
Redox-sensitive mesoporous silica nanoparticles functionalized with PEG through a disulfide bond linker for potential anticancer drug delivery
Huameng Gong,
Zhifei Xie,
Mingxing Liu*,
Hongda Zhu and
Honghao Sun*
School of Food and Pharmaceutical Engineering, Key Laboratory of Fermentation Engineering (Ministry of Education), Hubei Provincial Cooperative Innovation Center of Industrial Fermentation, Hubei University of Technology, Wuhan 430068, PR China. E-mail: lmxing@mail.hbut.edu.cn; sunhonghao@mail.hbut.edu.cn; Fax: +86 27 59750481; Tel: +86 27 59750482
First published on 3rd July 2015
Abstract
In this paper, redox-sensitive mesoporous silica nanoparticles functionalized with polyethylene glycol (PEG) through a disulfide bond linker (MSNs–SS–PEG) were successfully synthesized using silica nanoparticles modified with a thiol group (MSNs–SH) and thiol-functionalized methoxy polyethylene glycol (MeOPEG–SH). Meanwhile, the particle size, pore size and structural properties of these materials were characterized by transmission electron microscopy (TEM), field emission scanning electron microscopy (FE-SEM), dynamic light scattering (DLS), Fourier transform infrared spectroscopy (FT-IR), X-ray diffraction (XRD) and nitrogen adsorption–desorption measurements. Furthermore, the in vitro drug release behaviour of DOX-loaded MSNs–SS–PEG (DOX@MSNs–SS–PEG) was investigated. It was shown that DOX release was markedly accelerated with the increasing concentration of glutathione (GSH), while DOX was not released from the carrier materials in the absence of GSH. Cytotoxicity evaluation revealed the good biocompatibility of the blank nanoparticles and the DOX@MSNs–SS–PEG exhibited comparative anticancer activity to free DOX towards BEL-7402 cells. Therefore, the MSNs–SS–PEG might be a great potential carrier for anticancer drug delivery.
Introduction
In the past decade, ordered mesoporous silica nanoparticles (MSNs) with a series of advantages, such as tremendous surface area, adjustable pore diameter, regular pore structure and facile modification to both internal and external surfaces, have been considered as an ideal vehicle to deliver catalysts, drugs and other functional molecules.1–5 For example, Xu et al. reported a facile and effective method for preparation of hollow MSNs, which exhibited high loading capacity due to their unique porous properties and high surface areas as a platform for drug delivery.6 Meanwhile, owing to the good biocompatibility of MSNs, they can undergo endocytosis by cellular uptake.7,8 However, premature release from MSNs, which could cause serious side effects due to widespread distribution, is undesirable. Therefore, surface functionalization is particularly important for specific cell targeting or for pore gating. The MSNs, especially functionalized with stimuli responsive materials, could deliver a specific drug to the homologous target site. Based on the different signals in vivo or in vitro, such as pH,9,10 temperature,11–13 enzymes14,15 and reducing agents,16–18 the drugs could release in the target site in a more controlled way. Patra and co-workers have successfully encapsulated curcumin inside the micro/nanocapsules by using polymeric materials, inorganic salts and silica nanoparticles, which could realize the control release of curcumin through pH trigger.19,20 In addition, it would markedly enhance the specificity and efficacy of the cancer treatment to employ specific novel drugs along with functionalized MSNs.21 Tian and co-workers have reported that paclitaxel-loaded MSNs significantly increased apoptosis when cultured with MCF-7 cells compared to free drug molecules, which proved MSNs applying for drug delivery could improve the efficiency of drug molecules.22 So the MSNs have a potential application in the drug delivery systems.Polyethylene glycol (PEG) is widely used due to its good biocompatibility.23,24 It can form shield effect through wrapping up the ordered mesoporous materials, which renders them possible to escape the absorption of the reticuloendothelial system (RES).25,26 Moreover, drug carrier modified with PEG can prolong circulation time in vivo and improve the enhanced permeability and retention (EPR) effect.27–29
It was reported that the concentration of GSH in the tumor cells (2–8 mmol L−1) is almost 1000 fold higher than that of in the plasma environment (1–2 μmol L−1).30 The GSH could be responsive to the disulfide bond,31 so drug could be released from the functionalized materials in specific times in tumour cells. The unique feature has greatly promoted the development of redox-responsive control carriers for intracellular delivery of drugs. For example, Rosenholm and co-workers have synthesized a novel MSNs functionalized with a cleavable disulfide bond in the spacer arm and organic surface linkers bearing an amino group end by successive co-condensation and covalent modification to deliver short oligonucleotides.32 Herein, in our work, we designed a type of redox-sensitive mesoporous silica nanoparticles (MSNs–SS–PEG) by grafting PEG–SH onto the surface of MSNs–SH via the disulfide bond linker. There into, the MeOPEG (mol. wt 5000) modified with thiol group (PEG–SH) was achieved through Michael addition reaction. Meanwhile, the properties of these materials, the drug loading and release profile were investigated. Also, in vitro cytotoxicity to the BEL-7402 cells was evaluated in detail in our work.
