
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A rationally designed small molecule for identifying an in vivo link between metal–amyloid-β complexes and the pathogenesis of Alzheimer's disease†
Michael W.
Beck‡
ab,
Shin Bi
Oh‡
c,
Richard A.
Kerr
b,
Hyuck Jin
Lee
ab,
So Hee
Kim
c,
Sujeong
Kim
c,
Milim
Jang
a,
Brandon T.
Ruotolo
*b,
Joo-Yong
Lee
*cd and
Mi Hee
Lim
*ae
aDepartment of Chemistry, Ulsan National Institute of Science and Technology (UNIST), Ulsan 689-798, Republic of Korea. E-mail: mhlim@unist.ac.kr
bDepartment of Chemistry, University of Michigan, Ann Arbor, MI 48109-1055, USA. E-mail: bruotolo@umich.edu
cAsan Institute for Life Sciences, Asan Medical Center, Seoul 138-736, Republic of Korea. E-mail: jlee@amc.seoul.kr
dDepartment of Neurology, University of Ulsan College of Medicine, Seoul 138-736, Republic of Korea
eLife Sciences Institute, University of Michigan, Ann Arbor, Michigan 48109-2216, USA
First published on 27th January 2015
Abstract
Multiple factors, including amyloid-β (Aβ), metals, and reactive oxygen species (ROS), are involved in the development of Alzheimer's disease (AD). Metal ions can interact with Aβ species generating toxic oligomers and ROS in vitro; however, the involvement of metal–Aβ complexes in AD pathology in vivo remains unclear. To solve this uncertainty, we have developed a chemical tool (L2-b) that specifically targets metal–Aβ complexes and modulates their reactivity (i.e., metal–Aβ aggregation, toxic oligomer formation, and ROS production). Through the studies presented herein, we demonstrate that L2-b is able to specifically interact with metal–Aβ complexes over metal-free Aβ analogues, redirect metal–Aβ aggregation into off-pathway, nontoxic less structured Aβ aggregates, and diminish metal–Aβ-induced ROS production, overall mitigating metal–Aβ-triggered toxicity, confirmed by multidisciplinary approaches. L2-b is also verified to enter the brain in vivo with relative metabolic stability. Most importantly, upon treatment of 5XFAD AD mice with L2-b, (i) metal–Aβ complexes are targeted and modulated in the brain; (ii) amyloid pathology is reduced; and (iii) cognition deficits are significantly improved. To the best of our knowledge, by employing an in vivo chemical tool specifically prepared for investigating metal–Aβ complexes, we report for the first time experimental evidence that metal–Aβ complexes are related directly to AD pathogenesis.
Introduction
Alzheimer's disease (AD), a progressive neurodegenerative disease, is the most common form of dementia afflicting 24 million people worldwide.1 Despite AD being the sixth leading cause of death in the United States, there are currently no disease modifying treatments; approved therapies only offer symptomatic relief without having an effect on the underlying pathogenesis.1,2 Development of effective therapeutics has been hindered by the fact that AD pathogenesis is still poorly understood. Pathologically, AD is characterized by the accumulation of aggregated, misfolded proteins, such as amyloid-β (Aβ) peptides (two major forms exist, Aβ40 and Aβ42).3,4 The amyloid cascade hypothesis suggests that Aβ is the causative agent in AD;5 however, the etiology of AD can be multifactorial; of particular interest is the role of Aβ with other factors (i.e., metals) toward AD development.4,6–11High concentrations of Fe, Cu, and Zn (ca. low mM) are found within Aβ deposits in ex vivo tissues from the AD-afflicted brain.12,13 These metal ions are observed to coordinate to Aβ peptides in vitro forming metal–Aβ complexes which could direct toxicity via two possible pathways:4,6–11,14–19 (i) metals could influence the Aβ aggregation pathways leading to the generation and stabilization of toxic Aβ oligomers;4,7–9,14 (ii) redox active metal ions (i.e., Cu(I/II) and Fe(II/III)) associated with Aβ are shown to produce reactive oxygen species (ROS) under physiological conditions through Fenton-like reactions.4,6–11,16–19 Overproduction of ROS by metal–Aβ can result in oxidative stress and eventually neuronal death in the AD-affected brain. Although the reactivity of metal–Aβ (i.e., (i) metal–Aβ aggregation (toxic Aβ oligomer formation) and (ii) redox active metal–Aβ-triggered ROS generation, vide supra) has been indicated in vitro,4,6–11,14–19 the direct involvement of metal–Aβ complexes in AD pathogenesis in vivo is uncertain.
Metal chelating agents have shown that the interference of metal–Aβ interactions as well as the modulation of metal distribution in the brain could lead to an improvement in AD pathology.4,19–24 8-Hydroxyquinoline derivatives have been employed to regulate metal-related neurotoxicity in AD; some small molecules, including clioquinol (CQ) and PBT2, have indicated promising results for possible AD treatment in clinical trials.4,22,23 The effects of CQ and PBT2 are mainly from their ability to act as an ionophore to redistribute metal ions in the brain instead of directly disrupting metal–Aβ complexes;4,19,23–25 thus, these compounds would not be able to directly probe the relation between metal–Aβ complexes and AD pathogenesis. Therefore, chemical tools, termed as metamorphosizers, have been recently developed in order to (i) specifically target metal–Aβ complexes and (ii) alter the interaction between the metal and Aβ, consequently (iii) redirecting the toxic aggregation pathway of metal–Aβ into off-pathway, less toxic unstructured Aβ forms and (iv) reducing metal–Aβ-induced ROS production, which eventually alleviates metal–Aβ-linked toxicity.4,24
Herein, we demonstrate that a chemical tool (L2-b, Fig. 1a) stands out as being well suited in vivo for identifying the association of metal–Aβ40/Aβ42 with AD pathogenesis, through in vitro biochemical/biophysical/cytotoxicity/metabolism investigations, as well as in vivo brain uptake studies. Our in vivo tool specifically interacts with metal–Aβ over metal-free Aβ and generates a ternary L2-b–metal–Aβ complex causing structural compaction, as validated by mass spectrometry (MS) and ion mobility-mass spectrometry (IM-MS). Most significantly, we present the first report that the control of metal–Aβ interaction and reactivity by an in vivo chemical tool mitigates amyloid pathology and improves cognitive deficits in the 5XFAD AD mouse model. This robust AD mouse model develops severe amyloid pathology and cognitive decline at an early age through high expression of three familial mutant types of human amyloid precursor protein (hAPP; Swedish, Florida, and London) and two mutant forms of presenilin (PSEN1; M146L and L286V).26 Overall, our studies establish strong experimental evidence for an in vivo link between metal–Aβ and AD development, implying that targeting metal–Aβ complexes could be an effective strategy for the future development of new therapeutics.
