
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTarget profiling of an antimetastatic RAPTA agent by chemical proteomics: relevance to the mode of action†
Maria V.
Babak
ab,
Samuel M.
Meier
c,
Kilian V. M.
Huber
d,
Jóhannes
Reynisson
a,
Anton A.
Legin
b,
Michael A.
Jakupec
b,
Alexander
Roller
b,
Alexey
Stukalov
d,
Manuela
Gridling
d,
Keiryn L.
Bennett
d,
Jacques
Colinge
d,
Walter
Berger
e,
Paul J.
Dyson
f,
Giulio
Superti-Furga
d,
Bernhard K.
Keppler
b and
Christian G.
Hartinger
*a
aSchool of Chemical Sciences, University of Auckland, Private Bag 92019, Auckland 1142, New Zealand. E-mail: c.hartinger@auckland.ac.nz
bInstitute of Inorganic Chemistry, University of Vienna, Waehringer Str. 42, A-1090 Vienna, Austria
cInstitute of Analytical Chemistry, University of Vienna, Waehringer Str. 38, A-1090 Vienna, Austria
dCeMM Research Center for Molecular Medicine, Lazarettgasse 14, AKH BT 25.3, A-1090 Vienna, Austria
eDepartment of Medicine I, Institute of Cancer Research, Medical University Vienna, Borschkegasse 8a, A-1090 Vienna, Austria
fInstitut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland
First published on 9th February 2015
Abstract
The clinical development of anticancer metallodrugs is often hindered by the elusive nature of their molecular targets. To identify the molecular targets of an antimetastatic ruthenium organometallic complex based on 1,3,5-triaza-7-phosphaadamantane (RAPTA), we employed a chemical proteomic approach. The approach combines the design of an affinity probe featuring the pharmacophore with mass-spectrometry-based analysis of interacting proteins found in cancer cell lysates. The comparison of data sets obtained for cell lysates from cancer cells before and after treatment with a competitive binder suggests that RAPTA interacts with a number of cancer-related proteins, which may be responsible for the antiangiogenic and antimetastatic activity of RAPTA complexes. Notably, the proteins identified include the cytokines midkine, pleiotrophin and fibroblast growth factor-binding protein 3. We also detected guanine nucleotide-binding protein-like 3 and FAM32A, which is in line with the hypothesis that the antiproliferative activity of RAPTA compounds is due to induction of a G2/M arrest and histone proteins identified earlier as potential targets.
Introduction
The modes of action of organometallic anticancer ruthenium complexes, which are substantially different from commonly used platinum-based chemotherapeutics, account for the growing interest in this compound class.1–4 RAPTA complexes are a promising class of organometallic RuII compounds which inhibit processes related to metastasis in vitro and exhibit pronounced antimetastatic activity in vivo, but only low antiproliferative activity.5,6 The general formula of RAPTA compounds is [Ru(arene)(PTA)X2] (Fig. 1), where PTA = 1,3,5-triaza-7-phosphatricyclo-[3.3.1.1]decane and X = halogenide or biscarboxylate. With their cis-configured halogenido ligands resembling the cisplatin structure, DNA was initially considered to be the target.7,8 However, in recent years the focus of mode of action studies has shifted from investigations of RAPTA–DNA to RAPTA–protein interactions.9–11 It has been demonstrated that RAPTA compounds preferentially bind to proteins even in the presence of DNA, as shown in crystallographic and bioanalytical studies with the nucleosome core particle.12,13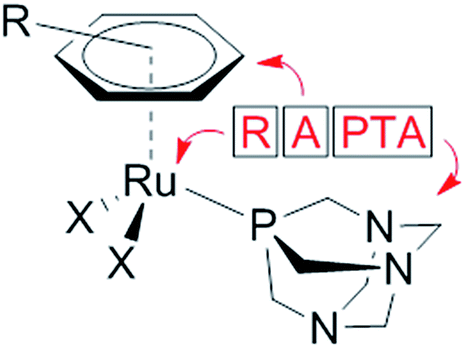 | ||
| Fig. 1 Schematic representation of the components of the RAPTA framework. | ||
Notably, the selection of the ligands determines the reactivity of organoruthenium compounds with biological targets. In contrast to the RAPTA derivatives, Ru(arene) complexes with a chelating ethylenediamine ligand bind preferentially to DNA.13 In addition, adduct formation of RAPTA with a range of isolated proteins has been demonstrated, and in some cases enzyme inhibition has been observed.9–11,14–17 In comparison to platinum compounds, RAPTA complexes tend to be more reactive towards proteins, but also display greater selectivity, even though both compound classes react with the same binding sites in proteins.14
Molecular profiling of the interactions of RAPTA compounds with their cellular targets would help to improve our understanding of their modes of action. The screening of putative RAPTA–protein interactions is laborious because of the enormous number of potential biomolecular targets in cells. Moreover, additional complexity arises because of the reactivity of metal complexes in aqueous systems due to hydrolysis. There is also the possibility that non-selective binding to any nucleophilic amino acid side chain donor atoms may occur. Therefore, a test system that does not distort the actual system too much is required, which, however, necessitates advanced analytical and molecular biology approaches.18 Recently, Messori et al. and Wolters et al. demonstrated the importance of using proteomic studies in the evaluation of cancer cell responses to RAPTA-T, [Ru(η6-toluene)(PTA)Cl2], treatment at the protein level.19,20 Wolters et al. employed multidimensional protein identification technology and identified 414 proteins out of which 74 proteins were further analyzed on their regulation profile,19 and histones were suggested to play an important role in the mode of action of RAPTA complexes.12 Messori et al. used 2-dimensional difference gel electrophoresis to monitor the changes in the expression of intracellular proteins upon exposure of cancer cells to RAPTA-T. In comparison to the control experiment, RAPTA-T did not induce significant modifications of protein expression profiles although a small number of up- and down-regulated proteins were detected.20 It is worth noting that in both cases substantial differences in the proteome profiles of cells treated with RAPTA compounds and those treated with platinum complexes were observed, highlighting their different modes of action.
In this paper, we describe the development of a chemical proteomic method (“drug pull-down”), involving affinity chromatography, shotgun proteomics and bioinformatics, to identify molecular targets of an antimetastatic RAPTA anticancer agent. To the best of our knowledge, such an approach is unprecedented for metal-based anticancer agents. The solid-phase functionalized with the RAPTA derivative was especially designed for this purpose.
Results and discussion
The molecular targets of metallodrugs are often elusive despite intensive analytical and biochemical efforts to identify them. This problem may partially be ascribed to the reactivity of metallodrugs in aqueous solution and the multitude of ligand exchange reactions that may occur depending on pH and concentrations of potential nucleophiles. Drug pull-down experiments allow the molecular targets of drugs to be identified. This approach has been applied to organic drugs; however, to the best of our knowledge immobilizing an organometallic anticancer agent is unprecedented and requires careful functionalization of the pharmacophore and selection of the experimental conditions.Experimental design
In order to establish the ‘natural’ target profile of RAPTA anticancer agents, we used a combination of drug affinity chromatography with RAPTA-modified beads, subsequent high-end mass spectrometry and bioinformatics. This approach is termed drug pull-down and the work-flow is depicted in Fig. 2.21 The natural state and environment of proteins, e.g. abundance, post-translational modifications, natural binding partners, etc. in the employed whole cell lysates are preserved.22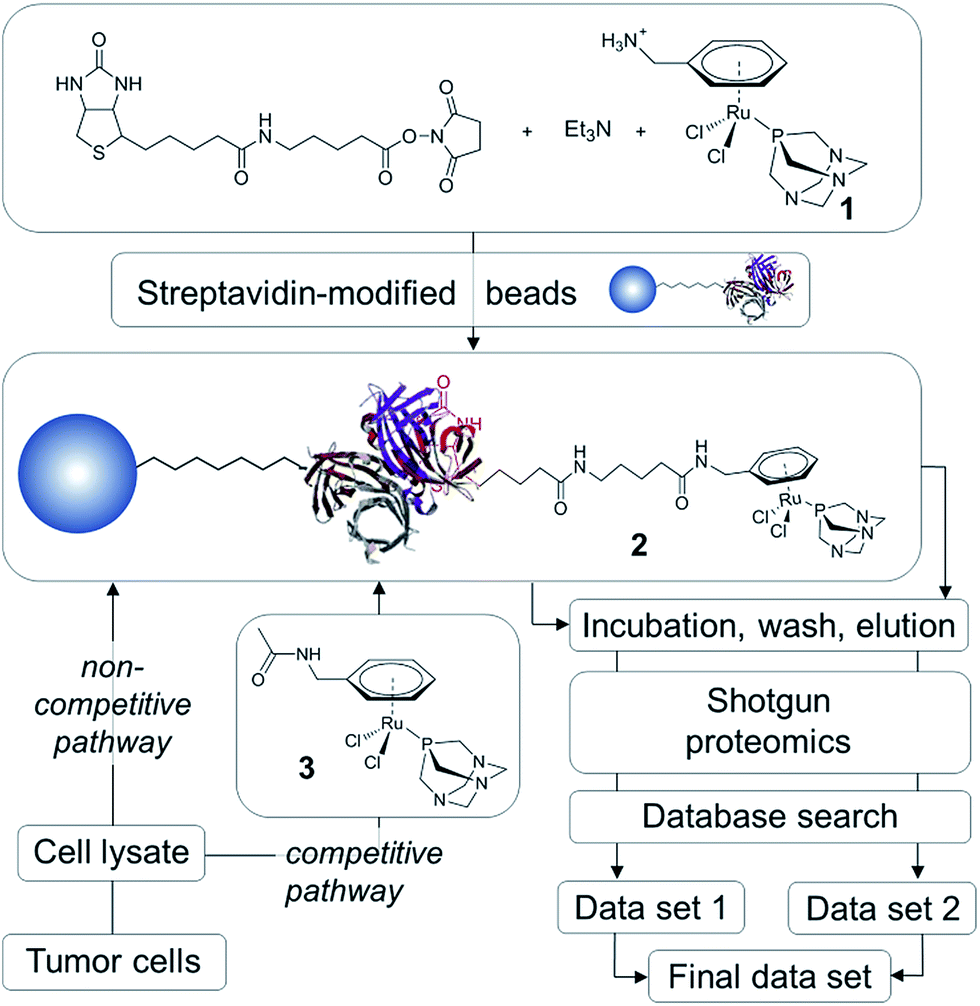 | ||
| Fig. 2 Schematic representation of the work-flow used in the metallodrug pull-down experiments. In the non-competitive pathway (data set 1) proteins can bind only to modified beads, whereas in the competitive pathway (data set 2) proteins can bind to modified beads and competitive binder 3. | ||
High-affinity binders may be identified by comparing the two routes of analysis, i.e., the non-competitive (data set 1) and competitive route (data set 2; Fig. 2). To obtain data set 1 in a non-competitive experiment, the beads loaded with the probe 2 are exposed to the cell lysate. In contrast, in the competitive experiment, the probe is in competition with the free drug analogue 3 for the preferred protein target. A significant reduction or even the complete disappearance of spectral counts during competitive reactions indicate high-affinity binders.
The traditional drug pull-down experiment involves covalent attachment of drug molecules or fragments to matrices such as N-hydroxysuccinimidyl-sepharose and requires extensive cleansing to block unreacted beads.22 Such cleansing is not possible using reactive metallodrugs as they are likely to undergo undesirable side reactions during the workflow. To avoid decomposition of the organometallic complex during immobilization onto the matrix, we employed a biotin/streptavidin approach, which draws on one of the largest known binding constants of K ∼ 1014 M−1 and has been used in biocatalysis.23 This self-assembly approach results in near quantitative functionalization of the beads with the RAPTA moiety, requiring minimal purification that could potentially deactivate the complex.
Since the chlorido ligands bound to the RuII center of RAPTA anticancer agents undergo hydrolysis and subsequent reaction with nucleophiles to form coordinative bonds to biological targets,9,10,12,16 the primary ligand sphere seems unsuitable for immobilization onto beads. Therefore, the arene ligand was functionalized with a primary amine and subsequently with biotin via an aminocaproic acid linker to yield 2 (Fig. 2). This compound was generated in situ and immobilized on streptavidin-modified beads. In order to perform competition experiments (competitive pathway in Fig. 2), 1 was converted into the non-immobilized acetyl derivative 3.
The functionalized RAPTA complexes 1 and 3 were synthesized using a similar procedure to one described in the literature24 by stirring PTA with the corresponding chlorido-bridged dinuclear ruthenium precursor in dry DMF for 3–4 h. In order to avoid undesirable coordination of the –NH2 group to the ruthenium center upon addition of base, benzylamine was chosen. It was reduced by Birch reduction and protonated prior to complexation with RuCl3 to give the dinuclear precursor [(η6-benzylammonium)RuCl2]2 chloride. Subsequently, complex 2 was obtained by stirring 1 with biotin-6-aminohexanoic acid-N-hydroxysuccinimide ester and triethylamine in dry DMF for 24 h. By employing 6-aminohexanoic acid as a linker, the distance between streptavidin and the reactive metal center was extended to allow sufficient flexibility and low steric demand so as not to impede reactions with target proteins. Functionalized complexes 1 and 3 were characterized by NMR spectroscopy, ESI mass spectrometry and elemental analysis (see Experimental section for full details). The presence of the functional groups did not significantly affect the 31P{1H} chemical shift being observed at −31 ppm (cf. −34.9 ppm for RAPTA-C,5i.e., [Ru(η6-p-cymene)(PTA)Cl2]). The signal corresponding to the protons in the –NH3+ group in 1 was detected at 8.39 ppm as a broad singlet, whereas the NH signal in 3 appeared as a well-resolved triplet at 8.30 ppm. Compound 2 was characterized in situ by high resolution nESI-Q-TOF MS and the pseudomolecular mass signal [M + H]+ was found with an accuracy of 5 ppm.
The molecular structures of 1 and 3 were established by single crystal X-ray diffraction analysis (Fig. 3). Crystals were grown by slow diffusion of diethyl ether into DMF at 277 K. Analysis of the bond lengths and angles confirmed structural similarity to RAPTA-C (Tables S1 and S2, ESI†).25 The distance between the ruthenium center and the centroid of the arene is similar among the three complexes (1.700, 1.699 and 1.692 Å for 1, 3 and RAPTA-C, respectively) indicating that methylamine derivatization does not alter the electron density on the metal. The Ru–P bond length (around 2.3 Å) and the Ru–Cl bond lengths in 1 and 3 range from 2.3966(4) to 2.4291(4) Å and are similar to those in RAPTA-C (2.412(3) Å and 2.429(2) Å), which underlines the retained geometry of the first coordination sphere upon derivatization of the arene. P–Ru–Cl angles vary between 82 and 87°, which is also in agreement with RAPTA-C (83–87°). Furthermore, the Cl1–Ru–Cl2 angle is around 88° in all complexes. This indicates that the RAPTA derivatives can be used as suitable structural models for drug pull-down experiments.
