
Light-emitting properties of donor–acceptor and donor–acceptor–donor dyes in solution, solid, and aggregated states: structure–property relationship of emission behavior†
Tsutomu Ishi-i*a,
Kei Ikedab,
Michiaki Ogawaa and
Yutarou Kusakakib
aDepartment of Biochemistry and Applied Chemistry, National Institute of Technology, Kurume College, 1-1-1 Komorino, Kurume 830-8555, Japan. E-mail: ishi-i@Kurume-nct.ac.jp
bMaterial Engineering Advanced Course, Advanced Engineering School, National Institute of Technology, Kurume College, 1-1-1, Komorino, Kurume, Fukuoka 830-8555, Japan
First published on 14th October 2015
Abstract
In this paper, we report a systematic study on the light-emitting behavior of a series of triphenylamine-based donor–acceptor-type dyes in the solution and solid states as well as in the aggregated state in polar aqueous media. The emission band shifted bathochromically along with the decrease in the fluorescence quantum yield as the solvent polarity was increased from nonpolar cyclohexane to polar DMF. In a THF/water medium, the emission was quenched in a low water volume, whereas the emission was recovered and increased in a high water volume. In a low water volume, the dye molecules exist in a monomeric form, and the fluorescence quenching increases with increasing water fraction, similar to that observed in the solvent-polarity-dependence study. In contrast, the dye molecules aggregated in a high water volume. This is probably because the inside of aggregates is less polar than the outside, thus preventing nonradiative deactivation and recovering the emission. This unusual emission was achieved by triphenylamine-based dyes containing a relatively strong acceptor moiety such as quinoxaline, benzothiadiazole, and thiadiazolopyridine, providing longer-wavelength red and near-IR emission. In the benzothiadiazole-based dyes, when the phenyl groups in the donor moieties were replaced with methyl groups, the fluorescence quantum yield decreased, indicating that the triphenylamine donor moiety is suitable for emission in the aggregated state. The nonplanar structure of triphenylamine disrupts an ordered packing and produces a less-ordered spherical aggregate, leading to an efficient light emission even in polar aqueous media.
Introduction
Fluorescent organic dyes have attracted much interest because of their potential applications in material sciences such as organic light-emitting diodes and organic solid lasers,1,2 as well as in biological sciences such as probes and sensors.3,4 In general, an efficient light emission is observed in relatively diluted solution state, whereas the efficiency of light emission decreases in the solid state as well as in a highly concentrated solution state, causing fluorescence quenching.5,6 The fluorescence quenching can be attributed to aggregates formed in the concentrated medium, favoring the formation of a nonradiative deactivation channel. Such a fluorescence quenching increases in dyes with a planar structure and an extended π-conjugation system, even though the dyes exhibit interesting longer-wavelength emission.7Alternatively, push–pull-type conjugated systems comprising electron-donating and electron-accepting moieties are the simplest systems for generating longer-wavelength emissions.8 With the increase in the strength of donor–acceptor conjugates, the emission band shifts bathochromically, producing red and near-infrared (near-IR) light emissions.9,10 The red and near-IR light emissions produced by such systems are very important for biological applications, because they lie in the biological optical window.11 However, the efficiency of longer-wavelength emission decreases in polar aqueous media, because a highly polarized excited state arising from the donor–acceptor characteristic increases the formation of a nonradiative deactivation channel.12,13
In a previous communication, we reported that red-light emission can be attributed to the aggregation of donor–acceptor dyes comprising benzothiadiazole acceptor and triphenylamine donor moieties even in a polar aqueous medium.14 The prevention of the highly polarized excited state due to aggregation also contributes to red-light emission,15 in addition to the prevention of intramolecular rotation as widely known in aggregation-induced emission.16–20 Recently, some donor–acceptor systems were developed for aggregation-induced emission, where, in fact, red and near-IR light emissions were achieved in an aqueous medium containing an organic solvent.21–28 An amino donor moiety was combined with an acceptor moiety such as cyano-substituted oligo(phenylene vinylenes),21,22 triazines,23 fluorenones,24 boron dipyrromethenes,25 fumaronitriles,26 acrylonitriles,27 and quinoline-malononitriles.28 Important information about aggregation-induced emission can be obtained, if such interesting light-emitting systems are systematically studied by screening the donor and acceptor moieties. Thus, more efforts should be devoted to systematic studies on elucidating the structure–property relationship of this interesting emission behavior. In this study, a series of novel donor–acceptor-type dyes were synthesized, and a library of donor–acceptor light-emitting systems was developed as follows: first, various benzothiadiazole–triphenylamine-based donor–acceptor–donor and donor–acceptor dyes were studied to generalize the light emission behavior in the aggregated state. To further expand the generality, additional π-electron spacers such as ethylene, acetylene, and benzene moieties were introduced between the triphenylamine donor and benzothiadiazole acceptor moieties. Second, the benzothiadiazole acceptor moiety was replaced with weak acceptor moieties such as benzene, naphthalene, and thiazolothiazole, as well as with strong acceptor moieties such as quinoxaline, fluorobenzothiadiazole, and thiadiazolopyridine. In this donor–acceptor library, the characteristics of donor–acceptor conjugate were determined based on the combined electrochemical and photochemical results. The emission behavior in the aggregated state was systematically studied based on the donor–acceptor strength. Finally, the donor moiety was screened. In the triphenylamine moiety, the phenyl group was replaced with a methyl group to evaluate the steric and electronic effects of the donor moiety. In this paper, we report a systematic study on the light-emitting behavior of various donor–acceptor-type dyes and propose a convenient strategy for generating longer-wavelength emission based on the aggregation of donor–acceptor-type dyes.
Results and discussion
Preparation of dyes
Novel dyes 5, 7, 8, 9, 10, 11, and 12 (Scheme 1) were prepared following the methods shown in Scheme 2; the coupling reactions of dibromides or diiodides with the corresponding boronic acid in the presence of a Pd(0) catalyst were the key reactions. The dyes were characterized by spectroscopic methods and elemental analysis. Dyes 1a–d,29 2,30 3,15 4,29 and 6 (ref. 29) were prepared following the methods reported previously. The dyes were fluorescent in both the solution and solid states. | ||
| Scheme 1 Structures of donor–acceptor–donor and donor–acceptor-type dyes. | ||
 | ||
| Scheme 2 Preparation of donor–acceptor–donor-type dyes. | ||
Electrochemical characteristics
The donor–acceptor characteristics of the dyes were investigated by cyclic voltammetry (Table 1 and Fig. S1, ESI†). In triphenylamine-based dyes 1a–d, 2, 3, and 7–12, a quasireversible oxidation potential was applied between 0.45 and 0.56 V (vs. Fc/Fc+); this oxidation potential is responsible for the oxidation of electron–donating triphenylamine moiety. Dye 1b bearing an ethylene spacer moiety showed the lack of reversibility in the oxidation process. The oxidation potentials shifted to negative potentials when the phenyl group of the donor moiety was replaced with a methyl group. The order of electron–donating property increased as follows: dye 6 (0.25 V, vs. Fc/Fc+) bearing dimethylamino groups > dye 5 (0.37 V) bearing methylphenylamino groups > dye 1a (0.47 V) bearing diphenylamino groups. No oxidation potential was observed in dye 4 lacking triphenylamine donor moieties.| Comp. | Ered1/2b (V, vs. Fc/Fc+) | Eox1/2b (V, vs. Fc/Fc+) | ELUMOc (eV) | EHOMOc (eV) | ΔEc (eV) |
|---|---|---|---|---|---|
| a Measured in dichloromethane (5.0 × 10−4 M) in the presence of tetrabutylammonium hexafluoroborate (0.1 M) at a scan rate of 100 mV s−1.b Ered1/2 is the half-wave reduction potential, and Eox1/2 is the half-wave oxidation potential.c HOMO and LUMO level were calculated using the HOMO level of ferrocene (−4.80 eV). ELUMO = −(4.80 + Eredonset). EHOMO = −(4.80 + Eoxoffset). ΔE = ELUMO − EHOMO.d The quasireversible oxidation potentials could not be observed. | |||||
| 1a | −1.90 | 0.47 | −2.85 | −5.33 | 2.48 |
| 1b | −1.77 | 0.28, 0.50d | −2.98 | — | — |
| 1c | −1.65 | 0.56 | −3.10 | −5.44 | 2.34 |
| 1d | −1.86 | 0.48 | −2.90 | −5.34 | 2.44 |
| 2 | −1.93 | 0.49 | −2.83 | −5.34 | 2.51 |
| 3 | −1.89 | 0.51 | −2.86 | −5.36 | 2.50 |
| 4 | −1.90 | — | −2.85 | — | — |
| 5 | −1.94 | 0.37 | −2.81 | −5.22 | 2.41 |
| 6 | −2.03 | 0.25 | −2.72 | −5.13 | 2.05 |
| 7 | — | 0.45 | — | −5.34 | — |
| 8 | — | 0.49 | — | −5.34 | — |
| 9 | −2.52 | 0.50 | −2.21 | −5.40 | 3.19 |
| 10 | −2.14 | 0.50 | −2.61 | −5.38 | 2.77 |
| 11 | −1.86 | 0.51 | −2.90 | −5.40 | 2.50 |
| 12 | −1.64 | 0.52 | −3.11 | −5.41 | 2.30 |
In addition to the oxidation potential, a quasireversible reduction potential was observed between −1.64 and −2.52 V (vs. Fc/Fc+) for dyes 1a–d, 2, 3, 5, 6, and 9–12 bearing a relatively strong acceptor moiety, indicating the donor–acceptor characteristic. In contrast, the reduction potential was not observed in dyes 7 and 8 bearing a benzene moiety and a naphthalene moiety, respectively. In the triphenylamine-based donor–acceptor–donor dyes, the order of electron–accepting property increased as follows: dye 12 (−1.64 V, vs. Fc/Fc+) bearing a thiadiazolopyridine moiety > dye 11 (−1.86 V) bearing a fluorobenzothiadiazole moiety ≥ dye 1a (−1.90 V) bearing a benzothiadiazole moiety > dye 10 (−2.14 V) bearing a quinoxaline moiety > dye 9 (−2.52 V) bearing a thiazolothiazole moiety > dye 7/8 bearing a benzene/naphthalene moiety.
The HOMO and LUMO energy levels and band gaps were estimated from the reduction and oxidation potentials (Table 1). Quinoxaline-, benzothiadiazole-, and thiadiazolopyridine-based dyes 1a, 10, 11, and 12 exhibited smaller band gaps (2.30 to 2.77 eV) than benzene-, naphthalene-, and thiazolothiazole-based dyes 7, 8, and 9 (>3.19 eV). Thus, the dyes can be divided into two groups: a strong donor–acceptor system (dyes 1a and 10–12) and a weak donor–acceptor system (dyes 7–9). Moreover, the band gap decreased from dye 1a (2.48 eV) to dyes 5 (2.41 eV) and 6 (2.05 eV) by replacing the phenyl group of the donor moieties with a methyl group.
Absorption and emission behavior
In the UV/vis absorption spectra, the donor–acceptor–donor and donor–acceptor dyes showed an absorption band at approximately 350–500 nm; this can be attributed to the intramolecular charge-transfer transition arising from the donor–acceptor–donor and donor–acceptor chromophores.29 The absorption band (λabs) shifted slightly according to the solvent polarity (cyclohexane, toluene, THF, dichloromethane (DCM), and DMF) (Table S1, ESI†). The results indicate that the ground state is hardly affected by the difference in the solvent polarity.On the other hand, the fluorescence spectra showed a strong dependence on solvent polarity. The emission band (λem) shifted bathochromically along with the decrease in the fluorescence quantum yield (ΦF) as the solvent polarity was increased (Fig. 1, S2 and S3, and Table S1, ESI†).25–27 When the benzothiadiazole–triphenylamine dye 1a was dissolved in nonpolar solvents such as cyclohexane and toluene, a yellow light was emitted at 557–592 nm with a high ΦF value of 0.80–0.85 (Fig. 1 and Table S1, ESI†). The emission band shifted to the longer-wavelength orange-red-light region at 614, 639, and 657 nm in THF, DCM, and DMF, respectively. Along with the bathochromic shift, the ΦF value decreased from 0.85 in cyclohexane to 0.36 and 0.22 in DCM and DMF, respectively (Table S1, ESI†). This decrease in fluorescence correlated with the increase in polarity, which facilitates the excited-state polarization of the molecule through a charge-transfer process.21 The polarized excited-state is stabilized by solvation owing to the polar solvent molecules.15
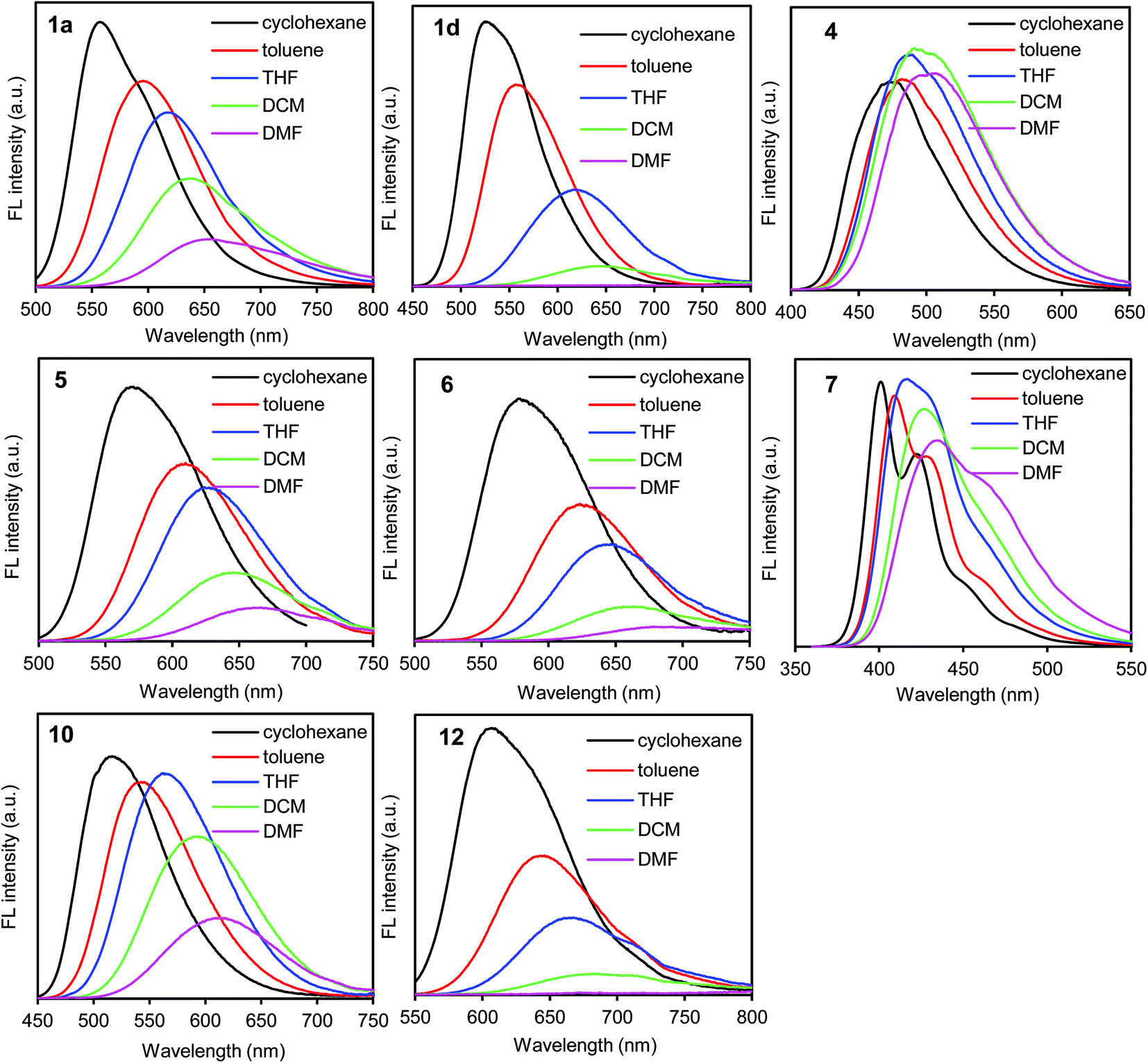 | ||
| Fig. 1 Fluorescence spectra of 1a (ex. 460 nm), 1d (ex. 420 nm), 4 (ex. 390 nm), 5 (ex. 450 nm), 6 (ex. 470 nm), 7 (ex. 360 nm), 10 (ex. 420 nm), and 12 (ex. 500 nm) in cyclohexane, toluene, THF, DCM, and DMF at 1.0 × 10−6 M. | ||
A similar trend of bathochromic shift along with the decrease in fluorescence was observed also in asymmetric donor–acceptor dyes 2 and 3 bearing one triphenylamine moiety and in donor–acceptor–donor dyes 1b–d bearing an additional π-electron spacer between the donor and acceptor moieties. In contrast, such a strong dependence on solvent polarity was not observed in dye 4 lacking triphenylamine donor moieties. The emission band shifted slightly in the 477–507 nm range (Fig. 1). The lacking donor moieties prevents the polarization of excited-state, leading to the slight solvent dependence. Significant polarity dependence was observed in dye 1d bearing an additional benzene π-spacer. The emission band shifted bathochromically from 527 nm in cyclohexane to 680 nm in DMF. The ΦF value was less than 0.01 in DMF. Compared to dye 1a, the bathochromic shift along with decrease in the emission of dye 1d increased significantly in polar media. The increased fluorescence can be attributed to twisted intramolecular charge transfer (TICT),25,31 as observed in the benzothiadiazole–triphenylamine dye bearing a benzene spacer.32 The difference in solvent-polarity dependence can be obtained from the Mataka–Lippert plot; the solvent effect can be evaluated using this plot from the dependence of the Stokes shift on the solvent polarity parameter.33 In the plot, the slope (13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700) of dye 1d is larger than that (8000) of dye 1a (Fig. S4, ESI†).
700) of dye 1d is larger than that (8000) of dye 1a (Fig. S4, ESI†).
In a polar DMF solution, the emission band shifted bathochromically from 434–522 nm in dyes 7–9 to 615–659 nm in dyes 1a, 10, and 11 in the triphenylamine-based donor–acceptor–donor dyes (Fig. 1, S2 and Table S1, ESI†). Along with the bathochromic shift, the ΦF value decreased from 0.55–0.60 in dyes 7–9 to 0.11–0.34 in dyes 1a, 10, and 11. In particular, a significant fluorescence quenching was observed in dye 12 bearing a strong acceptor thiadiazolopyridine moiety; the emission intensity was too weak to be detected. Thus, the solvent-polarity dependence increased as follows: dyes 12 > 11 ≥ 1a > 10 > 9 > 7/8. This trend is consistent with the electron-accepting property of the dyes as determined by cyclic voltammetry.
In the benzothiadiazole-based donor–acceptor–donor dyes 1a, 5, and 6, the solvent-polarity dependence increased by replacing the phenyl group of the donor moieties with a methyl group (Fig. 1, S2 and Table S1, ESI†). In DMF, the emission band shifted bathochromically from 657 nm in dye 1a to 666 and 679 nm in dyes 5 and 6, respectively, as predicted from the cyclic voltammetry data. A large fluorescence quenching (ΦFL = 0.05) was observed in dye 6 bearing strong electron-donating dimethylamino groups.
The emission observed in the solution state was observed also in the solid state with moderate ΦF values (Table S1, ESI†). This indicates that emission will occur even in aqueous systems upon the formation of aggregates.
Emission behavior in aggregated state
The emission in aqueous environments was studied using a THF/water mixture, in which the concentration of dye was maintained at 1 × 10−5 M, and the water fraction (vol%) was increased from 0% to 90%. All the dyes showed a change in the UV/vis absorption and fluorescence spectra depending on the water fraction (Fig. 2, 4, 6, and S5–S16, ESI†). At 0% water fraction (100% THF solution), dye 1a showed an orange-red emission at 614 nm with a high ΦF value of 0.71. As the water volume of the solution was increased to 60% water, the emission intensity decreased significantly to a ΦF value of 0.08 (Fig. 3c). In a low water volume, dye 1a existed in the monomeric form; the fluorescence quenching increased with solvent polarity, as observed in DMF. However, once the water fraction of the solution exceeded 70% water, the emission intensity recovered and increased gradually. Finally, the ΦF reached 0.80 with a 10-fold increase at the 90% water fraction (Fig. 3c). In a high water volume, the molecules of dye 1a started to aggregate. The inside of the aggregates is less polar than the outside, thus preventing the excited-state polarization and subsequent nonradiative deactivation. This leads to emission recovery.15 | ||
| Fig. 2 (a and b) UV/vis absorption and (c and d) fluorescence spectra of 1a in THF/water (10:0, 9:1, 8:2, 7:3, 6:4, 5:5, 4:6, 3:7, 2:8, and 1:9, (v/v)) at 1.0 × 10−5 M excited at 460 nm. | ||
 | ||
| Fig. 3 Plots of (a) the shift of the absorption band (Δλabs), (b) the shift of the emission band (Δλem), and (c) the fluorescence quantum yield versus the water fraction (vol%) of 1a, 1b, 1c, 1d, 2, and 3. In 1d, the data in the range of 60–70% water fraction was omitted because of precipitate formation. The absorption bands were detected at 460 nm for 1a, at 510 nm for 1b, at 479 nm for 1c, at 424 nm for 1d, at 436 nm for 2, and at 436 nm for 3 (at 0% water), and at 496 nm for 1a, at 537 nm for 1b, at 496 nm for 1c, at 443 nm for 1d, at 448 nm for 2, and at 462 nm for 3 (at 90% water). The emission bands were detected at 614 nm for 1a, at 640 nm for 1b, at 619 nm for 1c, at 617 nm for 1d, at 611 nm for 2, and at 613 nm for 3 (at 0% water), and at 610 nm for 1a, at 647 nm for 1b, at 622 nm for 1c, at 588 nm for 1d, at 601 nm for 2, and at 604 nm for 3 (at 90% water). | ||
The formation of aggregates in the emission recovery region was confirmed by the shifts in the absorption and emission bands. In dye 1a, the absorption band slightly changed to approximately 460–463 nm in the fluorescence quenching region (10–60% water fraction), indicating that the molecules of dye 1a existed as monomers (Fig. 3a). In the monomer region, the emission band shifted bathochromically from 614 nm to 639 nm due to the increase in polarity as observed in polar solvents such as DCM and DMF (Fig. 3b). On the other hand, in the emission recovery region (70–90% water fraction), the absorption band shifted bathochromically from 460 nm (60% water) to 496 nm (90% water) (Fig. 3a). The observed bathochromic shift (Δλabs) of 39 nm is most likely due to the formation of π-stacked aggregates with the J-type stacking mode, as predicted by the molecular exciton model.34 Moreover, a significant hypsochromic shift (Δλem) of the emission band was observed, from 639 nm (60% water) to 610 nm (90% water), despite an increase in the polarity (Fig. 3b). This Δλem value of 29 nm is most likely due to the local hydrophobic environment inside the aggregate structure. Thus, the fluorescence quenching was prevented by the aggregation, leading to the emission recovery and enhancement. The observed transition from the bathochromic to hypsochromic shift in the emission band is consistent with the change in the fluorescence quantum yields.
At 90% water fraction, the aggregate formation of dye 1a was confirmed and detected from the position of λFL at ∼610 nm. In the concentration range from 1 × 10−5 M to 1 × 10−10 M, the emission band hardly shifted. The fluorescence intensity at 610 nm correlated linearly with the concentration (Fig. S17 and S18, ESI†). The results indicate that the aggregates were maintained and did not dissociate into monomers even at a dilute concentration of 1 × 10−10 M.
A similar fluorescence emission behavior of aggregates was observed also in other triphenylamine–benzothiadiazole dyes 1b, 1c, and 1d bearing various π-spacers and also in asymmetric dyes 2 and 3 (Fig. 3 and S5–S9, ESI†). In these systems, the emission was quenched in a low water volume, whereas the emission was recovered and increased in a high water volume (Fig. 3c). In the emission recovery region, the bathochromic shift of the absorption band and the hypochromic shift of the emission band were observed also. Interestingly, dye 1d bearing a benzene pacer showed a large hypochromic shift of +29 nm at 90% water fraction, which is larger than those (−10 to +7 nm) of dyes 1a–c, 2, and 3. The hypochromic shift in dye 1d can be attributed to the TICT characteristic as indicated by the change in emission depending on solvent polarity. At the initial state of 0% water fraction, the emission band shifted to a longer-wavelength region because of the TICT effect, leading to a large hypochromic shift at 90% water fraction. Because of the bathochromic shift, the reddish-orange-light emission (at 627 nm) at 0% water fraction changed to an orange-light emission (at 588 nm) at 90% water fraction. The results indicate that the emission intensity as well as the emission color can be controlled by the combined effect of aggregation and TICT.
At 90% water fraction, dyes 1a, 1b, 1c, 1d, 2, and 3 showed red/orange-light emissions at 610 nm, 647 nm, 622 nm, 588 nm, 601 nm, and 604 nm (Fig. 8), with ΦF values of 0.80, 0.15, 0.28, 0.44, 0.54, and 0.47, respectively. The benzothiadiazole acceptor and triphenylamine donor moieties may play an important role in the emission of aggregates. Therefore, dye 4, without the triphenylamine donor moieties, as well as dyes 5 and 6 lacking the phenyl groups in the donor moieties were investigated (Fig. 4, 5, S10 and S11, ESI†).
 | ||
| Fig. 4 (a and b) UV/vis absorption and (c and d) fluorescence spectra of 4 in THF/water (10:0, 9:1, 8:2, 7:3, 6:4, 5:5, 4:6, 3:7, 2:8, and 1:9, (v/v)) at 1.0 × 10−5 M excited at 390 nm. | ||
 | ||
| Fig. 5 Plots of (a) the shift of the absorption band (Δλabs), (b) the shift of the emission band (Δλem), and (c) the fluorescence quantum yield versus the water fraction (vol%) of 1a, 4, 5, and 6. The absorption bands were detected at 460 nm for 1a, at 388 nm for 4, at 462 nm for 5, and at 476 nm for 6 (at 0% water), and at 496 nm for 1a, at 383 nm for 4, at 474 nm for 5, and at 529 nm for 6 (at 90% water). The emission bands were detected at 614 nm for 1a, at 489 nm for 4, at 625 nm for 5, and at 645 nm for 6 (at 0% water), and at 610 nm for 1a, at 499 nm for 4, at 625 nm for 5, and at 663 nm for 6 (at 90% water). | ||
In dye 4, the fluorescence emission system was divided into two regions of low and high water volumes, as observed in dyes 1a–d, 2, and 3. However, dye 4 showed a different light-emitting behavior. In a low water volume, the ΦF value remained unchanged at ∼0.8, whereas the ΦF value decreased significantly to 0.08 in a high water volume (Fig. 5c). Compared to dyes 1a–d, 2, and 3, the donor–acceptor effect in dye 4 was weakened due to the lack of triphenylamine donor moieties. The weak donor–acceptor effect also resulted in the light-emitting color of the 90% water fraction, a greenish-blue light emission with a λem of 499 nm (Fig. 8). The significant fluorescence quenching in a high water volume can be attributed to different aggregate morphologies as described below.