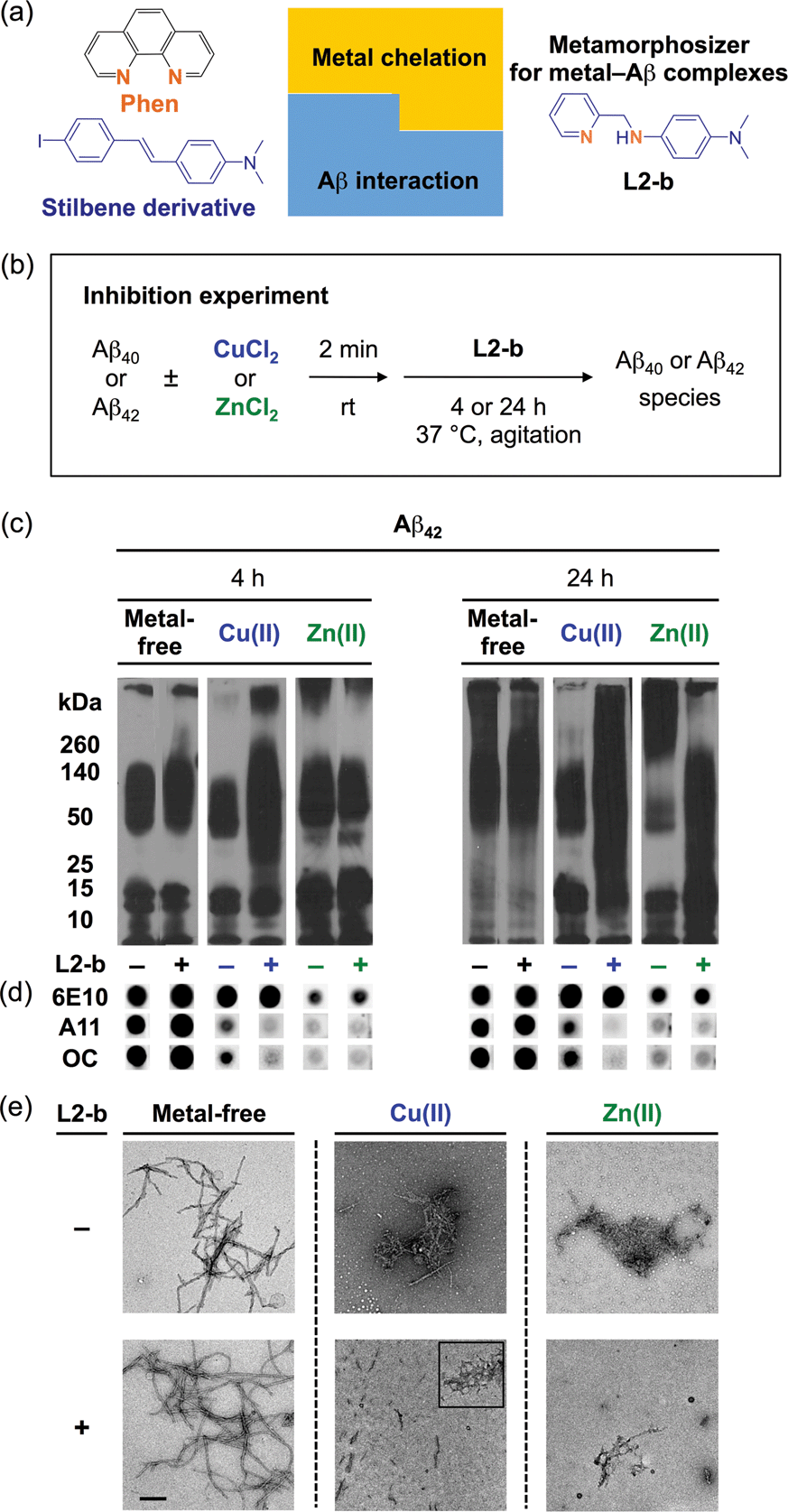 | ||
| Fig. 1 Design principle of L2-b and its effect on metal-free and metal-induced Aβ aggregation. (a) Design principle of L2-b, a metamorphosizer for metal–Aβ complexes: a metal binding site (orange) is incorporated into an Aβ interacting framework (blue). (b) Scheme showing the inhibition experiment: metal-free or metal-treated [CuCl2 (blue) or ZnCl2 (green)] Aβ40/Aβ42 was incubated with (+) or without (−) L2-b for 4 h (left) and 24 h (right). Conditions: [Aβ] = 25 μM; [Cu(II) or Zn(II)] = 25 μM; [L2-b] = 50 μM; pH 6.6 (for Cu(II) samples) or pH 7.4 (for metal-free and Zn(II) samples); 37 °C; constant agitation. (c) Analysis of the size distribution of the resultant Aβ42 species by gel electrophoresis and Western blotting with an anti-Aβ antibody (6E10). (d) Dot blot analysis of the resulting Aβ42 species employing 6E10, an anti-Aβ oligomer antibody (A11), and an anti-Aβ fibril antibody (OC). (e) TEM images of the 24 h incubated samples (scale bar = 200 nm). | ||
Results and discussion
Design principle and characterization of a chemical tool for investigating metal–Aβ complexes in vivo
L2-b (Fig. 1a) was designed to target metal–Aβ complexes and modulate their interaction/reactivity with subsequent reduction of toxicity,27 in order to determine whether they are connected with AD pathology. For in vivo applications, first, chemical tools for this purpose must have specificity toward metal–Aβ complexes in order to limit the disruption of other metalloproteins.4,24 This property can be imparted into small molecules by using inorganic chemistry concepts to allow specificity for disease-relevant metal ions (Fe(II/III), Cu(I/II), and Zn(II)), along with limiting the metal binding affinity (Kd) to ≥10−10 M, and by including structural components for Aβ interaction.4,24 To satisfy this aspect, L2-b (a bidentate ligand; Fig. 1a) was constructed upon incorporation of two nitrogen donor atoms (for metal chelation) into the structure of an Aβ aggregate imaging agent (stilbene derivative; for Aβ interaction),28 which could interact with metal–Aβ complexes (Fig. 1a).27L2-b is shown to have apparent Kd values of ca. 10−10 and 10−6 M for Cu(II) and Zn(II), respectively, and is relatively selective for Cu(II) over other biologically relevant bivalent ions.27 Secondly, the blood–brain barrier (BBB) permeability of L2-b is critical for applications in the brain, which was first predicted by considering Lipinski's rules of drug-likeness and observing calculated log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) BB values.27 Employing CD1 mice, in vivo brain uptake studies of L2-b newly confirmed its BBB penetration. L2-b (ca. 250 ng g−1) is observed to be available in the brain when administered by oral gavage (10 mg kg−1) to the mice (Table S1†). Thirdly, the metabolic stability of L2-b for in vivo applications was also verified utilizing human liver microsomes. Susceptibility of L2-b to metabolism is between 30 min and 120 min indicating that this compound has moderate metabolic stability, suggesting its suitability for use in vivo. Lastly, L2-b acts as an antioxidant as well as an inhibitor of Cu(I/II)– or Cu(I/II)–Aβ-induced ROS production as presented in previous studies.27,29 From our newly performed study using the Trolox equivalent antioxidant capacity in a cellular environment (i.e., murine neuroblastoma Neuro-2a (N2a) cell lysates),30L2-b exhibits a greater free radical scavenging capacity (2.3 ± 0.2) than Trolox (1.0 ± 0.1), a known antioxidant vitamin E analogue. Therefore, L2-b is clearly demonstrated to be viable for in vivo use as a chemical tool for exploring the association of metal–Aβ complexes with AD pathogenesis.
