
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Group 4 metal complexes bearing the aminoborane motif: origin of tandem ring-opening metathesis/vinyl-insertion polymerization†
M.
Wang
a,
G.
Xu
a,
D.
Wang
a,
Y.
Zou
a,
W.
Frey
b and
M. R.
Buchmeiser
*ac
aLehrstuhl für Makromolekulare Stoffe und Faserchemie, Institut für Polymerchemie, Universität Stuttgart, Pfaffenwaldring 55, D-70569 Stuttgart, