
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hyperbranched polydendrons: a new nanomaterials platform with tuneable permeation through model gut epithelium†
Fiona L.
Hatton
a,
Lee M.
Tatham
b,
Louise R.
Tidbury
b,
Pierre
Chambon
a,
Tao
He
c,
Andrew
Owen
*b and
Steven P.
Rannard
*a
aDepartment of Chemistry, University of Liverpool, Crown Street, L69 7ZD, UK. E-mail: srannard@liv.ac.uk
bDepartment of Molecular and Clinical Pharmacology, University of Liverpool, Block H, 70 Pembroke Place, Liverpool L69 3GF, UK
cInstitute of Chemical and Engineering Sciences, Agency for Science, Technology and Research (A*STAR), 1, Pesek Road, Jurong Island, 627833, Singapore
First published on 3rd October 2014
Abstract
The development of nanomaterials for advanced therapies requires the formation of versatile platforms that may be tuned to maximize beneficial attributes and minimize unwanted negative behaviour. Additionally, the optimum route of administration is a key consideration of any new treatment and much work has been focused on direct injection into the systemic circulation rather than oral delivery. Here we describe a new approach to polymeric nanoparticle design and present initial results showing the potential for tuneable permeation through a gut epithelium model. Through the use of mixed initiators and branched vinyl polymerization, a series of systematically varying branched polymers have been synthesized and nanoprecipitated. The surprisingly uniform structures have undergone preliminary pharmacological evaluation to establish low cytotoxicity and enhanced permeation through model intestinal epithelial cells. This presents potential opportunities for future developments that may allow oral dosing to result in circulating polymeric nanoparticles; behaviour that may prove clinically desirable to many non-terminal or chronic diseases that utilise nanomedicines but wish to avoid regular or repeated intravenous administration.
Introduction
Clinically successful nanomedicines predominantly utilize two general approaches: orally-dosed solid-drug nanoparticles (SDNs)1–3 and intravenous drug-nanocarriers.4–6 Oral dosing is globally the most patient-acceptable drug administration format, often requiring no intervention by trained healthcare workers, offering reduced costs and requiring lower levels of sterility.7,8 SDN strategies aim predominantly to enhance the low oral bioavailability observed with many drug classes which may force large clinically administered doses or lead to the potential failure of new drug candidates. Relatively recently, SDNs have also been clinically evaluated as depot injections to provide long-term (months) exposure from a single subcutaneous or intramuscular injection of particulate drugs.9–11 Injected drug-nanocarriers (sometimes described as drug delivery systems) are typically administered intravenously and offer enhanced systemic circulation, lower doses, reduction of side effects, and targeting/accumulation within cellular or tissue sites. Such benefits from many nanocarriers require the nanocarrier/drug vehicle to reach the systemic circulation12 as an intact nanoparticle whereas overcoming low bioavailability via SDNs may result solely from enhanced drug dissolution prior to, or during, permeation of the intestinal epithelium.1 Injections are not widely used clinically for chronic or non-terminal diseases and a clear, yet highly challenging, target for nanomedicine is the development of orally-dosed nanocarriers that can deliver drugs into the systemic circulation whilst maintaining their particulate form. Candidates such as polymeric micelles, polymer-drug conjugates, nanoprecipitates and nanoemulsions are undergoing considerable study in this respect,13–16 however, the synthetic/biological challenge and difficulties of in vivo detection within the blood, and during clearance, continue to act as a barrier to translation.Arguably the most diverse organic nanomaterial studies have involved ideally-branched dendrimers,17–19 and linear–dendritic polymer hybrids.20 Multi-step convergent or divergent dendrimer syntheses have led to branched polymer chemistries, from polyimines/polyamines21 and polycarbonates22 through to polyamides,23,24 polyamido-amines,25 polyesters,26 polyurethanes,27 and polycarbosilanes28 to name a selection, and dendritic polymer application29 has begun to mature in nanomedicine.30 Obstacles continue to exist for cost-effective applications including the synthetic complexity and molecular weight/physical size limitations. Although dendrimers possess uniform molecular weight distributions and well defined shape, the available size range has long been limited to <15 nm31 until very recent reports of generation 13 triazine-derived materials with molecular weights >8 × 106 g mol−1 and diameters of 30 nm.32
Many dendrimer materials also require lengthy syntheses that often utilize large reagent excesses. To provide broader materials options, dendrimer sub-units (dendrons) have been attached to linear polymer chain-ends or side-chains to form linear–dendritic hybrid block copolymers with multiple additional functionality and supramolecular assembly properties.20 Micellar structures of amphiphilic linear–dendritic copolymers (30–70 nm),26 and micelle aggregates (approx. 200 nm),33 have been reported after association of relatively small building blocks. The control of structures within the 30–250 nm size range is of interest in drug delivery (conjugated or encapsulated drug),19 with success reported for combinations of dendrons with linear polymers leading to greater flexibility and scope for molecular design and the direction of particle formation.
One-pot, branched vinyl copolymerization techniques34 have been shown to form nanoparticles in a single synthetic procedure35via a concerted propagation and interchain branching of linear polymer chains using low concentrations of bifunctional monomers. When used in conjunction with controlled radical polymerization techniques, high molecular weight branched amphiphilic block copolymers have been formed that produce spherical nanoparticles when dialysed using water.36 The introduction of multi-functional initiators have also allowed dumbbell and clover-leaf nanoparticles to be formed,37 however, the loss of chain end functional multiplicity distances these materials from ideal dendrimers or linear–dendritic hybrids.
Herein we combine the aspects of linear–dendritic hybrids and branched vinyl polymerization to provide high molecular weight dendron-functional macromolecules, termed hyperbranched-polydendrons (hyp-polydendrons).38 By utilizing mixed initiator systems, this new synthetic strategy allows a ready methodology to systematically control mixed surface functionality and form complex spherical nanoparticles. These materials have: (1) a unique control of surface functionality; (2) directed aqueous self-assembly; (3) low dispersity when used to form nanoparticles with controlled diameters (75–210 nm); (4) encapsulation capabilities; and (5) variable interaction with model gut epithelial monolayers. Our preliminary studies of interactions of hyp-polydendron nanoprecipitates with a gut epithelium model show potential value in further work to understand and optimize nanoparticle transport across the gut barrier and the benefits that might be achievable after oral dosing.