BB values.27 Employing CD1 mice, in vivo brain uptake studies of L2-b newly confirmed its BBB penetration. L2-b (ca. 250 ng g−1) is observed to be available in the brain when administered by oral gavage (10 mg kg−1) to the mice (Table S1†). Thirdly, the metabolic stability of L2-b for in vivo applications was also verified utilizing human liver microsomes. Susceptibility of L2-b to metabolism is between 30 min and 120 min indicating that this compound has moderate metabolic stability, suggesting its suitability for use in vivo. Lastly, L2-b acts as an antioxidant as well as an inhibitor of Cu(I/II)– or Cu(I/II)–Aβ-induced ROS production as presented in previous studies.27,29 From our newly performed study using the Trolox equivalent antioxidant capacity in a cellular environment (i.e., murine neuroblastoma Neuro-2a (N2a) cell lysates),30L2-b exhibits a greater free radical scavenging capacity (2.3 ± 0.2) than Trolox (1.0 ± 0.1), a known antioxidant vitamin E analogue. Therefore, L2-b is clearly demonstrated to be viable for in vivo use as a chemical tool for exploring the association of metal–Aβ complexes with AD pathogenesis.
Specific modulation of metal-induced over metal-free Aβ aggregation pathways in vitro
To elucidate whether L2-b could redirect metal–Aβ aggregation into off-pathway amorphous Aβ aggregates, suggested to be less toxic or nontoxic,31 while leaving metal-free Aβ cases unaffected, inhibition (Fig. 1b) and disaggregation (Fig. S1a†) experiments30 were performed employing Aβ40 and Aβ42, the two main Aβ forms found in the AD-affected brain. The influence of L2-b on both metal-free and metal-mediated Aβ aggregation was monitored at short and long incubation time points.32 Gel electrophoresis and Western blotting (gel/Western blot, utilizing an anti-Aβ antibody, 6E10)30 were conducted to determine the molecular weight (MW) distribution of the resulting Aβ aggregates. Dot blot analysis with an anti-Aβ oligomer antibody A1133 and an anti-Aβ fibril antibody OC,34 along with 6E10, was carried out to identify the type of Aβ species produced. Moreover, transmission electron microscopy (TEM) images were taken to visualize the morphologies of the resultant Aβ aggregates.30
Both the inhibition and disaggregation experiments indicate that L2-b does not modulate the aggregation pathways of both Aβ40 and Aβ42 under metal-free conditions after either short or long incubation periods. Nearly identical MW distributions of the Aβ species in the absence and presence of L2-b were observed in the gel/Western blots (Fig. 1c, S1b, and S2a†). The dot blots of the inhibition samples indicated A11 (oligomer)- and OC (fibril)-positive aggregates for metal-free Aβ40/Aβ42 even when treated with L2-b (Fig. 1d and S2b†). TEM images revealed that Aβ fibrils were mainly present in both the inhibition and disaggregation experiments of metal-free Aβ40/Aβ42 with and without L2-b after 24 h of incubation (Fig. 1e, S1c, and S2c†). Thus, metal-free Aβ aggregation is not noticeably influenced upon treatment with L2-b.
In contrast to the metal-free conditions, significantly noticeable changes in the metal [Cu(II) or Zn(II)]-induced Aβ40 and Aβ42 aggregation pathways by L2-b were observed compared to L2-b-untreated analogues. In both the inhibition and disaggregation experiments, after 24 h of incubation of the Cu(II)–Aβ species with L2-b, the resulting peptide species with a wide range of MWs were visualized by gel/Western blot (Fig. 1c, S1b, and S2a†). In the inhibition studies of both Aβ40 and Aβ42, as well as in the disaggregation experiment of Aβ42, Cu(II)–Aβ samples treated with L2-b even for 4 h also exhibited the distinct MW distribution of Aβ (Fig. 1c, S1b and S2a†). Distinguishably, L2-b was capable of limiting the formation of A11- and OC-positive Cu(II)-induced Aβ40/Aβ42 aggregates at both short and longer incubation times (Fig. 1d and S2b†). Morphologies of L2-b-incubated Cu(II)–Aβ, analyzed by TEM, displayed both narrower and shorter fibrils, as well as unstructured Aβ aggregates in the inhibition experiments (Fig. 1e and S2c†); while less dense, thinner fibrils were mainly observed in the disaggregation experiments (Fig. S1c†). In the case of Zn(II)–Aβ, L2-b could also transform the aggregation pathways (Fig. 1c, S1b, and S2a†). The TEM studies revealed L2-b-triggered, smaller amorphous Zn(II)–Aβ aggregates in both the inhibition and disaggregation experiments (Fig. 1e, S1c, and S2c†). Overall, L2-b is observed to redirect metal–Aβ aggregation mainly into unstructured Aβ aggregates that are generated via the off-pathway aggregation and are known to be less toxic or nontoxic.31 Thus, L2-b could be used as a chemical tool specific for such anti-amyloidogenic activity toward metal–Aβ complexes over metal-free Aβ in this manner.
Formation of structurally-compact complexes with metal–Aβ not metal-free Aβ in vitro
In order to explore the specific interaction of L2-b with metal–Aβ over metal-free Aβ, nanoelectrospray ionization-MS (nESI-MS) studies were employed (Fig. 2a). When metal-free Aβ40 was allowed to react with L2-b, no binding events were observed, even with a six fold excess of the ligand (Fig. 2a(ii)). In comparison, incubating a comparatively smaller concentration of L2-b with Aβ40 and Cu(II) promoted readily observed levels of complexes containing Aβ40, Cu(II), and L2-b approximately in the ratio 1:2:1, supporting the metal specific nature of the interaction (Fig. 2a(iv)). The formation of a ternary complex between L2-b and Cu(II)–Aβ40 is supported by the previously reported NMR studies of L2-b with Zn(II)–Aβ40 in solution.27
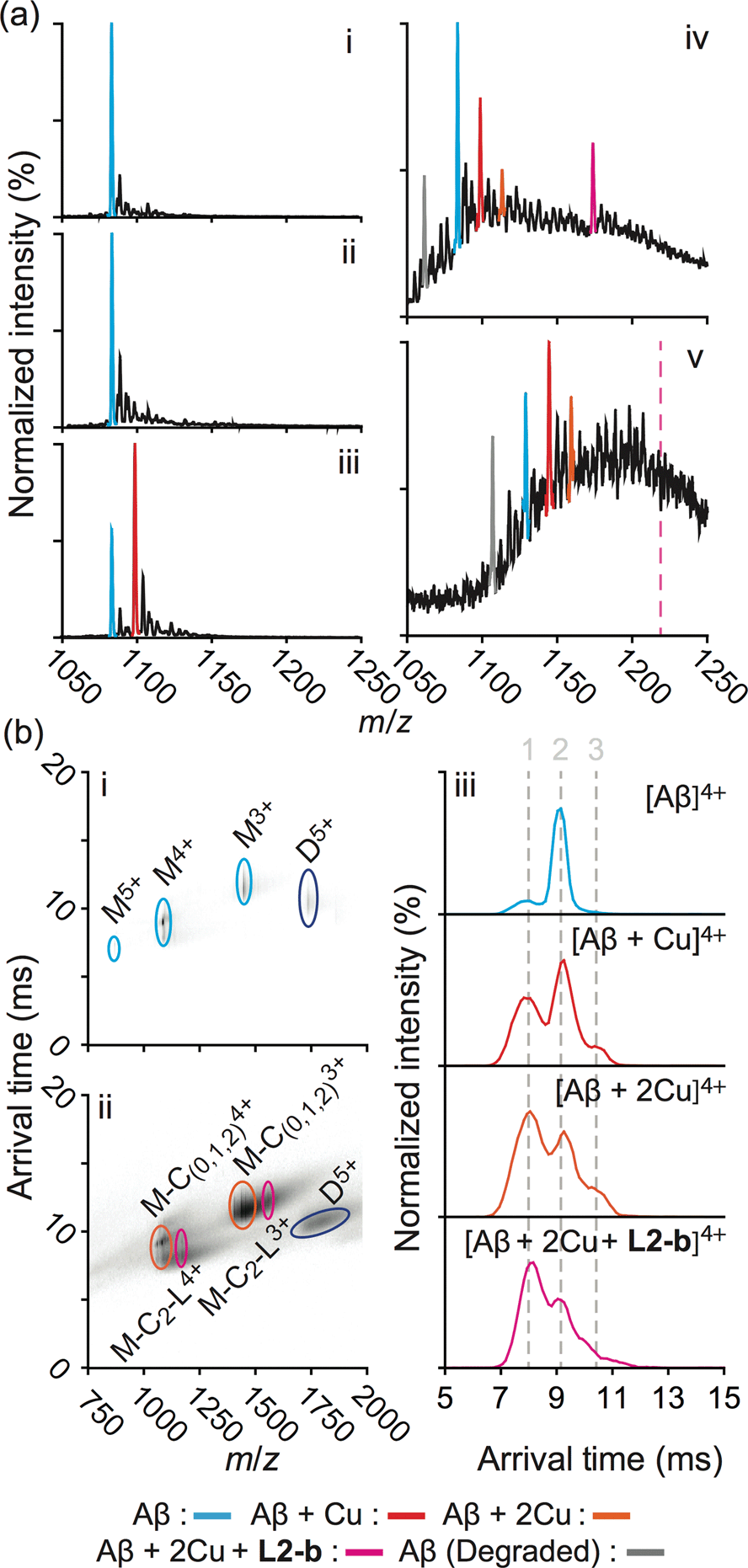 | ||
| Fig. 2 Mass spectrometric (MS) and ion mobility-mass spectrometric (IM-MS) analyses of Aβ in the presence of L2-b and/or Cu(II). (a) Comparison of incubated Aβ 4+ charge states in the samples containing (i) Aβ40 (18 μM) alone and Aβ40 co-incubated with (ii) excess L2-b (120 μM), (iii) Cu(II) (40 μM), and (iv) both L2-b (40 μM) and Cu(II) (40 μM) [(v) Aβ42 (pH 9, 18 μM) with L2-b (40 μM) and Cu(II) (40 μM) is also presented]. Consistent with data shown here, the gray signal represents a currently unidentified chemical modification of the N-terminus up to, and including, residue 5 (Fig. S3†) but not F4 (Fig. S4†). The projected location of the complex consisting of Aβ42, Cu(II), and L2-b in a ratio of 1:2:1 is indicated in pink. (b) IM-MS analysis of Aβ40 (18 μM) incubated in the (i) absence and (ii) presence of L2-b (40 μM) and Cu(II) (40 μM). Extracted arrival time distributions support the existence of three resolvable structural populations (Collision Cross Section (CCS) data, Table S2†). | ||
Additionally, another MS signal was observed. This signal corresponds to an intact molecular mass of 89.24 Da less than the full-length Aβ40 peptide in good agreement with ternary Aβ40–Cu(II)–L2-b complex formation (gray, Fig. 2a(iv)). Tandem MS data (Fig. S3†) and subsequent analysis of the fragment ions indicate that this new signal corresponds to a chemical modification within the first five residues of Aβ40 (D1A2E3F4R5). Given a mass measurement error of ±1 Da, and supporting L2-b binding experiments performed using an Aβ40 F4A sequence variant, as well as acetylated analogs (Fig. S4†), we can eliminate alterations to F4 as a source of the product observed and show that free primary amines are critical for binding and subsequent Aβ degradation. While no direct observations of the Cu(II)–L2-b-bound Aβ42 form were indicated by MS, the 89.24 Da mass loss product was detected (gray, Fig. 2a(v)) upon addition of both L2-b and Cu(II) to the samples, implying the generation of a transient ternary Aβ42–Cu(II)–L2-b complex of unknown stoichiometry. These Aβ40/Aβ42 fragmentation results also suggest that, as expected, Cu(II) likely binds to Aβ proximal to the site of L2-b attachment.4,7 In all cases, neither L2-b nor Cu(II) was detected in complex with the identified Aβ degradation product. Detailed structures of these ternary complexes will be the subject of future studies.