Next, the effect of a donor moiety on the aggregation and emission behaviors was evaluated. In dye 5 bearing methylphenylamino groups and dye 6 bearing dimethylamino groups, the transition from the initial to the final emission state via the fluorescence quenching state was observed, similar to that observed for the corresponding dye 1a bearing diphenylamino groups. At 90% water fraction, the emission band shifted bathochromically from 610 nm in 1a to 625 nm in 5 and to 663 in 6, by replacing the phenyl group of the donor moieties with a methyl group. The trend is consistent with the order of electron-donating characteristic as determined by cyclic voltammetry. Although an emission recovery was observed in a high water volume, the degree of emission recovery was different. Dye 5 showed a moderate recovery of ΦF from 0.59 (0% water fraction) to 0.49 (90%), whereas dye 6 showed only a weak recovery from 0.46 (0%) to 0.14 (90%) (Fig. 5c). The degree of emission recovery in dye 5 was similar to that (from 0.79 (0%) to 0.80 (90%)) of dye 1a. In addition to dye 1a, both dyes 4 and 5 formed aggregates in the emission recovery region, as shown by the bathochromic shift of the absorption band and the hypochromic shift of the emission band (Fig. 5a and b). However, a difference was observed in the hypochromic shift. As the water fraction was changed from 60% to 90%, large hypsochromic shifts of 29 and 30 nm in dyes 1a and 5 were achieved, respectively, whereas a small shift of 11 nm was observed in dye 6 (Fig. 5b). The aggregates in dye 6 bearing dimethylamino groups may have different morphologies and insufficient hydrophobic space inside the aggregate structure, leading to a low emission recovery. The results indicate that the diphenylamino (the triphenylamine moiety) and methylphenylamino groups contribute more to the emission recovery than the dimethylamino moiety. The superiority can be attributed to the nonplanarity of the donor moieties (the diphenylamino and methylphenylamino groups) as described below.
To generally develop this light-emission system, triphenylamine-based donor–acceptor–donor dyes 7–12 bearing various acceptor moieties were investigated in THF/water aqueous media (Fig. 6 and S12–16, ESI†). In the strong acceptor systems 10–12, emission recovery was also observed in a high water volume along with the bathochromic shifts of the absorption band and the hypsochromic shifts of the emission band, as observed in the benzothiadiazole acceptor system 1a (Fig. 6 and 7). In 90% water fraction, the emission band shifted bathochromically with the increase in the electron-accepting property as follows: 1a (610 nm) ≥ 11 (603 nm) > 10 (558 nm). In dye 12 bearing a thiadiazolopyridine acceptor moiety, the emission band was observed in the longest wavelength of 658 nm at 90% water fraction, thus extending the emission tail to the near-IR region. However, a relatively small ΦF value of 0.12 was obtained (Fig. 7c). The small value can be explained by the energy-gap law: the nonradiative deactivation rate increases exponentially with decreasing emission energy.35 Thus, it can be concluded that a turn-on-type change in emission was achieved by a triphenylamine-based dye system containing a strong acceptor moiety: the fluorescence-quenching OFF-state changed to the light-emitting ON-state upon the formation of aggregates.
 | ||
| Fig. 6 (a and b) UV/vis absorption and (c and d) fluorescence spectra of 7 in THF/water (10:0, 9:1, 8:2, 7:3, 6:4, 5:5, 4:6, 3:7, 2:8, and 1:9, (v/v)) at 1.0 × 10−5 M excited at 350 nm. | ||
 | ||
| Fig. 7 Plots of (a) the shift of the absorption band (Δλabs), (b) the shift of the emission band (Δλem), and (c) the fluorescence quantum yield versus the water fraction (vol%) of 7, 8, 9, 10, 11, and 12. In 11, the data at 60% water fraction was omitted because of precipitate formation. The absorption bands were detected at 356 nm for 7, at 343 nm for 8, at 426 nm for 9, at 412 nm for 10, at 455 nm for 11, and at 504 nm for 12 (at 0% water), and at 373 nm for 7, at 352 nm for 8, at 432 nm for 9, at 425 nm for 10, at 467 nm for 11, and at 523 nm for 12 (at 90% water). The emission bands were detected at 415 nm for 7, at 437 nm for 8, at 483 nm for 9, at 564 nm for 10, at 614 nm for 11, and at 665 nm for 12 (at 0% water), and at 415 nm for 7, at 436 nm for 8, at 489 nm for 9, at 558 nm for 10, at 603 nm for 11, and at 658 nm for 12 (at 90% water). | ||
On the other hand, weak acceptor systems 7–9 showed different emission behaviors (Fig. 7). In a low water volume, the emission intensity remained unchanged, because the highly polarized excited state could not be achieved by the weak donor–acceptor characteristic. The weak donor–acceptor effect was reflected also in the emitting colors: a blue-light emission with a λem of approximately 420–430 nm in dye 7, a blue-light emission of approximately 440–460 nm in dye 8, and a greenish-blue-light emission of approximately 480–500 nm in dye 9 were observed (Fig. 8). In a high water volume, the emission intensity decreased moderately (Fig. 7c). A similar behavior of unchanged emission and subsequent decrease in ΦF were also observed in dye 4 lacking triphenylamine donor moieties as described above. The difference between dyes 4 and 7–9 is the degree of decrease in ΦF. Compared to the large decrease in ΦF (ΦF = 0.08, 90% water fraction) in dye 4, a slight decrease in ΦF was observed in dyes 7–9. Thus, dyes 7–9 showed a moderate ΦF value even in a high water volume as follows: 0.25 in dye 7, 0.44 in dye 8, and 0.23 in dye 9 in 90% water fraction (Fig. 7c). These values are comparable to those of dyes 1a and 10–12 bearing strong acceptor moieties. However, a turn-on-type emission could not be achieved by the weak acceptor system because of the unchanged emission intensity in a low water volume. In the triphenylamine-based dyes, the strong donor–acceptor characteristic is very important for generating a turn-on-type emission in an aqueous environment.
 | ||
| Fig. 8 Fluorescence images of 1a–d, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, and 12 in THF/water (10:0, 9:1, 8:2, 7:3, 6:4, 5:5, 4:6, 3:7, 2:8, and 1:9, (v/v) from left to right) at 1.0 × 10−5 M under UV-light irradiation. | ||
As described above, the dyes have inherent emission behavior in the solid state (Table S1, ESI†). In the triphenylamine-based dyes, the ΦF values indicate a good relationship between the aggregated state (90% water fraction) and solid state (Fig. 9). For example, dyes 1a, 1d, 2, 3, 5, 8, and 11 with relatively large ΦF values in the solid state indicate relatively large ΦF values in 90% water fraction. It can be concluded that the inherent emission behavior in the solid state is preserved also in the aggregated state. In contrast, no relationship for ΦF value was observed in dye 4 lacking donor moieties. A small ΦF value of 0.08 was obtained in 90% water fraction; however, an excellent inherent emission behavior was observed in the solid state (ΦF = 0.92). On the other hand, dye 6 bearing dimethylamino groups showed a relatively good relationship for ΦF value, even though the inferior emission recovery in 90% water fraction indicated different aggregate morphologies.
 | ||
| Fig. 9 Plots of fluorescence quantum yields between the aggregated state (90% water fraction) and solid state in 1a–d, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, and 12. | ||
Aggregate analysis
The evidence for the formation of aggregates was obtained from dynamic light scattering (DLS) (Fig. S19 and S20, ESI†). In a high water volume, the donor–acceptor–donor and donor–acceptor dyes showed several hundred nanometer-sized aggregates. For example, above 70% water fraction, dye 1a showed particles with a size of 700–900 nm, whereas no particle was detected below 60% water fraction. In the dyes bearing a strong acceptor moiety (1a–d, 2, 3, 5, 6, and 10–12), the water fractions, where particles were formed, also showed emission recovery. The results indicate that the formation of aggregates results in light emission in an aqueous environment. The sizes of aggregates at 90% water fraction were 700, 270, 350, 250, 320, 390, 290, 870, 290, 290, and 380 nm for dyes 1a, 1b, 1c, 1d, 2, 3, 5, 6, 10, 11, and 12, respectively. Interestingly, dye 4 lacking a donor moiety also showed particles with a size of 800–1300 nm above 80% water fraction. Moreover, particles with a size of 320–640 nm were detected in weak acceptor systems 7–9 above 70% water fraction. Their particle-detected regions coincided with the emission-decreasing regions.The morphology of aggregates was observed by atomic force microscopy (AFM). In triphenylamine-based dyes 1a–d, 2, 3, and 7–12, a spherical aggregate morphology of several hundred nm size was observed (Fig. 10). The size of spherical aggregates is in good agreement with the DLS measurement (Fig. S19 and S20, ESI†). The spherical morphology can be attributed to a less-ordered aggregation owing to the nonplanar triphenylamine moieties, preventing an ordered packing of the molecules and thus producing an increased number of conformations of the molecules.36 The less-ordered aggregation was transformed to an ordered aggregation by replacing the phenyl group in the donor moieties with a methyl group. In dye 6 bearing dimethylamino groups, a crystalline-like corn-tassel-type aggregate was observed, probably composed of small aggregates. Thus, the aggregates lacking diphenylamino groups (triphenylamine moieties) have a different morphology and insufficient hydrophobic space inside the aggregate structure, leading to a low emission recovery. Furthermore, the crystallinity increased significantly in dye 4 lacking triphenylamine donor moieties, indicating a different plate form of aggregate (Fig. 10). The aggregation probably proceeded in an ordered manner due to the lack of triphenylamine moieties. Thus, it can be concluded that the triphenylamine moiety plays an important role in emission recovery.
 | ||
| Fig. 10 AFM images of 1a–d, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, and 12. The samples were prepared by drop-casting from the 1.0 × 10−4 M THF/water (1:9, v/v) solutions on freshly cleaved HOPG, followed by the vacuum evaporation. | ||
To investigate different aggregate morphologies, selected dyes (1a, 4, 6, and 12) were reprecipitated from concentrated THF/water solutions at 90% water fraction.37 In dye 4 lacking triphenylamine moieties, the X-ray diffraction (XRD) analysis showed a sharp reflection pattern, indicating the formation of crystalline aggregates (Fig. 11). The crystallinity was prevented by introducing a triphenylamine moiety in dyes 1a and 12, exhibiting a broad XRD pattern, indicating less-ordered aggregation (Fig. 11). Dye 6 bearing a dimethylamino group showed weak-intensity reflections, unlike the sharp XRD pattern observed in dye 4 and the broad XRD pattern observed in dyes 1a and 12. Therefore, the less-ordered aggregation arising from the nonplanar triphenylamine moieties is probably required for light emission in polar aqueous media.
 | ||
| Fig. 11 X-ray diffraction patterns of 1a, 4, 6, and 12. The samples were prepared by reprecipitation from THF/water (1:9, v/v) at 2.0 × 10−3 M. | ||
Conclusions
The light-emitting behavior in donor–acceptor and donor–acceptor–donor dyes in an aqueous environment was systematically studied. The dyes showed efficient light emission in a high water volume in THF/water media. The efficient light emission can be attributed to the aggregation of molecules in aqueous media. The aggregation efficiently prevents the fluorescence-quenching problem caused by the excited-state polarization arising from the donor–acceptor characteristic in polar aqueous media. The aggregates provide a less polar hydrophobic space inside the aggregate structure, thus avoiding excited-state polarization and subsequent fluorescence quenching. This increases emission efficiency. This light-emitting system can be developed using a series of benzothiadiazole–triphenylamine-based dyes, asymmetric donor–acceptor dyes, and symmetric donor–acceptor–donor dyes bearing an additional π-electron spacer between the donor and acceptor moieties. Furthermore, a generality of this approach was achieved by using a series of triphenylamine-based donor–acceptor–donor dyes bearing a strong acceptor moiety such as quinoxaline, fluorobenzothiadiazole, and thiadiazolopyridine. It is possible that a fluorescence-quenching OFF-state in a low water volume changes to a light-emitting ON-state upon the formation of aggregates in a high water volume. Such an interesting change in emission could not be obtained from triphenylamine-based donor–acceptor–donor dyes bearing a weak acceptor moiety such as benzene, naphthalene, and thiazolothiazole.The triphenylamine moiety plays an important role in the aggregation and emission behaviors. When the phenyl group in the donor moieties was replaced with a methyl group, the emission efficiency decreased. When a triphenylamine moiety was used as the donor moiety, the fluorescence-quenching problem arising from an ordered aggregation could be prevented. This is because the nonplanar structure of triphenylamine disrupts an ordered packing and produces a less-ordered spherical aggregate.
The emitted color can be controlled by tuning the acceptor moiety. With the increase in electron-accepting property, the emission band shifted to a longer wavelength, from the blue-light region to the red-light region. This red-light emission in aqueous media has great potential for biological applications, as the wavelength of the emission lies in the biological optical window. Moreover, this red-light emission can be generated by a two-photon excitation using a near-IR light source.29,30 The two-photon excitation is very useful for developing a biological system because of the increased penetration depth in tissues with less photodamage and pinpoint excitation by a focused light.38
We believe that this study provides a versatile strategy for achieving efficient light emission in an aqueous environment. The strategy is based on a simple combination of an electron-accepting moiety and an electron-donating amino moiety. Currently, new light-emitting systems are developed in our laboratory to improve the present strategy.
Experimental
General comments
All melting points are uncorrected. The IR spectra were recorded on a JASCO FT/IR-470 plus Fourier-transform infrared spectrometer and measured either as KBr pellets or on NaCl plates. The 1H NMR and 13C NMR spectra were obtained in CDCl3 using a JEOL JNM-LA 400 spectrometer. Residual solvent protons were used as the internal standard and chemical shifts (δ) are given relative to tetramethylsilane (TMS). The coupling constants (J) are reported in hertz (Hz). Elemental analysis was performed at the Elemental Analytical Center, Kyushu University. Fast atom bombardment mass spectrometry (FAB-MS) spectra were recorded using a JEOL JMS-70 mass spectrometer with m-nitrobenzyl alcohol (NBA) as the matrix. Analytical TLC was carried out on silica gel-coated on aluminum foil (Merck 60 F254). Column chromatography was carried out on silica gel (WAKO C300 or KANTO 60N). Gel permeation chromatography (GPC) was performed using a Japan Analytical Industry Co., Ltd LC-918 with a combination of JAIGEL-1H and JAIGEL-2H (20 × 600 mm) columns eluting with chloroform at a rate of 3.0 mL min−1.N-Methyldiphenylamine (13), bis(pinacolato)diboron, 1,4-diodobenzene (17), 4-diphenylaminophenylboronic acid (18), 1,4-dibromoanthracene (19), dithiooxamide (20), 4-(diphenylamino)benzaldehyde (21), and 4,7-dibromo-[1,2,5]thiadiazolo[3,4-c]pyridine (24) are commercially available and were used without purification. The benzothiadiazole dyes 1a–d,29 2,30 3,15 4,29 6,29 4,7-dibromo-2,1,3-benzothiadiazole (16),39 5,8-dibromoquinoxaline (22),40 and 4,7-dibromo-5-fluoro-2,1,3-benzothiadiazole (23)41 were prepared according to the same methods reported previously.
