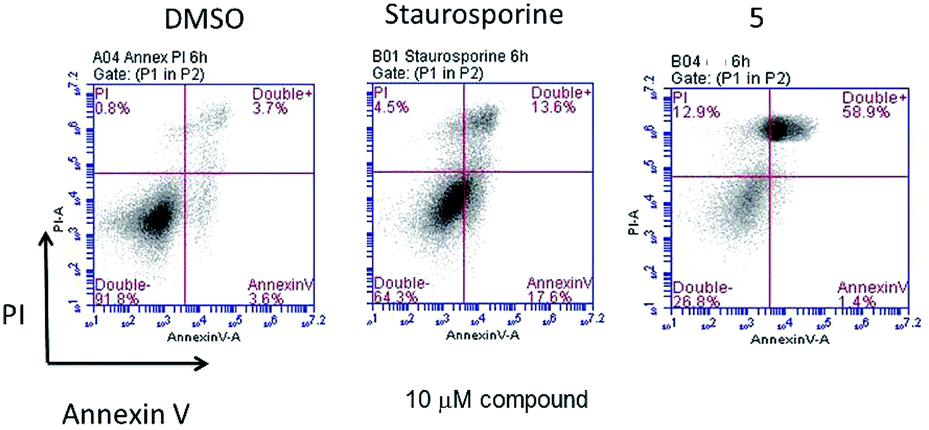 | ||
| Fig. 5 Cell death assays on Caki-1 cells induced by 5 (10 μM) measured by using two-colour flow cytometric analysis, after 6 h of incubation. 1% DMSO is vehicle control and staurosporine as positive control. | ||
Migration assays. In advanced tumors, increased cell migration is a hallmark of cancer cell invasion and metastasis.44,45 We therefore set out to evaluate the anti-invasive properties of the most active heterometallic titanocene–gold compounds described in this and previous work (compounds 331 and 5, respectively). Anti-invasive properties were evaluated by using a wound-healing scratch assay (details and explanations on the assay can be found on the ESI‡ section). Upon treatment with compound 3 [(η-C5H5)2Ti{OC(O)CH2PPh2AuCl}2], cell migration was slowed with 81% of the scratchleft uninvaded (Fig. 6). However, in highly metastatic Caki-1 cells treated with 0.1% DMSO control, only 19% of the scratch remained uninvaded. Treatment of these cells with 5 led to complete inhibition of migration (Fig. 6). Also, striking morphological changes did occur in cells treated with 5. We found that this compound caused distinct rounding of the cells and that after 12 hours of incubation there was a complete loss of cell–cell contacts. Those results clearly indicate anti-migratory properties for compounds 3 and 5. The cytostatic and anti-migratory phenotypes induced by new compound 5 warrants further exploration.
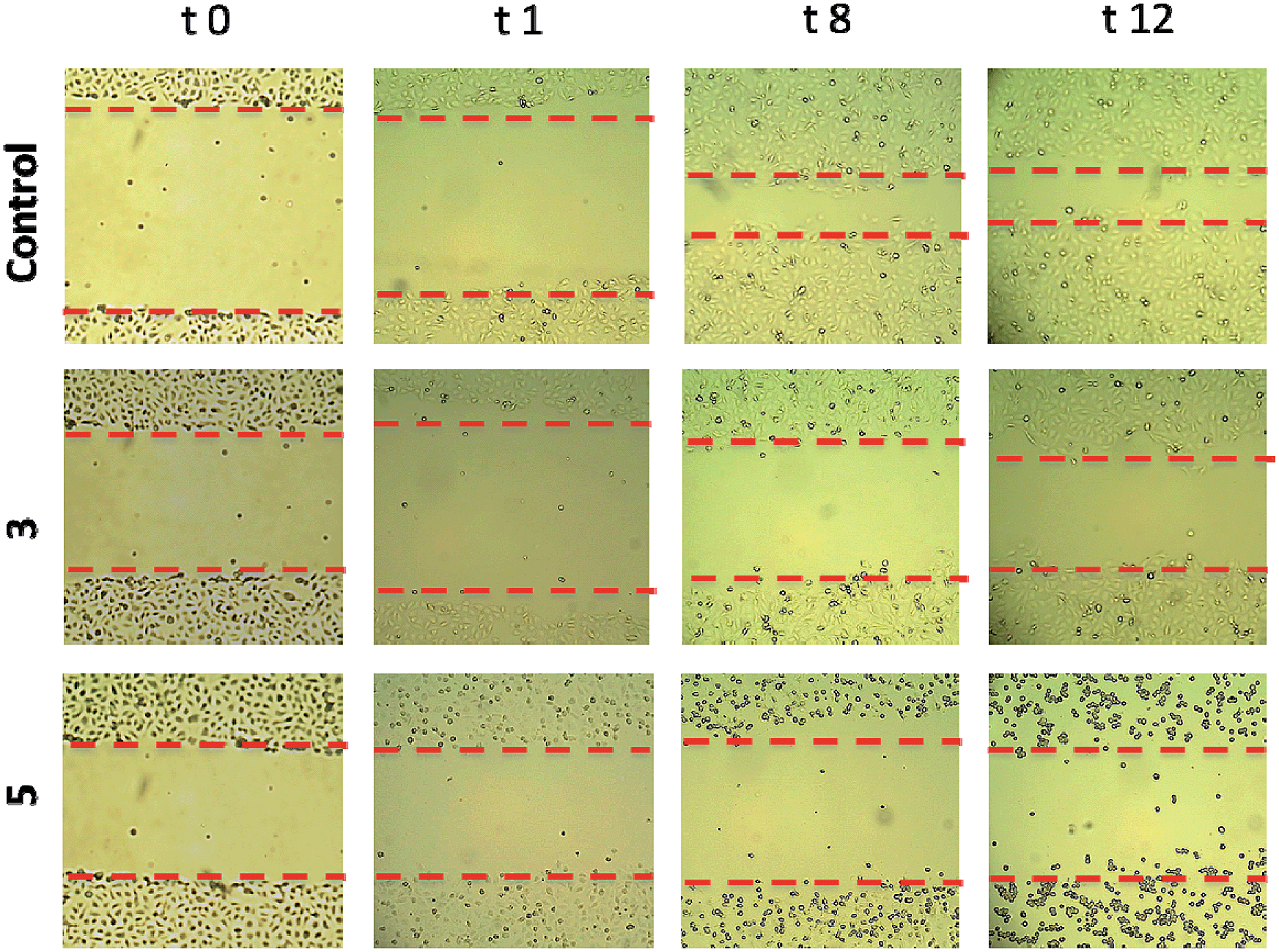 | ||
| Fig. 6 Scratch assay showing that compounds 3 and 5 interfere with Caki-1 migration. Panels show representative images of untreated cells at time points up to 12 hours (top row), or cells treated with 3 (middle row), and 5 (bottom row). The left column shows a representative image of untreated Caki-1 cells immediately post-scratch (t = 0 h). All other panels show Caki-1 cells at 1 hour (t = 1), 8 hours (t = 8) or 12 hours (t = 12) post-treatment. The graph represents results from measurements of the area of the scratch from 4 separate and randomly selected fields of view. | ||
Interactions with plasmid DNA
Since DNA replication is a key event for cell division, it is among critically important targets in cancer chemotherapy. Most cytotoxic platinum drugs form strong covalent bonds with DNA bases.46 However, a variety of platinum compounds act as DNA intercalators upon coordination to the appropriate ancillary ligands.47 DNA was believed to be the target for titanocene dichloride.38 Titanium accumulates in the cells in nuclear heterochromatin and, to a minor extent, in the nucleolus and ribosomes.48 Titanium–DNA adducts were detected in A2780 cells treated with TDC and this compound also inhibited DNA and RNA synthesis.49 However, most recent reports on titanocene dichloride indicate that at physiological pH it neither binds strongly to DNA nor suppresses DNA-processing enzymes.50 Titanocene Y was recently shown to interact weakly with DNA,51 consistent with the weak activity observed with previously described TiAu2 compounds (2 and 3).31 DNA-interactions were tested with heterometallic compounds 3 and 5, monometallic gold(I) derivatives 6 and 7, titanocene dichloride, or cisplatin by using plasmid (pBR322) DNA (Fig. 7). This plasmid has two main forms: OC (open circular or relaxed form, Form II) and CCC (covalently closed or supercoiled form, Form I). Agarose gel electrophoresis assays were performed whereby decreased electrophoretic mobility of both forms were taken as evidence of metal–DNA binding. Generally, the slower the mobility of supercoiled DNA (CCC, Form I), the greater the DNA unwinding produced by the drug.52 For example, binding of cisplatin to plasmid DNA results in decreased mobility of the CCC form and increased mobility of the OC form (see lanes a–d for cisplatin in Fig. 7). Treatment of plasmid DNA with increasing amounts of monometallic Au(I) compounds 4 and 6 or heterometallic TiAu derivatives 5 and 7 did not affect the mobility of the faster-running supercoiled form (Form I) even at the highest molar ratios (d). This result is in accordance with previously reported results on a titanocene–gold(I) phosphine derivative [(η5-C5H5)-(μ-η5:k1-C5H4PPh2)TiCl2]2Au]PF6 (Chart 2) and other TiAu2 derivatives described by us [(η-C5H5)2Ti{OC(O)RPPh2AuCl}2] (R = –CH2– 2 and R = –C6H4– 3).29,31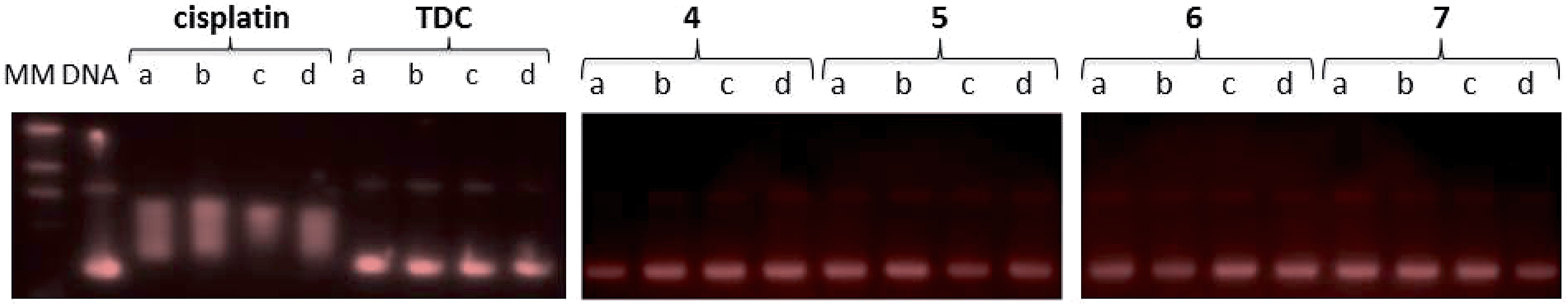 | ||
| Fig. 7 Electrophoresis mobility shift assays for cisplatin, titanocene dichloride, heterometallic TiAu compounds 5 and 7 and monometallic [Au(Hmba)(PR3)] 4 and 6 (see Experimental for details). DNA refers to untreated plasmid pBR322. (a, b, c and d) correspond to metal/DNAbp ratios of 0.25, 0.5, 1.0 and 2.0 respectively. | ||
Inhibition of thioredoxin reductase
Changes in cell anti-oxidant capacity are a characteristic of many chemo-resistant cancers, and overexpression of thioredoxin reductase (TrxR) in drug-resistant cells is part of a critical defense and survival mechanism of cisplatin-resistant cells, thus making this enzyme an important anti-cancer target53,54 A number of gold(I) compounds with anticancer properties are known to inhibit thioredoxin reductase isolated from human placenta,53 rat liver32,55–60 or isolated from treated cancer cells.34,61–63 One such example is the structurally related gold-thiolato-phosphane compound (Auranofin, AF) which inhibits 50% of the enzymatic activity of isolated TrxR at concentrations as low as 20 nM. AF also inhibits TrxR in human breast MCF-7 cancer cells (4 μM inhibits 18% total TrxR activity)62 and human ovarian A2780 cancer cells (10 μM inhibits 30% total activity).63 Therefore, we measured the activity of thioredoxin reductase in Caki-1 cells, following incubation with compounds 3, 5 and Auranofin was used as a positive control. We found thioredoxin reductase activity to be significantly lower in cells treated with 5 μM of either 3, 5 or Auranofin with an observed inhibition of 31.6%, 68.2%, and 74.1% respectively after a 5 hour incubation (Fig. 8).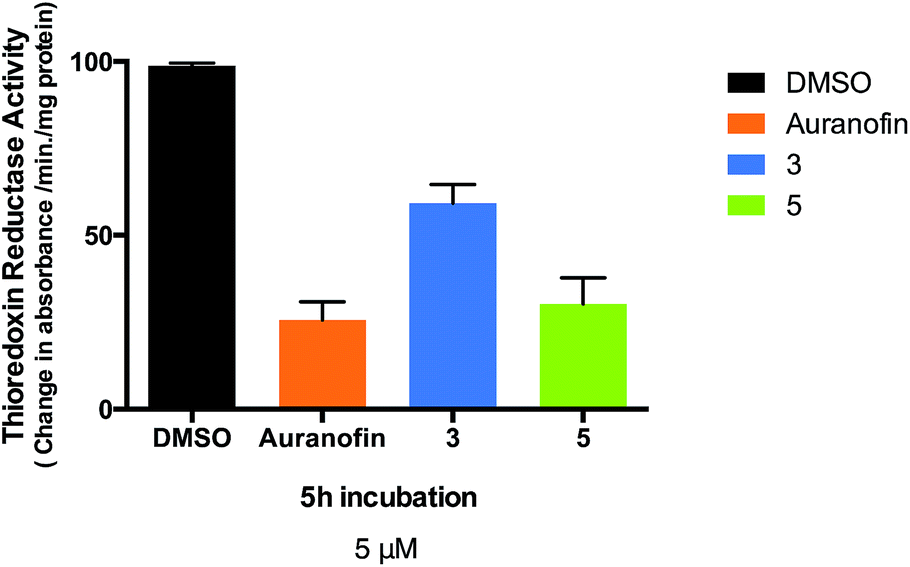 | ||
| Fig. 8 Thioredoxin reductase activity in 3, 5 or Auranofin treated Caki-1 cells. Activity of Caki-1 thioredoxin reductase from soluble whole cell lysates following incubation with 5 μM of 3, 5 or Auranofin for 5 hours. | ||
The half maximal inhibitory concentration (IC50) values were: 0.61 (±0.002) μM for AF, 4.75 (±0.048) μM for 3, and 0.88 (±0.008) μM for 5 after 12 hours. The similar values for AF and compound 5 imply that the two compounds have similar potencies in Caki-1 cells. However compound 5 is markedly more toxic to Caki-1 cells than Auranofin (IC50 = 3.08 ± 0.13 μM for AF vs. 0.12 ± 0.003 μM for 5) indicating that inhibition of TrxR is not the only factor involved in the cytotoxic mechanisms of these heterometallic compounds. The observed inhibitory effect of 3 and especially 5 on thioredoxin reductase activity in whole cell lysates indicates that this enzyme is a suitable selective target for cancer therapy with this type of compounds.
Studies with protein kinases
Protein kinases have an important role in oncogenesis and tumor progression, and therefore are frequently used as targets for designing anticancer drugs that have included organometallic complexes (Meggers and coworkers).64–66In an effort to gain new insights into the mechanism of action of heterometallic compounds, we performed immunoblotting assays on whole cell lysates of Caki-1 cells treated with compounds 3 or 5 to evaluate their effects on the expression levels of selected protein kinases, namely p90-RSK, AKT, and MAPKAPK3. Compound 5 produced dramatic decreases in expression of all three protein kinases by 12 hours post-treatment (Fig. 9A). In contrast, no loss of expression was noted following a similar treatment with compound 3 (Fig. 9B and ESI‡). These findings indicate that compound 5 acts through a transcriptional or translational mechanism after the first hour of treatment, thereby displacing direct drug–kinase interactions that we have observed in vitro (described in the ESI‡). Since all three enzymes have been implicated in promoting cell migration and therefore are strong targets for anti-metastasis drugs, these findings predict that compound 5 would be effective in controlling metastatic potential of renal tumors.
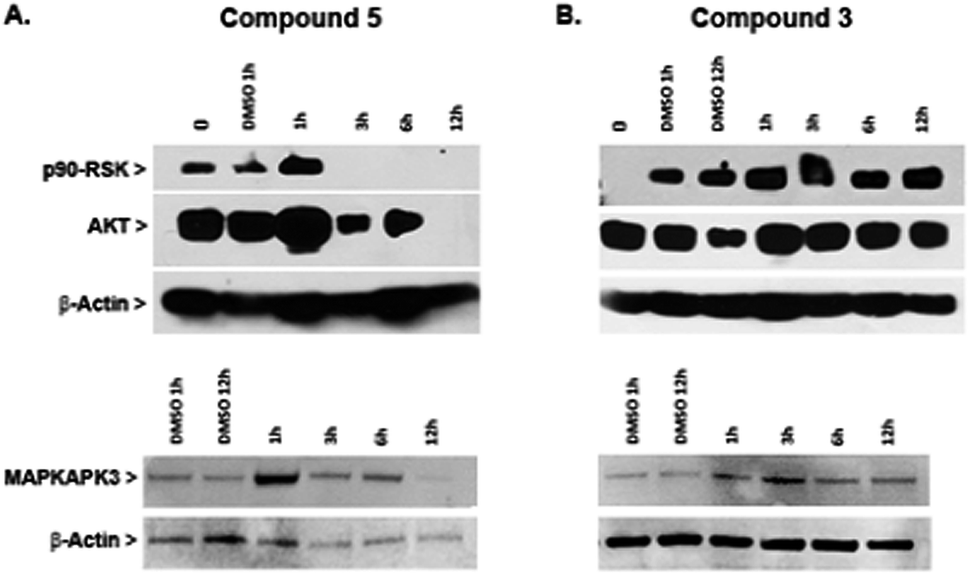 | ||
| Fig. 9 Decreased expression of p90-RSK, AKT, and MAPKAPK-3 in Caki-1 cells in response to compound 5. Cells were incubated with either 5, compound 3 or 0.1% DMSO for the indicated times, followed by cell lysis, and Western blot analysis. Blots were probed with anti-β-actin antibody as a control for protein loading. Samples from cells treated with compound 5 are shown in panel A, and samples from cells treated with compound 3 are shown in panel B. | ||
In summary, compound 5 inhibits thioredoxin reductase and causes decreased expression of pro-motility enzymes such as p90-RSK, AKT, and MAPKAPK3 in Caki-1 cells. In contrast, compound 3 has a more modest inhibitory effect on TrxR in Caki-1 cells but does not affect expression of the pro-motility enzymes. Nevertheless, compound 3 was found to reduce the secretion of IL-6 in Caki-1 cells thereby supporting the hypothesis that some MAPKAPK2/3 inhibition does occur.31 When Auranofin was tested as a negative control compound for TrRx inhibition studies, it was found to inhibit TrRx in the same range as compound 5 but failed to down-regulate MAKPAPK-3 in Caki-1 cells (Fig. S46 in ESI‡).