To study the molecular level structural dynamics by which L2-b redirects metal–Aβ aggregation pathways, IM-MS experiments of the complexes produced were performed. A comparison of the arrival time distributions of the metal-free Aβ40 form with the different ligated states supports an increasing level of structural compaction as additional components (i.e., Cu(II) and L2-b) associate with Aβ40 (Fig. 2b and Table S2†). Analyzing the arrival time distributions for all complex states presented, along with the nESI-MS data, our IM-MS investigations demonstrate that L2-b is capable of specifically interacting with Cu(II)-bound Aβ over metal-free Aβ, subsequently promoting a high level of structural compaction of the complex. This binding of L2-b to metal–Aβ with increased structural compactness could be a key property for the distinguishable reorganization of metal–Aβ aggregation pathways, similar to the previously suggested molecular level mode of action of EGCG toward metal–Aβ complexes which promotes the generation of nontoxic unstructured aggregates via off-pathway aggregation.24,31
Targeting and reacting with metal–Aβ complexes in living cells and in the brain of 5XFAD AD mice
The effect of L2-b on metal–Aβ40/Aβ42-induced toxicity was first examined using N2a cells, as an indication of its interaction with metal–Aβ complexes. An increase (ca. 10–20%) in cell viability for both Aβ40 and Aβ42 was displayed upon treatment of cells incubated with Cu(II) or Zn(II), Aβ, and L2-b (10 μM each; Fig. S5†). Moving forward, the ability of L2-b to penetrate the BBB and interact with metal–Aβ species in the brain was verified in the 5XFAD AD mouse model. Zn(II) found in Aβ plaques was visualized in the brain tissue slices by a fluorophore specific for Zn(II), 6-methoxy-(8-p-toluenesulfonamido)quinolone (TSQ; Fig. 3).35 Administration of L2-b to 5XFAD AD mice intraperitoneally for three weeks on a daily basis starting at the age of three months resulted in drastically diminished fluorescence of TSQ in the plaques (arrows shown in Fig. 3c). Additionally, in the hippocampal mossy fiber terminals, a Zn(II)-rich region in the brain,36 there was no difference in the fluorescence of TSQ between wild type and 5XFAD AD mice treated daily with the vehicle, whereas L2-b reduced fluorescence by ca. 13% (P < 0.05) in 5XFAD AD mice over the same time span (Fig. 3d). Thus, these in vivo studies suggest that L2-b is BBB permeable and can enter the brain to interact with intracerebral metals, including those found in Aβ plaques.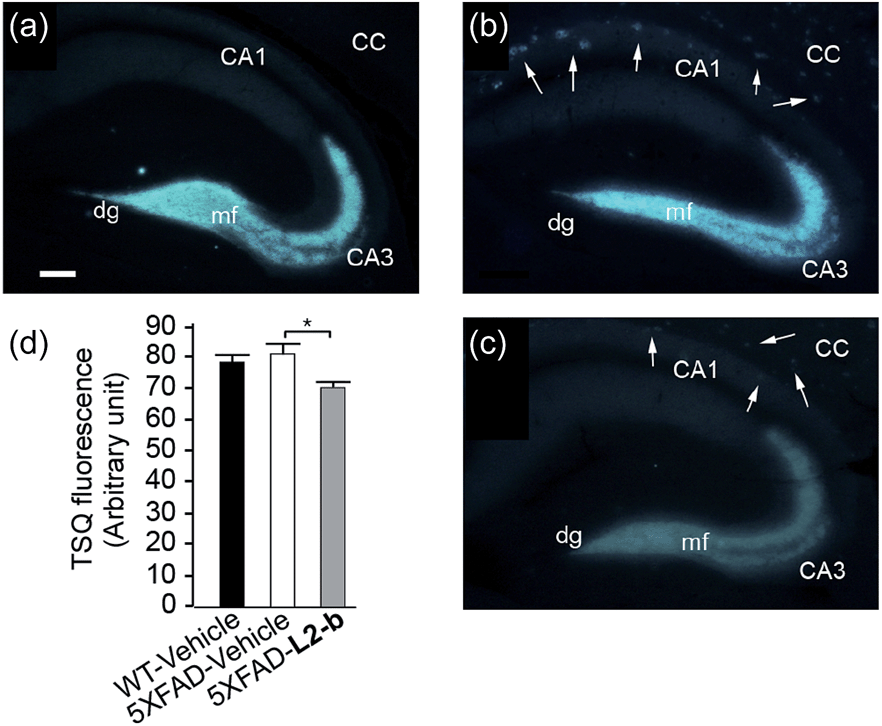 | ||
| Fig. 3 Levels of Zn(II) in the brain tissues of nontransgenic wild type (WT) and 5XFAD transgenic mice. Amounts of Zn(II) were determined using a fluorescent dye, 6-methoxy-(8-p-toluenesulfonamido)quinoline (TSQ), in the mossy fiber region (mf) of brains from (a) WT and (b and c) 5XFAD transgenic mice after intraperitoneal administration of (a and b) the vehicle or (c) L2-b (1 mg per kg per day) for three weeks beginning at three months of age (scale bar = 100 μm). The fluorescence response of TSQ was also shown in the zone of amyloid plaques in 5XFAD mice (shown by arrows; b and c). CC, corpus callosum; CA, cornu amonis; dg, dentate gyrus. (d) The fluorescence intensity of TSQ was quantified in the mossy fiber region (mf in a–c) of vehicle-treated WT (black bar; n = 6) and 5XFAD male mice (white bar; n = 10), or L2-b-treated 5XFAD male mice (gray bar; n = 9), where the measurement was performed using five sagittal sections selected randomly from each animal and denoted as an arbitrary unit of the TSQ fluorescence [mean ± standard error of the mean (S.E.M)]. *P < 0.05 by one-way analysis of variance (ANOVA). | ||
Reduction of amyloid pathology in 5XFAD AD mice
To identify the direct involvement of metal–Aβ complexes in amyloid pathology leading to improved cognition, the 5XFAD mouse model of AD26 was chosen. L2-b (1 mg kg−1) was injected into nontransgenic littermates (wild type) and 5XFAD AD mice via the intraperitoneal route for three weeks on a daily basis starting at the age of three months. All mice survived the consecutive treatments, which rarely caused changes in body weight (Table S3†). Necropsy of all major organs in L2-b-treated mice revealed no gross changes.The potential association of L2-b with amyloid pathology was investigated by first observing the amyloid plaque load in the brain tissue of 5XFAD AD mice. When the brain tissue slices of L2-b-administered 5XFAD AD mice were stained with an APP/Aβ-specific antibody (4G8) or a compact core amyloid plaque indicator (Congo red), it was found that the amyloid plaque burden was ameliorated (Fig. 4). Reduction (ca. 15%) of both the area of 4G8-immunoreactive deposits and the number of congophilic amyloid plaques was revealed in the cortex of L2-b-treated 5XFAD AD mice when compared to vehicle-treated 5XFAD AD mice (Fig. 4c and f). The changes in the amount of both Aβ40 and Aβ42 in the brain tissues of 5XFAD AD mice following L2-b administration were also assessed. Total amounts of Aβ peptides were analyzed by an enzyme-linked immunosorbent assay (ELISA) in sodium dodecyl sulfate (SDS)- and formic acid (FA)-soluble brain tissue lysates (Fig. 5a and b and S6†), as well as oligomeric and fibrillar Aβ aggregates in the phosphate buffered saline (PBS)-soluble fraction (Fig. 5c and d).37 Relative to vehicle-treated 5XFAD mice, the L2-b-treated 5XFAD mice showed diminished cerebral levels of both Aβ40 and Aβ42 in all fractions (ca. 15–20%, P < 0.05, Fig. 5a and b). Oligomeric and fibrillar Aβ species in the PBS fraction were additionally abated by 27% and 15%, respectively (P < 0.05, Fig. 5c and d). Similarly, the overall reduction of Aβ species was also indicated by gel/Western blot, where Aβ monomers and oligomers were noticeably decreased in brain tissue lysates from L2-b-treated 5XFAD AD mice (Fig. 5e). Together, these studies demonstrate that daily administration of L2-b to the AD model mitigates amyloid pathology in AD, including the load of amyloid plaque deposits and the levels of a wide range of conformations from monomers to fibrils.