Instrumentation
Cyclic voltammetry was performed with a BAS100B/W (CV 50W) electrochemical workstation, by using a cell equipped with glassy carbon as the working electrode, a platinum wire as the counter electrodes, and Ag/AgNO3 as the reference electrode, and at scan rate of 100 mV s−1. All electrochemical measurements were performed in a dichloromethane solution (5 × 10−4 M) containing 0.10 M tetrabutylammonium hexafluorophosphate. The UV/vis spectra were measured on a JASCO V-570 spectrophotometer in a 1.0 cm width quartz cell. Fluorescence spectra were measured on a HITACHI F-4500 fluorescence spectrophotometer in a 1.0 cm width quartz cell. Fluorescence quantum yields were estimated by using quinine bisulfate (0.55 in 1 N sulfuric acid) as the reference for 4, 7, and 8, fluorescein (0.97 in ethanol) as the reference for 1a, 1c, 1d, 2, 3, 5, 6, 9, 10, and 11, and rhodamine B (0.65 in ethanol) as the reference for 1b and 12. Absolute fluorescence quantum yield for solid samples was determined by Hamamatsu Photonics C9920-01 Absolute PL Quantum Yield Measurement System. Dynamic light scattering (DLS) was performed on Photal OTSUKA ELECTRONICS DLS-7000 with a 632 nm He–Ne laser source, using a fixed angle (90 °C). DLS experiments were performed at 25 °C in a 21 mm width quarts cell. Atomic force microscopy images were obtained on a SII SPA400 DFM (tapping mode) by using SI-DF20 type tips. X-ray diffraction measurements were performed on RIGAKU RINT-TTR III and carried out with Cu(Kα) radiation from an X-ray tube with a 0.5 × 10 mm2 filament operated at 50 kV × 300 mA (15 kW).Aggregate preparation
THF stock solutions including the dyes were prepared at 1.0 × 10−4 M. A portion (1 mL) of the stock solution was mixed with appropriate volumes of THF and distilled water (the total volume of 10 mL). In all samples, the concentration was kept at 1.0 × 10−5 M and only the water fraction (vol%) was changed from 0% to 90%.Preparation of compounds
:1, v/v) to give 15 in 34% yield (209 mg, 068 mmol) as a white solid: mp 83–85 °C; IR (KBr, cm−1) 3087, 3035, 2975, 2927, 2874, 2816, 1610, 1591, 1495, 1371, 1363 (νBO), 1349, 1323, 1315, 1268, 1195, 1144, 1132, 1095, 861, 822, 701, 656; 1H NMR (400 MHz, CDCl3) δ 1.33 (s, 12H, CH3), 3.34 (s, 3H, CH3), 6.88 (d, J = 8.6 Hz, 2H, ArH), 7.09 (t, J = 7.5 Hz, 1H, ArH), 7.15 (d, J = 7.5 Hz, 2H, ArH), 7.33 (t, J = 7.5 Hz, 2H, ArH), 7.67 (d, J = 8.6 Hz, 2H, ArH). 13C NMR (100 MHz, CDCl3) δ 24.84, 40.11, 83.36, 116.13, 123.49, 123.81, 129.42, 135.92, 148.31, 151.38; FAB-MS (positive, NBA) m/z 309 (M+). Anal. calcd for C19H24BNO2 (309.21): C, 73.80; H, 7.82; N, 3.50. Found: C, 73.68; H, 7.76; N, 3.55.:1, v/v) and to give 5 in 69% yield (103 mg, 0.21 mmol). An analytical sample was obtained by recrystallization from hexane/dichloromethane as red powder: mp 190–191 °C; IR (KBr, cm−1) 3057, 3035, 2924, 2820, 1604, 1592, 1517, 1495, 1480, 1348, 1253, 1195, 1137, 1112, 888, 816, 753, 697; 1H NMR (400 MHz, CDCl3) δ 3.40 (s, 6H, CH3), 7.06 (t, J = 7.5 Hz, 2H, ArH), 7.10 (d, J = 8.6 Hz, 4H, ArH), 7.18 (d, J = 7.5 Hz, 4H, ArH), 7.34 (t, J = 7.5 Hz, 4H, ArH), 7.71 (s, 2H, ArH), 7.88 (d, J = 8.6 Hz, 4H, ArH); 13C NMR (100 MHz, CDCl3) δ 40.22, 118.15, 122.71, 122.77, 127.09, 129.03, 129.38, 129.87, 132.13, 148.48, 148.97, 154.25; FAB-MS (positive, NBA) 498 m/z (M+). Anal. calcd for C36H26N4S (498.64): C, 77.08; H, 5.26; N, 11.24. Found: C, 77.03; H, 5.25; N, 11.21.:1, v/v) and by recrystallization from hexane/chloroform to give 7 in 60% yield (340 mg, 0.602 mmol) as pale yellow powder: mp 243–244 °C; IR (KBr, cm−1) 3060, 3030, 1589, 1488, 1327, 1276, 818, 749, 695; 1H NMR (400 MHz, CDCl3) δ 7.04 (t, J = 7.3 Hz, 4H, ArH), 7.14 (d, J = 7.3 Hz, 8H, ArH), 7.15 (d, J = 8.8 Hz, 4H, ArH), 7.27 (t, J = 7.3 Hz, 8H, ArH), 7.52 (d, J = 8.8 Hz, 4H, ArH), 7.63 (s, 4H, ArH); 13C NMR (100 MHz, CDCl3) δ 122.93, 123.89, 124.43, 126.91, 127.57, 129.28, 134.56, 139.00, 147.16, 147.65; FAB-MS (positive, NBA) m/z 564 (M+). Anal. calcd for C42H32N2 (564.72): C, 89.33; H, 5.71; N, 4.96. Found: C, 89.43; H, 5.70; N, 4.95.:1, v/v) and by GPC eluting with chloroform to give 8 in 44% yield (272 mg, 0.44 mmol). An analytical sample was obtained by recrystallization from hexane/chloroform as white powder: mp 252–254 °C; IR (KBr, cm−1) 3062, 3032, 1590, 1508, 1492, 1387, 1314, 1272, 751, 697; 1H NMR (400 MHz, CDCl3) δ 7.06 (t, J = 7.3 Hz, 4H, ArH), 7.20 (d, J = 7.3 Hz, 8H, ArH), 7.20 (d, J = 7.8 Hz, 4H, ArH), 7.31 (t, J = 7.3 Hz, 8H, ArH), 7.41 (d, J = 7.8 Hz, 4H, ArH), 7.46–7.48 (m, 2H, ArH), 7.48 (s, 2H, ArH), 8.06–8.10 (m, 2H, ArH); 13C NMR (100 MHz, CDCl3) δ 122.96, 123.26, 124.54, 125.72, 126.46, 129.31 (×2), 130.89, 132.04, 134.74, 139.24, 147.02, 147.78; FAB-MS (positive, NBA) m/z 614 (M+). Anal. calcd for C46H34N2 (614.78): C, 89.87; H, 5.57; N, 4.56. Found C, 89.84; H, 5.61; N, 4.53.:1, v/v) and by GPC eluting with chloroform to give 9 in 48% yield (151 mg, 0.24 mmol). An analytical sample was obtained by recrystallization from hexane/dichloromethane orange powder: mp 289–290 °C; IR (KBr, cm−1) 3066, 3034, 3018, 1590, 1492, 1443, 1277, 749, 692; 1H NMR (400 MHz, CDCl3) δ 7.08 (d, J = 8.8 Hz, 4H, ArH), 7.10 (t, J = 7.3 Hz 4H, ArH), 7.16 (d, J = 7.3 Hz, 8H, ArH), 7.31 (t, J = 7.3 Hz, 8H, ArH), 7.81 (d, J = 8.8 Hz, 4H, ArH), 13C NMR (100 MHz, CDCl3) δ 121.93, 123.98, 125.32, 127.13, 127.24, 129.49, 146.89, 149.96, 150.15, 168.36; FAB-MS (positive, NBA) m/z 628 (M+). Anal. calcd for C40H28N4S2·(628.81): C, 76.40; H, 4.49; N, 8.91. Found: C, 76.45; H, 4.49; N, 8.90.:2, v/v) and by GPC eluting with chloroform to give 10 in 61% yield (373 mg, 0.605 mmol). An analytical sample was obtained by recrystallization from hexane/dichloromethane as pale yellow powder: mp 242–244 °C; IR (KBr, cm−1) 3063, 3031, 1588, 1511, 1488, 1466, 1333, 1317, 1276, 753, 697; 1H NMR (400 MHz, CDCl3) δ 7.06 (t, J = 7.3 Hz, 4H, ArH), 7.16–7.24 (m, 12H, ArH), 7.29 (t, J = 7.3 Hz, 8H, ArH), 7.62 (d, J = 8.8 Hz, 4H, ArH), 7.87 (s, 2H, ArH), 8.90 (s, 2H, ArH); 13C NMR (100 MHz, CDCl3) δ 122.55, 123.13, 124.86, 129.30, 129.97, 131.41, 131.91, 139.38, 141.22, 143.92, 147.39, 147.61; FAB-MS (positive, NBA) m/z 616 (M+). Anal. calcd for C44H32N4 (616.75): C, 85.69; H, 5.23; N, 9.08. Found: C, 85.78; H, 5.22; N, 9.06.:1, v/v) and by GPC eluting with chloroform to give 11 in 80% yield (309 mg, 0.648 mmol). An analytical sample was obtained by recrystallization from hexane/dichloromethane as an orange solid: mp 253–257 °C; IR (KBr, cm−1) 3060, 3034, 3006, 1589, 1557, 1515, 1487, 1326, 1317, 1277, 1195, 1180, 1043, 885, 830, 754, 697, 527, 514; 1H NMR (400 MHz, CDCl3) δ 7.07 (t, J = 8.8 Hz, 4H, ArH), 7.09 (t, J = 8.8 Hz, 4H, ArH), 7.18–7.23 (m, 12H, ArH), 7.30 (t, J = 8.8, Hz, 4H, ArH), 7.32 (t, J = 8.8, Hz, 4H, ArH), 7.61 (d, J = 11.7 Hz, 1H, ArH), 7.72 (d, J = 8.8 Hz, 2H, ArH), 7.88 (d, J = 8.8 Hz, 2H, ArH); 13C NMR (100 MHz, CDCl3) δ 116.5 (d, J = 16.5 Hz), 118.7 (d, J = 32.1 Hz), 122.18, 122.46, 123.38, 123.56, 125.06, 125.10, 129.35, 129.41, 129.91 (×2), 131.30, 131.34, 132.9 (d, J = 10.7 Hz), 147.27, 147.40, 147.90, 148.59, 151.16, 155.27 (d, J = 10.7 Hz), 159.56 (d, J = 249.5 Hz); FAB-MS (positive, NBA) 640 m/z (M+). Anal. calcd for C42H29FN4S (640.77): C, 78.73; H, 4.56; N, 8.74. Found: C, 78.40; H, 4.51; N, 8.44.:1, v/v) and by GPC eluting with chloroform to give 12 in 65% yield (203 mg, 0.325 mmol). An analytical sample was obtained by recrystallization from hexane/dichloromethane as a red powder: mp 185–187 °C; IR (KBr, cm−1) 3060, 3033, 1589, 1487, 1454, 1334, 1316, 1275, 754, 697; 1H NMR (400 MHz, CDCl3) δ 7.08 (t, J = 7.3 Hz, 2H, ArH), 7.10 (t, J = 7.3 Hz, 2H, ArH), 7.17–7.25 (m, 12H, ArH), 7.27–7.34 (m, 8H, ArH), 7.90 (d, J = 8.8 Hz, 2H, ArH), 8.53 (d, J = 8.8 Hz, 2H, ArH), 8.76 (s, 1H, ArH); 13C NMR (100 MHz, CDCl3) δ 121.78, 122.83, 123.49, 123.85, 125.03, 125.41, 125.65, 128.00, 129.39, 129.43, 129.75, 130.07, 130.77, 142.17, 147.08, 147.35, 148.39, 149.75, 149.90, 151.09, 156.75; FAB-MS (positive, NBA) m/z 623 (M+). Anal. calcd for C41H29N5S (623.77): C, 78.95; H, 4.69; N, 11.23. Found: C, 78.81; H, 4.63; N, 11.21.Acknowledgements
We thank Professor Dr Teruo Shinmyozu (Kyushu University) for the CV measurements, Professor Dr Akira Harada and Dr Toshio Ishioka (Kyushu University) for the DLS measurements, and Professor Dr Hiroyuki Nakamura and Professor Dr Masumi Miyazaki (AIST) for the absolute fluorescence quantum yield measurements. This study was partially supported by Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Culture, Sports, and Technology of Japan (No. 26410105) and by the Cooperative Research Program of Network Joint Research Center for Materials and Devices (Institute for Materials Chemistry and Engineering, Kyushu University).Notes and references
- (a) U. Mitschke and P. Bäuerle, J. Mater. Chem., 2000, 10, 1471–1507 RSC; (b) A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed.