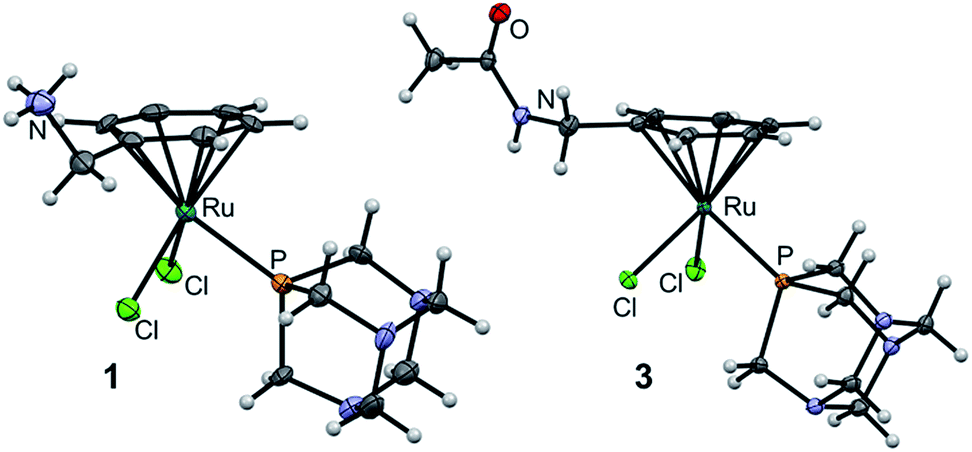 | ||
| Fig. 3 Molecular structures of 1 and 3 (one of two independent molecules) shown at the 50% probability level. For bond lengths and angles see ESI.† | ||
Immobilization of the pharmacophore and validation of its properties
In order to demonstrate that the modified RAPTA probe retains the chemical and biological properties of the parent RAPTA compound, a series of experiments were performed to elucidate the interactions with biomolecules.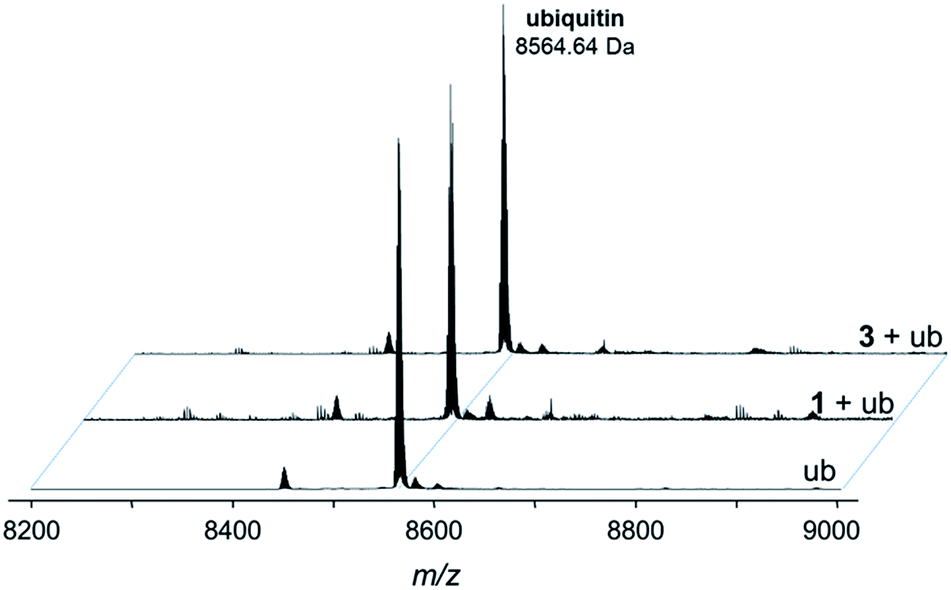 | ||
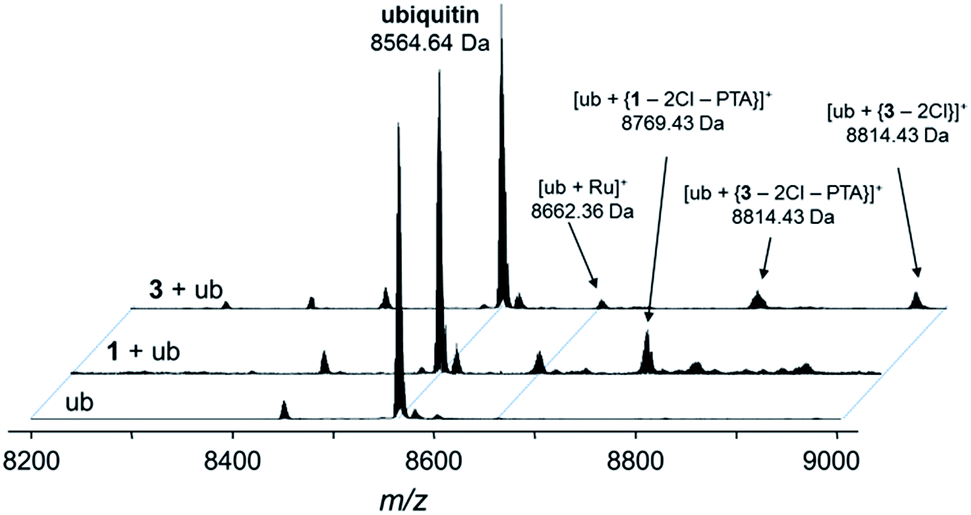
Fig. 4 Deconvoluted nESI-Q-TOF mass spectra of ubiquitin (8564.64 Da) and after incubation with 1 or 3 for 24 h. The mixture was incubated at a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 metal-to-protein ratio in tetramethylammonium acetate. :1 metal-to-protein ratio in tetramethylammonium acetate. | ||
 | ||
| Fig. 5 Deconvoluted nESI-Q-TOF mass spectra of ubiquitin (8564.64 Da) and after incubation with 1 or 3 for 48 h. The mixture was incubated at a 2:1 metal-to-protein ratio in water. | ||
The compounds form different low-abundance adducts, e.g. the [Ru(arene)(PTA)] fragment coordinated to ubiquitin in the case of 3 (ESI†). From these experiments, it is evident that the main form of interaction between the RAPTA derivatives and model proteins occurs via the substitution of the chlorido ligands. It should be noted that 1 contains a free amine that is charged at physiological pH, which is not the case for 2 or 3. The adduct types and abundances are similar independent of the arene substituent in case of ubiquitin (Fig. 4 and 5). Compound 1 containing the ammonium moiety on the arene is more reactive in the presence of the redox active cytochrome c and the adducts are mainly characterized by arene loss in slightly acidic milieu. Here, the relative abundance of the adducts are 36% and 24% for 1 and 3, respectively, in acidic milieu. In basic milieu, only 1 forms a monoadduct (22% relative abundance). It seems that the benzylammonium arene is slightly less stable than the acetylated analogue. However, the general reactivity of the RAPTA derivatives towards model proteins is therefore retained, although slightly lower compared to that of RAPTA-C.10
| Compound | IC50/μM | ||
|---|---|---|---|
| CH1 | SW480 | A549 | |
| 1 | 9.6 ± 1.2 | 358 ± 19 | >500 |
| 2 | 74 ± 6 | 216 ± 81 | >500 |
| 3 | 13 ± 1 | 357 ± 79 | >500 |
| RAPTA-C | 65 ± 15 | 170 ± 60 | >500 |
Drug pull-down experiments