Results and discussion
Synthesis of hyp-polydendrons with mixed functionality
To collectively overcome the synthetic complexity of dendrimer synthesis, and readily provide materials with the functional benefits of dendrimers, we recently reported the use of a dendron-derived initiator, 1, in the copper catalysed, branched vinyl atom transfer radical (ATRP) copolymerization of 2-hydroxypropyl methacrylate (HPMA), 2, and ethylene glycol dimethacrylate (EGDMA), 3, to form novel polymer architectures containing large numbers of dendrons at one end of each conjoined primary polymer chain38 (Fig. 1A). We have termed these materials hyp-polydendrons as they combine a highly branched vinyl polymer core with dendron-derived chain ends to form new hyperbranched linear–dendritic hybrid architectures.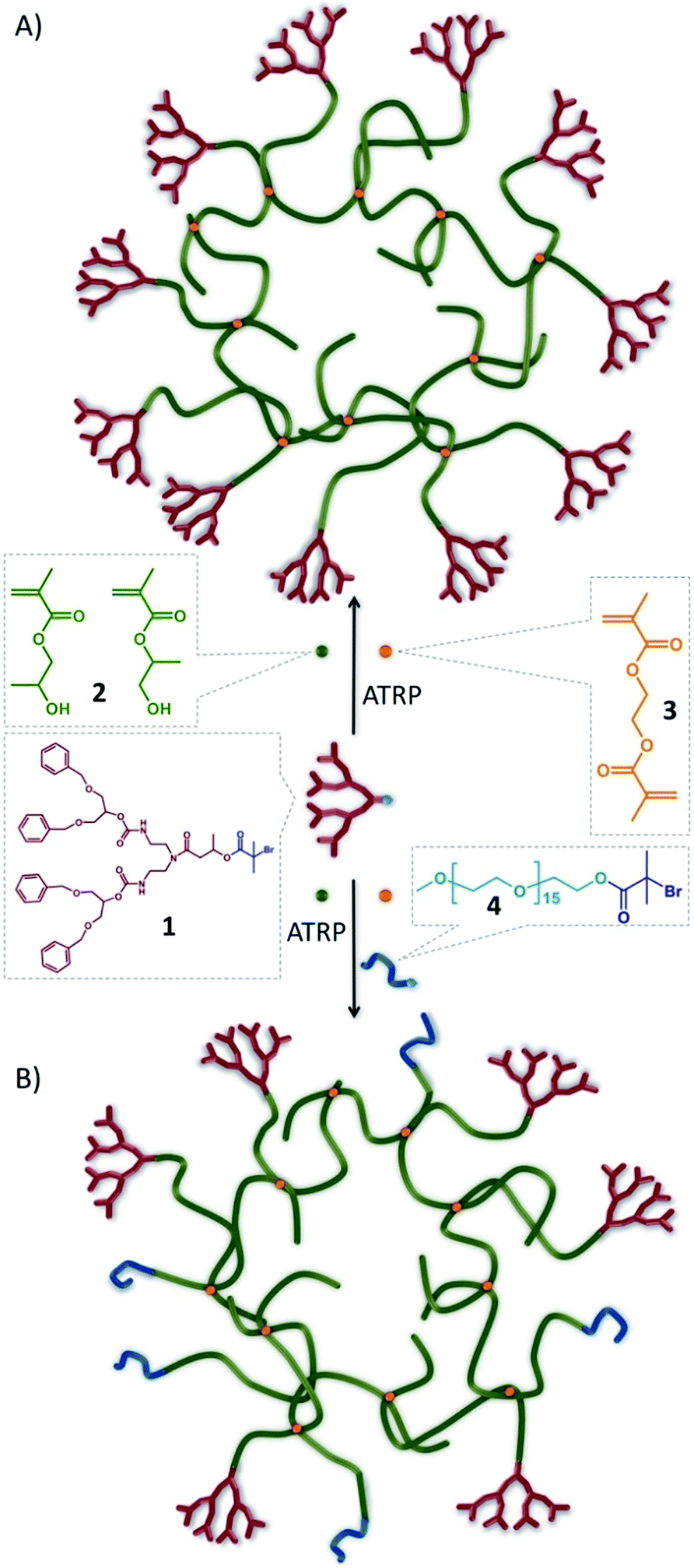 | ||
| Fig. 1 Schematic representation of idealised hyp-polydendron synthesis. Atom transfer radical copolymerization (ATRP) of 2-hydroxypropyl methacrylate, 2, and ethylene glycol dimethacrylate, 3, initiated by the functional G2 dendron, 1 to form (A) hyp-polydendrons; and (B) inclusion of a PEG16-initiator, 4, to form hyp-polydendrons with controlled and mixed surface functionality. | ||
In the absence of 3, dendron-initiated methanolic ATRP polymerizations of 2 (target number average degree of polymerization DPn = 50 monomer units), using the second generation (G2) dendron initiator, 1, led to high monomer conversion (>99%) and recovered polymers with a number average molecular weight (Mn) of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 g mol−1 and weight average molecular weight (Mw) of 16600 g mol−1, as determined by triple detection size exclusion chromatography (SEC; Table 1; Fig. S1 & S2†). In the presence of 3 (1:3 molar ratio of 1:0.8), polymerizations also reached high conversion (>99%) with no observable interference of the controlled radical polymerization kinetics (Fig. S3†), but polymers of considerably higher molecular weight (Mn = 90500 g mol−1; Mw = 1304000 g mol−1) were recovered (Table 1).
200 g mol−1 and weight average molecular weight (Mw) of 16600 g mol−1, as determined by triple detection size exclusion chromatography (SEC; Table 1; Fig. S1 & S2†). In the presence of 3 (1:3 molar ratio of 1:0.8), polymerizations also reached high conversion (>99%) with no observable interference of the controlled radical polymerization kinetics (Fig. S3†), but polymers of considerably higher molecular weight (Mn = 90500 g mol−1; Mw = 1304000 g mol−1) were recovered (Table 1).
| Initiator (mol fraction) | Target polymer composition | SEC | |||
|---|---|---|---|---|---|
|
a Linear polymerization.
b Branched polymerizations at total initiator:brancher ratio = 1:0.8.
c Branched polymerizations at total initiator:brancher ratio = 1:0.95.
|
|||||
| G2 | PEG16 | M n | M w | Đ (Mw/Mn) | |
| 1 | 0 | HPMA50a | 12200 |
16600 |
1.36 |
| 0.5 | 0.5 | HPMA50a | 13000 |
16900 |
1.30 |
| 0 | 1 | HPMA50a | 10600 |
16800 |
1.59 |
| 1 | 0 | HPMA50–EGDMA0.8b | 90500 |
1304000 |
14.40 |
| 0.9 | 0.1 | HPMA50–EGDMA0.8b | 68500 |
1495000 |
21.84 |
| 0.75 | 0.25 | HPMA50–EGDMA0.8b | 52400 |
987800 |
18.88 |
| 0.5 | 0.5 | HPMA50–EGDMA0.8b | 39400 |
480700 |
12.19 |
| 0.25 | 0.75 | HPMA50–EGDMA0.8b | 36200 |
315300 |
8.73 |
| 0.1 | 0.9 | HPMA50–EGDMA0.8b | 37700 |
286000 |
7.61 |
| 0 | 1 | HPMA50–EGDMA0.8b | 68100 |
296200 |
4.35 |
| 0.5 | 0.5 | HPMA50–EGDMA0.95c | 121900 |
1779000 |
14.60 |
| 0 | 1 | HPMA50–EGDMA0.95c | 74700 |
642700 |
8.60 |
Dendrimers with statistically mixed (divergent synthesis) and controlled mixing (convergent synthesis) of surface functionality have been reported previously.39–44 The hyp-polydendron synthesis strategy offers a unique opportunity for systematic incorporation of mixed functionalities through the mixing of ATRP initiators. Dendron-derived initiators guarantee one dendron on each primary polymer chain, but the introduction of varying ratios of a polyethylene glycol-derived (PEG16) ATRP initiator, 4, to 1 prior to copolymerization of 2 and 3 (Fig. 1B) has been undertaken. A range of branched polymerizations were conducted with 1:4 ratios of 100:0, 90:10, 75:25, 50:50, 25:75, 10:90 and 0:100, whilst maintaining the overall initiating species (based on tertiary bromide functionality) to brancher, (1 + 4):3, ratio of 1:0.8. Polymerization kinetic studies confirmed controlled radical polymerization, however, the achieved molecular weights (Mn and Mw) decreased with increasing content of 4. This is quite possibly due to an overall increase in initiation efficiency generating a decrease in the effective initiator:brancher ratio and subsequent lower branching; Mw values ranging between 1495000–296000 g mol−1 were observed across the range of initiator mixtures when utilising a (1 + 4):3 molar ratio of 1:0.8 (Table 1; Fig. S2†). Increased levels of 3 were tolerated within the polymerizations containing high concentrations of, 4 without the presence of gelation and Mw values between 2-fold and 4-fold higher were achievable at (1 + 4):3 molar ratio of 1:0.95 (Table 1); Mn values were also considerably increased.
Confirmation of the varying 1:4 ratio within the various hyp-polydendron structures was obtained by 1H nuclear magnetic resonance (NMR) spectroscopy (Fig. S4†). The calculated molar variation of G2 dendron and PEG16 end-groups were observed to follow theoretical concentrations closely (Fig. S5†); however, at higher target G2 dendron values, slightly lower dendron incorporation was observed, again a potential effect of the differing initiator efficiencies.
Aqueous nanoprecipitation and characterisation of hyp-polydendrons with mixed surface functionality
Nanoprecipitation of linear polymers45 has previously generated uniform spherical nanoparticles predominantly from biodegradable polymers.46 The nanoprecipitation mechanism is proposed to undergo a nucleation/aggregation process47 and a multitude of factors may be varied (solvent choice, polymer concentration, viscosity) to direct the formation of colloidally stable particles of varying size.48 We recently demonstrated the successful aqueous nanoprecipitation of branched polymer architectures49 and the self-assembly of hyp-polydendrons38 within organic solvent mixtures and water to form uniform, spherical, self-assembled nanostructures.Successful nanocarrier candidates (100–200 nm), formed by careful control of polymer structure and nanoprecipitation,50,51 have previously shown the potential for relatively high drug loadings. Chain-end modification of polyethylene glycol segments within amphiphilic A–B block copolymers by aptamers (Accurin™ technology) has also led to positive phase I evaluation of drug delivery to sites of prostate and lung cancer51,52 from injected nanoprecipitates with recently announced progression to phase II human studies. The seven branched polymers (one hyp-polydendron, five hyp-polydendrons with varying PEG16 content and one entirely PEG16 initiated hyp-polymer) were subjected to nanopreciptation46,49 under varying conditions (Table 2). A simple dropping method was employed using two initial polymer concentrations (CI) in a good solvent (10 or 5 mg mL−1 in tetrahydrofuran (THF)) and two dilution ranges (5 or 100-fold) into an antisolvent (water).
| G2–PEG16 molar ratio | Initial THF concentration (CI; mg mL−1) | Final H2O concentration (CF; mg mL−1) | D z (nm) | PdIa | ζ (mV) |
|---|---|---|---|---|---|
| a Measured by dynamic light scattering. | |||||
| 100:0 |
10 | 2 | 106 | 0.083 | −38.0 |
| 100:0 |
10 | 0.1 | 134 | 0.064 | −34.3 |
| 100:0 |
5 | 1 | 81 | 0.083 | −38.2 |
| 100:0 |
5 | 0.05 | 93 | 0.071 | −20.4 |
| 90:10 |
10 | 2 | 173 | 0.076 | −30.4 |
| 90:10 |
10 | 0.1 | 205 | 0.087 | −33.1 |
| 90:10 |
5 | 1 | 116 | 0.069 | −25.9 |
| 90:10 |
5 | 0.05 | 110 | 0.087 | −17.6 |
| 75:25 |
10 | 2 | 155 | 0.085 | −25.6 |
| 75:25 |
10 | 0.1 | 190 | 0.097 | −39.0 |
| 75:25 |
5 | 1 | 110 | 0.073 | −26.5 |
| 75:25 |
5 | 0.05 | 135 | 0.092 | −30.2 |
| 50:50 |
10 | 2 | 148 | 0.061 | −29.3 |
| 50:50 |
10 | 0.1 | 150 | 0.073 | −29.6 |
| 50:50 |
5 | 1 | 115 | 0.067 | −28.2 |
| 50:50 |
5 | 0.05 | 104 | 0.072 | −20.7 |
| 25:75 |
10 | 2 | 121 | 0.072 | −28.7 |
| 25:75 |
10 | 0.1 | 110 | 0.104 | −24.0 |
| 25:75 |
5 | 1 | 93 | 0.078 | −30.4 |
| 25:75 |
5 | 0.05 | 77 | 0.140 | −16.5 |
| 10:90 |
10 | 2 | 129 | 0.058 | −31.3 |
| 10:90 |
10 | 0.1 | 108 | 0.074 | −23.2 |
| 10:90 |
5 | 1 | 94 | 0.091 | −29.9 |
| 10:90 |
5 | 0.05 | 78 | 0.090 | −28.9 |
| 0:100 |
10 | 2 | 121 | 0.074 | −39.8 |
| 0:100 |
10 | 0.1 | 116 | 0.072 | −30.0 |
| 0:100 |
5 | 1 | 90 | 0.083 | −39.7 |
| 0:100 |
5 | 0.05 | 89 | 0.092 | −27.8 |
Subsequent organic solvent evaporation gave four aqueous nanoparticle dispersion concentrations (CF) for each material (2, 1, 0.1 and 0.05 mg mL−1), determined by the volume of the THF solution that was added to a fixed volume of water. Within each nanoprecipitation, the initial solvent droplet solution contains solvated branched polymer chains and, on addition to water, the THF-rich and water-rich phase boundary rapidly dilutes during diffusion of THF into the antisolvent (Fig. 2A). The branched vinyl polymer cores collapse to form nuclei that assemble to produce stabilized monodisperse nanoparticles within the aqueous phase.38 Characterisation was achieved using scanning electron (SEM) and atomic force microscopy (AFM; Fig. 2C and D) and dynamic light scattering (DLS; Fig. S6†). Unlike conventional nanoprecipitation, the assembled hyp-polydendron nanoparticles contain systematically varying peripheral dendrons and PEG16 chains at the nanoprecipitate surface (Fig. 2B(i)–(iii)), thereby opening considerable opportunities for future development.
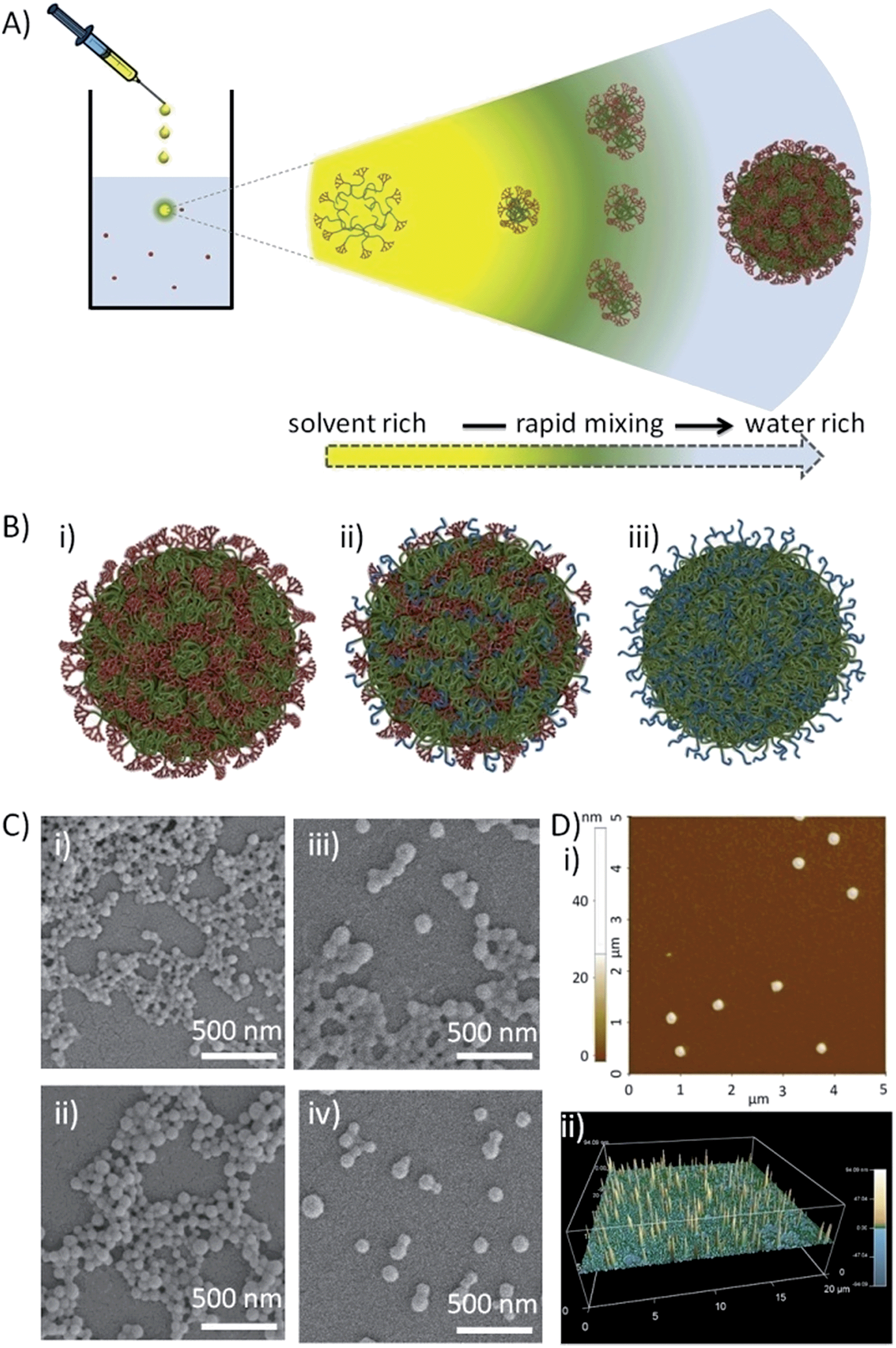 | ||
| Fig. 2 Nanoprecipitation of hyp-polydendrons. (A) Schematic of nano-precipitation from THF (good solvent) into water (poor solvent). Dissolved and expanded hyp-polydendrons collapse, forming self-assembling nuclei that form stable nanoparticles aqueous environments; (B) schematic representation of hyp-polydendron nanoprecipitates containing (i) 100% dendron initiator, (ii) a 50:50 molar ratio of G2 dendron–PEG16 initiators, and (iii) 100% PEG16 initiator; (C) scanning electron microscopy images of hyp-polydendron nanoprecipitates (initial concentration in THF = 5 mg mL−1, final aqueous concentration = 1 mg mL−1) with varying G2 dendron–PEG16 molar ratios: (i) 100:0, (ii) 90:10, (iii) 50:50, and (iv) 25:75; (D) atomic force microscopy of hyp-polydendron nanoprecipitate particles (initial concentration in THF = 5 mg mL−1, final aqueous concentration = 1 mg mL−1): (i) size image for nanoprecipitates generated from a G2 dendron–PEG16 molar ratio of 100:0, (ii) contour image of nanoprecipitates with a G2 dendron–PEG16 molar ratio of 25:75. | ||
The variation of nanoprecipitation conditions (polymer chemistry, polymer concentration and solvent:antisolvent dilution ratio) allowed control of the hyp-polydendron and hyp-polymer nanoprecipitate diameters (z-average diameters (Dz) from 77–205 nm, Table 2). Zeta potentials (ζ) were also measured and showed negatively charged particles (Table 2); these values are consistent with earlier unfunctionalised nanoprecipitates.49 Narrow size distributions, with polydispersity indices (PdI) ranging from 0.058 to 0.140 (Table 2), are highly surprising given the very broad molecular weight distribution of the hyp-polydendron materials, Table 1. It may be assumed that significant architectural diversity is produced across the molecular weight distribution during synthesis, with variation of branch point placement leading to different branching densities and flexibilities of the internal core structures. Mixed initiator polymerization leads to considerable additional polymer complexity, including complication of the inherent distributions of molecular weight, architecture, branching density and G2–PEG16 ratios. Despite this high level of complexity, the near monodisperse nanoprecipitates showed remarkably uniform behavior and stability, with little detectable difference when stored in water for nearly a year (Table S1†). Addition of THF to the aqueous nanoprecipitates led to swelling of the nanoparticles without dissolution, even at high levels of THF (>37.5% v/v) (Fig. S7b†), whilst dilution of the nanoparticles with water led to no appreciable changes over considerable concentration ranges (Fig. S7a†).