- (a) I. D. W. Samuel and G. A. Turnbull, Chem. Rev., 2007, 107, 1272–1295 CrossRef CAS PubMed; (b) M. D. McGehee and A. J. Heeger, Adv. Mater., 2000, 12, 1655–1668 CrossRef CAS.
- (a) K. E. Sapsford, L. Berti and I. L. Medintz, Angew. Chem., Int. Ed., 2006, 45, 4562–4588 CrossRef CAS PubMed; (b) J. Liu, Z. Cao and Y. Lu, Chem. Rev., 2009, 109, 1948–1998 CrossRef CAS PubMed; (c) H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620–2640 CrossRef CAS PubMed; (d) Y. Takaoka, A. Ojida and I. Hamachi, Angew. Chem., Int. Ed., 2013, 52, 4088–4106 CrossRef CAS PubMed; (e) R. W. Sinkeldam, N. J. Greco and Y. Tor, Chem. Rev., 2010, 110, 2579–2619 CrossRef CAS PubMed; (f) J. Han and K. Burgess, Chem. Rev., 2010, 110, 2709–2728 CrossRef CAS PubMed.
- (a) G. Zlokarnik, P. A. Negulescu, T. E. Knapp, L. Mere, N. Burres, L. Feng, M. Whitney, K. Roemer and R. Y. Tsien, Science, 1998, 279, 84–88 CrossRef CAS; (b) B. A. Griffin, S. R. Adams and R. Y. Tsien, Science, 1998, 281, 269–272 CrossRef CAS; (c) A. Ojida, Y. Mito-oka, M. Inoue and I. Hamachi, J. Am. Chem. Soc., 2006, 124, 6256–6258 CrossRef PubMed; (d) H. Nonaka, S. Fujishima, S. Uchinomiya, A. Ojida and I. Hamachi, J. Am. Chem. Soc., 2010, 132, 9301–9309 CrossRef CAS PubMed; (e) Y. Taniguchi, R. Kawaguchi and S. Sasaki, J. Am. Chem. Soc., 2011, 133, 7272–7275 CrossRef CAS PubMed; (f) T. Tamura, Y. Kioi, T. Miki, S. Tsukiji and I. Hamachi, J. Am. Chem. Soc., 2013, 135, 6782–6785 CrossRef CAS PubMed; (g) I. Takashima, M. Kinoshita, R. Kawagoe, S. Nakagawa, M. Sugimoto, I. Hamachi and A. Ojida, Chem.–Eur. J., 2014, 20, 2184–2192 CrossRef CAS PubMed; (h) S.-n. Uno, M. Kamiya, T. Yoshihara, K. Sugawara, K. Okabe, M. C. Tarhan, H. Fujita, T. Funatsu, Y. Okada, S. Tobita and Y. Urano, Nat. Chem., 2014, 6, 681–689 CAS.
- M. Shimizu and T. Hiyama, Chem.–Asian J., 2010, 5, 1516–1531 CrossRef CAS PubMed.
- (a) H. Langhals, T. Potrawa, H. Noth and G. Linti, Angew. Chem., Int. Ed. Engl., 1989, 28, 478–480 CrossRef PubMed; (b) H. Langhals, O. Krotz, K. Polborn and P. Mayer, Angew. Chem., Int. Ed., 2005, 44, 2427–2428 CrossRef CAS PubMed.
- F. Würthner, Chem. Commun., 2004, 1564–1579 RSC.
- C. T. Chen, Chem. Mater., 2004, 16, 4389–4400 CrossRef CAS.
- (a) K. R. Justin Thomas, J. T. Lin, Y.-T. Tao and C.-H. Chuen, Adv. Mater., 2002, 14, 822–826 CrossRef; (b) W.-C. Wu, H.-C. Yeh, L.-H. Chan and C.-T. Chen, Adv. Mater., 2002, 14, 1072–1075 CrossRef CAS; (c) C.-H. Zhao, A. Wakamiya, Y. Inukai and S. Yamaguchi, J. Am. Chem. Soc., 2006, 128, 15934–15935 CrossRef CAS PubMed; (d) A. Wakamiya, K. Mori and S. Yamaguchi, Angew. Chem., Int. Ed., 2007, 46, 4273–4276 CrossRef CAS PubMed; (e) E. Yamaguchi, C. Wang, A. Fukazawa, M. Taki, Y. Sato, T. Sasaki, M. Ueda, N. Sasaki, T. Higashiyama and S. Yamaguchi, Angew. Chem., Int. Ed., 2015, 54, 4539–4543 CrossRef CAS PubMed; (f) A. Purc, K. Sobczyk, Y. Sakagami, A. Ando, K. Kamada and D. T. Gryko, J. Mater. Chem. C, 2015, 3, 742–749 RSC.
- (a) G. Qian, Z. Zhong, M. Luo, D. Yu, Z. Zhang, Z. Y. Wang and D. Ma, Adv. Mater., 2009, 21, 111–116 CrossRef CAS PubMed; (b) G. Qian and Z. Y. Wang, Chem.–Asian J., 2010, 5, 1006–1029 CrossRef CAS PubMed; (c) M. Shimizu, R. Kaki, Y. Takeda, T. Hiyama, N. Nagai, H. Yamagishi and H. Furutani, Angew. Chem., Int. Ed., 2012, 51, 4095–4099 CrossRef CAS PubMed; (d) J. Massin, A. Charaf-Eddin, F. Appaix, Y. Bretonnière, D. Jacquemin, B. van der Sanden, C. Monnereau and C. Andrauda, Chem. Sci., 2013, 4, 2833–2843 RSC; (e) L. Yao, S. Zhang, R. Wang, W. Li, F. Shen, B. Yang and Y. Ma, Angew. Chem., Int. Ed., 2014, 53, 2119–2123 CrossRef CAS PubMed.
- V. J. Pansare, S. Hejazi, W. J. Faenza and R. K. Prud'homme, Chem. Mater., 2012, 24, 812–827 CrossRef CAS PubMed.
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- (a) F. D. Lewis and J.-S. Yang, J. Am. Chem. Soc., 1997, 119, 3834–3835 CrossRef CAS; (b) C. Bohne, H. Ihmels, M. Waidelich and C. Yihwa, J. Am. Chem. Soc., 2005, 127, 17158–17159 CrossRef CAS PubMed; (c) J. Do, J. Huh and E. Kim, Langmuir, 2009, 25, 9405–9412 CrossRef CAS PubMed; (d) A. Marini, A. Muñoz-Losa, A. Biancardi and B. Mennucci, J. Phys. Chem. B, 2010, 114, 17128–17135 CrossRef CAS PubMed; (e) Y. Niko, S. Kawauchi and G. Konishi, Chem.–Eur. J., 2013, 19, 9760–9765 CrossRef CAS PubMed; (f) A. Suzuki, N. Nemoto, I. Saito and Y. Saito, Org. Biomol. Chem., 2014, 12, 660–666 RSC.
- T. Ishi-i, K. Ikeda, Y. Kichise and M. Ogawa, Chem.–Asian J., 2012, 7, 1553–1557 CrossRef CAS PubMed.
- T. Ishi-i, I. Kitahara, S. Yamada, Y. Sanada, K. Sakurai, A. Tanaka, N. Hasebe, T. Yoshihara and S. Tobita, Org. Biomol. Chem., 2015, 15, 1818–1828 Search PubMed.
- (a) J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed; (b) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC; (c) J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- B.-K. An, S.-K. Kown, S.-D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410–14415 CrossRef CAS PubMed.
