
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymer brush functionalized SiO2 nanoparticle based Nafion nanocomposites: a novel avenue to low-humidity proton conducting membranes†
Aleeza
Farrukh
ac,
Fatima
Ashraf
b,
Anke
Kaltbeitzel
c,
Xiao
Ling
c,
Manfred
Wagner
c,
Hatice
Duran
 d,
Abdul
Ghaffar
b,
Habib
ur Rehman
a,
Sapun H.
Parekh
c,
Katrin F.
Domke
c and
Basit
Yameen‡
*a
d,
Abdul
Ghaffar
b,
Habib
ur Rehman
a,
Sapun H.
Parekh
c,
Katrin F.
Domke
c and
Basit
Yameen‡
*a
aDepartment of Chemistry, SBA School of Science and Engineering, Lahore University of Management Sciences, Lahore-54792, Pakistan. E-mail: basit.yameen@lums.edu.pk; yameen@mit.edu
bDepartment of Chemistry, University of Engineering and Technology Lahore, Pakistan
cMax-Planck-Institut für Polymerforschung, Ackermannweg 10, 55128 Mainz, Germany
dDepartment of Materials Science & Nanotechnology Engineering, TOBB University of Economics and Technology, Sogutozu Cad. 43, 06560 Ankara, Turkey
First published on 18th May 2015
Abstract
Polyelectrolyte membranes showing proton conductivity at moderate levels of relative humidity and temperatures are essential for the development of polyelectrolyte membrane fuel cells (PEMFCs). Herein, monomethoxy oligoethylene glycol methacrylate derived polymer brush functionalized silica nanoparticles (SiO2 NPs) are presented as humidifying-nanoadditives for the fabrication of Nafion nanocomposite membranes, exhibiting improved proton conductivities at moderate levels of relative humidity and temperatures. Polymer brush functionalized SiO2 NPs (SiO2-polymer-brush), fabricated via surface initiated atom transfer radical polymerization (SI-ATRP), are dispersed in the Nafion resin solution, and nanocomposite membranes (Nafion/SiO2-polymer-brush) are fabricated via solution casting. For comparison, composite membranes of Nafion are also prepared with bare SiO2 NPs. Spectroscopic measurements confirm the presence of polymer brushes in the final membranes and demonstrate increased water uptake in membranes with polymer brush-functionalized nanocomposite membranes. Electrochemical impedance analyses reveal that 1 wt% of functionalized SiO2 NPs is sufficient to achieve Nafion nanocomposite membranes with superior proton conductivities at ambient and moderately high temperatures over the entire range of relative humidity (RH). This study presents a facile avenue to membranes with superior proton conductivities under moderate levels of RH and temperature, and provides important insights into the scope of nanocomposite PEMs for fuel cell applications.
1. Introduction
Gradual depletion of natural energy resources necessitates the development of more sustainable and reliable energy conversion systems. Among the available alternative energy production technologies, fuel cells are recognized as environmentally benign, single step energy conversion systems.1,2 Polymer electrolyte membrane fuel cells (PEMFCs) constitute an important class of fuel cells that operate at moderate temperature ranges (60–120 °C) and are widely explored for applications in stationary and automotive devices.3–6Polyelectrolyte membranes (PEMs) are considered as the electrochemical hearts of the PEMFCs.7 Due to their high proton conductivities, Nafion based polymer membranes constituted by perfluorosulfonated backbones and pendant protogenic sulfonic acid moieties are considered as the gold standard for PEMFCs.8 Nafion conducts protons only under high relative humidity (RH) conditions and becomes essentially an insulator for protons in the dry state.9,10 Investigations of the microstructure and hydrophobic–hydrophilic phase separation in Nafion membranes have established that the presence of water filled, nanoscopic proton conducting channels is responsible for the high proton conductivity.8,11–15 Consequently, a high RH level is essential for keeping the nanoscopic hydrophilic channels hydrated and achieving the proton conductivity necessary for the operation of a PEMFC.16–19 The required RH level in an operating PEMFC is maintained by an external humidification system, which has implications for PEMFC efficiency and cost due to the additional space and heat requirements.20 The improvement in proton conductivity at low RH levels, even by small increments, can have a striking impact on the overall performance and real-life application of PEMFCs.21–24 Among the different strategies adopted to achieve high proton conductivity at low RH levels, incorporation of nano and micro scale hydrophilic or hygroscopic additives into the PEMs has shown promising results. Recently, Jiang and coworkers have reported an improvement in water retention ability and low RH proton conductivities of different PEMs by incorporating polymer microcapsule additives that are constituted by hydrophilic or hydrogen bond forming functionalities, such as carboxylic acid, sulfonic acid, pyridyl, imidazole, and zwitterionic groups.25–28 In another approach, the PEMs derived from inorganic/organic hybrid materials fabricated by employing various oxide nanoparticles (ZrO2, SiO2, TiO2 and SnO2) and metal–organic frameworks have been recently explored for inducing enhanced proton conductivity at low RH levels.17,19,29–34 This approach exploits the hydrogen bonding interaction of hygroscopic oxide nanoparticle (NP) surfaces with water molecules.35–38
Polyethylene glycol (PEG) based materials are also known for their hydrophilic and hygroscopic nature, and surface grafted PEG results in the formation of a water shell on material surfaces, making them resistant against biofouling. By employing a model system based on self-standing macroporous silicon membranes with highly ordered and monodisperse cylindrical nanochannels functionalized with polymer brushes,39–42 we have previously proposed and validated that the proton conductivity at low relative humidity levels can be improved by capitalizing on the hygroscopic and moisture retaining abilities of PEG.43–45 Inspired by these characteristics of PEG, and based on our previous findings, we anticipate that the incorporation of the hydrophilic and moisture retaining ethylene oxide moiety functionalized NP based additives would enhance the water retaining tendency and proton conductivity of PEMs at low RH. To prove this hypothesis, we present silica NPs functionalized with monomethoxy oligoethylene glycol methacrylate based polymer brushes as humidifying additives for enhancing the proton conductivity of the Nafion at moderate RH levels and temperatures. The choice of Nafion as PEM is based on the fact that despite extensive efforts employed in the search for alternative PEMs, Nafion still remains the PEM of choice for PEMFCs. Furthermore, the flexible nature of the Nafion membrane is also important for the mechanical stability of the active layer in a fuel cell assembly. The choice of polymer brush based additives is unique to our platform, which makes our strategy more resourceful when compared to the already explored PEMs. The nanocomposite membranes prepared by incorporating different percentages of polymer brush-functionalized silica NPs exhibited superior proton conductivities when compared to the reference pristine Nafion and Nafion nanocomposite membranes fabricated with silica NPs lacking the polymer brushes. The improvement in proton conductivity is particularly striking at moderate RH levels and temperatures. In the context of PEMFCs, this study highlights the potential of incorporating polymer brush-functionalized nanoparticles into standard Nafion materials to increase PEM performance.