Experimental
Materials
N-Cetyltrimethylammonium bromide (CTAB) (99%), tetraethyl orthosilicate (TEOS) (98%), (3-mercaptopropyl) trimethoxysilane (MPTMS) (95%), 2,2-dipyridyl disulfide (PyssPy) (98%), 1,3-dithiolpropane (DPP) (98%), acryloyl chloride (AC) (96%), and doxorubicin hydrochloride (DOX) (98%) were purchased from Aladdin Chemistry, Co. (Shanghai). Methoxy polyethylene glycol (MeOPEG, mol. wt 5000) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Aldrich. Nanopure water was deionized to 18.3 MΩ cm in a water purification system (Human up 900). All the chemicals were analytical grade and used without further treatment.Measurements
1H NMR spectra were recorded on a Bruker AM-400 NMR spectrometer in dimethyl sulfoxide (DMSO). Fourier transform infrared (FT-IR) spectra were recorded on a Nicolet Nexus 470. Malvern Zetasizer Nano ZS90 was used to determine the average particle size. Powder X-ray diffraction patterns (XRD) were collected with an X-ray diffractometer (Bruker D8 ADVANCE) using CuKα radiation (2θ: 1.8°–10°; step size: 0.005°). Particle morphologies were observed by using a JEOL JSM-5510LV Field Emission Scanning Electron Microscopy (FE-SEM) with a 20 kV acceleration voltage. The pore structures were determined by nitrogen sorption isotherms at 77 K using a micromeritics ASAP 2020M sorptometer. The surface areas were calculated by the Brunauer–Emmett–Teller (BET) method, and the pore size distributions were calculated by the Barrett–Joyner–Halenda (BJH) method. Transmission electron microscopy (TEM) images were obtained on a JEOL JEM-2100F transmission electron microscope, and samples for TEM measurements were made by casting one drop of the sample's ethanol solution on carbon-coated copper grids.Synthesis of MSNs–SH
MSNs–SH was prepared by the co-condensation method.33 N-Cetyltrimethylammonium bromide (CTAB, 0.25 g) was dissolved in the 120 mL of nanopure water. NaOH (aq) (0.875 mL, 2.0 M) was introduced to the CTAB solution at 55 °C. After vigorous stirring for 1 h, tetraethoxysilane (TEOS, 1.25 mL, 5.47 mmol) was added dropwise to the CTAB solution under vigorous stirring, followed by the dropwise addition of (3-mercaptopropyl) trimethoxysilane (MPTMS, 0.24 mL, 1.23 mmol). The mixture was allowed to stir for 2 h to generate a white precipitate. The solid product was filtered, washed with nanopure water and methanol, and dried under high vacuum at 45 °C for 12 h. The CTAB surfactant was removed by refluxing the material (1.0 g) in 1.0 mL of HCl (37.4%) and 100 mL of methanol for 12 h.Synthesis of MeOPEG–SH
MeOPEG–AC–DPP (MeOPEG–SH) was synthesized on the basis of the previously reported.34,35 MeOPEG (62.5 g, 12.5 mmol OH) was distilled in 100 mL toluene under nitrogen, removing about 90 mL of toluene by azeotropic distillation. The solution was cooled in the room temperature bath under nitrogen. Anhydrous dichloromethane (50 mL) was added into the solution. The 2.65 mL of anhydrous triethylamine (19.0 mmol) was added dropwise with stirring, followed by the dropwise addition of 1.6 mL of acryloyl chloride (AC) (19.0 mmol). The reaction was carried out at 25 °C for 24 h in the dark under nitrogen. The solution was filtered through paper until clear, followed by precipitation in diethyl ether. The methoxypolyethylene glycol–acrylate (MeOPEG–AC) was collected by filtration and dried under vacuum at 35 °C for 12 h. The obtained product was then dissolved in 100 mL deionized water. The pH was adjusted to pH 6 with sodium hydroxide, and extracted three times with 100 mL dichloromethane. The dichloromethane washes were combined, and the product was precipitated in diethyl ether and dried under vacuum at 35 °C for 12 h.A 100 mL three-necked flask equipped with a magnetic stirrer and a 50 mL dropping funnel under a nitrogen atmosphere, were charged with 1,3-propanedithiol (DPP) (3.5 g, 32.4 mmol) and methanol (10 mL). Then, MeOPEG–AC (15.3 g, 2.8 mmol) in dry methanol (30 mL) was added dropwise. The reaction was allowed to proceed at 25 °C for 24 h. The product (MeOPEG–SH) was isolated by precipitation into cold diethyl ether, filtration, and dried under vacuum at 35 °C for 12 h.