Based on these findings, the complex was prepared in situ and incubated with streptavidin beads (30 min, 4 °C). The bead slurry was centrifuged and washed with lysis buffer. DMSO, which is typically used for the reaction of organic molecules in pull-down experiments with beads, turned out to be detrimental for the immobilization of the metallodrug and was replaced by DMF. Incubation of 1 and 3 in DMSO resulted in partial loss of the arene moiety as evidenced by ESI-MS and 1H NMR spectroscopy stability experiments (Fig. 6, S5 and S6†). Upon incubation of 1 in DMSO-d6 for more than 3 h, a new set of signals was detected in the 1H NMR spectrum in the aromatic region (around 7.4–7.5 ppm) indicating the release of the arene (Fig. S6†). After 24 h, the intensity of these signals significantly increased and a second set of peaks in the PTA region was detected. These observations were confirmed by ESI-ion trap MS. After 3 h the mass signals corresponding to 1 completely disappeared and a new signal at m/z 527.81 was detected, which may be assigned to [Ru(DMSO)3(OH)2(PTA) + H]+ (Fig. 6 and S5†). It should be noted that the p-cymene ring in RAPTA-C is considerably more stable under similar conditions.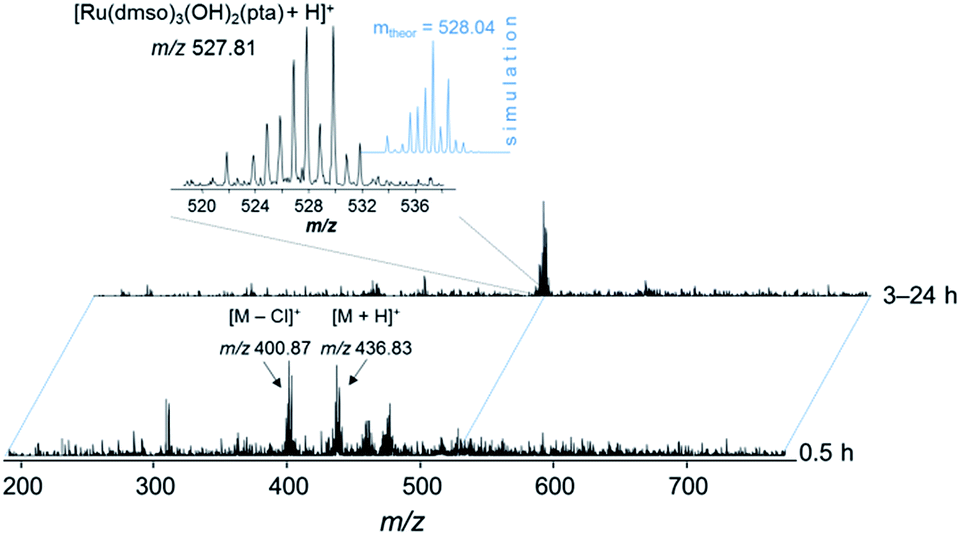 | ||
| Fig. 6 ESI-IT mass spectra of 1 in DMSO. The solution was diluted with water–methanol (1:1) prior to injection into the mass spectrometer. | ||
In DMF, however, the arene moiety of the RAPTA complex was sufficiently stable for at least 24 h (Fig. S7†). Therefore, the incubation of the probe was conducted in DMF (Fig. 2). Additionally, the behavior of complexes 1–3 in water (Fig. S8–S11†) was assayed by ESI-MS, which confirmed their stability during the washing steps of the drug pull-down experiments. However, even partial arene cleavage should not yield false positive data, as 3 as the competitive binder behaves very similarly to 2.
The incubation of the probe was followed by centrifugation, an additional washing step and subsequent incubation of the beads with CH1 lysates rather than treating live cells with the biotin-functionalized pharmacophore. This circumvents issues that may be arising by adding the biotin group to a small molecule and therefore changing the biological properties of the pharmacophore, including the uptake and distribution in the cell. This may lead to accumulation in cell compartments different to the original drug, which would impact the analysis. After centrifugation, the beads were washed with lysis buffer and HEPES, respectively. High chloride concentrations, as in the lysis buffer, are known to slow down any ligand exchange reactions of RAPTA compounds and should preserve the pharmacophore in the desired form.28,29 To elute bound proteins, beads were treated with elution buffer (50% urea, 50% formic acid). All eluates were separated by on-line HPLC and analyzed by MS. The acquired raw MS data files were converted into Mascot generic format (mgf) files and the resultant peak lists were searched against the human SwissProt database. Identified and validated proteins were merged and grouped according to shared peptides (see Experimental section).