Conversely, studies of nanoprecipitates derived from the analogous unbranched linear–dendritic hybrids that comprise the primary chain structure of the hyp-polydendrons, and PEG16-(HPMA)50 analogues (Table 1), led to broad size distributions after nanoprecipitation and increasing Dz and PdI (bimodal size distributions in some cases) over extended periods, with visual precipitation (Fig. S8†).
Nile Red (NR) and pyrene encapsulation was accomplished by co-dissolving the fluorescent dyes into the branched polymer–THF solution of (0.1% w/w relative to polymer) prior to nanoprecipitation. These dyes were chosen due to their environment-dependent fluorescence and the ability to report on differences in lipophilicity (NR) or polarity (pyrene) within the core of the nanoprecipitates; no appreciable changes to Dz (Fig. S9 and S10†) were observed with encapsulated dye. NR emission (630 nm) was measured after excitation at 552 nm and was clearly dependent on the G2 dendron–PEG16 ratio (Fig. 3A).
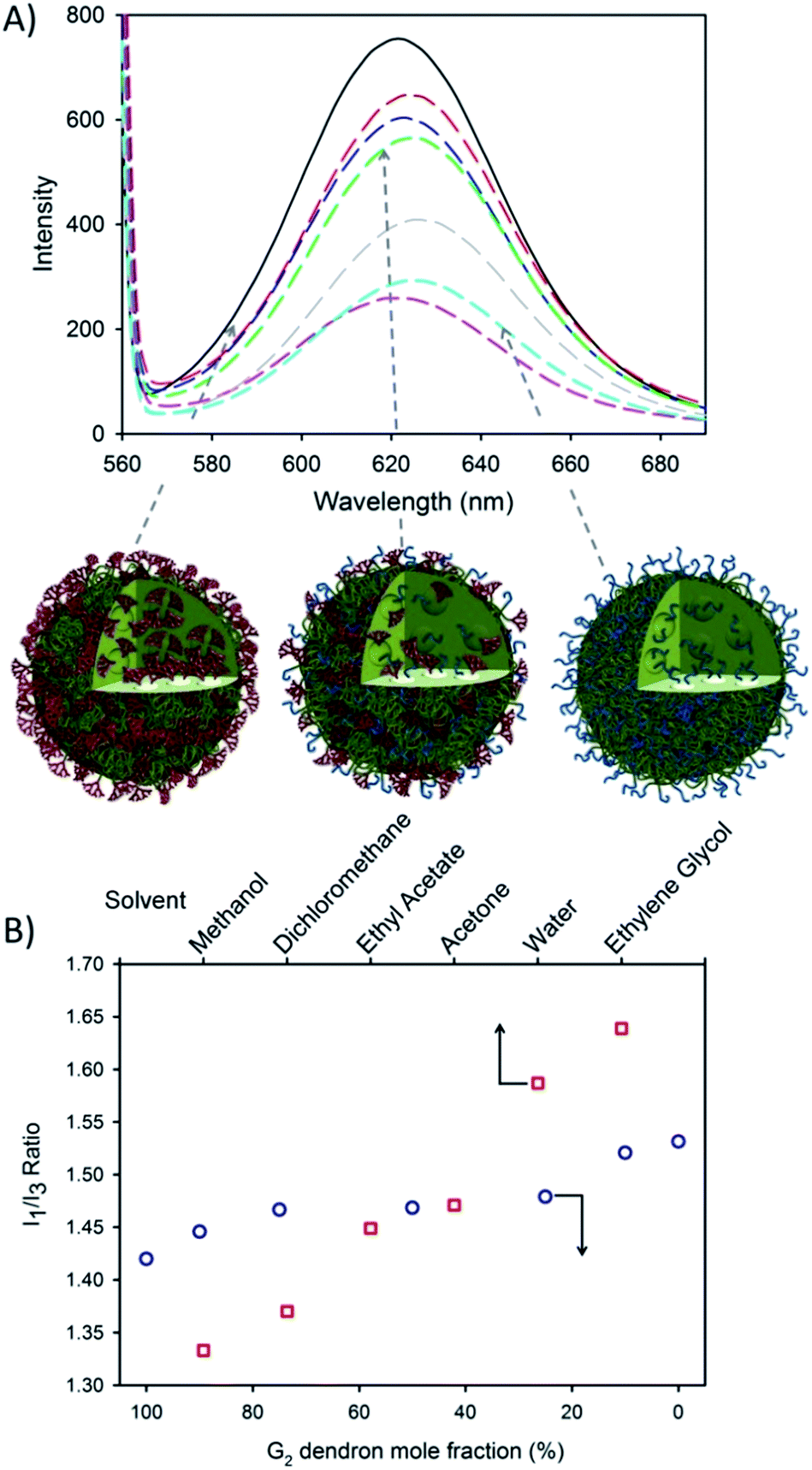 | ||
| Fig. 3 Encapsulation of hydrophobic guest molecules during nanoprecipitation of hyp-polydendrons with varying G2 dendron–PEG16 content. (A) Fluorimetric analysis of hyp-polydendron nanoprecipitates containing Nile Red produced from varying G2 dendron–PEG16 initiator ratios of 100:0 (solid black), 90:10 (red dash), 75:25 (blue dash), 50:50 (green dash), 27:75 (pink dash), 10:90 (grey dash) and 0:100 (cyan dash); (B) comparison of the observed I1/I3 ratio of pyrene within varying solvents (open red squares)53,54 and encapsulated within hyp-polydendrons synthesized using different G2 dendron–PEG16 initiator ratios (open blue circles). | ||
This suggests the control of the environment within the hyp-polydendron nanoprecipitates whilst simultaneously modifying the expression of surface functionality. This is consistent with an assembly model involving aggregation of individual collapsed hyp-polydendron nuclei as complete entrapment of hyp-polydendrons within the nanoprecipitate would result (Fig. 3A), leading to internalization of varying internal ratios of G2 dendron and PEG16.