Effects on tumor growth in vivo with compounds 3 and 5
We therefore chose the dose of 7.5 mg per kg per day for 3 and 3 mg per kg per day for 5 every other day to conduct the subsequent in vivo trials. The choice of 3 and 5 for a subsequent in vivo trial was based on their selectivity in vitro against renal cancer cell lines when compared to non-tumorigenic human kidney cell lines (HEK-293T and RPTC) and their favourable preliminary toxicity profile on C57BL/6 mice.
In the group treated with 3 we observed no decrease in tumor size, neither did we observe any hindrance in tumor growth from the starting volume between day 1 and day 28 of treatment (after a total of 14 doses). For this compound we observed a 90% increase in tumor volume (see Fig. S47 in ESI‡). For the group treated with 5 we observed an impressive decrease in tumor size (shrinkage) of 67% from the starting volume between day 1 and day 28 of treatment (after a total of 14 doses). In contrast, in the control vehicle-treated group we observed 106% increase in tumor volume between day 1 and day 28 of treatment (see Table 3 and Fig. 10). We note that there was no weight loss in mice treated with treated with either 3 or 5 by this protocol.
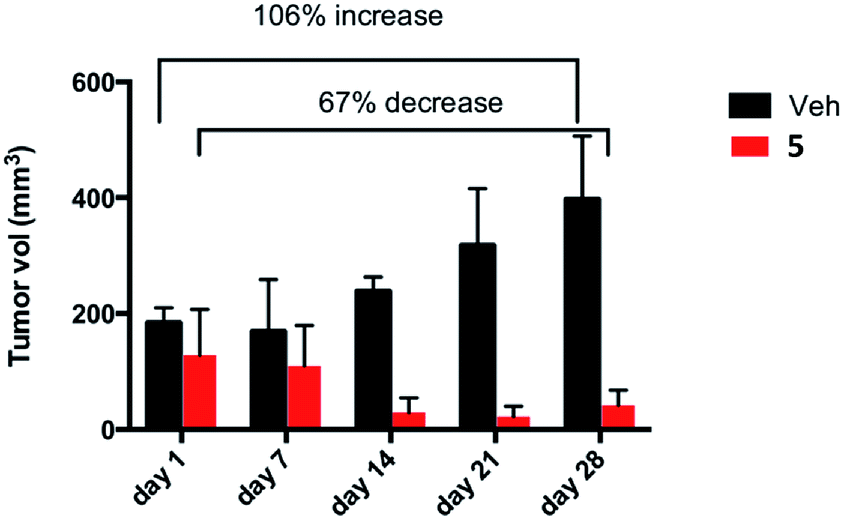 | ||
| Fig. 10 Percent reduction of tumour burden in a cohort of 18 female NOD.CB17-Prkdc scid/J mice inoculated subcutaneously with 8 × 106 Caki-1 cells. The treatment started when tumors were palpable (6 mm diameter). 6 mice were treated with compound 5 (red bars), and 6 were treated with the vehicle 100 μL normal saline (0.9% NaCl) (black bars). Compound 5 was administered in the amount of 3 mg per kg per every other day. | ||
The results clearly indicate that compound 5 is extremely efficient in vivo since it not only inhibits tumor growth but it decreases the size of the tumors by 67%, while compound 3 has no notable effect on tumor size.
Pharmacokinetic study
Content of compound 3 and compound 5 in blood and tissues was calculated from their gold content and normalized to the extraction efficiency of gold in the respective tissues and blood. The pharmacokinetic profile of compound 3 in mice (Fig. 11A) is summarized in Table 4.
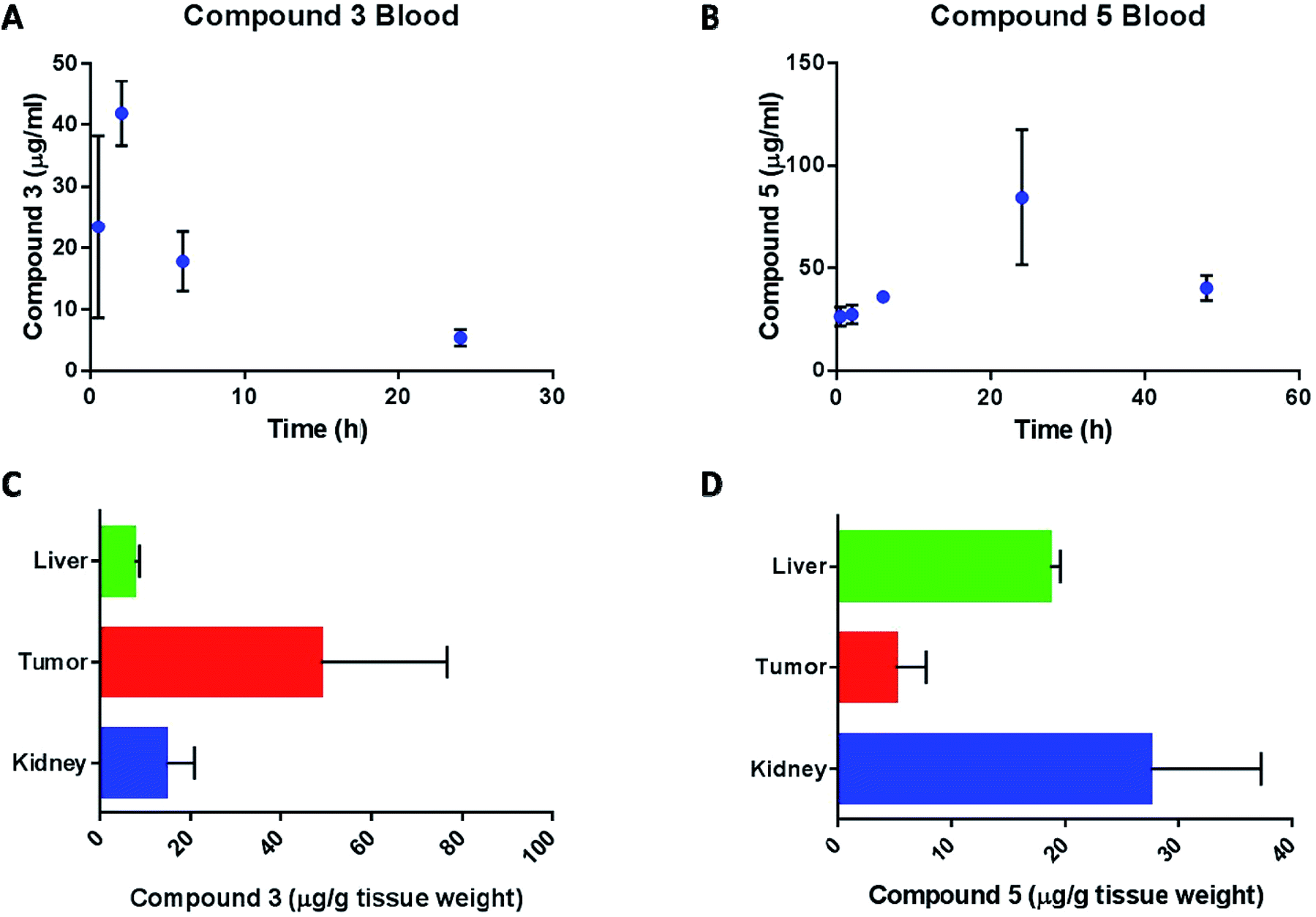 | ||
| Fig. 11 Concentration of compounds 3 (A) and 5 (B) in plasma at various intervals after the first dose; data represents mean ± SD. N = 3. Content of compounds 3 (C) and 5 (D) in tissues at the end of efficacy study; data represents mean ± SD; N = 3; P < 0.05. | ||
| Pharmacokinetic parameters | Values |
|---|---|
| K abs | 1.3 h−1 |
| K e | 0.1 h−1 |
| t 1/2 e | 7.4 h |
| t 1/2 abs | 0.6 h |
| t max | 2.2 h |
| C max | 37.7 μg mL−1 |
The compound was absorbed quickly into plasma (t1/2 abs = 0.55 h) and the peak plasma concentration was reached after 2 hours of dosing. The drug was eliminated slowly from the blood compartment with an elimination half-life (t1/2 e) of about 7.5 hours. The concentration of gold in the blood at 6 hours after the last dose of compound 3, was 24.1 ± 2.6 μg mL−1, which is lower (P < 0.01) than the Cmax after the first dose and not significantly different (P > 0.1) from the blood concentration at 6 hours after the first dose. This suggests that compound 3 doesn't accumulate in the blood. On the other hand, the pharmacokinetic profile of compound 5 (Fig. 11B) was characterized by a slow absorption with a constant increase in blood levels of the drug up to 24 hours. At the time of the second dose (48 hours after the first dose), the blood concentration of compound 5 was 40.4 ± 6.0 μg mL−1. At the end of the study, gold content in liver, kidney and tumor was determined for compound 3 (Fig. 11C) and compound 5 (Fig. 11D) treatments. The level of gold in liver and kidney was less than 15 μg g−1 tissue weight while the tumor concentration was about 50 μg g−1. The high level in tumor suggests enhanced tumor accumulation of compound 3. On the other hand, compound 5 concentration in the liver and kidney were significantly higher than tumor. As mentioned above, pharmacokinetic analysis of compound 3 in blood was performed based on a non-compartment model. Blood level of compound 3 after 48 hours of dosing was determined to be 45 μg mL−1 which was much greater than the blood level at 24 hours (5.5 μg mL−1). Since the drug was already at the end of the elimination phase at 24 hours, the high level at 48 hour was considered as an outlier and was not used in the determination of the pharmacokinetic parameters for compound 3. The blood concentration of compound 5 increased steadily up to 24 hours and then decreased at 48 hours. We speculate that the dosing regimen of the animals (every 48 hours) did not allow for complete elimination of compound 5 from the animals and thus we could not estimate the pharmacokinetic profile of this compound. However, since the multiple dosing demonstrated the superior efficacy of the compound in tumor bearing mice, we hypothesize that the drug effectiveness against tumor is due to the combination of several factors such as higher accumulation of the drug in the tumor tissues, longer half-life of the drug in the body and accumulation of drug in kidney tissue. Future studies will be aimed at estimating the elimination phase of the compound by dosing the animals with single dose.
We believe that the impressive tumor reduction shown by 5, its selectivity for kidney cancer cells in vitro, and low toxicity in mice in vivo warrant future experiments in vivo to better determine the ideal dosage and get more accurate pharmacokinetic data.
Experimental
General and instrumentation for the characterization and stability studies of the new compounds
NMR spectra were recorded in a Bruker AV400 1H NMR at 400 MHz, 13C{1H} NMR at 100.6 MHz and 31P{1H} NMR at 161.9 MHz. Chemical shifts (δ) are given in ppm using CDCl3 as the solvent, unless otherwise stated. 1H and 13C{1H} NMR resonances were measured relative to solvent peaks considering tetramethylsilane = 0 ppm, and 31P{1H} NMR was externally referenced to H3PO4 (85%). Coupling constants J are given in hertz. IR spectra (4000250 cm−1) were recorded on a Nicolet 6700 Fourier transform infrared spectrophotometer on solid state (ATR accessory). Elemental analyses were performed on a Perkin-Elmer 2400 CHNS/O series II analyzer. Mass spectra (electrospray ionization, ESI) were performed on a Waters Q-Tof Ultima. Stability studies were performed in a Cary 100 Bio UV-visible spectrophotometer. The pH was measured in an OAKTON pH conductivity meter in 1:99 DMSO/H2O solutions.
Synthesis
All compounds involving titanium centers were prepared and handled with rigorous exclusion of air and moisture under a nitrogen atmosphere by using standard nitrogen/vacuum manifold and Schlenk techniques. Solvents were purified by use of a PureSolv purification unit from Innovative Technology, Inc. Titanocene dichloride was purchased from Aldrich and 4-mercaptobenzoic acid from TCI America Inc. and used without further purification. Complex 4,31 [AuCl(tht)]70 and [AuCl(MPPF)]71 were prepared as previously reported.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), δ 148.08 (s, 4-C6H4), δ 134.21 (d, 2JPC = 13.8 Hz, o-PPh3), δ 131.88 (s, 2-C6H4), δ 131.80 (s, p-PPh3), δ 129.29 (d, 1JPC = 49.6 Hz, ipso-PPh3), δ 129.28 (d, 3JPC = 11.5 Hz, m-PPh3), δ 128.23 (s, 3-C6H4), δ 125.30 (s, 1-C6H4), δ 114.35 (s, C5H5), δ 44.14 (s, CH3). IR (cm−1): 3050 w (Cp), 1631 vs. (νasym CO2), 1435 s (Cp), 1298 vs. (νsym CO2), 1167 m (Cp), 815 s (Cp). pH 5 (5 × 10−5 M in 1:99 DMSO:H2O) = 5.13. For comparison, pH of previously described compound 434 was evaluated (5 × 10−5 M in 1:99 DMSO:H2O) = 4.90.
:1 EtOH/H2O mixture giving rise to a pale yellow solution that was stirred at RT for 10 minutes becoming colorless. The addition of [AuCl(MPPF)] (0.426 g, 0.70 mmol) led to thick yellow suspension that was stirred for 1 hour at RT. After solvents removal, the crude was washed with water (3 × 5 mL) and a 9:1 n-hexane/Et2O mixture (3 × 10 mL). Complex 10 was then isolated as a fine yellow solid in 89% yield (0.455 g). Crystals of 6 were obtained from a solution of 6 in CH2Cl2 layered with n-hexane at RT as orange prisms. Anal. calc. for C29H24AuFeO2PS (720.35): C, 48.35; H, 3.36; S, 4.45. Found: C, 48.46; H, 3.37; S, 4.60. 31P{1H} NMR (CDCl3): δ 34.04. 1H NMR (CDCl3): δ 10.58 (1H, s, COOH), δ 7.87 (2H, d, 3JHH = 8.3 Hz, 2-C6H4), δ 7.77 (2H, d, 3JHH = 8.3 Hz, 3-C6H4), δ 7.63 (4H, m, PPh2), δ 7.50 (6H, m PPh2), δ 4.60 (2H, s, C5H4), δ 4.41 (2H, s, C5H4), δ 4.20 (5H, s, C5H5). 13C{1H} NMR (CDCl3): δ 170.89 (s, CO), δ 152.47 (s, 4-C6H4), δ 133.58 (d, 2JPC = 13.8 Hz, o-PPh2), δ 132.14 (s, 2-C6H4), δ 131.47 (d, 4JPC = 2.4 Hz, p-PPh2), δ 131.46 (d, 1JPC = 58.7 Hz, ipso-PPh2), δ 129.78 (s, 3-C6H4), δ 128.94 (d, 3JPC = 11.9 Hz, m-PPh2), δ 123.77 (s, 1-C6H4), δ 73.46 (d, 2JPC = 13.8 Hz, C5H4), 72.52 (d, 3JPC = 8.7 Hz, C5H4), 70.06 (s, C5H5). IR (cm−1): 3069 w (OH), 1674 vs. (νasym CO2), 1416 vs. (νsym CO2). pH 6 (5 × 10−5 M in 1:99 DMSO:H2O) = 4.99.
O), δ 148.23 (s, 4-C6H4), δ 133.60 (d, 2JPC = 13.7 Hz, o-PPh2), δ 132.02 (s, 2-C6H4), δ 131.41 (d, 4JPC = 1.7 Hz, p-PPh2), δ 129.57 (s, 3-C6H4), δ 128.89 (d, 3JPC = 11.4 Hz, m-PPh2), δ 128.64 (d, 1JPC = 81.5 Hz, ipso-PPh2), δ 125.31 (s, 1-C6H4), δ 114.34 (s, C5H5), δ 73.47 (d, 2JPC = 13.8 Hz, C5H4), 72.48 (d, 3JPC = 8.6 Hz, C5H4), 70.07 (s, C5H5), δ 44.14 (s, CH3). IR (cm−1): 3086 w (Cp), 1633 vs. (νasym CO2), 1479 s (Cp), 1299 vs. (νsym CO2), 1168 m (Cp), 817 s (Cp). pH 7 (5 × 10−5 M in 1:99 DMSO:H2O) = 5.21.
O), δ 148.08 (s, 4-C6H4), δ 134.21 (d, 2JPC = 13.8 Hz, o-PPh3), δ 131.88 (s, 2-C6H4), δ 131.80 (s, p-PPh3), δ 129.29 (d, 1JPC = 49.6 Hz, ipso-PPh3), δ 129.28 (d, 3JPC = 11.5 Hz, m-PPh3), δ 128.23 (s, 3-C6H4), δ 125.30 (s, 1-C6H4), δ 114.35 (s, C5H5), δ 44.14 (s, CH3). IR (cm−1): 3050 w (Cp), 1631 vs. (νasym CO2), 1435 s (Cp), 1298 vs. (νsym CO2), 1167 m (Cp), 815 s (Cp). pH 5 (5 × 10−5 M in 1:99 DMSO:H2O) = 5.13. For comparison, pH of previously described compound 434 was evaluated (5 × 10−5 M in 1:99 DMSO:H2O) = 4.90.
:1 EtOH/H2O mixture giving rise to a pale yellow solution that was stirred at RT for 10 minutes becoming colorless. The addition of [AuCl(MPPF)] (0.426 g, 0.70 mmol) led to thick yellow suspension that was stirred for 1 hour at RT. After solvents removal, the crude was washed with water (3 × 5 mL) and a 9:1 n-hexane/Et2O mixture (3 × 10 mL). Complex 10 was then isolated as a fine yellow solid in 89% yield (0.455 g). Crystals of 6 were obtained from a solution of 6 in CH2Cl2 layered with n-hexane at RT as orange prisms. Anal. calc. for C29H24AuFeO2PS (720.35): C, 48.35; H, 3.36; S, 4.45. Found: C, 48.46; H, 3.37; S, 4.60. 31P{1H} NMR (CDCl3): δ 34.04. 1H NMR (CDCl3): δ 10.58 (1H, s, COOH), δ 7.87 (2H, d, 3JHH = 8.3 Hz, 2-C6H4), δ 7.77 (2H, d, 3JHH = 8.3 Hz, 3-C6H4), δ 7.63 (4H, m, PPh2), δ 7.50 (6H, m PPh2), δ 4.60 (2H, s, C5H4), δ 4.41 (2H, s, C5H4), δ 4.20 (5H, s, C5H5). 13C{1H} NMR (CDCl3): δ 170.89 (s, CO), δ 152.47 (s, 4-C6H4), δ 133.58 (d, 2JPC = 13.8 Hz, o-PPh2), δ 132.14 (s, 2-C6H4), δ 131.47 (d, 4JPC = 2.4 Hz, p-PPh2), δ 131.46 (d, 1JPC = 58.7 Hz, ipso-PPh2), δ 129.78 (s, 3-C6H4), δ 128.94 (d, 3JPC = 11.9 Hz, m-PPh2), δ 123.77 (s, 1-C6H4), δ 73.46 (d, 2JPC = 13.8 Hz, C5H4), 72.52 (d, 3JPC = 8.7 Hz, C5H4), 70.06 (s, C5H5). IR (cm−1): 3069 w (OH), 1674 vs. (νasym CO2), 1416 vs. (νsym CO2). pH 6 (5 × 10−5 M in 1:99 DMSO:H2O) = 4.99.
O), δ 148.23 (s, 4-C6H4), δ 133.60 (d, 2JPC = 13.7 Hz, o-PPh2), δ 132.02 (s, 2-C6H4), δ 131.41 (d, 4JPC = 1.7 Hz, p-PPh2), δ 129.57 (s, 3-C6H4), δ 128.89 (d, 3JPC = 11.4 Hz, m-PPh2), δ 128.64 (d, 1JPC = 81.5 Hz, ipso-PPh2), δ 125.31 (s, 1-C6H4), δ 114.34 (s, C5H5), δ 73.47 (d, 2JPC = 13.8 Hz, C5H4), 72.48 (d, 3JPC = 8.6 Hz, C5H4), 70.07 (s, C5H5), δ 44.14 (s, CH3). IR (cm−1): 3086 w (Cp), 1633 vs. (νasym CO2), 1479 s (Cp), 1299 vs. (νsym CO2), 1168 m (Cp), 817 s (Cp). pH 7 (5 × 10−5 M in 1:99 DMSO:H2O) = 5.21.
Crystal structure determination
Single crystals of 6 (orange prisms with approximate dimensions 0.25 × 0.23 × 0.23 mm) were mounted on a glass fiber in a random orientation. Data collection was performed at room temperature on a Kappa CCD diffractometer using graphite monochromated Mo-Kα radiation (l = 0.71073 Å). Space group assignments were based on systematic absences, E statistics and successful refinement of the structures. The structures were solved by direct methods with the aid of successive difference Fourier maps and were refined using the SHELXTL 6.1 software package. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were assigned to ideal positions and refined using a riding model (ESI‡).DFT calculations
The calculations have been performed using the hybrid density functional method B3LYP,72,73 as implemented in Gaussian09.74 Geometries were optimized with the 6-311G(d) basis set for the P and S elements, the 6-31G(d,p) basis set for the C, N, P, S, and H elements and the SDD pseudopotential for the titanium, iron and gold metal centers.75,76 Frequency calculations have been done at the same level of theory as the geometry optimizations to confirm the nature of the stationary points.Cell culture
Human renal cell carcinoma lines A498, Caki-1 and UO31, as well as the human prostate carcinoma cell lines DU145 and PC3 were newly obtained for these studies from the American Type Culture Collection (ATCC) (Manassas, Virginia, USA) and cultured in Roswell Park Memorial Institute (RPMI-1640) (Mediatech Inc., Manassas, VA) media containing 10% fetal bovine serum (FBS, Life Technologies, Grand Island, NY), 1% Minimum Essential Media (MEM) nonessential amino acids (NEAA, Mediatech) and 1% penicillin–streptomycin (PenStrep, Mediatech). HEK-293T cells were newly purchased from ATCC (Manassas, Virginia, USA) and maintained in Dulbecco's modified Eagle's medium (DMEM) (Mediatech) supplemented with 10% FBS, 1% NEAA and 1% PenStrep. Normal human renal epithelial cells (RPTC) were purchased from Lifeline Cell Technology (Lifeline Cell Technology, Frederick, MD, USA) and maintained in Lifeline's Renal Life Medium from Lifeline Cell Technology supplemented with 2.4 mM L-glutamine, 5 μg mL−1 rh insulin, 1.0 nM epinephrine, 10 nM triiodothyronine, 0.1 μg mL−1 hydrocortisone hemisuccinate, 10 ng mL−1 rhEGF, 0.50% FBS, 5 μg mL−1 transferrin PS. All cells were cultured at 37 °C and 5% CO2 in a humidified incubator.Cell viability assay
Cells were seeded at a concentration of 5000 cells/90 μL per well of either RPMI or DMEM without phenol red and without antibiotics, supplemented with 10% FBS and 2 mM L-glutamine into tissue culture grade 96-well flat bottom microplates (BioLite Microwell Plate, Fisher Scientific, Waltham, Massachusetts, USA) and grown for 24 h at 37 °C in a humidified incubator. Afterwards, the intermediate dilutions of the compounds were added to the wells (10 μL) to obtain a final concentration ranging from 0.1 to 200 μM, and the cells were incubated for 24 h or 72 h. Following 24 h or 72 h drug exposure, 50 μL per well of 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT) (Roche Diagnostics, Indianapolis, Indiana, USA) labelling mixture was added to the cells at a final concentration of 0.3 mg mL−1 and incubated for 4 h at 37 °C in a humidified incubator. The optical absorbance of each well in a 96-well plate was quantified using BioTek ELx808 absorbance microplate reader (BioTek Winooski, VT) set at 450 nm wavelength. The percentage of surviving cells was calculated from the ratio of absorbance of treated to untreated cells. The IC50 value was calculated as the concentration reducing the proliferation of the cells by 50% and is presented as a mean (±S.E.) of at least two independent experiments each with triplicate measurements.Stability of compound 5in vitro and co-localization of Ti/Au metals in Caki-1 cells
Caki-1 cells were incubated with 1 μM and 5 μM compound 5 for 24 hours. Post-incubation, the cells were washed twice with cold PBS and lysed with cell lysis buffer comprising of 1% (v/v) Triton-X-100, 25 mM HEPES, 100 mM NaCl, 1 mM EDTA, 10% (v/v) glycerol and protease and phosphatase inhibitors. Lysates from untreated cells incubated with media supplemented with DMSO for same duration, were used as controls. Gold and titanium content in the cell lysates was determined using ICP-MS. One hundred μL of lysates were transferred into a glass vials and 1 mL of concentrated acid mix (comprising of 75% of 16 N nitric acid and 25% of 12 N hydrochloric acid) was added. The mixture was heated at 90 °C for 5 hours. After cooling, the samples were diluted with water, 40 ppb of Indium internal standard was added and analyzed in a Thermo Scientific XSERIES 2 ICP-MS with ESI PC3 Peltier cooled spray chamber with SC-FAST injection loop and SC-4 autosampler. All the elements were analyzed using He/H2 collision-reaction mode. One micromole of compound 5 was processed similarly to determine the extraction efficiency of gold and titanium. The protein content of the cell lysates were determined using bicinchoninic acid based protein assay kit (Thermo Scientific). The final levels of either Ti or Au were normalized to the cellular protein levels.Annexin V/PI assay
Confluent Caki-1 cells were treated with either 10 μM of 5, 0.1% DMSO or 5 μM of staurosporine for 6 h (Fig. 5). After incubation, cells were trypsinized with 0.25% trypsin without EDTA (Life Technologies, NY, USA) and stained for extracellular phosphatidylserine expression using FITC conjugated annexin V to label early apoptotic cells and co-stained with propidium iodide (PI) to identify necrotic cells according to the manufacturer instructions for the dyes (BD Biosciences, San Jose, CA). Stained cells were analyzed by flow cytometry using Accuri C6 software (BD Biosciences, San Jose, CA, USA).Cell migration
For wound healing assays, Caki-1 cells were seeded in 6 cm dishes at a density of 0.8 × 106 in complete media, then grown to confluence for 24 hours at 37 °C (5% CO2), then cross-shape wounds/scratches were performed in the monolayer using a sterile 10 μL pipette tip. Following the treatment of confluent cells with 5 μM compound 3 or 5 or 0.1% DMSO as control, images of wounds were captured by light microscopy using the Labomed TCM 400 Inverted Research Microscope (Abo America Inc., Fremont, CA) and a digital Moticam 10 camera(Fisher Scientific Pittsberg, PA) immediately after scratching (T0), after 1 hour (T1) and after 12 h (T12). To quantify cell migration, the area of the initial wound (T0) is compared with the area of the healing wound at three time points after the scratch is introduced. Percent migration = [area of original wound − area of wound during healing]/area of original wound] × 100.77Mobility shift assay
10 μL aliquots of pBR322 plasmid DNA (20 μg mL−1) in buffer (5 mM Tris/HCl, 50 mM NaClO4, pH = 7.39) were incubated with different concentrations of the compounds (4–7, and titanocene dichloride) (in the range 0.25 and 4.0 metal complex:DNA bp) at 37 °C for 20 h in the dark. Samples of free DNA and cisplatin–DNA were prepared as controls. After the incubation period, the samples were loaded onto the 1% agarose gel. The samples were separated by electrophoresis for 1.5 h at 80 V in Tris–acetate/EDTA buffer (TAE). Afterwards, the gel was stained for 30 min with a solution of GelRed nucleic acid stain.
Thioredoxin reductase inhibition studies in Caki-1 cells
For thioredoxin reductase activity assays, whole cell lysates was obtained from Caki-1 cells treated in vitro with either 0.1 μM, 0.5 μM, 1 μM, or 5 μM of 3, 5 or Auranofin or 1% DMSO. After 5, 12 or 24 hours of treatment incubation cells were washed three times in PBS, and lysed by douncing using scrapers and sheer force though syringe with a 34 gauge needle in assay buffer (Abcam Thioredoxin Reductase Assay kit, ab83463) added to 1 mM protease inhibitor cocktail (Abcam, ab65621). The lysates were centrifuged at 10000 × g for 15 minutes at 4 °C to isolate insoluble material. The total protein concentrations of soluble lysates were measured using the BCA Protein Assay (Life Technologies). The soluble lysates were incubated for 20 minutes in assay buffer before adding DTNB (5,5′-dithiobis(2-nitrobenzoic)acid), and measuring activity at 1 minute intervals for 30 minutes using the BioTek fluorescence microplate reader (BioTek U.S., Winooski, VT) set at λ = 412 nm. Lysates were tested in duplicate. TrxR activity was calculated based on the linear amount of TNB produced per minute per mg of total protein and corrected for background activity from enzymes other than TrxR in the lysates.
Kinase inhibition studies
In vitro profiling of a 35 selected member protein kinase panel (see ESI‡) was performed at Reaction Biology Corporation using the “HotSpot” assay platform. Briefly, specific kinase/substrate pairs along with required cofactors78 were prepared in reaction buffer; 20 mM Hepes pH 7.5, 10 mM MgCl2, 1 mM EGTA, 0.02% Brij35, 0.02 mg mL−1 BSA, 0.1 mM Na3VO4, 2 mM DTT, 1% DMSO. Compounds were delivered into the reaction, followed ∼20 min later by addition of a mixture of ATP (Sigma) and 33P-ATP (Perkin Elmer) to a final concentration 10 μM. Reactions were carried out at 25 °C for 120 min, followed by spotting of the reactions onto P81 ion exchange filter paper (Whatman). Unbound phosphate was removed by extensive washing of filters in 0.75% phosphoric acid. After subtraction of background derived from control reactions containing inactive enzyme, kinase activity data were expressed as the percent remaining kinase activity in test samples compared to vehicle (dimethyl sulfoxide) reactions. IC50 values and curve fits were obtained using Prism (GraphPad Software).Cell lysis and immunoblotting
Caki-1 cells were seeded into 10 cm dishes at a density of 2.2 × 106 and incubated for 24 hours. Following the treatment of confluent cells with 5 μM of compound 3 or 5 and 0.1% DMSO as control for either 1 hour, 3 hours, 6 hours or 12 hours, the cultures were washed with cold PBS, and the cells were harvested by scraping with a rubber cell scraper. After microcentrifuging for 5 minutes at 4 °C, pellets were treated with ice-cold lysis buffer with cell extraction buffer (BioSource, Camarillo, CA) containing 0.1% protease inhibitors and 1% phosphatase inhibitors cocktail (Fisher Scientific, Pittsburg, PA) for 10 minutes, then sonicated for 10 seconds three times, and placed on ice. The lysed cells were centrifuged at 10000 × g at 4 °C for 10 minutes to remove the pellets representing the insoluble cell fraction.
The protein concentration was determined with BCA protein dye reagent (Pierce, Fisher Scientific Pittsberg, PA). Samples were denatured with 5× SDS sample buffer 0.25% bromophenol blue, 0.5 M dithiothreitol (DTT), 50% glycerol 50% (v/v), 10% sodium dodecyl sulfate (SDS) followed by heating at 95 °C for 5 minutes. Proteins were separated by SDS-PAGE, transferred to a PVDF membrane (Millipore Corp.), and probed with MAPKAPK-3 antibody (Cell Signaling Technology, Danvers, MA, USA), as well as β-actin antibody (Cell Signaling Technology, Danvers, MA, USA). Immunoreactive bands of primary antibodies were detected using HRP goat anti-rabbit and secondary antibodies were detected with peroxidase-conjugated secondary antibody and detected by chemiluminescence (Pierce Biotechnology, Rockford, IL).
Determination of lethal and maximum tolerated doses (LD and MTD) in mice
The preliminary toxicity testing of compound 5 was performed in C57BL/6 female mice 6 to 8 weeks of age, maintained in accordance with institutional guidelines at the University of Hawaii Cancer Center (UHCC) governing the care of laboratory animals (IACUC number: A3423-01). To determine the lethal dose, mice were treated for 5 consecutive days at dosages ranging from 1.5 mg per kg per day to 6 mg per kg per day. We used one mouse per dose. Mice were weighed every 48 hours, and sacrificed 24 hours after the last dose. The compounds were administered in a solution of 0.5% DMSO and 99.5% normal saline (0.9% NaCl) (G-Biosciences, St. Louis, MO, U.S.A) once daily by subcutaneous injection. In order to determine the maximum tolerated dose (MTD) the animals were monitored by trained individuals for pain and distress as appropriate for the animal, under conditions and by procedures established by the UHCC vivarium veterinarian staff and research personnel (BTE). The maximum tolerated dose (MTD) was determined by observing the progression of the mice treated at doses below the lethal dose. Body weights, changes in behaviour and sign of distress were recorded. The dose at which neither debilitating effects nor sign of distress were observed was set as the MTD. More specifically the signs for distress monitored were: (1) decreased food and water consumption; (2) weight loss (more than 20% loss in body weight or dropping at or below 18 g) was consistent with significant distress and mice exhibiting such weight loss were euthanized; (3) abnormal posture/positioning (e.g., head-pressing, hunched back); (4) unkempt appearance (erected, matted, or dull haircoat); (5) self-mutilation, gnawing at limbs; (6) excessive self-imposed isolation/hiding. The MTD dose was confirmed by treating a cohort of 3 mice per compound and one control group, every other day for 14 days with the MTD dose. One group of mice was treated with the solvent (negative control). During the trial the mice did not exhibit any signs of distress.Study of the effects of 2 in Caki-1 xenografts in mice
12 female NOD.CB17-Prkdc scid/J (non-obese diabetic–severe combined immunodeficiency) from Jackson Laboratory (Bar Harbor, ME and Sacramento, CA, USA) were used for the xenograft experiment (ages 8 to 12 weeks and weighing 19–22 g) were used. Each mouse received 8 × 106 tumor cells subcutaneously without anesthesia. Exponentially growing Caki-1 human kidney cancer cells were suspended in 1:1 ratio 50 μL phosphate-buffered saline (PBS; pH 7.4) plus 50 μL of matrigel (BD Biosciences, San Jose, CA, USA) were injected subcutaneously on both left and right flank of each mice. The diameter of the tumors was measured once weekly using an electronic digital caliper and the tumor volume (TV) was calculated according to the empirical equation TV = (a)(b2) × π/6 where a = longest dimension; b = largest dimension orthogonal to a. The median volumes of each group were normalized to the initial tumor volume resulting in the relative tumor volume. Each group of six Caki-1-transplanted animals received compound 3 (7.5 mg per kg per every other day), compound 5 (3 mg per kg per every other day) or vehicle (0.1% DMSO in 0.9% NaCl) intraperitoneally (i.p.). Treatment started when tumors were palpable (about 6 mm diameter). Mice were randomized to treatment groups based on their starting tumour burden at 12 weeks of age to ensure equivalent distribution between the 3 groups. At trial end-point the mice were sacrificed and tumors measured again after excision and then processed for further analysis. Histological as well as biochemical evaluations of blood, liver, spleen, and kidney, were conducted. Tumor volumes were graphed for 3 and 5 treated mice compared to vehicle-treated mice, based on weekly external digital caliper measurements.
Bio-distribution: determination of gold and titanium content in the organs and plasma
Female NOD.CB17-Prkdc scid/J bearing subcutaneous Caki-1 tumors and treated with subcutaneous injection of either compound # 3 (7.5 mg per kg per 48 hours) or compound 5 (3 mg per kg per 48 hours) were used for pharmacokinetic distribution of the drug in blood and other tissues. Blood was collected by retro-orbital bleeding into heparinized blood collection vials on ice at time intervals of 30 minutes, 2 hours, 6 hours, 24 hours, and 48 hours after the first dose. The blood samples were centrifuged at 2800 rpm at 4 °C for 15 min and the supernatant plasma was transferred into 1.5 mL micro-centrifuge tubes and stored at −80 °C until further analysis.Gold and titanium content was determined using ICP-MS. In brief, fifty microliters of plasma was transferred into a glass vial and 1 mL of concentrated acid mix (comprising of 75% of 16 N nitric acid and 25% of 12 N hydrochloric acid) was added. The mixture was heated at 90 °C for 5 hours. After cooling, the samples were diluted with water, 40 ppb of Indium internal standard was added and analyzed in a Thermo Scientific XSERIES 2 ICP-MS with ESI PC3 Peltier cooled spray chamber with SC-FAST injection loop and SC-4 autosampler. All the elements were analyzed using He/H2 collision-reaction mode. Plasma from control mice was spiked with the test compounds to determine the extraction efficiency of gold and titanium.
At the end of the study, liver, kidney and tumor of the animals were harvested, weighed and transferred into glass vials. One mL of water was added to each samples and subjected to ultrasonic tissue disruption at 15 W power for 1 minute. The tissue homogenates were frozen at −80 °C for 2 hours and lyophilized. The lyophilized product was heated at 90 °C with the concentrated acid mix (described above) for 5 hours, cooled, diluted with water and analyzed for titanium and gold by ICP-MS. Pharmacokinetic estimates were obtained from the plasma concentration–time profiles by non-compartmental analysis.
Conclusions
In conclusion we have described the preparation and promising antitumor properties of a new, unusually stable titanocene heterometallic compound containing a methyl group and a carboxylate ligand (mba = −S–C6H4–COO−) bound to a gold(I)-triphenylphosphane fragment through a thiolate group [(η-C5H5)2TiMe(μ-mba)Au(PPh3)] (5). This compound is able to block renal cancer growth both in vitro and in vivo by pathway(s) that involve the inhibition of thioredoxin reductase and decreased expression of proteins kinases known to drive cell migration. Preliminary evidence indicates that compound 5 may have appreciable anti-invasive properties. In addition its robustness has been demonstrated in cellular uptake experiments on Caki-1 cells by co-localization of Ti and Au metals in a 1:1 ratio. The impressive tumor reduction (67%) observed in the in vivo trial with mice warrants further in vivo studies to better determine the minimum most effective dosage and to get more accurate pharmacokinetic data. Studies on the mechanism(s) of this compound and effects on other cancers are under way. Overall, our results strongly support the efficacy of multifunctional heterometallic compounds, thus highlighting their great chemotherapeutic potential.
Acknowledgements
We thank the National Cancer Institute (NCI) for grant 1SC1CA182844 (M. C.) and the National Institute of General Medical Sciences (NIGMS) for grant RO1GM088266-A1 (J. W. R.). M. C. is very grateful to Mr and Mrs Leonard and Claire Tow and the Tow Foundation for a travel fellowship to perform some experiments at the University of Hawaii Cancer Center. We thank Dr Chris Farrar (University of Hawaii Cancer Center) for assistance with the flow cytometry experiments. We thank Prof. Jayanth Panyam from the Center for Translational Drug Delivery at the University of Minnesota for helpful discussions about the pharmacokinetic studies and Dr Rick Knurr from the Geochemical Lab in the Department of Earth Sciences at the University of Minnesota for assistance with ICP-MS analysis. We thank the CUNY HPCC which is operated by the College of Staten Island and funded, in part, by grants from the City of New York, State of New York, CUNY Research Foundation, and National Science Foundation Grants CNS-0958379, CNS-0855217 and ACI 1126113.Notes and references
- S. Medici, M. Peana, V. M. Murchi, J. I. Lachowicz, G. Crisponi and M. A. Zoroddu, Coord. Chem. Rev., 2015, 284, 329 CrossRef CAS PubMed.
- A. M. Thayer, Chem. Eng. News, 2010, 88, 24 Search PubMed.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573 CrossRef CAS PubMed.
- W. E. Berdel, H. J. Schmoll, M. E. Scheulen, A. Korfel, M. F. Knoche, A. Harstrick, F. Bach and G. Sa, J. Cancer Res. Clin. Oncol., 1994, 120, R172 Search PubMed.
- P. M. Abeysinghe and M. M. Harding, Dalton Trans., 2007, 32, 3474 RSC and refs. therein.
- U. Olszewski and G. Hamilton, Anti-Cancer Agents Med. Chem., 2010, 10, 320 Search PubMed and refs. therein.
- G. Lummen, H. Sperling, H. Lubolt, T. Otto and H. Rubben, Cancer Chemother. Pharmacol., 1998, 42, 415 CrossRef CAS PubMed.
- N. Kroger, U. R. Kleeberg, K. Mross, L. Edler, G. Sab and D. Hossfeld, Onkologie, 2000, 23, 288 Search PubMed.
- K. Strohfeldt and M. Tacke, Chem. Soc. Rev., 2008, 37, 1174 RSC.
- Recent selected example: U. Olszewski, A. Deally, M. Tacke and G. Hamilton, Neoplasia, 2012, 14, 813 CrossRef CAS and refs. therein.
- J. H. Bannon, I. Fitchner, A. O'Neill, C. Pampillón, N. J. Sweeney, K. Strohfeldt, R. W. Watson, M. Tacke and M. M. Mc Gee, Br. J. Cancer, 2007, 97, 1234 CrossRef CAS PubMed.
- I. Fichtner, C. Pampillón, N. J. Sweeney, K. Strohfeldt and M. Tacke, Anti-Cancer Drugs, 2006, 17, 333 CrossRef CAS PubMed.
- I. Fichtner, D. Behrens, J. Claffey, A. Deally, B. Gleeson, S. Patil, H. Weber and M. Tacke, Lett. Drug Des. Discovery, 2011, 8, 302 CrossRef CAS.
- W. Walther, I. Fichtner, A. Deally, M. Hogan and M. Tacke, Lett. Drug Des. Discovery, 2013, 10, 375 CrossRef CAS.
- B. Bertrand and A. Casini, Dalton Trans., 2014, 4209 RSC.
- S. Nobili, E. Mini, I. Landini, C. Gabbiani, A. Casini and L. Messori, Med. Res. Rev., 2010, 30, 550 CAS.
- A. Casini, C. Hartinger, C. Gabbiani, E. Mini, P. J. Dyson, B. K. Keppler and L. Messori, J. Inorg. Biochem., 2008, 102, 564 CrossRef CAS PubMed.
- S. J. Berners-Price and A. Filipovska, Metallomics, 2011, 3, 863 RSC.
- L. Dalla Via, C. Nardon and D. Fregona, Future Med. Chem., 2012, 4, 525 CrossRef CAS PubMed.
- A. Casini, G. Kelter, C. Gabbiani, M. A. Cinellu, G. Minghetti, D. Fregona, H.-H. Fiebig and L. Messori, JBIC, J. Biol. Inorg. Chem., 2009, 14, 1139 CrossRef CAS PubMed.
- F. Magherini, A. Modesti, L. Bini, M. Puglia, I. Landini, S. Nobili, E. Mini, M. A. Cinellu, C. Gabbiani and L. Messori, JBIC, J. Biol. Inorg. Chem., 2010, 15, 573 CrossRef CAS PubMed.
- Y. Zhu, B. R. Cameron, R. Mosi, V. Anastassov, J. Cox, L. Qin, Z. Santucci, M. Metz, R. T. Skerlj and S. P. Fricker, J. Inorg. Biochem., 2011, 105, 754 CrossRef CAS PubMed.
- E. Erdogan, T. Lamark, M. Stallings-Mann, L. Jamieson, M. Pellecchia, E. A. Thompson, T. Johansen and A. P. Fields, J. Biol. Chem., 2006, 281, 28450 CrossRef CAS PubMed.
- M. Stallings-Mann, L. Jamieson, R. P. Regala, C. Weems, N. R. Murray and A. P. Fileds, Cancer Res., 2006, 66, 1767 CrossRef CAS PubMed.
- K. I. Jeon, M. S. Biun and D. M. Jue, Exp. Mol. Med., 2003, 35, 61 CrossRef CAS PubMed.
- M. Serratrice, F. Edafe, F. Mendes, R. Scopelliti, S. M. Zakeeruddin, M. Gratzel, I. Santos, M. A. Cinellu and A. Casini, Dalton Trans., 2012, 3287 RSC.
- F. Mendes, M. Groessl, A. A. Nazarov, Y. O. Tsybin, G. Sava, I. Santos, P. J. Dyson and A. Casini, J. Med. Chem., 2011, 54, 2196 CrossRef CAS PubMed.
- M. Yang, A. J. Pickard, X. Quiao, M. J. Gueble, C. S. Day, G. L. Kucera and U. Bierbach, Inorg. Chem., 2015, 54, 3316 CrossRef CAS PubMed.
- M. Wenzel, B. Bertrand, M.-J. Eymin, V. Comte, J. A. Harvey, P. Richard, M. Groessl, O. Zava, H. Amrouche, P. D. Harvey, P. Le Gendre, M. Picquet and A. Casini, Inorg. Chem., 2011, 50, 9472 CrossRef CAS PubMed.
- J. F. González-Pantoja, M. Stern, A. A. Jarzecki, E. Royo, E. Robles-Escajeda, A. Varela-Ramirez, R. J. Aguilera and M. Contel, Inorg. Chem., 2011, 50, 11099 CrossRef PubMed.
- J. Fernández-Gallardo, B. T. Elie, J. Florian, J. Sulzmaier, M. Sanaú, J. W. Ramos and M. Contel, Organometallics, 2014, 33, 6669 CrossRef PubMed.
- M. P. Rogibello, L. Messori, G. Marcon, M. A. Cinellu, M. Bragadin, A. Folda, G. Scutari and A. Bindoli, J. Inorg. Biochem., 2004, 98, 1634 CrossRef PubMed.
- A. Bindoli, M. P. Rigobello, G. Scutari, C. Gabbiani, A. Casini and L. Messori, Coord. Chem. Rev., 2009, 253, 1692 CrossRef CAS PubMed.
- L. Ortego, F. Cardoso, S. Martins, M. F. Fillat, A. Laguna, M. Meireles, M. D. Villacampa and M. C. Gimeno, J. Inorg. Biochem., 2014, 130, 32 CrossRef CAS PubMed.
- U. Helmstedt, S. Lebedkin, T. Hocher, S. Blaurock and E. Hey-Hawkins, Inorg. Chem., 2008, 47, 5815 CrossRef CAS PubMed.
- D. de Vos, D. R. Smyth and E. R. T. Tiekink, Met.-Based Drugs, 2002, 8, 303 CrossRef PubMed.
- J. D. E. T. Wilton-Ely, A. Schier, N. W. Mitzel and H. Schmidbaur, Dalton Trans., 2001, 7, 1058 RSC.
- E. Barreiro, J. S. Casas, M. D. Couce, A. Sanchez, A. Sanchez-Gonzalez, J. Sordo, J. M. Varela and E. M. V. Lopez, J. Inorg. Biochem., 2008, 102, 184 CrossRef CAS PubMed.
- G. B. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227 CrossRef CAS.
- D. Martiınez, M. Motevalli and M. Watkinson, Dalton Trans., 2010, 446 RSC.
- X. Che and L. Zhou, J. Mol. Struct., 2010, 940, 45 CrossRef PubMed.
- N. J. Sweeney, O. Mendoza, H. Muller-Bunz, C. Pampillon, F.-J. K. Rehmann, K. Strohfeldt and M. Tacke, J. Organomet. Chem., 2005, 690, 4537 CrossRef CAS PubMed.
- L. Vela, M. Contel, L. Palomera, G. Azaceta and I. Marzo, J. Inorg. Biochem., 2011, 105, 1306 CrossRef CAS PubMed.
- K. A. Moutasim, M. L. Nystrom and G. J. Thomas, Methods Mol. Biol., 2011, 731, 333 CAS.
- K. I. Hulkower and R. L. Herber, Pharmaceutics, 2011, 3, 107 CrossRef CAS PubMed.
- J. C. Dabrowiak, Metals in medicine, John Wiley and Sons, Ltd., Chichester, UK, 2009, ch. 4, p. 109 Search PubMed.
- H.-K. Liu and P. Sadler, Acc. Chem. Res., 2011, 44, 349 CrossRef CAS PubMed.
- P. Kopf-Maier, W. Wagner and E. Liss, J. Cancer Res. Clin. Oncol., 1982, 103, 145 CrossRef CAS.
- C. V. Christodoulou, A. G. Eliopuulos, L. S. Young, L. Hodgkins, D. R. Ferry and D. J. Kerr, Br. J. Cancer, 1998, 77, 2088 CrossRef CAS PubMed.
- M. Rivera, E. Gabano, S. Baracco and D. Osella, Inorg. Chim. Acta, 2009, 362, 1303 CrossRef PubMed and refs. therein.
- G. Lally, A. Deally, F. Hackenberg, S. J. Quinn and M. Tacke, Lett. Drug Des. Discovery, 2013, 10, 675 CrossRef CAS.
- K. Fox, Drug-DNA Interact. Protocols. Methods in Mol. Biol., Humana Press Inc, Totowa, NJ, 1997 Search PubMed.
- H.-V. Lim, S. Hong, W. Jin, S. Lim, S.-J. Kim, H.-J. Kang, E.-H. Park, K. Ahn and C.-J. Lim, Exp. Mol. Med., 2005, 37, 497 CrossRef CAS PubMed.
- C. Marzano, V. Gandin, A. Folda, G. Scutari, A. Bindoli and A. P. Rigobello, Free Radical Biol. Med., 2007, 42, 872 CrossRef CAS PubMed.
- S. Gromer, L. D. Arscott, C. H. Williams Jr, R. H. Schirmer and K. Becker, J. Biol. Chem., 1998, 273, 20096 CrossRef CAS PubMed.
- R. Rubbiani, I. Kitanovic, H. Alborzinia, S. Can, A. Kitanovic, L. A. Onambele, M. Stefanopoulou, Y. Geldmacher, W. S. Sheldrick, G. Wolber, A. Prokop, S. Wolf and I. Ott, J. Med. Chem., 2010, 53, 8608 CrossRef CAS PubMed.
- A. Meyer, C. P. Bagowski, M. Kokoschka, M. Stefanopoulou, H. Alborzinia, S. Can, D. H. Vlecken, W. S. Shledrick, S. Wolfl and I. Ott, Angew. Chem., Int. Ed., 2012, 51, 8895 CrossRef CAS PubMed.
- F. Di Sarra, B. Fresch, R. Bini, G. Saielli and A. Bagno, Eur. J. Inorg. Chem., 2013, 2718 CrossRef CAS PubMed.
- D. Zhang, Z. Xu, J. Yuan, Y.-X. Zhao, Z.-Y. Quiao, Y. J. Gao, G.-A. Yu, J. Li and H. Wang, J. Med. Chem., 2014, 57, 8132 CrossRef CAS PubMed.
- T. V. Serebryanskaya, A. S. Lyakhov, L. S. Ivashkevich, J. Schur, C. Frias, A. Prokop and I. Ott, Dalton Trans., 2015, 1161 RSC.
- E. Schuh, C. Pfluger, A. Citta, A. Folda, M. P. Rigobello, A. Bindoli, A. Casini and F. Mohr, J. Med. Chem., 2012, 55, 5518 CrossRef CAS PubMed.
- C. Liu, Z. Liu, M. Li, X. Li, Y.-S. Wong, S.-M. Ngai, W. Zheng, Y. Zhang and T. Chen, PLoS One, 2013, 8, e53945 CAS.
- B. Bertrand, A. de Almeida, E. P. M. van der Burgt, M. Piquet, A. Citta, A. Folda, M. P. Rigobello, P. Le Gendre, E. Bodio and A. Casini, Eur. J. Inorg. Chem., 2014, 4532 CrossRef CAS PubMed.
- L. Feng, Y. Geisselbrecht, S. Blanck, A. Wilbuer, G. E. Atilla-Gokcumen, P. Filippakopoulos, K. Kraling, M. A. Celik, K. Harms, J. Maksimoska, R. Marmorstein, G. Frenking, S. Knapp, L.-O. Essen and E. Meggers, J. Am. Chem. Soc., 2011, 133, 5976 CrossRef CAS PubMed.
- E. Meggers, Angew. Chem., Int. Ed., 2011, 50, 2442 CrossRef CAS PubMed.
- S. P. Mulcahy and E. Meggers, Top. Organomet. Chem., 2010, 32, 141 CrossRef CAS.
- L. Geraets, A. G. Oomen, P. Krystek, N. R. Jacobsen, H. Wallin, M. Laurentie, H. W. Verharen, E. F. Brandon and W. H. de Jong, Part. Fibre Toxicol., 2014, 11, 30 CrossRef PubMed.
- E. I. Hamilton, M. J. Minski and J. J. Cleary, Sci. Total Environ., 1972, 1, 1 CrossRef CAS.
- S. M. Sprague, J. T. Carrick, B. W. Wilkinson and G. H. Mayor, J. Radioanal. Chem., 1979, 52, 421 CrossRef CAS.
- R. Uson, A. Laguna and M. Laguna, Inorg. Synth., 1989, 26, 85 CrossRef CAS PubMed.
- G. P. Sollott, H. E. Mertwoy, S. Portnoy and J. L. Snead, J. Org. Chem., 1963, 28, 1090 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.2, Gaussian Inc., Wallingford CT, 2004 Search PubMed.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866 CrossRef CAS PubMed.
- D. Andrae, U. Haussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123 CrossRef CAS.
- C. C. Liang, A. Y. Park and J. L. Guan, Nat. Protoc., 2007, 2, 329 CrossRef CAS PubMed.
- T. Anastassiadis, S. W. Deacon, K. Devarajanm, H. Ma and J. R. Peterson, Nat. Biotechnol., 2011, 29, 1039n CrossRef PubMed.
Footnotes |
| † This paper is dedicated to Prof. Roberto Sánchez-Delgado, great mentor and excellent friend, on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available: Stability studies of the new compounds by NMR, UV-vis spectroscopy and MS spectrometry, crystallographic data for compound 6, DFT calculations for compounds 4–7, IC50 values in human renal cells at both 24 and 72 h, details on migration studies, TrxR inhibition studies for 3, 5 and AF at different times, inhibition studies of compound 5 against a panel of 35 protein kinases, effects of AF on MAPKAPK-3 in Caki-1 cells, effects of compound 3 in Caki-1 mouse xenografts. CCDC 1400886. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc01753j |
| § These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2015 |