2. Experimental
2.1 Materials
Tetraethylorthosilicate (TEOS), (3-aminopropyl) triethoxysilane (APTES), α-bromoisobutyryl bromide, dry dichloromethane (DCM), monomethoxy oligo(ethylene glycol) methacrylate (MeOEGMA), 2,2′-bipyridine (BiPy), copper(II) bromide (CuBr2), copper(I) chloride (CuCl), 20 wt% Nafion resin solution (Nafion® DE 2020), ethylenediaminetetraacetic acid (EDTA), methanol and ethanol were used as received from Sigma-Aldrich, Germany. Hydrochloric acid (HCl, Riedel-de Haën, Germany), aqueous ammonium hydroxide solution (35%, BDH AnalaR, UK), and sodium hydroxide (NaOH, Fisher) were used as received. Triethylamine (TEA, Aldrich) was dried by refluxing overnight with calcium hydride followed by distillation and was stored under nitrogen.2.2 Preparation of APTES functionalized silica particles (SiO2-NH2)
Amine functionalized silica NPs were prepared by a previously reported method.46 Briefly, TEOS (3.8 mL, 17 mmol) was added to a mixture of aqueous ammonium hydroxide solution (35%, 5.7 mL) and distilled ethanol (114 mL) and stirred overnight at room temperature. APTES (2.1 mL, 9.4 mmol) was added to the above solution and further stirred overnight at room temperature. Finally the particles were separated by centrifugation at 4000 rpm for 20 min, followed by washing three times with ethanol.2.3 Immobilization of ATRP initiator (SiO2-Br)
The ATRP initiator was immobilized by adapting a previously described method.37 Briefly, degassed solution of TEA (200 μL, 1.2 mmol) and α-bromoisobutyryl bromide (150 μL, 1.2 mmol) in DCM (15 mL) was injected over degassed nanoparticles (1 g) under an inert atmosphere and stirred at room temperature for 2.5 h. The particles were purified by centrifugation at 4000 rpm for 20 min, followed by redispersing twice each in ethanol and DCM.2.4 Fabrication of polyMeOEGMA brushes (SiO2-polymer brush)
MeOEGMA (5 mL, 17 mmol) was dissolved in water (10 mL), followed by addition of 2,2′-bipyridine (BiPy) (73 mg, 0.462 mmol) and Cu(II)Br2 (4 mg, 0.018 mmol). After degassing for 1 h, Cu(I)Cl (19 mg, 0.19 mmol) was added, and the reaction mixture was further degassed for 15 min. The above solution was transferred via a syringe under an inert environment to SiO2-Br silica NPs (1 g) and polymerization was carried out for 40 min at 30 °C. The reaction mixture was acidified by 0.1 M HCl, followed by treatment with 0.06 M Na2EDTA and purified by dialysis against water (molecular-weight cut-off (MWCO = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400).
400).
2.5 Preparation of proton conducting membranes
Polymer-brush functionalized SiO2 NPs (0.5, 1, 2, 4 wt%) were diluted with 1 mL, 30% water:ethanol solution and sonicated for 1 min, followed by gradual addition of Nafion solution while stirring. The suspension was further sonicated until clear uniform solution formation, followed by casting on glass Petri plates. The plates were placed under vacuum for 30 min at 90 °C and left under a constant flow of nitrogen for 24 h at 90 °C. The 110 μm (±15 μm) thick membranes were dried in a vacuum oven for 24 h at 50 °C, followed by leaching with water for 12 h. The membranes were dried at 50 °C for 48 h under vacuum and finally treated with 0.1 M HCl for 3 h. For comparison, Nafion reference membranes and Nafion composite membranes with pristine SiO2 NPs were casted following the above described procedure.
2.6 Water uptake capacity
The water uptake capacity was determined by submerging membranes in deionized water for 8 h followed by drying at 50 °C under vacuum for 8 h. The difference in weight of wet and dry membranes was recorded and the water uptake capacity was determined by the following expression:45 | (1) |
2.7 Methanol diffusion
For the study of methanol diffusion two 90° elbow shaped glass pipes (area A = 2.324 cm2) were connected to form a U-shaped profile. The membrane was fixed between these tubes by screw clamps thus vertically separating the cell into two compartments. One reservoir (“s = supply side”) was filled with V = 22.5 mL 3 molar methanol aqueous solution, the other reservoir (“d = diffusion side”) was filled with the same volume of deionized water. Both compartments were constantly agitated with a magnetic stirrer. Repeatedly 0.6 mL samples were taken from both sides and investigated by 1H-NMR studies to determine the methanol concentration cD(t) and cS(t) of the compartments as a function of time. The concentration was evaluated using the integral ratio of the methyl and water proton signals, including the contribution of the methanol hydroxyl group to the water signal. To check the reliability of this method a 0.03 molar methanol aqueous solution was investigated, the deviation between measured and calculated NMR-ratios being less than 1%. In our experiment neither supply concentrations nor volumes (V) were constant over the investigated period. However, by iteration both effects can be mathematically included. For a membrane with permeability P and thickness L the time dependent methanol concentrations can be calculated over the time interval between sampling (ti < t < ti+1):| cD(t) = 0.5(cS(ti) − cD(ti))(1 − exp−(2PA/LVi)t) + cD(ti) | (2) |
| cS(t) = 0.5(cS(ti) − cD(ti)) + 0.5(cS(ti) + cD(ti))(1 − exp−(2PA/LVi)t) | (3) |
To account for a variation of the membrane thickness L over its area, the thickness was measured at 11 different positions and the effective thickness was calculated by averaging over the inverse thickness. Effective and mean thicknesses deviate by less than 2%.