Further investigation of the internal nanoparticle environment was conducted via pyrene encapsulation. A similar variation of the internal nanoparticle environment was observed within the fine structure of pyrene fluorescence after excitation at 335 nm (Fig. S11†). The ratio of the intensities of the first and third vibrational bands (I1/I3) varied from 1.42–1.53, similar to pyrene fluorescence observed in solvents with polarities ranging from dichloromethane (I1/I3 = 1.37) to water (I1/I3 = 1.59) (Fig. 3B).53,54 The observed modification of the nanoparticle internal environment allows the potential for tuning of nanoparticle properties via initiator chemistry and/or monomer choice, providing considerable scope for tuning of physical properties. Similarly, modification of surface chemistry of the nanoprecipitate may also alter cellular or tissue interactions and this was investigated further with respect to model gut epithelium.
Model epithelial permeation studies using aqueous hyp-polydendron nanoprecipitates
There are many clinically-utilized nanocarrier therapies benefiting patients globally, across diseases including cancer (e.g. Doxil and Myocet), fungal infection (e.g. Abelcet and AmBisome), hepatitis (e.g. Epaxal and Pegintron) and meningitis (e.g. DepoCyt).55 Dendrimers have been utilized as DNA transfection reagents (e.g. SuperFECT) and cancer/cardiac diagnostics (Stratus CS Acute Care Assay), whilst only limited dendrimer-derived therapeutics are being sought (e.g. VivaGel®).55Nanocarriers are rarely dosed orally,56 but benefits may accrue in chronic diseases (e.g. HIV therapy or psychiatric disorders) where long-term daily dosing is required and repeated injection is not optimal for patients. The provision of an orally-administered nanocarrier, delivering nanoparticles to the circulation, may also provide cellular or tissue targeting benefits for acute diseases. Our aim within this study was to evaluate the potential for permeation through a gut epithelium model; therefore, a preliminary in vitro pharmacological evaluation of the behaviour of hyp-polydendron nanoprecipitates was conducted.
Cytotoxicity of the seven aqueous nanoprecipitates was evaluated against the human epithelial colorectal adenocarcinoma (Caco-2) cell line (Fig. 4A) and no appreciable cytotoxicity was observed at achievable concentrations in assays assessing either adenosine triphosphate or 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) turnover (Fig. S12a–d†); as such the determination of an IC50 value for each polymer was not possible, indicating very low cytotoxicity towards Caco-2 cells. In the absence of obvious cytotoxicity to the gut model cell line, the transcellular permeation of hyp-polydendron nanoprecipitates (with encapsulated NR) was studied across differentiated Caco-2 monolayers as a model of absorption through the intestinal epithelium (Fig. 4A).57
 | ||
| Fig. 4 Preliminary evaluation of oral dosing potential for hyp-polydendron nanoprecipitates with varying G2 dendron–PEG16 content. (A) Schematic representation of hyp-polydendron nanoprecipitates passing from (i) apical (top) compartment (modelling the gut side) of a differentiated Caco-2 cell monolayer through to the basolateral (bottom) compartment (modelling the blood side) to evaluate potential for intestinal epithelium permeation, and (ii) movement of hyp-polydendron nanoprecipitates from the basolateral to the apical side of the monolayer; (B) comparison of permeation of a hyp-polydendron (10:90 G2 dendron–PEG16 ratio) nanoprecipitate (blue squares) and aqueous/DMSO solution (<1% v/v) of Nile Red (red circles) through a Caco-2 monolayer – B > A permeation (open symbols); A > B permeation (closed symbols) (C) apparent permeability (Papp) of Nile Red when encapsulated in hyp-polydendron nanoprecipitates of varying G2 dendron–PEG16molar ratios (open green triangles) and as an aqueous/DMSO solution (<1% v/v) (red line). | ||
Assessment of the apparent permeability (Papp) of a substance is widely studied using this model, including the screening of discovery compounds within the pharmaceutical industry. Papp is a measurement of the permeation (flux) through the Caco-2 monolayer after normalization for the surface area of the membrane and the concentration of the study substance within the media.58 Although not fully representative of the in vivo material behaviour, this model uses a human cell line and provides indicative data to allow further development.
NR fluorescence monitoring was used to determine hyp-polydendron passage from the apical compartment of the transwell plate to the basolateral well (A > B; Fig. 4A(i)) which models gut to systemic circulation permeation. In addition, permeation studies from the basolateral to the apical compartment (B > A; Fig. 4A(ii)) was assessed. The Papp ratio is calculated as the ratio of (A > B)/(B > A), providing a relative indication of apparent oral absorption in the presence of active transport proteins (e.g.P-glycoprotein) known to limit oral bioavailability of many compounds. In all studies the confluence of the Caco-2 cell monolayer was assessed to ensure that an intact membrane was present throughout the experiments (see ESI†).
The A > B permeation of the hyp-polydendron nanoprecipitates with encapsulated NR was approximately an order of magnitude higher than observed B > A permeation in all cases (Table 3; Fig. 4B). Variable behavior of the various nanoprecipitates was also noted over the 4 hours study exposure with materials comprising 75%, 50% and 0% G2 dendron showing a steady increase in fluorescence within the receiver compartment, suggesting a continual permeation through the Caco-2 membrane (Fig. S13†). Surprisingly, Papp ratios across the materials were all considerably higher than the aqueous/DMSO solution of NR (Table 3) and were highly correlated with the molar content of dendron; decreasing linearly with decreasing dendron content (simple linear regression, r2 = 0.78, P = 0.02; Fig. 4C). This data can also be interpreted as a possible impact of increasing PEG16 content leading to a decreasing overall permeation through the Caco-2 monolayers. However, the factors affecting the observed Papp ratio are clearly not straightforward as seen from the non-linear variation of either A > B or B > A permeation as the nanoprecipitate chemical composition varied, Table 3.