Synthesis of MSNs–SS–PEG
The synthesis route of MSNs–SS–PEG was shown in Fig. 1A. The MSNs–SS–PEG was synthesized according to the similar reported procedures.36 MSNs–SH (100 mg) was added in a solution of 2,2-dipyridyl disulfide (PyssPy) (48 mg) in 100 mL phosphate buffer solution (PBS, 10 mM, pH 7.4). After stirring at 25 °C for 12 h, the 2-pyridinyldisulfanylpropyl functionalized silica products were isolated and washed with methanol and PBS buffer. The purified materials were introduced to the 100 mL PBS solution of MeOPEG–SH (128 mg, 24.8 mmol). The mixture was stirred at 25 °C for 24 h, then separated by centrifugation (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 20 min). The product (MSNs–SS–PEG) washed with methanol and PBS buffer, and dried under high vacuum at 45 °C for 12 h.
000 rpm, 20 min). The product (MSNs–SS–PEG) washed with methanol and PBS buffer, and dried under high vacuum at 45 °C for 12 h.
 | ||
| Fig. 1 (A) Synthesis route and (B) the diagrammatic sketch of mesoporous silica nanoparticles functionalized with PEG through a disulfide bond linker (MSNs–SS–PEG). | ||
Preparation of DOX@MSNs–SS–PEG
Typically, DOX (8 mg) as a model drug was dissolved in 30 mL of PBS. Then MSNs–SH (50 mg) was added in the above solution. The mixture was sonicated for 30 min, and stirred at 30 °C for 24 h. The precipitate was centrifuged, washed extensively with PBS to remove the redundant DOX on the surface of MSNs–SH. Subsequently, the above precipitate were added in the 10 mL PBS solution of PyssPy (50 mg), and stirred at 25 °C for 12 h. The precipitate was centrifuged and washed with PBS. The obtained product was introduced to the 30 mL PBS solution of MeOPEG–SH (65 mg). The mixture was stirred at 25 °C for 24 h, then separated by centrifugation (12000 rpm, 10 min). The precipitate (DOX@MSNs–SS–PEG) washed with methanol and PBS, and dried under high vacuum at 45 °C for 12 h.
The centrifugate was collected and measured by UV-vis spectrophotometer at 480 nm to calculate the drug loading content and entrapment efficiency of MSNs–SS–PEG by the following equations.
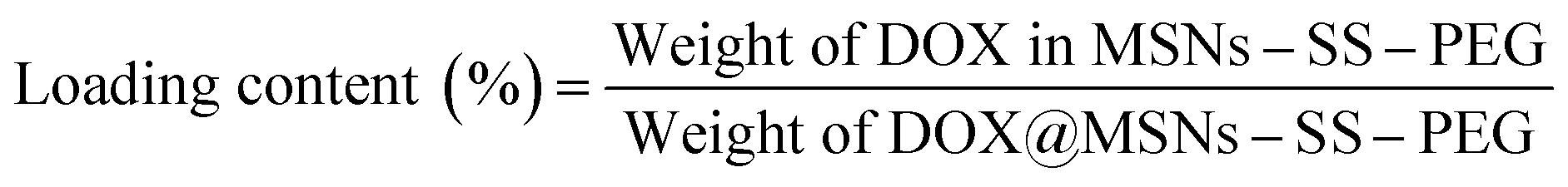

In vitro drug release
The release of DOX from MSNs–SS–PEG was evaluated by dialysis method. The DOX@MSNs–SS–PEG (8 mg) was dispersed in PBS with different concentration of GSH (0 mM, 4 mM, 7 mM, and 10 mM) under ultrasonic, and the dispersion was transferred to a dialysis bag. Then the bag was immersed in 150 mL of PBS, shaking at 150 rpm (37 °C). At predetermined time intervals, 2 mL of PBS was taken out from the system and immediately supplemented with an equal volume of fresh PBS. The release content of DOX was analysed by UV-vis spectrophotometer at 480 nm.Cytotoxicity test
The cytotoxicity effect of free DOX, MSNs–SS–PEG and DOX@MSNs–SS–PEG was evaluated by MTT assay against BEL-7402 cells (a human hepatoma cell line). The BEL-7402 cells were cultured and prepared as previous work.37 The BEL-7402 cells were seeded in a 96-well plates with the density of 1 × 104 cells per well and incubated at 37 °C for 24 h. Then the samples with a series of predetermined concentration were prepared and added into the experimental well for 24 h. After that, MTT solution (20 μL, 5 mg mL−1 in PBS) was added and the plate was incubated for another 4 h. In order to dissolve the formazan crystals, the medium was removed and replaced with DMSO (150 μL) in each well. Finally, the plates were shaken for 10 min, and the absorbance was measured by microplate reader at 490 nm. The percentage of cell viability was calculated relative to negative control (media alone). Three replicates were performed for each sample and the mean values were used as the final data.Results and discussion
Synthesis and characterization
To confirm the successful conjugation of –SH on to the surface of PEG, the FT-IR spectra of the MeOPEG, the MeOPEG–AC, and the MeOPEG–SH was shown in Fig. 2. Compared with MeOPEG, a new absorption peak appeared at 1736 cm−1 was attributed to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in Fig. 2B. While the primary carbonyl absorption peak at 1736 cm−1 was remained in Fig. 2C, a new absorption peak at 2560 cm−1 was ascribed to the vibration of –SH. These results demonstrated that the –SH had successfully coupled with MeOPEG.