 | ||
| Fig. 4 Effect of daily treatments with L2-b for three weeks on the amyloid deposits in the brains of 5XFAD male mice. Representative microscopic images of (a and b) 4G8-immunostained or (d and e) Congo red stained brain sections of 5XFAD mice, which were given daily (a and d) the vehicle or (b and e) L2-b (1 mg per kg per day) via intraperitoneal injection for three weeks starting at three months of age (magnification = 40×; scale bar = 100 μm). Inset in (d) and (e): enlarged micrographs of congophilic amyloid plaques in the cortical area (magnification, 400×; hip, hippocampus; ctx, cortex). To evaluate the amyloid pathology of the vehicle (black bars; n = 5)- or L2-b (gray bars; n = 7)-treated male 5XFAD mice, (c) the load of 4G8-immunoreactive amyloid deposits and (f) the number of congophilic amyloid plaques in the cortex were measured in five brain sections taken from each animal. *P < 0.05 by one-way ANOVA. | ||
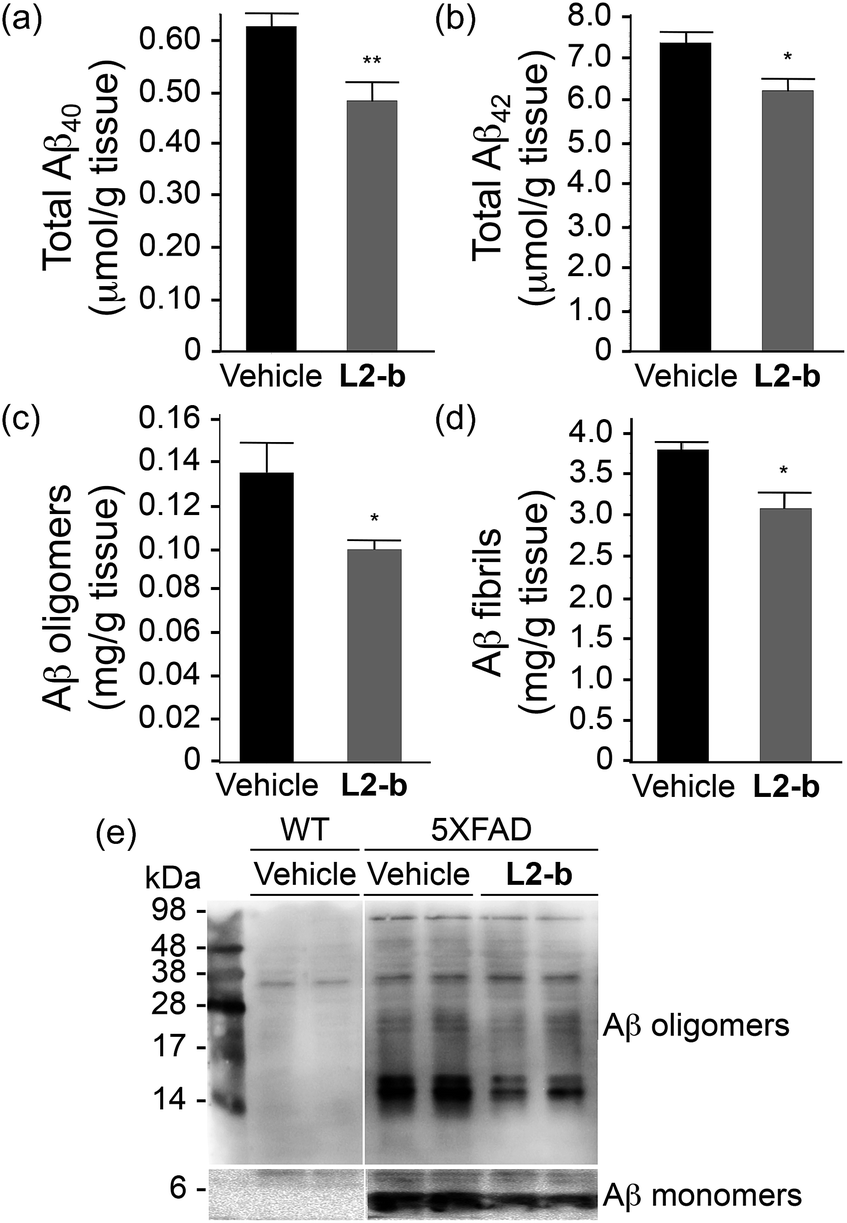 | ||
| Fig. 5 Levels of Aβ in whole brain tissues of three-month-old male 5XFAD mice. The amounts of (a) total Aβ40, (b) total Aβ42, (c) PBS-soluble Aβ oligomers, and (d) Aβ fibrils were assessed using ELISA after three weeks of treatment with vehicle (black bars; n = 5) or L2-b (1 mg per kg per day; gray bars; n = 7). Bars denote the levels of Aβ, which were calculated from three independent experiments and expressed as values per gram of tissue. *P < 0.05 or **P < 0.01 by one-way ANOVA. (e) 4–20% (lower panels) and 16.5% (upper panels) tris–glycine gel/Western blot analyses were performed to visualize the Aβ monomers and aggregates, respectively, in the brain tissue lysates of wild type (WT; left panels) and 5XFAD male mice (right panels). | ||
Cognitive improvement in 5XFAD AD mice
Investigation of behavioral performance was carried out by administering L2-b to 5XFAD AD mice which suffer from deficits in learning and memory capabilities as amyloid pathology progresses.26 The Morris water maze was used to evaluate different aspects of spatial learning and memory in three-month-old 5XFAD AD mice.26 The wild type mice, which were consecutively injected with vehicle during the experimental period, normally took shorter times upon repetition of the training trial to find the escape platform, located in the northwest (NW) quadrant (Fig. 6a and b). In contrast, vehicle-treated 5XFAD AD mice spent longer times searching for and reaching the platform indicating they had difficulties with learning and memory (Fig. 6). Administration of L2-b to 5XFAD AD mice led to a remarkable improvement in the performance of the task. L2-b-treated 5XFAD AD mice were capable of finding the target in a comparable time to the wild type mice displaying significantly better memory and learning abilities than their untreated 5XFAD AD littermates (P < 0.05, Fig. 6a and b). Additionally, L2-b-treated 5XFAD AD mice took a more direct and easier path than the vehicle-treated 5XFAD AD mice to search for the platform (P < 0.05, Fig. 6c). Therefore, L2-b, a chemical reagent specific for metal–Aβ, ameliorates cognitive defects in the AD mouse model, along with the attenuation of amyloid pathology. These overall in vivo observations and results indicate that metal–Aβ complexes could be directly linked to AD pathogenesis.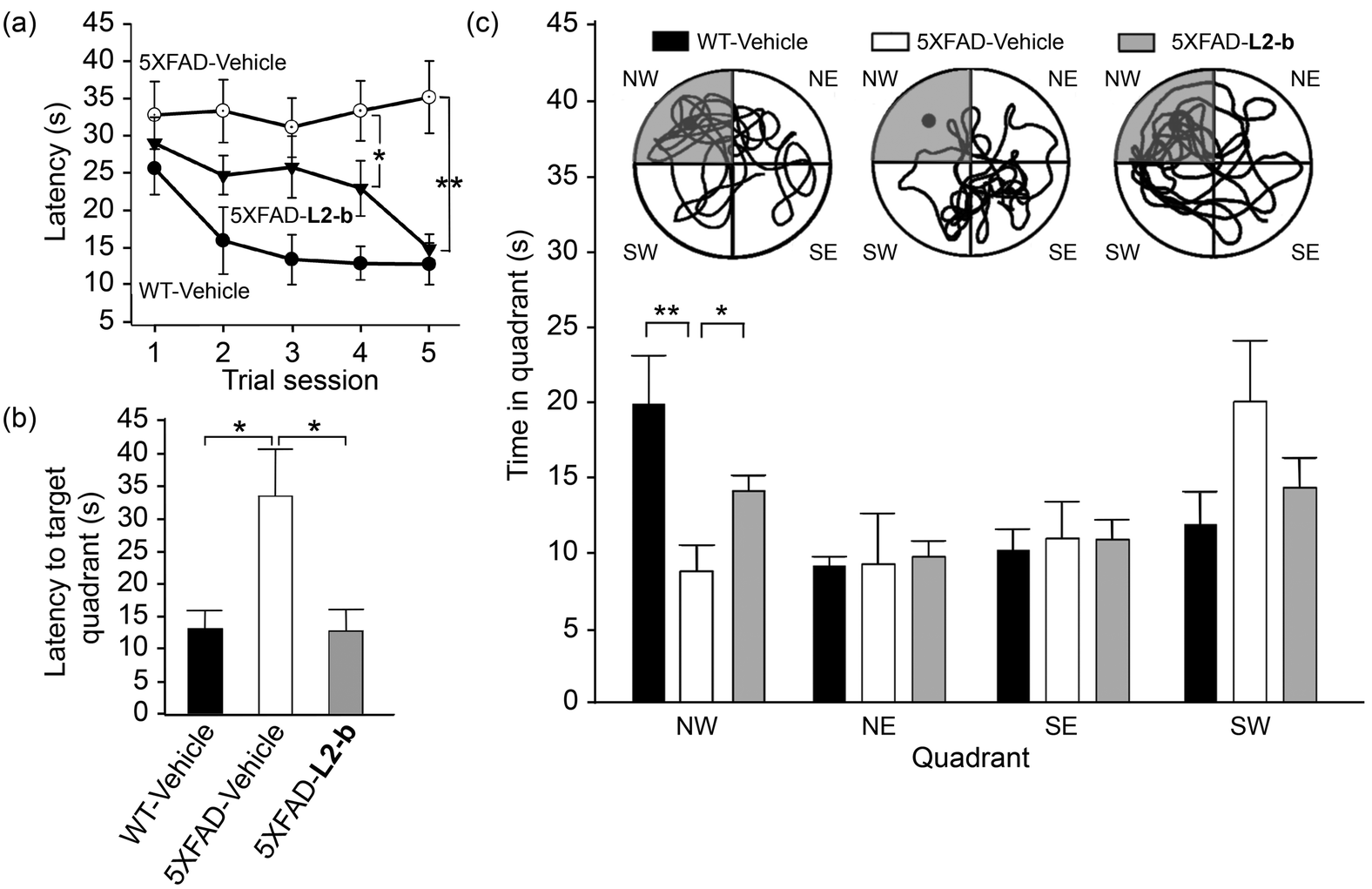 | ||