The permeability P was determined by minimizing the sum of squared residuals using eqn (3).
For comparison with the literature, a commercial Nafion 117 sample was subject to the same testing. The measured permeabilities of Nafion 117 were in good accordance with literature reports.47
2.8 Proton conductivity
The in-plane proton conductivity of the membranes was measured by impedance spectroscopy in a two-electrode geometry using an SI 1260 impedance/gain-phase analyzer and a Novocontrol broadband dielectric converter. Humidity and temperature were controlled by a climate chamber (Binder KBF 240). From the Cole–Cole and Bode plots, the specific conductivity of the membranes was evaluated. The reported proton conductivities are averaged from two independent proton conductivity measurements.2.9 Characterization
FTIR spectra were recorded on an ATR-FTIR (Alpha Bruker, Germany) spectrometer, with 100 scans. X-ray photoelectron spectroscopy (XPS) was carried out using a Thermo Scientific K-Alpha. The Mg Kα (1253.6 eV) X-ray source was operated at 300 W. A pass energy of 117.40 eV was used for the survey scans. The spectra were recorded using a 60° take off angle relative to the surface normal. Transmission electron microscopy (TEM) images were obtained for silica nanoparticles by using an FEI Tecnai G2 F30 instrument. Samples were prepared by drop casting two to three drops of particle dispersions in ethanol onto a carbon coated copper TEM grid. To obtain a representative cross section of the hybrid membrane films, TEM samples were prepared using a cryo-Leica Ultra-microtome (EMUC6 + EMFC6) −100 °C. A 45 degree cryo-diamond knife was used. The hybrid film approach step of the ultra-microtome was constant at 0.2 μm with a constant speed of 10 mm s−1. During each cycle, samples with 30–50 nm thickness were obtained. The films were collected in a boat. The boat was filled up with a 2.3 M saccharose solution in order to prevent water from freezing. Scanning electron microscopy (SEM) imaging was carried out on a FEI Quanta 200 FEG SEM with 4 nm resolution. The electron acceleration voltage used was around 10 kV. Thermogravimetric analysis (TGA) was performed on SDTQ600 (TA Instruments, United States) under air atmosphere.Optical microscopy was carried out using confocal laser scanning microscopy on a Leica TCP SP5. A stack of 318 images (168 nm per slice) was recorded in reflection mode using 458 nm excitation. The images were deconvoluted and visualized in isosurface mode with Huygens Software.
For coherent anti-Stokes Raman scattering (CARS) spectroscopy, dry membranes were washed with MilliQ water (18.2 MΩ) in an ultrasonic bath. Spectra from fully hydrated membranes were obtained by immersing the sonicated membranes in MilliQ water for at least one week. For dried membranes, the hydrated membranes were left to dry in a standard laboratory ambient environment for three days after which spectra were recorded. All samples were sandwiched between standard glass coverslips and glass slides for CARS acquisition. A dual-output laser source (Leukos-CARS, Leukos, Limoges, France) provides the pump and Stokes beams. The pump is a passively Q-switched 1064 nm microchip laser, with less than 1 ns pulses at a 32 kHz repetition rate. The Stokes beam is provided by a supercontinuum fibre with a bandwidth from 1.1 μm to 1.9 μm. The Stokes and pump beams are polarization matched, overlapped at a dichroic mirror (LP02-1064RU-25, Semrock), and introduced into a modified inverted microscope (Eclipse Ti-U, Nikon). The beams are focused onto the sample by an objective (LCPlan N, 100×/0.85 IR, Olympus), and the forward CARS signal is collected with a 10× magnification, 0.25 NA air objective (Newport). The full power of both the pump/probe (30 mW) and Stokes (30 mW) beams was used in these experiments.
The samples were raster scanned across the focal volume with x–y steps of 0.5 × 0.5 μm in plane with piezo driven stages (Nano-PDQ 375 HS, Mad City Labs), and a full CARS spectrum was obtained at each location (pixel) with an acquisition time of 100 ms on a CCD (Newton DU920P-BR-DD, Andor). For each sample, 101 × 101 spatial pixels were scanned. The CARS data are acquired in custom software written in LabView (National Instruments) and saved for offline processing with Igor Pro (Wavemetrics).
Each raw CARS spectrum was converted into a Raman-like spectrum using a modified Kramers-Kronig phase-retrieval transform, with the CARS spectrum from glass serving as the non-resonant background.48 After transformation, a 5th order polynomial was used to fit a slowly varying error phase to regions of the spectra without resonances. This fit was subtracted from the transformed data, which yielded the final resonantly retrieved CARS (Raman-like) data presented here. For all data shown here, the fingerprint (500 to 1800 cm−1) and water (1900 to 4000 cm−1) regions of the spectra are processed independently. This was done to minimize the error phase from the transformed CARS spectra. Finally, the processed CARS spectra shown here were averaged from 49 spatial pixels.
3. Results and discussion
3.1 Synthesis and characterization
Amine functionalized silica NPs (SiO2-NH2) were synthesized by the hydrolysis of TEOS under basic conditions and in situ functionalization with APTES. The reaction of surface amino groups of SiO2-NH2 NPs with α-bromoisobutyryl bromide (BIBr) produced ATRP initiator functionalized SiO2 NPs (SiO2-Br). The subsequent surface functionalization of silica NPs with poly(monomethoxy oligoethylene glycol methacrylate), poly(MeOEGMA), polymer brush was achieved by employing aqueous surface initiated ATRP (SI-ATRP) at 30 °C in the presence of the bipyridine, CuCl and CuBr2 catalyst system (Scheme 1).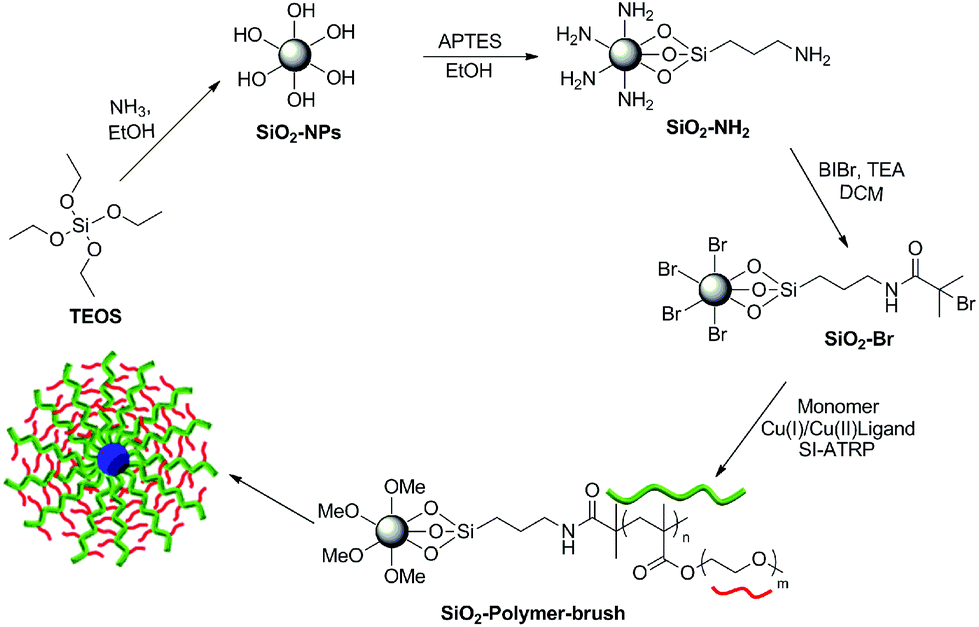 | ||
| Scheme 1 Schematic illustration of the reaction pathway employed for the synthesis and polymer brush functionalization of the silica NPs. | ||
The attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopic analysis revealed an absorption band at 1724 cm−1 only for the poly(MeOEGMA) brush functionalized SiO2 NPs (SiO2-polymer-brush), which corresponds to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of the pendant ester linkages present in the poly(MeOEGMA) brush (Fig. 1a). The absorption band corresponding to the stretching vibration of the aliphatic C–H bond around 2800–2900 cm−1 was clearly evident in the case of polymer-brush functionalized SiO2 NPs, which reflects the incorporation of a higher C–H content at the surface as a result of poly(MeOEGMA) brush functionalization. The characteristic absorption band for Si–O–Si stretching vibration (1046 cm−1) was also evident in the FTIR spectrum of the SiO2-polymer-brush. All the surface chemical modifications as depicted in Scheme 1 were further ascertained by XPS. The signals for the Si 2p orbital at 100 eV and Si 2s orbital at 154 eV originating from the silica contents of the NPs were clearly observable in the survey scan of the SiO2-NH2 NPs (Fig. 1b). The signals at 400 eV and 285 eV corresponding to the N 1s and C 1s orbitals validated the functionalization of SiO2 NPs with APTES. The subsequent immobilization of the ATRP initiator was ascertained by the signals at 70 eV for Br 3d orbitals and 185 eV for Br 3p orbitals. The effective growth of the poly(MeOEGMA) brush from the surface of SiO2-Br via SI-ATRP resulted in the masking of nitrogen and bromine signals (Fig. 1c). The polymer brush functionalization of SiO2 NPs was further confirmed by the fact that the O/C ratio (0.425) deduced from the XPS derived surface element composition of the SiO2-polymer-brush was in good agreement with the theoretical O/C (0.428) ratio for poly(MeOEGMA). Furthermore, the high resolution C 1s scan of the SiO2-polymer-brush NPs could be the curve fit for the ester (O–CO, 288.4 eV), ether (C–O–C, 285.9 eV) and aliphatic (C–H and C–C, 284.3 eV) carbon constituents of the poly(MeOEGMA) brush (Fig. 1d). These characterization methods established the successful fabrication of poly(MeOEGMA) brush functionalized SiO2 NPs.