O in Fig. 2B. While the primary carbonyl absorption peak at 1736 cm−1 was remained in Fig. 2C, a new absorption peak at 2560 cm−1 was ascribed to the vibration of –SH. These results demonstrated that the –SH had successfully coupled with MeOPEG.
 | ||
| Fig. 2 FT-IR spectra of MeOPEG (A), MeOPEG–AC (B), MeOPEG–SH (C), MSNs (D), MSNs–SH (E), MSNs–SS–PEG (F). | ||
Besides, a comparison of the FT-IR spectra of the MSNs, the MSNs–SH, and the MSNs–SS–PEG was shown in Fig. 2. The characteristic peaks showed at 1080 cm−1, 800 cm−1 and 960 cm−1 in Fig. 2D could be ascribed to the vibrations of asymmetric stretching Si–O–Si, symmetric stretching Si–O–Si and stretching vibrations of Si–OH groups, respectively. In Fig. 2E, an additional adsorption peak showed at 2555 cm−1, which was ascribed to the stretching vibration of –SH, confirmed that the thiol group was successfully modified on the MSNs. After grafting with MeOPEG–SH, the typical vibration bands of silica remained and the stretching vibration of CO at 1736 cm−1 appeared in Fig. 2F, indicated the successful grafting of MeOPEG–SH on the surface of MSNs via a disulfide bond.
The structures of MSNs–SH and MSNs–SS–PEG were also determined by 1H NMR analysis (Fig. 3). As displayed in Fig. 3A, the presence of peak at δ 1.06 and δ 2.51 corresponded to the protons of –SH and –OH of MSNs–SH, respectively, which indicated the structure of the thiol-modified silica nanoparticles. In Fig. 3B, the peak at δ 3.43 could be ascribed to the proton of the methoxy in MSNs–SS–PEG. Meanwhile, the proton peak of –OH was maintained and the proton peak of –SH was disappeared in MSNs–SS–PEG. These results suggested that the MeOPEG–SH successfully grafted onto the surface of MSNs–SH via the disulfide bond linker. In addition, the other peaks appeared at δ 2.55–δ 4.2 could be attributed to the methylene protons of MSNs–SS–PEG.
 | ||
| Fig. 3 1H NMR spectra of MSNs–SH (A) and MSNs–SS–PEG (B) in DMSO. | ||
Dynamic light scattering (DLS) was used to determine the average particle size and the zeta potential of the silica materials. As indicated in Table 1, the average size of the MSNs–SH and MSNs–SS–PEG were 321.9 nm and 241.5 nm, respectively. Compared with the conventional mesoporous materials, which is limited for applications in drug delivery due to their relatively large particle sizes in the micrometer range,38 the nanoscale particles we obtained could be more helpful to enhance the drug stability. In addition, the polydispersity index (PDI) of MSNs–SH and MSNs–SS–PEG were 0.25 and 0.16, respectively. Compared with MSNs–SH, both the particle size and the PDI of MSNs–SS–PEG were smaller and lower than that of MSNs–SH, which indicated that MSNs functionalized with PEG via disulfide bond were well dispersed in the aqueous solution without significant aggregation. Besides, the zeta potential for the silica nanomaterials in aqueous solution was also measured. As listed in Table 1, the zeta potential of MSNs–SH and MSNs–SS–PEG were −35.5 mV and −31.3 mV, respectively. These results suggested that there were no significant differences between MSNs–SH and MSNs–SS–PEG. In addition, no matter whether functionalized with PEG, both the zeta potential of the silica materials was negative. This phenomenon could be attributed to the negative external and internal surface of the silica materials, which could facilitate the loading of cationic drugs through electrostatic interactions. And the anionic surface of the nanoparticle was reported to be more compatible with blood compared with their cationic counterparts.39
| Material | Particle size (nm) | PDI | Zeta potential (mv) |
|---|---|---|---|
| MSNs–SH | 321.9 | 0.25 | −35.5 |
| MSNs–SS–PEG | 241.5 | 0.16 | −31.3 |
Transmission electron microscopy (TEM) was carried out to characterize the morphology and pore structure of silica materials. As presented in Fig. 4A, the MSN–SH exhibited a relatively uniform spherical shape with the mean diameter of about 200 nm, and more interestingly, the order 2D-hexagonal porosity was obviously presented. After grafting PEG onto the surface of the MSNs, the PEG shell surrounding the core of silica nanomaterial was clearly observed and the morphology nearly maintained unchanged in Fig. 4B.
 | ||
| Fig. 4 TEM images of MSNs–SH (A) and MSNs–SS–PEG (B). | ||
To further investigate the morphology of the synthesized silica materials, Field Emission Scanning Electron Microscope (FE-SEM) was also utilized. As shown in Fig. 5, all the silica materials were uniform spherical shape with an average diameter of about 200 nm, which was corresponding to the results of TEM. Therefore, it could be concluded that the PEG functionalization had no significant effect on the morphology of silica nanomaterials. In this study, the average size determined by DLS was larger than that of TEM and SEM, which could be attributed to the hydration layer when determined by DLS in aqueous condition. Similar results were also observed in other works.40,41
 | ||
| Fig. 5 SEM images of MSNs–SH (A) and MSNs–SS–PEG (B). | ||
The mesoporous structure of the silica nanoparticles was also characterized by X-ray diffraction (XRD). In the low 2θ region, three characteristic diffraction peaks appeared at about 2.5°, 4° and 4.8° were obviously displayed in Fig. 6A, which was coincident with the characteristic diffraction patterns of MCM-41.40 However, in Fig. 6B, after the conjugation of the PEG chains, the characteristic diffraction peaks at 4° and 4.8° disappeared and the XRD pattern of MSNs–SS–PEG only exhibited a weak diffraction peak at 2.5°. This phenomenon may be attributed to the mesoporous channels blocked by the presence of the PEG chains.
 | ||
| Fig. 6 XRD patterns of MSNs–SH (A) and MSNs–SS–PEG (B). | ||
The surface area, pore volume and pore size distribution of the obtained silica nanoparticles were measured by nitrogen adsorption–desorption techniques. As depicted in Fig. 7, the nitrogen adsorption–desorption isotherm of the nanoparticles was similarly consistent with the type of IV nitrogen adsorption–desorption patterns, which illustrated that the nanoparticles possessed uniform mesoporous structure. Besides, the value of the surface area, pore volume and pore size distribution of MSNs–SH and MSNs–SS–PEG was concluded in Table 2. The surface area of the nanoparticles dramatically decreased from 1249 m2 g−1 to 336 m2 g−1 after the conjugation of the PEG chains. Correspondingly, the pore volume of the nanoparticles decreased from 0.9 cm3 g−1 to 0.37 cm3 g−1. These results could be attributed to the blocked pore channels after grafting PEG onto the surface of MSNs–SH.
 | ||
| Fig. 7 N2 adsorption–desorption isotherms of MSNs–SH (A) and MSNs–SS–PEG (B). | ||
| Material | Surface area (m2 g−1) | Pore volume (cm3 g−1) | Pore size (nm) |
|---|---|---|---|
| MSNs–SH | 1249 | 0.90 | 2.9 |
| MSNs–SS–PEG | 336 | 0.37 | 4.5 |
Drug loading and release
The DOX could be effectively loaded into the pore of silica nanoparticles due to the electrostatic interactions between the DOX and silica nanoparticles. DOX loading content and entrapment efficiency of MSNs–SS–PEG determined by UV-vis spectrophotometer were 12.3 wt% and 88.2 wt%, respectively. The obtained results indicated that there was high entrapment efficiency for the drug carrier material.The in vitro cumulative drug release amounts of DOX@MSNs–SS–PEG were investigated in PBS with different concentrations of GSH at 37 °C (pH 7.4 and pH 5.0). As shown in Fig. 8, in the absence of GSH, the drug was effectively wrapped by the polyethylene glycol without leakage at pH 7.4 within 24 h, which could decrease the side effect due to the premature release before reaching the target site. However, the cumulative release amounts of DOX slightly increased at pH 5.0 compared with pH 7.4 without GSH. The DOX might be easy to diffuse from the carrier at low pH due to the decreased electrostatic interaction between the DOX and the MSNs. Nevertheless, as the curves displayed, the main factor which triggered DOX release from the MSNs was the concentration of GSH. After 24 h, the cumulative release amounts of DOX at pH 7.4 were 22.8% at 4 mM GSH, 39.7% at 7 mM GSH and 52.3% at 10 mM GSH, respectively. So the release of DOX from MSNs–SS–PEG was much faster with the increasing concentration of GSH. The redox-dependent drug release behavior might be ascribed to the cleavage of the disulfide bond. This phenomenon also demonstrated that the mesoporous silica nanoparticles based on disulfide bond possessed the good redox response property. It's worth noting that the released amount of DOX from the carrier raised significantly in presence of 10 mM GSH at pH 5.0 which was similar to the tumor cell environment, reaching 62.3% within 24 h. This character made the MSNs–SS–PEG more ideal to be a drug delivery carrier with the acid environment in tumor cells.