| Fig. 6 Learning and memory abilities of three-month-old male wild type (WT) and 5XFAD male mice treated with vehicle (black and white bars) and L2-b (gray), measured using the Morris water maze task. (a) The escape latency time was counted every day during the period of the 21st–25th daily treatments of either vehicle or L2-b and the probe trials were performed on the day of the final 25th treatment to measure (b) how quickly the mice reach and (c) how long they spend in the target quadrant (NW, highlighted in gray; circles show images of the representative tracks of the mice in the water maze). *P < 0.05 or **P < 0.01 by one-way ANOVA (n = 6, 13, and 14 for vehicle-treated WT and vehicle-/L2-b-treated 5XFAD mice, respectively). | ||
Conclusions
In summary, for the first time, experimental evidence affirms that metal–Aβ complexes can be directly associated with AD pathogenesis, by applying the first in vivo chemical tool which specifically targets metal–Aβ complexes and ameliorates metal–Aβ reactivity (i.e., metal–Aβ aggregation, formation of toxic oligomers, and ROS production) in 5XFAD AD mice. Our findings presented herein demonstrate the feasibility of developing small molecules as in vivo chemical tools for studying metal–Aβ. In addition, our studies indicate that research efforts toward understanding metal–Aβ-induced pathological pathways and identifying interrelated partners with metal–Aβ in AD onset and progression at the molecular level should continue to be made. The current and future outcomes, obtained from metal–Aβ-involved AD research, can open new directions for our long-term goal, the discovery of effective drugs for this fatal neurological disorder.Acknowledgements
This work was supported by the Ruth K. Broad Biomedical Foundation, the 2013 Research Fund (Project Number 1.130068.01) of Ulsan National Institute of Science and Technology (UNIST), and the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) (NRF-2014R1A2A2A01004877) (to M.H.L.); the Korea Healthcare Technology R&D Project, Ministry for Health and Welfare, Republic of Korea (A092042) and the Basic Science Research Program, National Research Foundation of Korea, Ministry of Education, Republic of Korea (NRF-2012R1A1A2006801) (to J.-Y.L.); the University of Michigan Protein Folding Disease Initiative (to B.T.R. and M.H.L.).Notes and references
- K. Fargo and L. Bleiler, Alzheimer's Dementia, 2014, 10, e47–e92 CrossRef PubMed.
- A. Corbett, J. Pickett, A. Burns, J. Corcoran, S. B. Dunnett, P. Edison, J. J. Hagan, C. Holmes, E. Jones, C. Katona, I. Kearns, P. Kehoe, A. Mudher, A. Passmore, N. Shepherd, F. Walsh and C. Ballard, Nat. Rev. Drug Discovery, 2012, 11, 833–846 CrossRef CAS PubMed.
- R. Jakob-Roetne and H. Jacobsen, Angew. Chem., Int. Ed., 2009, 48, 3030–3059 CrossRef CAS PubMed.
- M. W. Beck, A. S. Pithadia, A. S. DeToma, K. J. Korshavn and M. H. Lim, in Ligand Design in Medicinal Inorganic Chemistry, ed. T. Storr, Wiley, Chichester, 2014, ch. 10, pp. 256–286 Search PubMed.
- J. A. Hardy and G. A. Higgins, Science, 1992, 256, 184–185 CAS.