O stretching vibration of the pendant ester linkages present in the poly(MeOEGMA) brush (Fig. 1a). The absorption band corresponding to the stretching vibration of the aliphatic C–H bond around 2800–2900 cm−1 was clearly evident in the case of polymer-brush functionalized SiO2 NPs, which reflects the incorporation of a higher C–H content at the surface as a result of poly(MeOEGMA) brush functionalization. The characteristic absorption band for Si–O–Si stretching vibration (1046 cm−1) was also evident in the FTIR spectrum of the SiO2-polymer-brush. All the surface chemical modifications as depicted in Scheme 1 were further ascertained by XPS. The signals for the Si 2p orbital at 100 eV and Si 2s orbital at 154 eV originating from the silica contents of the NPs were clearly observable in the survey scan of the SiO2-NH2 NPs (Fig. 1b). The signals at 400 eV and 285 eV corresponding to the N 1s and C 1s orbitals validated the functionalization of SiO2 NPs with APTES. The subsequent immobilization of the ATRP initiator was ascertained by the signals at 70 eV for Br 3d orbitals and 185 eV for Br 3p orbitals. The effective growth of the poly(MeOEGMA) brush from the surface of SiO2-Br via SI-ATRP resulted in the masking of nitrogen and bromine signals (Fig. 1c). The polymer brush functionalization of SiO2 NPs was further confirmed by the fact that the O/C ratio (0.425) deduced from the XPS derived surface element composition of the SiO2-polymer-brush was in good agreement with the theoretical O/C (0.428) ratio for poly(MeOEGMA). Furthermore, the high resolution C 1s scan of the SiO2-polymer-brush NPs could be the curve fit for the ester (O–CO, 288.4 eV), ether (C–O–C, 285.9 eV) and aliphatic (C–H and C–C, 284.3 eV) carbon constituents of the poly(MeOEGMA) brush (Fig. 1d). These characterization methods established the successful fabrication of poly(MeOEGMA) brush functionalized SiO2 NPs.
 | ||
| Fig. 1 ATR-FTIR of SiO2-Br and SiO2-Polymer-brush (a), and XPS analysis of SiO2-NH2 and SiO2-BI (b), SiO2-Polymer-brush (c), and C 1s high resolution scan of SiO2-Polymer-brush (d). | ||
The imaging of SiO2-NH2 NPs using scanning electron microscopy (SEM) and transmission electron microscopy (TEM) revealed spherical NPs with an average diameter of ∼250 nm (Fig. 2a and b). Compared to the SiO2-NH2 NPs (Fig. 2c), the high resolution TEM (HRTEM) images of polymer brush functionalized SiO2 NPs revealed a homogeneous lower contrast layer covering the SiO2 NP surface, presumably due to poly(MeOEGMA) (Fig. 2d). The poly(MeOEGMA) brush functionalized SiO2 NPs 1 wt% were then dispersed in a commercially available Nafion resin solution (20 wt% in lower aliphatic alcohols and water), which was solution casted in glass Petri plates under an inert atmosphere at 90 °C to give nanocomposite membranes. For the sake of comparison, nanocomposite membranes without NPs and with pristine SiO2 NPs were also fabricated under the identical conditions. Microtome TEM images of the resulting nanocomposite membranes revealed that the NPs homogeneously dispersed in the membrane (Fig. 2f–h). In the case of the polymer-brush functionalized silica NP based Nafion nanocomposite membrane, the uniform polymer brush layer covering the surface of NPs can be observed (Fig. 2h).
 | ||
| Fig. 2 SEM image of SiO2-NH2 NPs (a). TEM image of SiO2-NH2 NPs (b) and HRTEM images of SiO2-NH2 NPs (c). HRTEM of SiO2-Polymer brush (d). Microtome TEM images of Nafion reference membrane (e), Nafion/pristine SiO2 NPs (f), Nafion/SiO2-NH2 NPs nanocomposite membrane (g), and Nafion/SiO2-Polymer-brush nanocomposite membrane (h). | ||
The distribution of SiO2-polymer-brush NPs in the Nafion membrane was further investigated by confocal laser scanning microscopy (refer to the video provided as a part of the ESI†). The NPs reflect incoming light due to the difference of the refractive index with respect to the embedding medium. To confirm the assignment of the reflected signal to NPs, the corresponding Nafion reference film was investigated at the same, and with even more sensitive settings, which did not show any scattering objects. The elongation of the spherical NPs in the axial direction is an artifact of the anisotropic resolution of confocal microscopes.
Fabricated membranes were further investigated by coherent anti-Stokes Raman scattering (CARS) spectroscopy. CARS spectra of the Nafion reference, Nafion/SiO2, and Nafion/SiO2-polymer-brush (1 wt%) membranes were taken under both dry and fully hydrated conditions. Fig. 3a shows processed CARS (Raman-like) spectra after drying the membranes under ambient conditions for three days, which have all been normalized to the CF2 vibration band at 735 cm−1. The CH-region of the vibrational spectrum (2800 to 3100 cm−1) of the Nafion/SiO2-polymer-brush samples shows a significant increase in band intensity compared to the Nafion and Nafion/SiO2 reference spectra. The additional Raman band intensity corresponds to the CH2 asymmetric (2943 cm−1) and CH3 asymmetric (2980 cm−1) modes, presumably from the polymer brush layer. For the fully hydrated membranes (Fig. 3b), the CH resonances of the polymer brush, despite being partly obscured by strong water resonances, are visible and confirm the presence of the polymer-brush layer upon SiO2-polymer-brush nanoparticle incorporation into the Nafion membrane.
 | ||
| Fig. 3 Processed CARS spectra of Nafion reference and Nafion nanocomposite membranes in dry (a) and fully hydrated states (b). In order to compare different spectra, each spectrum is normalized by the corresponding intensity of the CF2 vibration at 735 cm−1. | ||
The Raman-like spectra allow direct quantification of the relative amount of absorbed water per CF2 group by integration of water band intensity (3060–3900 cm−1), IH2O. Spectral analysis shows that the nanoparticle-modified membranes retain higher amounts of water after three days of ambient drying relative to pristine Nafion (R = IH2O,X/IH2O,Nafion), where X is (Nafion/SiO2) or (Nafion/SiO2-polymer-brush). For (Nafion/SiO2) and (Nafion/SiO2-polymer-brush), R = 1 and 1.45, respectively. Under fully hydrated conditions, the Nafion/SiO2-polymer-brush membrane also shows the strongest water absorption, (R = 1.56) compared to Nafion/SiO2 (R = 1.36), which is consistent with the hygroscopic character of oligomeric ethylene glycol pendant groups. The processed CARS spectra provide direct spectroscopic evidence that the water absorption and retention abilities of the tested membranes decrease in the following order: Nafion/SiO2-polymer brush > Nafion/SiO2 > Nafion.