 | ||
| Fig. 8 The in vitro DOX release from DOX@MSNs–SS–PEG at different GSH concentrations in PBS at pH 5.0 and 7.4, 37 °C. | ||
In vitro cell cytotoxicity
The in vitro cell cytotoxicity of MSNs–SS–PEG to BEL-7402 cells was investigated by MTT assay. It was seen from Fig. 9A that the MSNs–SS–PEG showed no obvious cytotoxicity effect on the BEL-7402 cells at 0.1–100 μg mL−1 after incubation for 24 h. As the concentration of MSNs–SS–PEG was as high as 500 μg mL−1, the cell viability was about 22% after incubation for 24 h. These results demonstrated that the MSNs–SS–PEG were nontoxic at low concentration and certain toxic at high concentration. It was reported that the concentration of the mesoporous nanoparticles as drug platforms to kill cancer cells effectively was lower than 10 μg mL−1.42 Fig. 9B showed the in vitro cellular cytotoxicity of free DOX and DOX@MSNs–SS–PEG to BEL-7402 cells in the different DOX concentration. The cytotoxicity of free DOX and DOX@MSNs–SS–PEG increased with the increase of DOX concentration. It was also seen that the cytotoxicity of DOX@MSNs–SS–PEG was not markedly different with free DOX in the same concentration (≤0.5 μg mL−1). It was important to note that at the concentration of 0.5 μg mL−1, the cell viabilities of DOX and DOX@MSNs–SS–PEG were 42% and 40%, respectively. And when the concentration was 1 μg mL−1 and 10 μg mL−1, the toxicity of DOX@MSNs–SS–PEG was lower than that of free DOX obviously. These results suggested that MSNs–SS–PEG were highly biocompatible and suitable to use as the drug carriers in control drug delivery system.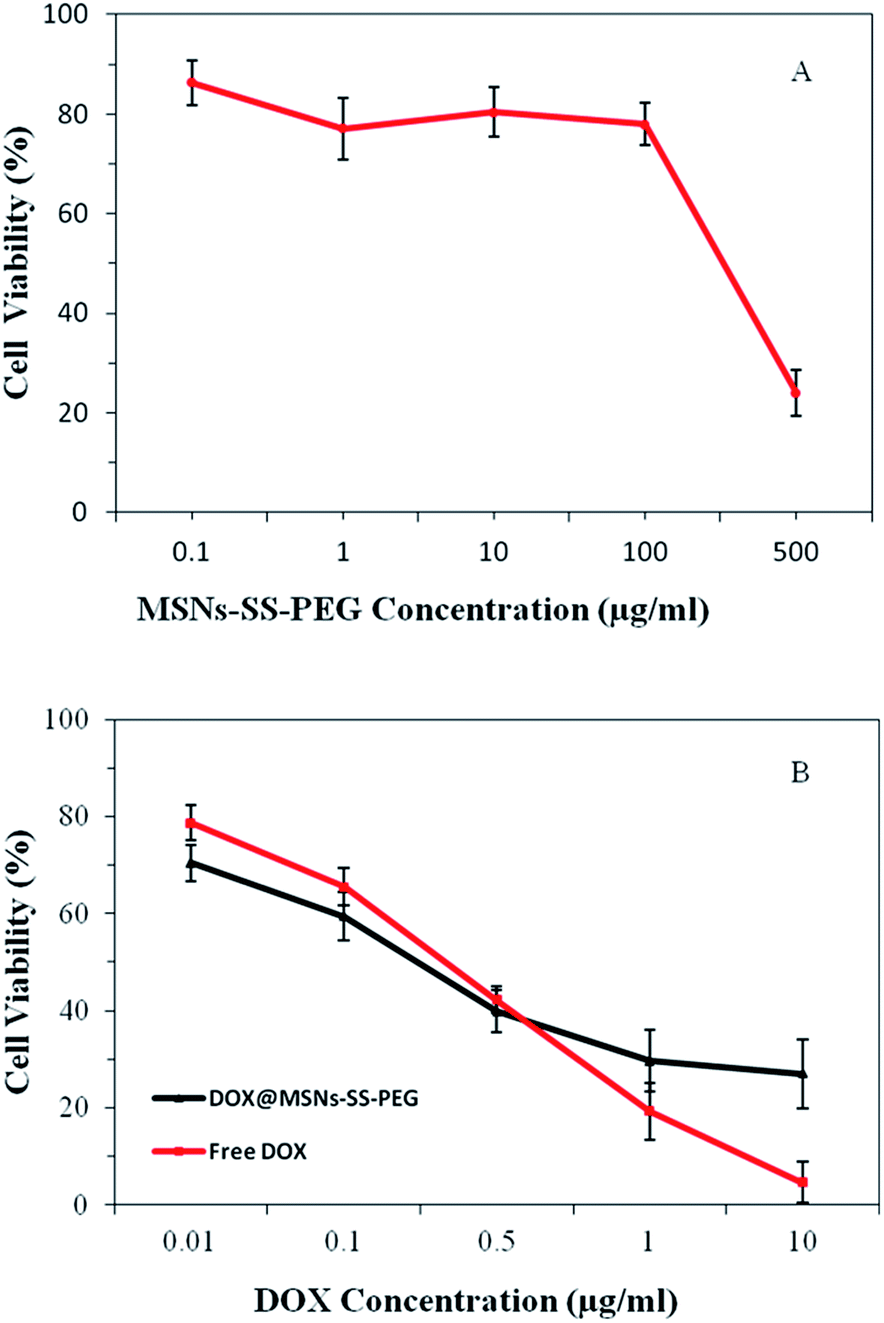 | ||
| Fig. 9 The in vitro cytotoxicity studies of MSNs–SS–PEG (A), free DOX and DOX@MSNs–SS–PEG (B) on BEL-7402 cells incubated for 24 h. | ||
Conclusions
In summary, a type of redox-sensitive mesoporous silica nanoparticles functionalized with PEG through a disulfide bond was successfully prepared. The MSNs–SS–PEG showed the stable and intact order 2D-hexagonal micropore structure with an average diameter of about 200 nm, and emerged the high specific surface areas and large pore volumes. The release of DOX from MSNs–SS–PEG was increased with the increasing concentration of GSH. The redox-dependent drug release behavior might be ascribed to the cleavage of the disulfide bond. So, MSNs–SS–PEG exhibited the good redox-sensitive property. The cytotoxicity test demonstrated that MSNs–SS–PEG was highly biocompatible and the DOX loaded nanoparticles possessed comparative anticancer activity with the free DOX towards BEL-7402 cells. These results demonstrated that MSNs–SS–PEG would be a great potential carrier for anticancer drug delivery.Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 81201197 and 21401051). We thank Prof. Xincai Xiao of the Analytic and Testing Center of South-Central University for Nationalities for the measurements of TEM and SEM and Prof. Qingzhi Wu of the Analytic and Testing Center of Wuhan University of Technology for the measurements of XRD and Nitrogen adsorption–desorption.References
- F. Q. Tang, L. L. Li and D. Chen, Adv. Mater., 2012, 24, 1504–1534 CrossRef CAS PubMed.
- B. G. Trewyn, I. I. Slowing, S. Giri, H. T. Chen and V. S. Y. Lin, Acc. Chem. Res., 2007, 40, 846–853 CrossRef CAS PubMed.
- X. X. Sun, Y. N. Zhao, V. S. Y. Lin, I. I. Slowing and B. G. Trewyn, J. Am. Chem. Soc., 2011, 133, 18554–18557 CrossRef CAS PubMed.
- Y. Zhao, B. G. Trewyn, I. I. Slowing and V. S. Y. Lin, J. Am. Chem. Soc., 2009, 131, 8398–8400 CrossRef CAS PubMed.
- F. Torney, B. G. Trewyn, V. S. Y. Lin and K. Wang, Nat. Nanotechnol., 2007, 2, 295–300 CrossRef CAS PubMed.
- H. J. Xu, H. J. Zhang, D. H. Wang, L. Wu, X. W. Liu and Z. Jiao, J. Colloid Interface Sci., 2015, 451, 101–107 CrossRef CAS PubMed.
- L. Chen, Z. Zhang, X. M. Yao, X. F. Chen and X. S. Chen, Microporous Mesoporous Mater., 2015, 201, 169–175 CrossRef CAS PubMed.
- A. Agostini, L. Mondragón, A. Bernardos, R. Martínez-Máñez, M. D. Marcos, F. Sancenón, J. Soto, A. Costero, C. Manguan-García, R. Perona, M. Moreno-Torres, R. Aparicio-Sanchis and J. R. Murguía, Angew. Chem., Int. Ed., 2012, 51, 10556–10560 CrossRef CAS PubMed.
- L. Du, S. Liao, H. A. Khatib, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 15136–15142 CrossRef CAS PubMed.
- F. Muharnmad, M. Y. Guo, W. X. Qi, F. X. Sun, A. F. Wang, Y. J. Guo and G. S. Zhu, J. Am. Chem. Soc., 2011, 133, 8778–8781 CrossRef PubMed.
- X. X. Hu, X. H. Hao, Y. Wu, J. C. Zhang, X. N. Zhang, P. C. Wang, G. Z. Zou and X. J. Liang, J. Mater. Chem. B, 2013, 1, 1109–1118 RSC.
- C. Choi, S. Y. Chae and J. W. Nah, Polymer, 2006, 47, 4571–4580 CrossRef CAS PubMed.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS PubMed.
- C. Park, H. Kim, S. Kim and C. Kim, J. Am. Chem. Soc., 2009, 131, 16614–16615 CrossRef CAS PubMed.
- A. Schlossbauer, J. Kecht and T. Bein, Angew. Chem., Int. Ed., 2009, 48, 3092–3095 CrossRef CAS PubMed.
- H. Kim, S. Kim, C. Park, H. Lee, H. J. Park and C. Kim, Adv. Mater., 2010, 22, 4280–4283 CrossRef CAS PubMed.
- Q. Zhang, F. Liu, K. T. Nguyen, X. Ma, X. Wang, B. Xing and Y. Zhao, Adv. Funct. Mater., 2012, 22, 5144–5156 CrossRef CAS PubMed.
