
Triple helical collagen-like peptide interactions with selected polyphenolic compounds
Abstract
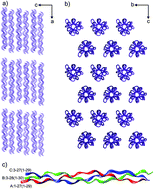
Because collagen is the most abundant component of connective tissue, it is an excellent biomaterial in numerous medical applications. However, the utility of collagen is limited by its low mechanical strength in aqueous solutions and its susceptibility to proteolytic degradation in vivo. To improve the physical properties of collagen and to enhance its chemical resistance, it is necessary to stabilize its structure through chemical or physical modifications. In this study, we analyzed the interactions of a model molecule, a synthetic triple helical collagen-like peptide, with polyphenols such as curcumin, rutin, quercetin, naringin, and hypericin. Interactions between the peptide and polyphenolic compounds were analyzed using various techniques. The layer-by-layer assembly processes of a gold surface using the peptide and polyphenols was performed via surface plasmon resonance (SPR), atomic force microscopy (AFM), and ellipsometry. SPR screening of polyphenols was conducted in real time to select compounds that bind to the collagen-like peptide and could thus be applied to the stabilization of collagen. Selected polyphenols, especially naringin and hypericin, demonstrated notable binding to the peptide. To determine the nature of these interactions, experiments were supplemented with crystallographic studies and molecular docking of plant metabolites and collagen-like peptides.
 Please wait while we load your content...
Please wait while we load your content...