| G2–PEG16 molar ratio | P app (cm s−1) | P app ratio (A > B)/(B > A) | |
|---|---|---|---|
| Apical > Basolateral (A > B) | Basolateral > Apical (B > A) | ||
| a <1% v/v DMSO/water solution. | |||
| 100:0 |
1.763 × 10−5 | 1.538 × 10−6 | 11.4605 |
| 75:25 |
2.613 × 10−5 | 2.056 × 10−6 | 12.7123 |
| 50:50 |
5.271 × 10−5 | 5.555 × 10−6 | 9.4872 |
| 25:75 |
4.135 × 10−5 | 4.684 × 10−6 | 8.8279 |
| 10:90 |
4.042 × 10−4 | 4.580 × 10−5 | 8.8255 |
| 0:100 |
2.060 × 10−5 | 3.188 × 10−6 | 6.4626 |
| Aq. Nile Reda | 2.371 × 10−5 | 6.384 × 10−6 | 3.7140 |
The mechanisms that underpin this correlation are not immediately obvious and further investigation is required to understand the relationship. The increased presence of short PEG16 chains appears to have a negative impact on the Papp ratio within this series of materials; however, the hyp-polymer (without G2 dendron) still provided a noticeable enhancement of permeation. These exploratory data indicate the potential for further tuning of behaviour and the production of orally dosed materials that deliver benefits as systemically circulating nanoparticles carrying encapsulated drugs.
Conclusions
In summary, a strategy has been presented to synthesize a novel complex macromolecular architecture, hyperbranched polydendrons, with controlled surface functionality. Despite the non-uniform nature of the high molecular weight materials, monodisperse nanoprecipitates of tuneable size have been produced that are stable in water and encapsulate hydrophobic materials with control of internal nanoprecipitate environment. Exploratory studies have indicated the potential to mediate permeation through model human gut epithelium. This strategy offers synthetic ease and provides a route to the relatively simple formation of nanoparticles. Our ongoing research is focusing on manipulation of the structural components of the hyp-polydendron architecture to introduce additional chemical and physical functionality that is tuneable to specific pharmacological ambitions such as oral delivery and targeting.Acknowledgements
The authors wish to thank the Engineering and Physical Sciences Research Council for a PhD studentship (FL) and grant funding (EP/I038721/1) that underpinned the research. The University of Liverpool and the Centre for Materials Discovery at Liverpool is also gratefully acknowledged for access to scanning electron microscopy.Notes and references
- J.-U. A. H. Junghanns and R. H. Müller, Int. J. Nanomed., 2008, 3, 295–310 CAS.
- B. E. Rabinow, Nat. Rev. Drug Discovery, 2004, 3, 785–796 CrossRef CAS PubMed.
- T. O. McDonald, M. Giardiello, P. Martin, M. Siccardi, N. J. Liptrott, D. Smith, P. Roberts, P. Curley, A. Schipani, S. H. Khoo, J. Long, A. J. Foster, S. P. Rannard and A. Owen, Adv. Healthcare Mater., 2014, 3, 400–411 CrossRef CAS PubMed.
- W. T. Al-Jamal and K. Kostarelos, Acc. Chem. Res., 2011, 44, 1094–1104 CrossRef CAS PubMed.
- R. Duncan, Curr. Opin. Biotechnol., 2011, 22, 492–501 CrossRef CAS PubMed.
- C. E. Ashley, E. C. Carnes, G. K. Phillips, D. Padilla, P. N. Durfee, P. A. Brown, T. N. Hanna, J. Liu, B. Phillilps, M. B. Carter, N. J. Carroll, X. Jiang, D. R. Dunphy, C. L. Willman, D. N. Petsev, D. G. Evans, A. N. Parikh, B. Chackerian, W. Wharton, D. S. Peabody and C. J. Brinker, Nat. Mater., 2011, 10, 389–397 CrossRef CAS PubMed.
- F. E. Stuurman, B. Nuijen, J. H. Beijnen and J. H. M. Schellens, Clin. Pharmacokinet., 2013, 52, 399–414 CrossRef CAS PubMed.
- M. D. N. Siddiqui, G. Garg and P. K. Sharma, Adv. Biol. Res., 2011, 5, 291–303 Search PubMed.
- (a) M. Boffito, A. Jackson, A. Owen and S. Becker, Drugs, 2014, 74, 7–13 CrossRef CAS PubMed; (b) M. N. Samtani, A. Vermeulen and K. Stuyckens, Clin. Pharmacokinet., 2009, 48, 585–600 CrossRef CAS PubMed.
- G. van't Klooster, E. Hoeben, H. Borghys, A. Looszova, M.-P. Bouche, F. van Velsen and L. Baert, Antimicrob. Agents Chemother., 2010, 54, 2042–2050 CrossRef PubMed.
- W. Spreen, S. Min, S. L. Ford, S. Chen, Y. Lou, M. Bomar, M. St Clair, S. Piscitelli and T. Fujiwara, HIV Clin. Trials, 2013, 14, 192–203 CrossRef CAS PubMed.
- M. C. Branco and J. P. Schneider, Acta Biomater., 2009, 5, 817–831 CrossRef CAS PubMed.
- G. Gaucher, P. Satturwar, M.-C. Jones, A. Furtos and J.-C. Leroux, Eur. J. Pharm. Biopharm., 2010, 76, 147–158 CrossRef CAS PubMed.
- L. Bromberg, J. Controlled Release, 2008, 128, 99–112 CrossRef CAS PubMed.
- I. Pepić, J. Lovrić and J. Filipović-Grčić, Eur. J. Pharm. Sci., 2013, 50, 42–55 CrossRef PubMed.
- L. Mei, Z. Zhang, L. Zhao, L. Huang, X.-L. Yang, J. Tang and S.-S. Feng, Adv. Drug Delivery Rev., 2013, 65, 880–890 CrossRef CAS PubMed.
- S. M. Grayson and J. M. J. Fréchet, Chem. Rev., 2001, 101, 3819–3867 CrossRef CAS PubMed.
- L. Röglin, E. H. M. Lempens and E. W. Meijer, Angew. Chem., Int. Ed., 2011, 50, 102–112 CrossRef PubMed.
- C. C. Lee, J. A. MacKay, J. M. J. Fréchet and F. Szoka, Nat. Biotechnol., 2005, 23, 1517–1526 CrossRef CAS PubMed.
- F. Wurm and H. Frey, Prog. Polym. Sci., 2011, 36, 1–52 CrossRef CAS PubMed.
- T. M. Hermans, M. A. C. Broeren, N. Gomopoulos, P. van der Schoot, M. H. P. van Genderen, N. A. J. M. Sommerdijk, G. Fytas and E. W. Meijer, Nat. Nanotechnol., 2009, 4, 721–726 CrossRef CAS PubMed.
- S. P. Rannard and N. J. Davis, J. Am. Chem. Soc., 2000, 122, 11729–11730 CrossRef CAS.