- H. Y. He, H. H. Kuang, L. S. Yan, F. B. Meng, Z. G. Xie, X. B. Jing and Y. B. Huang, Phys. Chem. Chem. Phys., 2013, 15, 14210–14218 RSC.
- D. Patra and F. Sleem, Anal. Chim. Acta, 2013, 795, 60–68 CrossRef CAS PubMed.
- M. Mouslmani, J. M. Rosenholm, N. Prabhakar, M. Peurla, E. Baydoun and D. Patra, RSC Adv., 2015, 5, 18740–18750 RSC.
- R. Senthilkumar, D. S. Karaman, P. Paul, E. M. Björk, M. Odén, J. E. Eriksson and J. M. Rosenholm, Biomater. Sci., 2015, 3, 103–111 RSC.
- L. J. Jia, J. Y. Shen, Z. Y. Li, D. R. Zhang, Q. Zhang, G. P. Liu, D. D. Zheng and X. N. Tian, Int. J. Pharm., 2013, 445, 12–19 CrossRef CAS PubMed.
- L. S. Wang, L. C. Wu, S. Y. Lu, L. L. Chang, I. T. Teng, C. M. Yang and J. A. Ho, ACS Nano, 2010, 4, 4371–4379 CrossRef CAS PubMed.
- Q. F. Zhao, C. Wang, Y. Liu, J. H. Wang, Y. K. Gao, X. J. Zhang, T. Y. Jiang and S. L. Wang, Int. J. Pharm., 2014, 477, 613–622 CrossRef CAS PubMed.
- W. Feng, W. Nie, C. L. He, X. J. Zhou, L. Chen, K. X. Qiu, W. Z. Wang and Z. Q. Yin, ACS Appl. Mater. Interfaces, 2014, 6, 8447–8460 CAS.
- T. M. Allen, C. Hansen, F. J. Martin, C. Redemann and A. F. Yau-Young, Biochim. Biophys. Acta, 1991, 1066, 29–36 CrossRef CAS.
- Q. J. He, J. M. Zhang, J. L. Shi, Z. Y. Zhu, L. X. Zhang, W. B. Bu, L. M. Guo and Y. Chen, Biomaterials, 2010, 31, 1085–1092 CrossRef CAS PubMed.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS.
- L. E. van Vlerken, T. K. Vyas and M. M. Amiji, Pharm. Res., 2007, 24, 1405–1414 CrossRef CAS PubMed.
- R. Hong, G. Han, J. M. Fernández, B. j. Kim, N. S. Forbes and V. M. Rotello, J. Am. Chem. Soc., 2006, 128, 1078–1079 CrossRef CAS PubMed.
- L. Flohé, Biochim. Biophys. Acta, 2013, 1830, 3139–3142 CrossRef PubMed.
- J. X. Zhang, M. Niemelä, J. Westermarck and J. M. Rosenholm, Dalton Trans., 2014, 43, 4115–4126 RSC.
- C. Y. Lai, B. G. Trewyn, D. M. Jeftinija, K. Jeftinija, S. Xu, S. Jeftinija and V. S. Y. Lin, J. Am. Chem. Soc., 2003, 125, 4451–4459 CrossRef CAS PubMed.
- Y. M. Xu, F. H. Meng, R. Cheng and Z. Y. Zhong, Macromol. Biosci., 2009, 9, 1254–1261 CrossRef CAS PubMed.
- D. L. Elbert and J. A. Hubbell, Biomacromolecules, 2001, 2, 430–441 CrossRef CAS PubMed.
- R. Mortera, J. Vivero-Escoto, I. I. Slowing, E. Garrone, B. Onida and V. S. Y. Lin, Chem. Commun., 2009, 22, 3219–3221 RSC.
- Y. B. Guo, Y. J. Zhang, J. F. Li, Y. Zhang, Y. F. Lu, X. T. Jiang, X. He, H. J. Ma, S. An and C. Jiang, ACS Appl. Mater. Interfaces, 2015, 7, 5444–5453 CAS.
- C. Argyo, V. Weiss, C. Brauchle and T. Bein, Chem. Mater., 2014, 26, 435–451 CrossRef CAS.
- C. A. Fernandez, N. J. Baumhover, J. T. Duskey, S. Kharghariz, K. Kizzire, M. D. Ericson and K. G. Rice, Gene Ther., 2011, 18, 23–37 CrossRef CAS PubMed.
- V. Cauda, C. Argyo and T. Bein, J. Mater. Chem., 2010, 20, 8693–8699 RSC.
- H. J. Wang, P. Q. Zhao, X. F. Liang, X. Q. Gong, T. Song, R. F. Niu and J. Chang, Biomaterials, 2010, 31, 4129–4138 CrossRef CAS PubMed.
- M. Shi, J. Lu and M. S. Shoichet, J. Mater. Chem., 2009, 19, 5485–5498 RSC.
| This journal is © The Royal Society of Chemistry 2015 |