
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unravelling the pH-dependence of a molecular photocatalytic system for hydrogen production†
Anna
Reynal
*ab,
Ernest
Pastor
a,
Manuela A.
Gross
c,
Shababa
Selim
a,
Erwin
Reisner
*c and
James R.
Durrant
*a
aDepartment of Chemistry, Imperial College London, Exhibition Road, London SW7 2AZ, UK. E-mail: j.durrant@imperial.ac.uk
bSchool of Chemistry, Newcastle University, Newcastle Upon Tyne, NE1 7RU, UK. E-mail: anna.reynal@ncl.ac.uk
cChristian Doppler Laboratory for Sustainable SynGas Chemistry, Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: reisner@ch.cam.ac.uk
First published on 28th May 2015
Abstract
Photocatalytic systems for the reduction of aqueous protons are strongly pH-dependent, but the origin of this dependency is still not fully understood. We have studied the effect of different degrees of acidity on the electron transfer dynamics and catalysis taking place in a homogeneous photocatalytic system composed of a phosphonated ruthenium tris(bipyridine) dye (RuP) and a nickel bis(diphosphine) electrocatalyst (NiP) in an aqueous ascorbic acid solution. Our approach is based on transient absorption spectroscopy studies of the efficiency of photo-reduction of RuP and NiP correlated with pH-dependent photocatalytic H2 production and the degree of catalyst protonation. The influence of these factors results in an observed optimum photoactivity at pH 4.5 for the RuP–NiP system. The electron transfer from photo-reduced RuP to NiP is efficient and independent of the pH value of the medium. At pH <4.5, the efficiency of the system is limited by the yield of RuP photo-reduction by the sacrificial electron donor, ascorbic acid. At pH >4.5, the efficiency of the system is limited by the poor protonation of NiP, which inhibits its ability to reduce protons to hydrogen. We have therefore developed a rational strategy utilising transient absorption spectroscopy combined with bulk pH titration, electrocatalytic and photocatalytic experiments to disentangle the complex pH-dependent activity of the homogenous RuP–NiP photocatalytic system, which can be widely applied to other photocatalytic systems.
Introduction
The photochemical production of H2 from water is a rapidly expanding research field that aims to store solar energy in a chemical fuel.1 From the viewpoint of sustainability and economic viability, this proton reduction reaction should be carried out in aqueous conditions and use stable and Earth abundant materials.2 Current investigations for solar H2 synthesis include molecular dyes and electrocatalysts based on nickel, iron and cobalt, either in solution or immobilised onto the surface of a semiconductor.3–12 These photocatalytic systems typically require the use of sacrificial chemical reductants to provide the electrons to regenerate the oxidised dye following proton reduction.The efficiency of H2 evolving photo- and electrocatalytic systems is typically strongly pH dependent.13–16 Understanding the origins of this pH dependence is critical to guide further system development and optimisation. In particular, it is essential to determine whether such pH dependencies derive from the availability of protons to the molecular catalyst, from the function of the molecular light-harvesting unit or from the sacrificial electron donor.
We have recently reported a homogeneous photocatalytic system based on a molecular ruthenium photosensitiser (RuP) and a nickel catalyst (NiP) capable of producing H2 in pure water with a quantum efficiency near 10% in the presence of ascorbic acid (AA) as a sacrificial electron donor (Fig. 1).17 In this system, the electron transfer from the photoreduced dye (RuP−) to NiP takes place following reductive quenching of the photoexcited dye in the presence of the sacrificial agent, AA (Scheme 1). Under visible light irradiation, optimum performance of this photocatalytic system was observed at pH 4.5. In contrast, when used as an electrocatalyst, the proton reduction efficiency of the NiP catalyst was observed to increase towards more acidic pH.17 This pH dependence is typical of this type of nickel-based molecular electrocatalysts, and has been attributed to the presence of pendant amines with low pKa, which are thought to act as a proton relay between the solvent and the metal centre.13,18–20
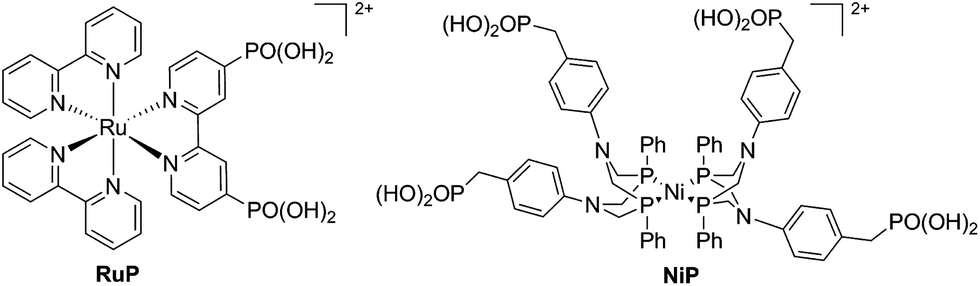 | ||
| Fig. 1 Molecular structures of the phosphonated ruthenium dye (RuP) and the nickel H2 evolution catalyst (NiP). The bromide counter ions have been omitted for clarity. | ||
 | ||
| Scheme 1 Schematic representation of the reductive electron transfer mechanism between RuP and NiP in the presence of ascorbic acid as sacrificial electron donor. | ||
Studies reporting the dependence of H2 evolution on the acidity of the aqueous media for molecular photocatalytic systems have typically focused on the overall system efficiency as a function of pH.13–15,21 Reaction mechanisms, where studied, have been addressed through theoretical calculations and experimental techniques such as nuclear magnetic resonance spectroscopy, electrochemistry and steady state spectroscopy;7,22–24 and to a lesser extent, time-resolved absorption spectroscopy.15,24–34 Herein, we report on the influence of the solution acidity on the formation of the photo-reduced RuP− species, the electron transfer kinetics between the optically active RuP− and NiP, as well as the pH dependence of H2 evolution observed in electrochemical and bulk photocatalytic experiments. We have employed transient absorption spectroscopy, combined with electrochemical experiments, to determine the working principles of this photocatalytic system. The correlation of these results allowed us to determine the pH-dependent rate-limiting steps in the photocatalytic system and give a rational explanation for the observed optimal activity at pH 4.5, as well as to provide a timescale for the electron transfer (ET) reactions between the sacrificial electron donor, the dye and the catalyst. Experimental details are described in the ESI.†
Results and discussion
At pH 4.5, photoexcitation of RuP in the presence of AA leads to the efficient formation of RuP− within t50% ∼ 250 ns through a reductive quenching mechanism, with a quantum yield estimated from transient emission studies of approximately 70%.17 The reduced photosensitiser RuP− shows a transient absorption peak at λ = 500 nm with a lifetime (t50%, calculations detailed in Fig. S1†) of 500–700 μs (Fig. 2).35 The yield of RuP− produced at different pH values can be determined from the initial amplitude (at ∼10 μs) of this RuP− transient absorption signal at λ = 500 nm. It is apparent (Fig. 2, inset) that this assay of the yield of RuP− increases with increasing pH, reaching a maximum at pH = 5. This behaviour can be explained by the different reactivity of two protonation states of ascorbic acid present in the pH range studied herein. At low pH, ascorbic acid exists primarily in its undissociated form H2A (pKa = 4.17), whereas the monoprotic ascorbate anion (HA−) predominates at higher pH values (pKa = 11.57). The ascorbate anion is a stronger reducing agent than its protonated form, and thus the reductive quenching of the excited dye, RuP*, is favoured at pH >4, where HA− is the dominating species.36–38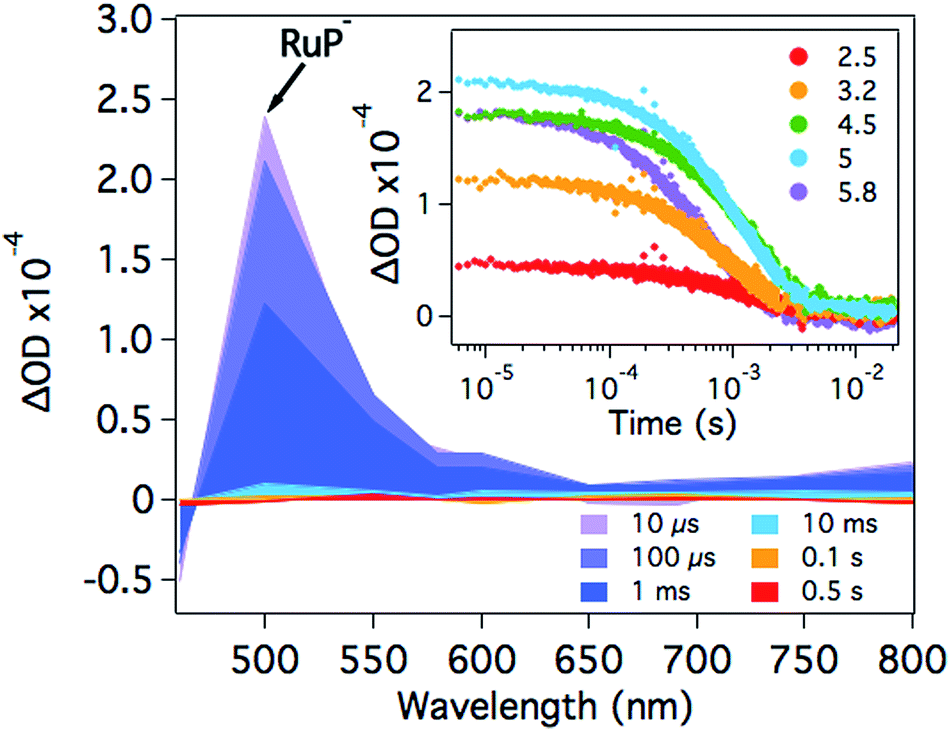 | ||
| Fig. 2 Transient absorption spectra of RuP (4 μM) in the presence of AA (0.1 M) at pH 4.5 as a function of time delay. The inset shows the corresponding kinetics probed at λ = 500 nm in the pH range between 2.5 and 5.8. The samples were excited at λ = 355 nm. | ||
After the formation of RuP−, electrons should be transferred from the reduced dye to the catalyst. In the presence of NiP, the positive transient absorption signal corresponding to RuP− absorption at λ = 500 nm is rapidly quenched (within 50–100 μs on the range of pH values studied herein), leading to the appearance of a negative signal at longer timescales (500 μs to 1 s; Fig. 3 and S2†). This negative signal is assigned to electron transfer from RuP− to NiP, resulting in bleaching of ground state absorption of NiP.17 This bleach is not observed in the absence of either RuP or NiP (see for example Fig. 2 and S3†), suggesting that it is due to intermolecular electron transfer (ET) between RuP− and NiP (rather than the direct photoexcitation of NiP). The fast electron transfer kinetics between RuP− and NiP at all studied pH values suggests that this process is not limiting the catalytic activity of NiP (Fig. S2†). However, the long-lived transient absorption bleach signal corresponding to reduced NiP indicates that the subsequent protonation step is more likely to be the rate limiting reaction. We can also estimate the yield of NiP reduced by RuP− from the amplitude of the bleach (Fig. 3 inset). Thus, a greater negative signal indicates the reduction of more NiP due to ET from RuP−. It is apparent that the yield of reduced NiP increases as the pH is increased, reaching a maximum at pH = 5.
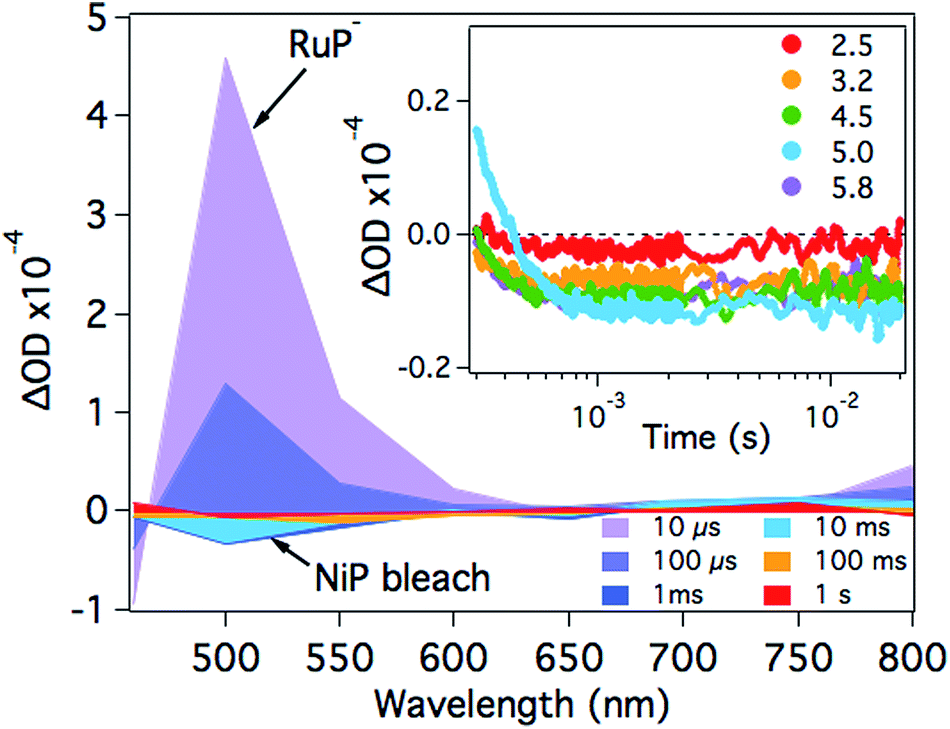 | ||
| Fig. 3 Transient absorption spectra of a RuP (4 μM) and NiP (8 μM) mixture in the presence of AA (0.1 M) at pH 4.5 as a function of time delay. The inset shows the time profile of the negative signal monitored at λ = 500 nm, assigned to the loss of ground state absorption of NiP in the pH range studied. The samples were excited at λ = 355 nm. | ||
Fig. 4 compares the pH dependence of the 500 nm transient absorption bleach signal assigned to the yield of reduced NiP (blue circles) and the TOFNiP(H2) per catalyst molecule of the system (red squares) determined from bulk photocatalysis experiments reported previously (see ESI†).17 Also shown in Fig. 4 is the ratio of reduced NiP per RuP− (black triangles, calculations detailed in the ESI†). It is apparent that whilst both the TOF and yield of reduced NiP are strongly pH dependent, the ratio of reduced NiP/RuP− is independent of pH. Thus, our results suggest that the yield of reduction of NiP by RuP− is pH independent. In contrast, from pH 2 to 4.5, both the NiP reduction yield and the TOFNiP increase. As the efficiency of electron transfer from RuP− to NiP is pH independent, the increase in the yield of reduced NiP with higher pH can be assigned directly to the increased efficiency of RuP− formation due to the pH dependence of the electron donating function of the ascorbic acid as discussed above. It is also striking from Fig. 4 that at pH >4.5, the TOFNiP rapidly decreases despite the yield of reduced NiP remaining high. Such a sharp maximum in the pH dependence of TOFcatalyst has also been observed in many other photocatalytic systems.14,15,17,25,39,40
 | ||
| Fig. 4 TOFNiP(H2) of a homogeneous AA (0.1 M) aqueous solution at different pH values, containing RuP (0.3 μmol, 133 μM) and NiP (0.1 μmol, 44 μM; red squares). Transient absorption signal amplitudes of the NiP bleach at 1 ms (absolute values, blue circles) and transient absorption amplitude ratios of NiP at 1 ms and RuP− at 10 μs (black triangles). | ||
As the yield of reduced catalyst is approximately constant between pH 4.5 and 6, the drop on hydrogen generation towards neutral pH is strongly indicative of a decreasing activity in proton reduction catalysed by NiP. The exact catalytic mechanism for proton reduction using nickel bis(diphosphine) catalysts is still not fully elucidated, with little evidence of the catalytic intermediates in aqueous media.41,42 Although protonation of the reduced Ni species may in principle occur at the pendant amines of the ligand or directly at the Ni metal centre, DFT calculations support protonation of the amines.41 This agrees with the dependence of the electrocatalytic activity on acid concentration of bis(diphosphine) nickel electrocatalysts, which has been explained by the presence of pendant amines in the second coordination sphere. These amines with a relatively low pKa have been suggested to act as proton relays between the solvent and the metal centre.13,18,19,43 Although these studies were mainly performed in pure organic solvents or aqueous-organic solvent mixtures in the presence of strong acids, the electrocatalytic proton reduction activity of NiP was observed to increase towards more acidic pH.17 In this article, we detail the dependence of the catalytic activity of NiP on pH in pure water.
In order to further investigate the drop in the H2 production yield of the photocatalytic system towards neutral pH, the protonation state of NiP at different pH values was studied. The titration of NiP with NaOH (0.1 M) shows two equivalence points, at pH ∼5 and pH ∼9 (Fig. 5). In agreement with previous reports, these processes are assigned to the deprotonation of the pendant amines and the second deprotonation of the phosphonic acid groups, respectively.20,44 The assignment of the deprotonation of the amines is further confirmed by the presence of only one equivalence point at pH ∼5 for the titration of an analogous bis(diphosphine) nickel complex where the phopsphonic acid substituents are protected with ethyl ester groups (NiPEt) (Fig. S4†). A pKa ∼3 is calculated from the Henderson–Hasselbach equation for the pendant amines in the ligand with an equivalence point at pH ∼5 (see ESI† for details), meaning that at pH >5, the amines are largely deprotonated. Since these amines are considered to play an important role as proton relays between the solvent and the nickel metal centre,18,19 it is likely that, at less acidic media, the catalytic efficiency is limited by a poor degree of protonation of the pendant amines of the catalyst, which inhibits the ability of NiP to reduce protons to H2. It is worth noting that the photosensitiser employed in our studies contains phosphonic acid substituents. This dye was chosen for consistency and to allow for direct comparison with our previous studies.17 The pKa values of RuP have been reported to be ∼1 and ∼12, suggesting that the buffer capacity of RuP within the pH range employed in this study is limited.45,46
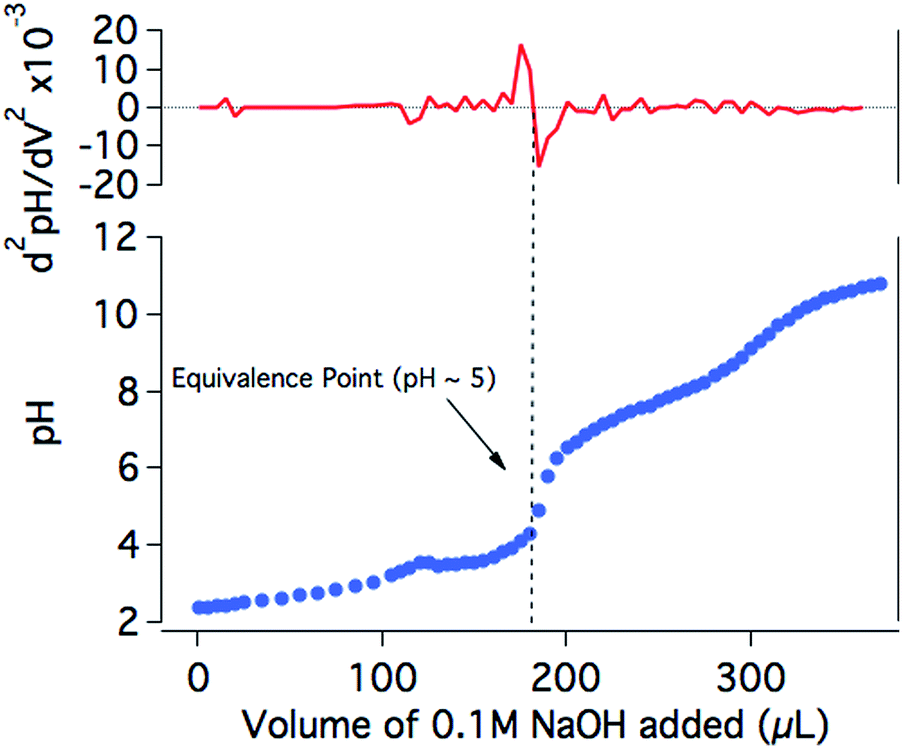 | ||
| Fig. 5 Titration of NiP (0.57 mM) in KCl (0.1 M) with NaOH (0.1 M; blue trace) and the second derivative of the pH with respect to the added volume (red trace). NiP is less soluble below pH 3 and dissolves completely upon the addition of approximately 120 μL of NaOH. | ||
Our results match well with the strong pH-dependencies reported with other proton reduction photocatalytic systems that employ either AA, triethanolamine (TEOA) or ethylenediaminetetraacetic acid (EDTA) as sacrificial electron donors.14,15 In acidic media, the sacrificial electron donor molecules become protonated, resulting in a poor electron-donating ability due to the anodic shift of the reduction potential.4,17 Hence our studies show that the optimum pH of active homogeneous proton reduction systems is a compromise between electron donating ability of the sacrificial agent and the optimum working environment for the catalyst.
Conclusions
In summary, we have used transient absorption spectroscopy, combined with titration studies, electrochemistry and bulk photocatalytic experiments, to study the pH-dependence of the electron transfer reactions of a ruthenium-based photosensitiser and a nickel bis(diphosphine) catalyst for the production of H2 under visible light irradiation. Our results suggest that the yield and kinetics of the electron transfer from the sensitiser to the catalyst are independent of the pH. However, at pH <4.5, the catalysis is limited by the number of RuP− molecules available to reduce the catalyst due to the poor reducing character of undissociated AA. In contrast, at less acidic pH, low TOFNiP(H2) are observed despite the large concentration of RuP− molecules available to reduce NiP. Titration studies of NiP with NaOH show that at pH >5, the amines are largely deprotonated and electrochemical studies confirm the lower activity at such pH values.17 Since these amines have been reported to play an important role as proton relays between the solvent and the nickel metal centre, it is likely that the catalytic efficiency is limited by the lack of protonated amines in the nickel catalyst. In the wider context, our studies suggest that the pH of photocatalytic systems using a sacrificial agent has to be adjusted to match the pH at which the dye is effectively reduced by the sacrificial electron donor and the pH at which the catalyst can be efficiently protonated. We have also demonstrated how transient absorption spectroscopy, bulk photocatalytic and titration studies and electrochemical experiments can be combined for a rational analysis of limiting factors in a homogeneous photocatalytic system.Author contributions
E.P., A.R. and S.S. conducted the spectroscopic experiments. J.R.D. and A.R. designed the experiments. M.A.G. synthesised the compounds and carried out the titration experiments. M.A.G. and E.R. developed the photocatalytic system. E.P., A.R., M.A.G., E.R. and J.R.D. wrote the paper.Acknowledgements
Financial support from the ERC (291482, Intersolar to J. D.), the EPSRC (EP/H00338X/2 to E. R., DTG scholarship to E. P.), the European Commission Marie Curie CIG (303650, PhotoCO2 to A. R.), the Christian Doppler Research Association (Austrian Federal Ministry of Science, Research and Economy and National Foundation for Research, Technology and Development), and the OMV Group (to E. R.) is gratefully acknowledged.Notes and references
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum, S. Ardo, H. Wang, E. Miller and T. F. Jaramillo, Energy Environ. Sci., 2013, 6, 1983–2002 CAS
.
- P. Du and R. Eisenberg, Energy Environ. Sci., 2012, 5, 6012–6021 CAS
- W. T. Eckenhoff and R. Eisenberg, Dalton Trans., 2012, 41, 13004–13021 RSC
- F. Lakadamyali, A. Reynal, M. Kato, J. R. Durrant and E. Reisner, Chem. - Eur. J., 2012, 18, 15464–15475 CrossRef CAS PubMed
- A. Krawicz, J. Yang, E. Anzenberg, J. Yano, I. D. Sharp and G. F. Moore, J. Am. Chem. Soc., 2013, 135, 11861–11868 CrossRef CAS PubMed
- G. F. Moore and I. D. Sharp, J. Phys. Chem. Lett., 2013, 4, 568–572 CrossRef CAS
- D. L. DuBois, Inorg. Chem., 2014, 53, 3935–3960 CrossRef CAS PubMed
- H. S. Ahn, T. C. Davenport and T. D. Tilley, Chem. Commun., 2014, 50, 3834–3837 RSC
- J. R. McKone, S. C. Marinescu, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Chem. Sci., 2014, 5, 865–878 RSC
- M. Wang, K. Han, S. Zhang and L. Sun, Coord. Chem. Rev., 2015, 287, 1–14 CrossRef CAS PubMed
- A. Reynal, F. Lakadamyali, M. A. Gross, E. Reisner and J. R. Durrant, Energy Environ. Sci., 2013, 6, 3291–3300 CAS
- F. Lakadamyali and E. Reisner, Chem. Commun., 2011, 47, 1695–1697 RSC
- S. Horvath, L. E. Fernandez, A. M. Appel and S. Hammes-Schiffer, Inorg. Chem., 2013, 52, 3643–3652 CrossRef CAS PubMed
- S. Fukuzumi, T. Kobayashi and T. Suenobu, Angew. Chem., Int. Ed., 2011, 50, 728–731 CrossRef CAS PubMed
- T. Stoll, M. Gennari, I. Serrano, J. Fortage, J. Chauvin, F. Odobel, M. Rebarz, O. Poizat, M. Sliwa, A. Deronzier and M.-N. Collomb, Chem. - Eur. J., 2013, 19, 782–792 CrossRef CAS PubMed
- A. M. Appel, D. H. Pool, M. O'Hagan, W. J. Shaw, J. Y. Yang, M. Rakowski DuBois, D. L. DuBois and R. M. Bullock, ACS Catal., 2011, 1, 777–785 CrossRef CAS
- M. A. Gross, A. Reynal, J. R. Durrant and E. Reisner, J. Am. Chem. Soc., 2014, 136, 356–366 CrossRef CAS PubMed
- U. J. Kilgore, J. A. S. Roberts, D. H. Pool, A. M. Appel, M. P. Stewart, M. R. DuBois, W. G. Dougherty, W. S. Kassel, R. M. Bullock and D. L. DuBois, J. Am. Chem. Soc., 2011, 133, 5861–5872 CrossRef CAS PubMed
- M. Rakowski DuBois and D. L. DuBois, C. R. Chim., 2008, 11, 805–817 CrossRef PubMed
- A. Dutta, S. Lense, J. Hou, M. H. Engelhard, J. A. S. Roberts and W. J. Shaw, J. Am. Chem. Soc., 2013, 135, 18490–18496 CrossRef CAS PubMed
- P. Du, K. Knowles and R. Eisenberg, J. Am. Chem. Soc., 2008, 130, 12576–12577 CrossRef CAS PubMed
- A. D. Wilson, R. K. Shoemaker, A. Miedaner, J. T. Muckerman, D. L. DuBois and M. Rakowski DuBois, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6951–6956 CrossRef CAS PubMed
- M. O'Hagan, M.-H. Ho, J. Y. Yang, A. M. Appel, M. Rakowski DuBois, S. Raugei, W. J. Shaw, D. L. DuBois and R. M. Bullock, J. Am. Chem. Soc., 2012, 134, 19409–19424 CrossRef PubMed
- C. D. Windle, E. Pastor, A. Reynal, A. C. Whitwood, Y. Vaynzof, J. R. Durrant, R. N. Perutz and E. Reisner, Chem. - Eur. J., 2015, 21, 3746–3754 CrossRef CAS PubMed
- M. Guttentag, A. Rodenberg, R. Kopelent, B. Probst, C. Buchwalder, M. Brandstätter, P. Hamm and R. Alberto, Eur. J. Inorg. Chem., 2012, 59–64 CrossRef CAS PubMed
- E. Pastor, F. M. Pesci, A. Reynal, A. D. Handoko, M. Guo, X. An, A. J. Cowan, D. R. Klug, J. R. Durrant and J. Tang, Phys. Chem. Chem. Phys., 2014, 16, 5922–5926 RSC
- B. H. Solis and S. Hammes-Schiffer, Inorg. Chem., 2014, 53, 6427–6443 CrossRef CAS PubMed
- A. Rodenberg, M. Orazietti, B. Probst, C. Bachmann, R. Alberto, K. K. Baldridge and P. Hamm, Inorg. Chem., 2015, 54, 646–657 CrossRef CAS PubMed
- D. E. Polyansky, D. Cabelli, J. T. Muckerman, T. Fukushima, K. Tanaka and E. Fujita, Inorg. Chem., 2008, 47, 3958–3968 CrossRef CAS PubMed
- A. Lewandowska-Andralojc, T. Baine, X. Zhao, J. T. Muckerman, E. Fujita and D. E. Polyansky, Inorg. Chem., 2015, 54, 4310–4321 CrossRef CAS PubMed
- J. L. Dempsey, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2010, 132, 16774–16776 CrossRef CAS PubMed
- J. L. Dempsey, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2010, 132, 1060–1065 CrossRef CAS PubMed
- D. Streich, Y. Astuti, M. Orlandi, L. Schwartz, R. Lomoth, L. Hammarström and S. Ott, Chem. - Eur. J., 2010, 16, 60–63 CrossRef CAS PubMed
- W. M. Singh, M. Mirmohades, R. T. Jane, T. A. White, L. Hammarström, A. Thapper, R. Lomoth and S. Ott, Chem. Commun., 2013, 49, 8638–8640 RSC
- C. Creutz and N. Sutin, J. Am. Chem. Soc., 1976, 98, 6384–6385 CrossRef CAS
- D. H. Macartney and N. Sutin, Inorg. Chim. Acta, 1983, 74, 221–228 CrossRef CAS
- M. P. McLaughlin, T. M. McCormick, R. Eisenberg and P. L. Holland, Chem. Commun., 2011, 47, 7989–7991 RSC
- T. W. Birch and L. J. Harris, Biochem. J., 1933, 27, 595–599 CAS
- Z. Han, F. Qiu, R. Eisenberg, P. L. Holland and T. D. Krauss, Science, 2012, 338, 1321–1324 CrossRef CAS PubMed
- W. R. McNamara, Z. Han, P. J. Alperin, W. W. Brennessel, P. L. Holland and R. Eisenberg, J. Am. Chem. Soc., 2011, 133, 15368–15371 CrossRef CAS PubMed
- A. Kochem, F. Neese and M. van Gastel, J. Phys. Chem. C, 2014, 118, 2350–2360 CAS
- E. B. Hulley, K. D. Welch, A. M. Appel, D. L. DuBois and R. M. Bullock, J. Am. Chem. Soc., 2013, 135, 11736–11739 CrossRef CAS PubMed
- R. M. Bullock, A. M. Appel and M. L. Helm, Chem. Commun., 2014, 50, 3125–3143 RSC
- K. Swierczek, A. S. Pandey, J. W. Peters and A. C. Hengge, J. Med. Chem., 2003, 46, 3703–3708 CrossRef CAS PubMed
- H. Park, E. Bae, J.-J. Lee, J. Park and W. Choi, J. Phys. Chem. B, 2006, 110, 8740–8749 CrossRef CAS PubMed
- M. Montalti, S. Wadhwa, W. Y. Kim, R. A. Kipp and R. H. Schmehl, Inorg. Chem., 2000, 39, 76–84 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc01349f |
| This journal is © The Royal Society of Chemistry 2015 |