- S. Rannard, N. Davis and H. McFarland, Polym. Int., 2000, 49, 1002–1006 CrossRef CAS.
- F. Aulenta, M. G. B. Drew, A. Foster, W. Hayes, S. Rannard, D. W. Thornthwaite, D. R. Worrall and T. G. A. Youngs, J. Org. Chem., 2005, 70, 63–78 CrossRef CAS PubMed.
- M. S. Diallo, L. Balogh, A. Shafagati, J. H. Johnson, W. A. Goddard and D. A. Tomalia, Environ. Sci. Technol., 1999, 33, 820–824 CrossRef CAS.
- A. Carlmark, E. Malmström and M. Malkoch, Chem. Soc. Rev., 2013, 42, 5858–5879 RSC.
- A. Stoddart, W. J. Feast and S. P. Rannard, Soft Matter, 2012, 8, 1096–1108 RSC.
- P. Ortega, M. J. Serramia, M. A. Munoz-Fernandez, F. J. de la Mata and R. Gomez, Tetrahedron, 2010, 66, 3326–3331 CrossRef CAS PubMed.
- D. Astruc, E. Boisselier and C. Ornelas, Chem. Rev., 2010, 110, 1857–1959 CrossRef CAS PubMed.
- J. M. Oliveira, A. J. Salgado, N. Sousa, J. F. Mano and R. L. Reis, Prog. Polym. Sci., 2010, 35, 1163–1194 CrossRef CAS PubMed.
- A. R. Menjoge, R. M. Kannan and D. A. Tomalia, Drug Discovery Today, 2010, 15, 171–185 CrossRef CAS PubMed.
- J. Lim, M. Kostiainen, J. Maly, V. C. P. da Costa, O. Annunziata, G. M. Pavan and E. E. Simanek, J. Am. Chem. Soc., 2013, 135, 4660–4663 CrossRef CAS PubMed.
- Y. Chang and C. Kim, J. Polym. Sci., Part A-1: Polym. Chem., 2001, 39, 918–926 CrossRef CAS.
- N. O'Brien, A. McKee, D. C. Sherrington, A. T. Slark and A. Titterton, Polymer, 2000, 41, 6027–6031 CrossRef.
- J. V. M. Weaver, S. P. Rannard and A. I. Cooper, Angew. Chem., Int. Ed., 2009, 48, 2131–2136 CrossRef CAS PubMed.
- T. He, D. J. Adams, M. F. Butler, C. T. Yeoh, A. I. Cooper and S. P. Rannard, Angew. Chem., Int. Ed., 2007, 46, 9243–9247 CrossRef CAS PubMed.
- T. He, D. J. Adams, M. F. Butler, A. I. Cooper and S. P. Rannard, J. Am. Chem. Soc., 2009, 131, 1495–1501 CrossRef CAS PubMed.
- F. L. Hatton, P. Chambon, T. O. McDonald, A. Owen and S. P. Rannard, Chem. Sci., 2014, 5, 1844–1853 RSC.
- R. B. Kolhatkar, K. M. Kitchens, P. W. Swaan and H. Ghandehari, Bioconjugate Chem., 2007, 18, 2054–2060 CrossRef CAS PubMed.
- H. Willcock, A. I. Cooper, D. J. Adams and S. P. Rannard, Chem. Commun., 2009, 3095–3097 RSC.
- B. Huang, J. F. Kukowska-Latallo, S. Tang, H. Zong, K. B. Johnson, A. Desai, C. L. Gordon, P. R. Leroueil and J. R. Baker, Bioorg. Med. Chem. Lett., 2012, 22, 3152–3156 CrossRef CAS PubMed.
- C. J. Hawker and J. M. J. Fréchet, J. Am. Chem. Soc., 1992, 114, 8405–8413 CrossRef CAS.
- K. Yoon, P. Goyal and M. Weck, Org. Lett., 2007, 9, 2051–2054 CrossRef CAS PubMed.
- P. Antoni, Y. Hed, A. Nordberg, D. Nystrom, H. von Holst, A. Hult and M. Malkoch, Angew. Chem., Int. Ed., 2009, 48, 2126–2130 CrossRef CAS PubMed.
- O. Thioune, H. Fessi, J. P. Devissaguet and F. Puisieux, Int. J. Pharm., 1997, 146, 233–238 CrossRef CAS.
- S. Schubert, J. T. Delaney and U. S. Schubert, Soft Matter, 2011, 7, 1581–1588 RSC.
- V. K. Lamer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847–4854 CrossRef CAS.
- C. Zhang, V. J. Pansare, R. K. Prud'homme and R. D. Priestly, Soft Matter, 2012, 8, 86–93 RSC.
- R. A. Slater, T. O. McDonald, D. J. Adams, E. R. Draper, J. V. M. Weaver and S. P. Rannard, Soft Matter, 2012, 8, 9816–9827 RSC.
- P. M. Valencia, P. A. Basto, L. Zhang, M. Rhee, R. Langer, O. C. Farokhzad and R. Karnik, ACS Nano, 2010, 4, 1671–1679 CrossRef CAS PubMed.
- J. Cheng, B. A. Teply, I. Sherifi, J. Sung, G. Luther, F. X. Gu, E. Levy-Nissenbaum, A. F. Radovic-Moreno, R. Langer and O. C. Farokhzad, Biomaterials, 2007, 28, 869–876 CrossRef CAS PubMed.
- J. Shi, Z. Xiao, N. Kamaly and O. C. Farokhzad, Acc. Chem. Res., 2011, 44, 1123–1134 CrossRef CAS PubMed.
- D. C. Dong and M. A. Winnik, Photochem. Photobiol., 1982, 35, 17–21 CrossRef CAS PubMed.
- K. Kalyanasundaram and J. K. Thomas, J. Am. Chem. Soc., 1977, 99, 2039–2044 CrossRef CAS.
- R. Duncan and R. Gaspar, Mol. Pharm., 2011, 8, 2101–2141 CrossRef CAS PubMed.
- S. Maher, T. W. Leonard, J. Jacobsen and D. J. Brayden, Adv. Drug Delivery Rev., 2009, 61, 1427–1449 CrossRef CAS PubMed.
- L. Profit, V. A. Eagling and D. J. Back, AIDS, 1999, 13, 1623–1627 CrossRef CAS PubMed.
- P. Palumbo, U. Picchini, B. Beck, J. van Gelder, N. Delbar and A. DeGaetano, J. Pharmacokinet. Pharmacodyn., 2008, 35, 235–248 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, full experimental details and characterisation. See DOI: 10.1039/c4sc02889a |
| This journal is © The Royal Society of Chemistry 2015 |