3.2 Evaluation of the proton conducting ability of nanocomposite membranes
The nanocomposite PEMs were solution casted by dispersing different amounts (wt%) of pristine SiO2 NPs, and SiO2-polymer-brush NPs in commercial 20 wt% Nafion resin solution (available acid capacity = meq. g−1 ≥ 1.00, total acid capacity = 1.03–1.12 meq.). The dry state thickness of the resulting membranes was around 110 μm (±15 μm). The fabricated membranes were thermally stable (Fig. S6†) as determined by the thermogravimetric analysis (TGA) and did not show any sign of brittleness (Fig. S7†). A detailed proton conductivity study was carried out on the nanocomposite membranes containing 0.5 to 4 wt% of NPs, and a considerable increase in proton conductivity was observed for the nanocomposite membranes containing SiO2-polymer brush NPs (ESI, Table S1, and Fig. S1–S3†). Just 1 wt% of the NPs was sufficient to achieve the highest proton conductivity, while a further increase in the NP content resulted in decreased proton conductivity. At ambient temperature (25 °C) and at a moderately high temperature (55 °C), the nanocomposite membranes with 1 wt% of SiO2-polymer-brush NPs (Nafion/SiO2-polymer-brush-1%) exhibited proton conductivity remarkably higher than the Nafion reference sample over the entire range of RH (Fig. 4). At 25 °C and 20% RH the Nafion/SiO2-polymer-brush-1% showed a proton conductivity of 3.4 mS cm−1 that is ∼11 times higher than the proton conductivity of the Nafion membrane (0.3 mS cm−1) under identical conditions. At 25 °C, the proton conductivities of all the membranes increased with the increase in RH, and at 80% RH the Nafion/SiO2-polymer-brush-1% nanocomposite membrane exhibited ∼4 times higher proton conductivity (95.4 mS cm−1) than the proton conductivity of the Nafion membrane (24.5 mS cm−1). The superior proton conducting characteristics of the Nafion/SiO2-polymer-brush-1% nanocomposite membrane were also maintained at the moderately high temperature of 55 °C. The proton conductivity of the Nafion/SiO2-polymer-brush-1% nanocomposite membrane (9.03 mS cm−1) was ∼5.7 times higher than the proton conductivity of Nafion (1.59 mS cm−1) at 55 °C and 20% RH. The proton conductivity of the Nafion/SiO2-polymer-brush-1% nanocomposite membrane reached a maximum value of 157.2 mS cm−1 at 55 °C and 80% RH, which is ∼3 times higher than the proton conductivity of Nafion (52.9 mS cm−1) under identical conditions. Nafion/SiO2-polymer-brush nanocomposite membranes also showed superior proton conductivity characteristics when compared to the Nafion/SiO2 membranes containing identical wt% of NP additives. At lower temperatures the proton conductivity profiles were comparable, however a particularly striking difference was observed at moderately high temperatures. For instance at 55 °C, the proton conductivity of the Nafion/SiO2-polymer-brush-1% nanocomposite membrane was ∼4 (at 20% RH) and ∼1.2 (at 80% RH) times higher than the proton conductivities of Nafion/SiO2 nanocomposite membranes. This highlights the proton conductivity enhancing influence of the poly(MeOEGMA) polymer brush. To assess the stability of the equilibrium proton conductivity, the conductivities of Nafion and Nafion/SiO2-polymer-brush 1% were monitored as a function of time at a constant 5% RH and 55 °C (Fig. S4†). Fitting and extrapolation were done with a logarithmic function. There is no boundary value for a log function, so the value after 20 h was taken as the equilibrium value. Waiting much longer would not significantly change the conductivity. Furthermore, proton conductivities measured as a function of increasing and decreasing RH values resulted in a very small hysteresis (Fig. S5†). These studies reveal that the proton conducting characteristics of the membranes are fairly stable. The activation energy data are also provided in the ESI (Table S2†). | ||
| Fig. 4 Influence of humidity on conductivity of Nafion and Nafion/SiO2-Polymer-brush nanocomposite PEMs at 25 °C (a), at 55 °C (b). | ||
The superior proton conducting characteristics of the Nafion/SiO2-polymer-brush nanocomposite membrane in comparison with Nafion over the entire RH range stem from the higher water uptake tendency of poly(MeOEGMA). The Nafion exhibited a water uptake of ∼12%, whereas the water uptake for the Nafion/SiO2-polymer-brush nanocomposite membrane containing 1 wt% of particles was found to be around 18%. The water uptake for the nanocomposite membranes containing 1 wt% of bare SiO2 NPs was around 14%. These results are quantitatively similar to those obtained from the CARS measurements, confirming the relatively increased water uptake in polymer-brush functionalized composite membranes compared to pristine Nafion.
In order to assess the effect of nanoparticle additives on the methanol diffusion across the Nafion membrane, we also evaluated the methanol permeability of our nanocomposite membranes. The methanol permeability of the Nafion/SiO2-polymer-brush 1% nanocomposite (2.25 × 10−6 cm2 s−1) was comparable to Nafion 117 (2.09 × 10−6 cm2 s−1) (Fig. 5), which suggests that the polymer-brush grafted SiO2 NPs increase the proton conductivity of Nafion without affecting its methanol permeability.