Bioinformatic analysis of the datasets obtained from drug pull-down experiments resulted in the identification of a total of 184 proteins. To reduce the signal-to-noise ratio without reducing the sensitivity of the pull-down protocol, competition experiments with the acetylated RAPTA analogue 3, which mimics the conjugated complex 1, were included in the workflow. For competition experiments, cell lysates were pre-incubated with 3 before incubation with the affinity matrix. Complex 3 and immobilized 2 compete for the same set of proteins. When analyzed by MS, this results in a significantly reduced abundance of high-affinity binders and their interactors in the purified sample. When the two independent data sets are compared, the complete disappearance of the targets or a significant reduction of the MS spectral counts (i.e., the number of mass spectra recorded for a peptide) for the competitive pull-down, is suggestive of binding. This method is sensitive and reliable with the limitation that high-abundance proteins could also be targets of the drug.30 A threshold of at least 2-fold increase in spectral counts from competitive to regular pull-down was considered to be significant (Tables 2 and S3†) and 29 of 184 proteins (16%) successfully fulfilled this criterion. In general, the spectral counts for the enriched proteins were low compared to the overall counts and sequence coverage for those was less than 10% in most cases.
| Protein type | Gene | Name | Drug pull-down (data set 1)a | Competition experiment (data set 2)a |
|---|---|---|---|---|
| a The numbers indicate the spectral counts. b Decrease of spectral counts by a factor of 1.5. | ||||
| Extracellular growth factor | MK_HUMAN | Midkine | 6 | 2 |
| PTN_HUMAN | Pleiotrophin | 5 | 3b | |
| FGFP3_HUMAN | Fibroblast growth factor-binding protein 3 | 2 | 0 | |
| Cell cycle-regulating | GNL3_HUMAN | Guanine nucleotide-binding protein-like 3 | 2 | 0 |
| FA32A_HUMAN | Protein FAM32A | 4 | 0 | |
| VIR_HUMAN | Protein virilizer homolog | 3 | 0 | |
| CGBP1_HUMAN | CGG triplet repeat-binding protein 1 | 2 | 0 | |
| Histone-related | H31_HUMAN | Histone H3.1 | 4 | 1 |
| FBRL_HUMAN | rRNA 2′-O-methyltransferase fibrillarin | 2 | 0 | |
| CGBP1_HUMAN | CGG triplet repeat-binding protein 1 | 2 | 0 | |
| Ribosomal | RS20_HUMAN | 40S ribosomal protein S20 | 2 | 0 |
| RRP1B_HUMAN | Ribosomal RNA processing protein 1 homolog B | 4 | 1 | |
Pull-down data analysis
A significant amount of the 184 proteins identified in total comprised ribosomal proteins (18) and zinc finger proteins (16), which are “frequent hitters”, i.e. high-abundance proteins frequently observed in pull-down experiments. Notably, comparison of the dataset with that of the competition experiment eliminated most of them and only 5 ribosomal proteins and 2 zinc finger proteins were target proteins for RAPTA. Interestingly, several proteins identified in the MudPIT analysis by Wolters et al. (e.g., mitochondrial ATP synthase subunit O (ATPO), stress-70 protein (GRP75), myosin light chain 6B (MYL6B)) and heat shock proteins were also observed in our drug pull-down experiments.19 However, they were considered non-specific targets of RAPTA complexes, since the number of spectral counts did not decrease during competitive experiments. Out of the 29 enriched proteins, 15 proteins were found to be cancer-related (see Table 2). We analyzed the subcellular localization and biological function of these proteins (Table S4†). According to the UniProtKB database, these proteins are localized in the nucleus, cytoplasm, particulate fraction and extracellular region. Such widespread localization of proteins is in agreement with the results of subcellular fractionation experiments of the parent compound and indicates the dynamics of intracellular processes.19 These proteins may be specific targets of RAPTA complexes and may explain their in vitro antimetastatic properties as well as other effects such as the inhibition of angiogenesis.Summary and conclusions
Identification of cellular target proteins is a major challenge in drug development. With the exception of DNA targeting complexes the cellular targets of metal-based anticancer agents are widely unknown. Since RAPTA complexes are at the forefront of medicinal, organometallic chemistry, an understanding of their targets would facilitate further drug development. RAPTA compounds are able to interact with a wide range of different proteins, as expected from a relatively simple metal complex, undergoing ligand exchange reactions with nucleophilic amino acid donor atoms such as glutamate, lysine and histidine.12 This broad action is, nonetheless, important, as it may lead to a widespread modification of cellular proteins with effects on resistance and sensitizing phenomena. To learn more about the cellular targets of RAPTA compounds we developed a metallodrug pull-down assay to profile their molecular targets. For this purpose, a RAPTA-analogue suitable for immobilization on streptavidin beads was prepared by attaching biotin via a linker to the coordinated η6-arene. The immobilized metallodrug-analogue was exposed to cancer cell lysates, and the metal-binding proteins were identified by high resolution MS.Using this method 15 cancer-related proteins were identified which can be associated with the observed antimetastatic, antiangiogenic and antiproliferative activity of RAPTA agents. In particular, MK, PTN and FGF3 are angiogenesis and metastasis-related effectors. We were also able to identify proteins related to cell cycle regulation, i.e. GNL3, CGBP1, FAM32A and VIR. Some of the hits are proteins that have been proposed earlier as potential targets in complementary experiments, such as histone proteins, confirming the suitability of the approach. This methodology has broad applicability beyond RAPTA complexes and enables, for the first time, the direct identification of intracellular interactions of metallodrugs with proteins.
Contributions
Maria V. Babak performed the syntheses, Samuel M. Meier recorded the mass spectra, Kilian V. M. Huber conducted the pull-down experiment, Jóhannes Reynisson ran the docking experiments, Anton A. Legin and Michael A. Jakupec did the cell biological assays, Alexander Roller recorded the X-ray diffraction data, Alexey Stukalov, Manuela Gridling, Keiryn L. Bennett and Jacques Colinge collected the proteomics data, Walter Berger was involved in the cell biological assays, Paul J. Dyson contributed to the compound design, and Giulio Superti-Furga, Bernhard K. Keppler and Christian G. Hartinger designed the project. All authors wrote their respective parts of the paper and they were compiled by Maria V. Babak, Samuel M. Meier and Christian G. Hartinger.Abbreviations
Abbreviations of the proteins are listed in Table 2.| DMF | Dimethylformamide |
| DMSO | Dimethylsulfoxide |
| MTT | 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide |
| MudPIT | Multidimensional protein identification technology |
| PTA | 1,3,5-Triaza-7-phosphatricyclo-[3.3.1.1]decane |
| RAPTA-C | [Ru(η6-p-cymene)(PTA)Cl2 |
| RAPTA-T | [Ru(η6-toluene)(PTA)Cl2 |
| RP | Ribosomal proteins |
| SEC-ICP-MS | Size-exclusion chromatography inductively coupled plasma mass spectrometry |
Acknowledgements
This work was supported by the Platform Austria for Chemical Biology of the Genome Research Program Austria (GEN-AU, BMWF-70.081/0018-II/1a/2008), and COST CM1105. We gratefully acknowledge Prof. Vladimir Arion for the refinement of X-ray diffraction data and Prof. Markus Galanski for recording the 2D NMR spectra. We also thank the Mass Spectrometry Center, University of Vienna, for access to the nESI-Q-TOF MS instrument.Notes and references
- E. A. Hillard and G. Jaouen, Organometallics, 2011, 30, 20–27 CrossRef CAS.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed.
- G. Sava, G. Jaouen, E. A. Hillard and A. Bergamo, Dalton Trans., 2012, 41, 8226–8234 RSC.
- C. G. Hartinger, N. Metzler-Nolte and P. J. Dyson, Organometallics, 2012, 31, 5677–5685 CrossRef CAS.
- C. Scolaro, A. Bergamo, L. Brescacin, R. Delfino, M. Cocchietto, G. Laurenczy, T. J. Geldbach, G. Sava and P. J. Dyson, J. Med. Chem., 2005, 48, 4161–4171 CrossRef CAS PubMed.
- A. Bergamo, A. Masi, P. J. Dyson and G. Sava, Int. J. Oncol., 2008, 33, 1281–1289 CAS.
- A. Dorcier, P. J. Dyson, C. Gossens, U. Rothlisberger, R. Scopelliti and I. Tavernelli, Organometallics, 2005, 24, 2114–2123 CrossRef CAS.
- A. Dorcier, W. H. Ang, S. Bolano, L. Gonsalvi, L. Juillerat-Jeannerat, G. Laurenczy, M. Peruzzini, A. D. Phillips, F. Zanobini and P. J. Dyson, Organometallics, 2006, 25, 4090–4096 CrossRef CAS.
- C. Scolaro, A. B. Chaplin, C. G. Hartinger, A. Bergamo, M. Cocchietto, B. K. Keppler, G. Sava and P. J. Dyson, Dalton Trans., 2007, 5065–5072 RSC.
- A. Casini, G. Mastrobuoni, W. H. Ang, C. Gabbiani, G. Pieraccini, G. Moneti, P. J. Dyson and L. Messori, ChemMedChem, 2007, 2, 631–635 CrossRef CAS PubMed.
- A. Casini, C. Gabbiani, F. Sorrentino, M. P. Rigobello, A. Bindoli, T. J. Geldbach, A. Marrone, N. Re, C. G. Hartinger, P. J. Dyson and L. Messori, J. Med. Chem., 2008, 51, 6773–6781 CrossRef CAS PubMed.
- B. Wu, M. S. Ong, M. Groessl, Z. Adhireksan, C. G. Hartinger, P. J. Dyson and C. A. Davey, Chem.–Eur. J., 2011, 17, 3562–3566 CrossRef CAS PubMed.
- Z. Adhireksan, G. E. Davey, P. Campomanes, M. Groessl, C. M. Clavel, H. Yu, A. A. Nazarov, C. H. F. Yeo, W. H. Ang, P. Dröge, U. Rothlisberger, P. J. Dyson and C. A. Davey, Nat. Commun., 2014, 5 DOI:10.1038/ncomms4462.
- A. Casini, C. Gabbiani, E. Michelucci, G. Pieraccini, G. Moneti, P. J. Dyson and L. Messori, JBIC, J. Biol. Inorg. Chem., 2009, 14, 761–770 CrossRef CAS PubMed.
- A. Casini, A. Karotki, C. Gabbiani, F. Rugi, M. Vasak, L. Messori and P. J. Dyson, Metallomics, 2009, 1, 434–441 RSC.
- M. Groessl, M. Terenghi, A. Casini, L. Elviri, R. Lobinski and P. J. Dyson, J. Anal. At. Spectrom., 2010, 25, 305–313 RSC.
- F. Mendes, M. Groessl, A. A. Nazarov, Y. O. Tsybin, G. Sava, I. Santos, P. J. Dyson and A. Casini, J. Med. Chem., 2011, 54, 2196–2206 CrossRef CAS PubMed.
- M. Groessl and C. G. Hartinger, Anal. Bioanal. Chem., 2013, 405, 1791–1808 CrossRef CAS PubMed.
- D. A. Wolters, M. Stefanopoulou, P. J. Dyson and M. Groessl, Metallomics, 2012, 4, 1185–1196 RSC.
- F. Guidi, A. Modesti, I. Landini, S. Nobili, E. Mini, L. Bini, M. Puglia, A. Casini, P. J. Dyson, C. Gabbiani and L. Messori, J. Inorg. Biochem., 2013, 118, 94–99 CrossRef CAS PubMed.
- G. Superti-Furga, K. Huber and G. Winter, in Designing Multi-Target Drugs, ed. J. R. Morphy and C. J. Harris, The Royal Society of Chemistry, Cambridge, UK, 2012, vol. 21, ch. 7, pp. 94–110 Search PubMed.
- U. Rix, M. Gridling and G. Superti-Furga, Methods Mol. Biol., 2012, 803, 25–38 CAS.
- C. Letondor, A. Pordea, N. Humbert, A. Ivanova, S. Mazurek, M. Novic and T. R. Ward, J. Am. Chem. Soc., 2006, 128, 8320–8328 CrossRef CAS PubMed.
- C. Scolaro, T. J. Geldbach, S. Rochat, A. Dorcier, C. Gossens, A. Bergamo, M. Cocchietto, I. Tavernelli, G. Sava, U. Rothlisberger and P. J. Dyson, Organometallics, 2006, 25, 756–765 CrossRef CAS.
- C. S. Allardyce, P. J. Dyson, D. J. Ellis and S. L. Heath, Chem. Commun., 2001, 1396–1397 RSC.
- I. Le Trong, Z. Wang, D. E. Hyre, T. P. Lybrand, P. S. Stayton and R. E. Stenkamp, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 813–821 CrossRef CAS PubMed.
- M. Groessl, C. G. Hartinger, P. J. Dyson and B. K. Keppler, J. Inorg. Biochem., 2008, 102, 1060–1065 CrossRef CAS PubMed.
- B. Serli, E. Zangrando, T. Gianferrara, C. Scolaro, P. J. Dyson, A. Bergamo and E. Alessio, Eur. J. Inorg. Chem., 2005, 3423–3434 CrossRef CAS.
- C. Scolaro, C. G. Hartinger, C. S. Allardyce, B. K. Keppler and P. J. Dyson, J. Inorg. Biochem., 2008, 102, 1743–1748 CrossRef CAS PubMed.
- P. Fadden, K. H. Huang, J. M. Veal, P. M. Steed, A. F. Barabasz, B. Foley, M. Hu, J. M. Partridge, J. Rice, A. Scott, L. G. Dubois, T. A. Freed, S. M. A. Rehder, T. E. Barta, P. F. Hughes, A. Ommen, W. Ma, E. D. Smith, S. A. Woodward, J. Eaves, G. J. Hanson, L. Hinkley, M. Jenks, M. Lewis, J. Otto, G. J. Pronk, K. Verleysen, T. A. Haystead and S. E. Hall, Chem. Biol., 2010, 17, 686–694 CrossRef CAS PubMed.
- A. Bergamo, A. Masi, P. J. Dyson and G. Sava, Int. J. Oncol., 2008, 33, 1281–1289 CAS.
- T. Muramatsu, Proc. Jpn. Acad., Ser. B, 2010, 86, 410–425 CrossRef CAS.
- T. Muramatsu, J. Biochem., 2002, 132, 359–371 CrossRef CAS.
- P. Tao, D. Xu, S. Lin, G.-L. Ouyang, Y. Chang, Q. Chen, Y. Yuan, X. Zhuo, Q. Luo, J. Li, B. Li, L. Ruan, Q. Li and Z. Li, Cancer Lett., 2007, 253, 60–67 CrossRef CAS PubMed.
- T. Muramatsu, J. Biochem., 2002, 132, 359–371 CrossRef CAS.
- P. Nowak-Sliwinska, B. J. R. van, A. Casini, A. A. Nazarov, G. Wagnieres, D. B. H. van, P. J. Dyson and A. W. Griffioen, J. Med. Chem., 2011, 54, 3895–3902 CrossRef CAS PubMed.
- W. Zhang, Y. Chen, M. R. Swift, E. Tassi, D. C. Stylianou, K. A. Gibby, A. T. Riegel and A. Wellstein, J. Biol. Chem., 2008, 283, 28329–28337 CrossRef CAS PubMed.
- C. Han, X. Zhang, W. Xu, W. Wang, H. Qian and Y. Chen, Int. J. Mol. Med., 2005, 16, 205–213 CAS.
- S. Chatterjee, S. Kundu, A. Bhattacharyya, C. G. Hartinger and P. J. Dyson, JBIC, J. Biol. Inorg. Chem., 2008, 13, 1149–1155 CrossRef CAS PubMed.
- K. Horiuchi, T. Kawamura, H. Iwanari, R. Ohashi, M. Naito, T. Kodama and T. Hamakubo, J. Biol. Chem., 2013, 288, 33292–33302 CrossRef CAS PubMed.
- U. Singh, P. Roswall, L. Uhrbom and B. Westermark, BMC Mol. Biol., 2011, 12, 28 CrossRef CAS PubMed.
- U. Singh and B. Westermark, Exp. Cell Res., 2011, 317, 143–150 CrossRef CAS PubMed.
- U. Singh, V. Maturi, R. E. Jones, Y. Paulsson, D. M. Baird and B. Westermark, Cell Cycle, 2014, 13, 96–105 CrossRef CAS PubMed.
- X. Chen, H. Zhang, J. P. Aravindakshan, W. H. Gotlieb and M. R. Sairam, Oncogene, 2011, 30, 2874–2887 CrossRef CAS PubMed.
- B. Wu, M. S. Ong, M. Groessl, Z. Adhireksan, C. G. Hartinger, P. J. Dyson and C. A. Davey, Chem.–Eur. J., 2011, 17, 3562–3566 CrossRef CAS PubMed.
- P. Gomulak and J. Blasiak, Postepy Biochem., 2012, 58, 292–301 CAS.
- M.-D. Lai and J. Xu, Curr. Genomics, 2007, 8, 43–49 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, crystallographic, biological and mass spectrometric data, full list of proteins detected by chemical proteomics. CCDC 972667 and 972668. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc03905j |
| This journal is © The Royal Society of Chemistry 2015 |