- R. Deans, J. Kim, M. R. Machacek and T. M. Swager, J. Am. Chem. Soc., 2000, 122, 8565–8566 CrossRef CAS.
- (a) J. Liang, B. Z. Tang and B. Liu, Chem. Soc. Rev., 2015, 44, 2798–2811 RSC; (b) Z. Zhao, D. Liu, F. Mahtab, L. Xin, Z. Shen, Y. Yu, C. Y. K. Chan, P. Lu, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, B. Yang, Y. Ma and B. Z. Tang, Chem.–Eur. J., 2011, 17, 5998–6008 CrossRef CAS PubMed; (c) J. Wang, J. Mei, R. Hu, J. Zhi Sun, A. Qin and B. Z. Tang, J. Am. Chem. Soc., 2012, 134, 9956–9966 CrossRef CAS PubMed; (d) W. Qin, D. Ding, J. Liu, W. Z. Yuan, Y. Hu, B. Liu and B. Z. Tang, Adv. Funct. Mater., 2012, 22, 771–779 CrossRef CAS PubMed; (e) Y. Yuan, C.-J. Zhang, M. Gao, R. Zhang, B. Zhong Tang and B. Liu, Angew. Chem., Int. Ed., 2015, 54, 1780–1786 CrossRef CAS PubMed; (f) N. Zhao, Q. Gong, R. X. Zhang, J. Yang, Z. Y. Huang, N. Li and B. Z. Tang, J. Mater. Chem. C, 2015, 3, 839–8402 Search PubMed.
- (a) T. Sanji, K. Shiraishi, M. Nakamura and M. Tanaka, Chem.–Asian J., 2010, 5, 817–824 CrossRef CAS PubMed; (b) T. Hirose, K. Higashiguchi and K. Matsuda, Chem.–Asian J., 2011, 6, 1057–1063 CrossRef CAS PubMed; (c) T. Noguchi, T. Shiraki, A. Dawn, Y. Tsuchiya, L. T. N. Lien, T. Yamamoto and S. Shinkai, Chem. Commun., 2012, 48, 8090–8092 RSC; (d) S. Kamino, A. Muranaka, M. Murakami, A. Tatsumi, N. Nagaoka, Y. Shirasaki, K. Watanabe, K. Yoshida, J. Horigome, S. Komeda, M. Uchiyama and S. Enomoto, Phys. Chem. Chem. Phys., 2013, 15, 2131–2144 RSC; (e) R. Yoshii, A. Nagai, K. Tanaka and Y. Chujo, Chem.–Eur. J., 2013, 19, 4506–4512 CrossRef CAS PubMed; (f) B. Xu, J. Zhang, H. Fang, S. Ma, Q. Chen, H. Sun, C. Im and W. Tian, Polym. Chem., 2014, 5, 479–488 RSC.
- (a) B.-R. Gao, H.-Y. Wang, Y.-W. Hao, L.-M. Fu, H.-H. Fang, Y. Jiang, L. Wang, Q.-D. Chen, H. Xia, L.-Y. Pan, Y.-G. Ma and H.-B. Sun, J. Phys. Chem. B, 2010, 114, 128–134 CrossRef CAS PubMed; (b) B.-R. Gao, H.-Y. Wang, Z.-Y. Yang, H. Wang, L. Wang, Y. Jiang, Y.-W. Hao, Q.-D. Chen, Y.-P. Li, Y.-G. Ma and H.-B. Sun, J. Phys. Chem. C, 2011, 115, 16150–16154 CrossRef CAS.
- B. Wang, Y. Wang, J. Hua, Y. Jiang, J. Huang, S. Qian and H. Tian, Chem.–Eur. J., 2011, 17, 2647–2655 CrossRef CAS PubMed.
- (a) Z. Ning, Z. Chen, Q. Zhang, Y. Yan, S. Qian, Y. Cao and H. Tian, Adv. Funct. Mater., 2007, 17, 3799–3807 CrossRef CAS PubMed; (b) Y. Jiang, Y. Wang, J. Hua, J. Tang, B. Li, S. Qian and H. Tian, Chem. Commun., 2010, 46, 4689–4691 RSC.
- Y. Liu, X. Tao, F. Wang, X. Dang, D. Zou, Y. Ren and M. Jiang, J. Phys. Chem. C, 2008, 112, 3975–3981 CAS.
- R. Hu, E. Lager, A. Aguilar-Aguilar, J. Liu, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, Y. Zhong, K. S. Wong, E. Peña-Cabrera and B. Z. Tang, J. Phys. Chem. C, 2009, 113, 15845–15853 CAS.
- (a) X. Y. Shen, W. Z. Yuan, Y. Liu, Q. Zhao, P. Lu, Y. Ma, I. D. Williams, A. Qin, J. Zhi Sun and B. Z. Tang, J. Phys. Chem. C, 2012, 116, 10541–10547 CrossRef CAS; (b) X. Y. Shen, Y. J. Wang, E. Zhao, W. Z. Yuan, Y. Liu, P. Lu, A. Qin, Y. Ma, J. Z. Sun and B. Z. Tang, J. Phys. Chem. C, 2013, 117, 7334–7347 CrossRef CAS.
- W. Z. Yuan, Y. Gong, S. Chen, X. Y. Shen, J. W. Y. Lam, P. Lu, Y. Lu, Z. Wang, R. Hu, N. Xie, H. S. Kwok, Y. Zhang, J. Z. Sun and B. Z. Tang, Chem. Mater., 2012, 24, 1518–1528 CrossRef CAS.
- A. Shao, Y. Xie, S. Zhu, Z. Guo, S. Zhu, J. Guo, P. Shi, T. D. James, H. Tian and W.-H. Zhu, Angew. Chem., Int. Ed., 2015, 54, 7275–7280 CrossRef CAS PubMed.
- S. Kato, T. Matsumoto, M. Shigeiwa, H. Gorohmaru, S. Maeda, T. Ishi-i and S. Mataka, Chem.–Eur. J., 2006, 12, 2303–2317 CrossRef CAS PubMed.
- T. Ishi-i, Y. Taguri, S. Kato, M. Shigeiwa, H. Gorohmaru, S. Maeda and S. Mataka, J. Mater. Chem., 2007, 17, 3341–3346 RSC.
- (a) W. Ritter, Angew. Chem., Int. Ed. Engl., 1986, 25, 971–988 CrossRef PubMed; (b) Z. R. Grabowski and K. Rotkiewicz, Chem. Rev., 2003, 103, 3899–4031 CrossRef PubMed.
- S. D. A. Sandanayaka, Y. Araki, Y. Taguri, T. Ishi-i, S. Mataka and O. Ito, J. Phys. Chem. B, 2005, 109, 22502–22512 CrossRef PubMed.
- (a) E. Z. Lippert, Z. Naturforsch., A: Phys. Sci., 1955, 10, 541–545 Search PubMed; (b) N. Mataga, Y. Kaifu and M. Koizumi, Bull. Chem. Soc. Jpn., 1956, 29, 465–470 CrossRef CAS.
- M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CrossRef CAS.
- J. V. Caspar, E. M. Kober, B. P. Sullivan and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 630–632 CrossRef CAS.
- P. Strohriegl and J. V. Gazulevicius, Adv. Mater., 2002, 14, 1439–1452 CrossRef CAS.
- For dye 1a, fluorescence spectrum of the reprecipitated solid sample was measured and the data was compared to that of the aggregate solution (THF/water, 90% water). The fluorescence spectrum (610 nm) of the solid sample was similar to that (610 nm) of the aggregate solution. The result indicates that the aggregate structure formed in THF/water media is preserved in the solid state.
- (a) S. R. Marder, Chem. Commun., 2006, 131–134 RSC; (b) G. S. He, L.-S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245–1330 CrossRef CAS PubMed; (c) S. Sumalekshmy and C. J. Fahrni, Chem. Mater., 2010, 23, 483–500 CrossRef.
- K. Pilgram, M. Zupan and R. Skiles, J. Heterocycl. Chem., 1970, 7, 629–633 CrossRef CAS PubMed.
- M. J. Edelmann, J.-M. Raimundo, N. F. Utesch, F. Diederich, G. Boudon, J.-P. Gisselbrecht and M. Gross, Helv. Chim. Acta, 2002, 85, 2195–2213 CrossRef CAS.
- (a) T. S. Poll, J. A. Love, T.-Q. Nguyen and G. C. Bazan, Adv. Mater., 2012, 24, 3646–3649 CrossRef PubMed; (b) S. Albrecht, S. Janietz, W. Schindler, J. Frisch, J. Kurpiers, S. Inal, P. Pingel, K. Fostiropoulos, N. Koch and D. Neher, J. Am. Chem. Soc., 2012, 134, 14932–14944 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The cyclic voltammograms, the fluorescence spectra in various solvents, the fluorescence images in various solvents, the plots of the Stokes shift against the solvent polarity parameter, the UV/vis and fluorescence spectra in THF/water, and the DLS charts in THF/water. See DOI: 10.1039/c5ra18231j |
| This journal is © The Royal Society of Chemistry 2015 |