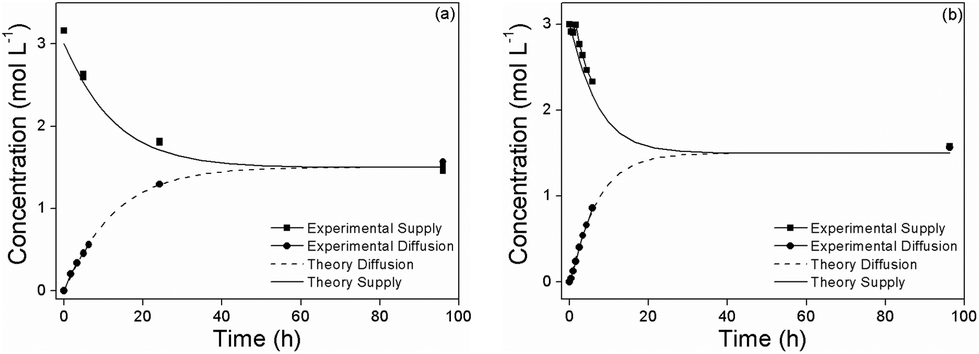 | ||
| Fig. 5 Methanol permeability of Nafion (a), and Nafion/SiO2-Polymer-brush-1% (b) membranes. | ||
4. Conclusions
A facile avenue to high proton conducting membranes under moderate levels of RH and temperature is presented. The oligoethylene glycol moieties were demonstrated to improve the proton conductivity of Nafion at moderate levels of RH and temperatures. Poly(MeOEGMA) polymer brush-functionalized SiO2 NPs fabricated via SI-ATRP were employed as conductivity enhancing additives. The nanocomposite membranes were prepared by a simple solution casting method. The proton conductivity studies showed that 1 wt% of the poly(MeOEGMA) polymer brush-functionalized SiO2 NP additive was enough to improve the proton conducting characteristics of Nafion. The nanocomposite Nafion membrane with 1 wt% of polymer brush-functionalized SiO2 NP additive showed ∼11 times higher proton conductivity at 20% RH and 25 °C, whereas at the same temperature and 80% RH, the proton conductivity of the nanocomposite membrane was ∼4 times higher than that of Nafion. The proton conductivities of all the membranes increased with an increase in temperature while the proton conductivity of nanocomposite membranes derived from polymer brush functionalized SiO2 NPs was always superior to that of Nafion. At 55 °C and 20% RH, the proton conductivity of the polymer brush-functionalized SiO2 NP derived Nafion nanocomposite membrane was ∼5.7 times higher than that of Nafion. At this higher temperature and under 80% RH, the nanocomposite membrane showed ∼3 times higher proton conductivity than Nafion. When compared to the Nafion nanocomposite membranes fabricated by using bare SiO2 NPs as additives, striking differences were observed at moderately higher temperature (55 °C), where nanocomposite membranes derived from polymer brush functionalized SiO2 NPs exhibited ∼4 (at 20% RH) and ∼1.2 (at 80% RH) times higher proton conductivity. The presented results are of significant relevance for the development of Nafion based nanocomposite membranes with high proton conductivities at moderate levels of RH and temperatures.Acknowledgements
B.Y. acknowledges funding from The Higher Education Commission (HEC) of Pakistan (project no. 20-1740/R&D/10/3368 and 20-1799/R&D/10-5302) and the LUMS Startup Grant. The research leading to these results has received funding from the ‘People’ Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme FP7/2007-2013/ under REA grant agreement no. [607842]. K.F.D. acknowledges generous support through the Emmy Noether program of the German Research Foundation (DFG) #DO 1691/1-1, and S.H.P. acknowledges funding from the Marie Curie Foundation #CIG322284.Notes and references
- M. Jacobson, W. Colella and D. Golden, Science, 2005, 308, 1901–1905 CrossRef CAS PubMed.
- B. C. Steele and A. Heinzel, Nature, 2001, 414, 345–352 CrossRef CAS PubMed.
- R. Kannan, B. A. Kakade and V. K. Pillai, Angew. Chem., Int. Ed., 2008, 47, 2653–2656 CrossRef CAS PubMed.
- T. Bock, H. Möhwald and R. Mülhaupt, Macromol. Chem. Phys., 2007, 208, 1324–1340 CrossRef CAS PubMed.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4270 CrossRef CAS.
- Y.-J. Wang, J. Qiao, R. Baker and J. Zhang, Chem. Soc. Rev., 2013, 42, 5768–5787 RSC.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- T. Wood and J. Badyal, ACS Appl. Mater. Interfaces, 2012, 4, 1675–1682 CAS.
- S. M. Haile, D. A. Boysen, C. R. Chisholm and R. B. Merle, Nature, 2001, 410, 910–913 CrossRef CAS PubMed.
- K.-D. Kreuer, S. J. Paddison, E. Spohr and M. Schuster, Chem. Rev., 2004, 104, 4637–4678 CrossRef CAS.
- O. Diat and G. Gebel, Nat. Mater., 2008, 7, 13–14 CrossRef CAS PubMed.
- K. Schmidt-Rohr and Q. Chen, Nat. Mater., 2007, 7, 75–83 CrossRef PubMed.
- C.-H. Chia, Z. Wu, C.-H. Wu, R.-H. Cheng and S. Ding, J. Mater. Chem., 2012, 22, 22440–22445 RSC.
- D. A. Bussian, J. R. O'Dea, H. Metiu and S. K. Buratto, Nano Lett., 2007, 7, 227–232 CrossRef CAS PubMed.
- Q. Li, R. He, J. O. Jensen and N. J. Bjerrum, Chem. Mater., 2003, 15, 4896–4915 CrossRef CAS.
- H. Zhang and P. K. Shen, Chem. Rev., 2012, 112, 2780–2832 CrossRef CAS PubMed.
- C. Laberty-Robert, K. Vallé, F. Pereira and C. Sanchez, Chem. Soc. Rev., 2011, 40, 961–1005 RSC.
- S. Pingá Jiang, Chem. Commun., 2013, 49, 6537–6539 RSC.
- X. Liang, F. Zhang, W. Feng, X. Zou, C. Zhao, H. Na, C. Liu, F. Sun and G. Zhu, Chem. Sci., 2013, 4, 983–992 RSC.
- R. Borup, J. Meyers, B. Pivovar, Y. S. Kim, R. Mukundan, N. Garland, D. Myers, M. Wilson, F. Garzon and D. Wood, Chem. Rev., 2007, 107, 3904–3951 CrossRef CAS PubMed.
- K. Miyatake and M. Watanabe, Electrochemistry, 2005, 73, 12–19 CAS.
- M. A. Hickner, H. Ghassemi, Y. S. Kim, B. R. Einsla and J. E. McGrath, Chem. Rev., 2004, 104, 4587–4612 CrossRef CAS.
- A. Noda, M. A. B. H. Susan, K. Kudo, S. Mitsushima, K. Hayamizu and M. Watanabe, J. Phys. Chem. B, 2003, 107, 4024–4033 CrossRef CAS.
- M. F. Schuster and W. H. Meyer, Annu. Rev. Mater. Res., 2003, 33, 233–261 CrossRef CAS.
- J. Wang, X. Yue, Z. Zhang, Z. Yang, Y. Li, H. Zhang, X. Yang, H. Wu and Z. Jiang, Adv. Funct. Mater., 2012, 22, 4539–4546 CrossRef CAS PubMed.
- J. Wang, H. Zhang, X. Yang, S. Jiang, W. Lv, Z. Jiang and S. Z. Qiao, Adv. Funct. Mater., 2011, 21, 971–978 CrossRef CAS PubMed.
- J. Wang, Z. Zhang, X. Yue, L. Nie, G. He, H. Wu and Z. Jiang, J. Mater. Chem. A, 2013, 1, 2267–2277 CAS.
- G. He, Z. Li, Y. Li, Z. Li, H. Wu, X. Yang and Z. Jiang, ACS Appl. Mater. Interfaces, 2014, 6, 5362–5366 CAS.
- V. Maneeratana, J. D. Bass, T. Azaïs, A. Patissier, K. Vallé, M. Maréchal, G. Gebel, C. Laberty-Robert and C. Sanchez, Adv. Funct. Mater., 2013, 23, 2872–2880 CrossRef CAS PubMed.
- N. H. Jalani, K. Dunn and R. Datta, Electrochim. Acta, 2005, 51, 553–560 CrossRef CAS PubMed.
- H. L. Tang and M. Pan, J. Phys. Chem. C, 2008, 112, 11556–11568 CAS.
- S. Horike, D. Umeyama and S. Kitagawa, Acc. Chem. Res., 2013, 46, 2376–2384 CrossRef CAS PubMed.
- D. Umeyama, S. Horike, M. Inukai, Y. Hijikata and S. Kitagawa, Angew. Chem., Int. Ed., 2011, 50, 11706–11709 CrossRef CAS PubMed.
- S. S. Nagarkar, S. M. Unni, A. Sharma, S. Kurungot and S. K. Ghosh, Angew. Chem., Int. Ed., 2013, 53, 2638–2642 CrossRef PubMed.
- N. Kamaly, Z. Xiao, P. M. Valencia, A. F. Radovic-Moreno and O. C. Farokhzad, Chem. Soc. Rev., 2012, 41, 2971–3010 RSC.
- B. Yameen, W. I. Choi, C. Vilos, A. Swami, J. Shi and O. C. Farokhzad, J. Controlled Release, 2014, 190, 485–499 CrossRef CAS PubMed.
- B. Yameen, M. Álvarez, O. Azzaroni, U. Jonas and W. Knoll, Langmuir, 2009, 25, 6214–6220 CrossRef CAS PubMed.
- A. Yousaf, A. Farrukh, Z. Oluz, E. Tuncel, H. Duran, S. Y. Doğan, T. Tekinay, H. ur Rehman and B. Yameen, React. Funct. Polym., 2014, 83, 70–75 CrossRef CAS PubMed.
- R. Barbey, L. Lavanant, D. Paripovic, N. Schuwer, C. Sugnaux, S. Tugulu and H.-A. Klok, Chem. Rev., 2009, 109, 5437–5527 CrossRef CAS PubMed.
- O. Azzaroni, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3225–3258 CrossRef CAS PubMed.
- T. Chen, R. Ferris, J. Zhang, R. Ducker and S. Zauscher, Prog. Polym. Sci., 2010, 35, 94–112 CrossRef CAS PubMed.
- A. Farrukh, A. Akram, A. Ghaffar, S. Hanif, A. Hamid, H. Duran and B. Yameen, ACS Appl. Mater. Interfaces, 2013, 5, 3784–3793 CAS.
- B. Yameen, A. Kaltbeitzel, A. Langner, H. Duran, F. Müller, U. Gösele, O. Azzaroni and W. Knoll, J. Am. Chem. Soc., 2008, 130, 13140–13144 CrossRef CAS PubMed.
- B. Yameen, A. Kaltbeitzel, A. Langer, F. Müller, U. Gösele, W. Knoll and O. Azzaroni, Angew. Chem., Int. Ed., 2009, 48, 3124–3128 CrossRef CAS PubMed.
- B. Yameen, A. Kaltbeitzel, G. Glasser, A. Langner, F. Müller, U. Gösele, W. Knoll and O. Azzaroni, ACS Appl. Mater. Interfaces, 2010, 2, 279–287 CAS.
- V. Mahalingam, S. Onclin, M. Péter, B. J. Ravoo, J. Huskens and D. N. Reinhoudt, Langmuir, 2004, 20, 11756–11762 CrossRef CAS PubMed.
- S. Molla, V. Compan, S. Luis Lafuente and J. Prats, Fuel Cells, 2011, 11, 897–906 CrossRef CAS PubMed.
- Y. Liu, Y. J. Lee and M. T. Cicerone, Opt. Lett., 2009, 34, 1363–1365 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5py00514k |
| ‡ Laboratory of Nanomedicine and Biomaterials, Harvard Medical School, 75 Francis St., Boston, MA 02115, and The David H. Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: yameen@mit.edu. |
| This journal is © The Royal Society of Chemistry 2015 |