- K. J. Barnham, C. L. Masters and A. I. Bush, Nat. Rev. Drug Discovery, 2004, 3, 205–214 CrossRef CAS PubMed.
- K. P. Kepp, Chem. Rev., 2012, 112, 5193–5239 CrossRef CAS PubMed.
- M. G. Savelieff, S. Lee, Y. Liu and M. H. Lim, ACS Chem. Biol., 2013, 8, 856–865 CrossRef CAS PubMed.
- H. J. Lee, K. J. Korshavn, A. Kochi, J. S. Derrick and M. H. Lim, Chem. Soc. Rev., 2014, 43, 6672–6682 RSC.
- M. A. Greenough, J. Camakaris and A. I. Bush, Neurochem. Int., 2013, 62, 540–555 CrossRef CAS PubMed.
- A. S. DeToma, S. Salamekh, A. Ramamoorthy and M. H. Lim, Chem. Soc. Rev., 2012, 41, 608–621 RSC.
- M. A. Lovell, J. D. Robertson, W. J. Teesdale, J. L. Campbell and W. R. Markesbery, J. Neurol. Sci., 1998, 158, 47–52 CrossRef CAS.
- E. L. Que, D. W. Domaille and C. J. Chang, Chem. Rev., 2008, 108, 1517–1549 CrossRef CAS PubMed.
- J. H. Viles, Coord. Chem. Rev., 2012, 256, 2271–2284 CrossRef CAS PubMed.
- T. R. Young, A. Kirchner, A. G. Wedd and Z. Xiao, Metallomics, 2014, 6, 505–517 RSC.
- S. Ayton, P. Lei and A. I. Bush, Free Radical Biol. Med., 2013, 62, 76–89 CrossRef CAS PubMed.
- A. S. Pithadia and M. H. Lim, Curr. Opin. Chem. Biol., 2012, 16, 67–73 CrossRef CAS PubMed.
- P. Faller, C. Hureau and G. La Penna, Acc. Chem. Res., 2014, 47, 2252–2259 CrossRef CAS PubMed.
- P. J. Crouch, A. R. White and A. I. Bush, FEBS J., 2007, 274, 3775–3783 CrossRef CAS PubMed.
- C. Rodríguez-Rodríguez, M. Telpoukhovskaia and C. Orvig, Coord. Chem. Rev., 2012, 256, 2308–2332 CrossRef PubMed.
- L. R. Perez and K. J. Franz, Dalton Trans., 2010, 39, 2177–2187 RSC.
- C. W. Ritchie, A. I. Bush, A. Mackinnon, S. Macfarlane, M. Mastwyk, L. MacGregor, L. Kiers, R. Cherny, Q.-X. Li, A. Tammer, D. Carrington, C. Mavros, I. Volitakis, M. Xilinas, D. Ames, S. Davis, K. Beyreuther, R. E. Tanzi and C. L. Masters, Arch. Neurol., 2003, 60, 1685–1691 CrossRef PubMed.
- P. A. Adlard, R. A. Cherny, D. I. Finkelstein, E. Gautier, E. Robb, M. Cortes, I. Volitakis, X. Liu, J. P. Smith, K. Perez, K. Laughton, Q.-X. Li, S. A. Charman, J. A. Nicolazzo, S. Wilkins, K. Deleva, T. Lynch, G. Kok, C. W. Ritchie, R. E. Tanzi, R. Cappai, C. L. Masters, K. J. Barnham and A. I. Bush, Neuron, 2008, 59, 43–55 CrossRef CAS PubMed.
- M. G. Savelieff, A. S. DeToma, J. S. Derrick and M. H. Lim, Acc. Chem. Res., 2014, 47, 2475–2482 CrossRef CAS PubMed.
- A. M. Mancino, S. S. Hindo, A. Kochi and M. H. Lim, Inorg. Chem., 2009, 48, 9596–9598 CrossRef CAS PubMed.
- H. Oakley, S. L. Cole, S. Logan, E. Maus, P. Shao, J. Craft, A. Guillozet-Bongaarts, M. Ohno, J. Disterhoft, L. V. Eldik, R. Berry and R. Vassar, J. Neurosci., 2006, 26, 10129–10140 CrossRef CAS PubMed.
- J.-S. Choi, J. J. Braymer, R. P. R. Nanga, A. Ramamoorthy and M. H. Lim, Proc. Natl. Acad. Sci. USA, 2010, 107, 21990–21995 CrossRef CAS PubMed.
- H. F. Kung, C.-W. Lee, Z.-P. Zhuang, M.-P. Kung, C. Hou and K. Plössl, J. Am. Chem. Soc., 2001, 123, 12740–12741 CrossRef CAS.
- M. G. Savelieff, Y. Liu, R. R. P. Senthamarai, K. J. Korshavn, H. J. Lee, A. Ramamoorthy and M. H. Lim, Chem. Commun., 2014, 50, 5301–5303 RSC.
- S. Lee, X. Zheng, J. Krishnamoorthy, M. G. Savelieff, H. M. Park, J. R. Brender, J. H. Kim, J. S. Derrick, A. Kochi, H. J. Lee, C. Kim, A. Ramamoorthy, M. T. Bowers and M. H. Lim, J. Am. Chem. Soc., 2014, 136, 299–310 CrossRef CAS PubMed.
- S.-J. Hyung, A. S. DeToma, J. R. Brender, S. Lee, S. Vivekanandan, A. Kochi, J.-S. Choi, A. Ramamoorthy, B. T. Ruotolo and M. H. Lim, Proc. Natl. Acad. Sci. USA, 2013, 110, 3743–3748 CrossRef CAS PubMed.
- As presented in ref. 27, the initial aggregation studies using L2-b were conducted only for metal-induced Aβ40 aggregation at one incubation time (i.e., 24 h).
- R. Kayed, E. Head, J. L. Thompson, T. M. McIntire, S. C. Milton, C. W. Cotman and C. G. Glabe, Science, 2003, 300, 486–489 CrossRef CAS PubMed.
- R. Kayed, E. Head, F. Sarsoza, T. Saing, C. W. Cotman, M. Necula, L. Margol, J. Wu, L. Breydo, J. L. Thompson, S. Rasool, T. Gurlo, P. Butler and C. G. Glabe, Mol. Neurodegener., 2007, 2, 18–28 CrossRef PubMed.
- C. J. Frederickson, E. J. Kasarskis, D. Ringo and R. E. Frederickson, J. Neurosci. Methods, 1987, 20, 91–103 CrossRef CAS.
- J.-Y. Lee, J. S. Kim, H.-R. Byun, R. D. Palmiter and J.-Y. Koh, Brain Res., 2011, 1418, 12–22 CrossRef CAS PubMed.
- S. B. Oh, C. J. Byun, J.-H. Yun, D.-G. Jo, P. Carmeliet, J.-Y. Koh and J.-Y. Lee, Neurobiol. Aging, 2014, 35, 511–519 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, Tables S1–S3, and Fig. S1–S6. See DOI: 10.1039/c4sc03239j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |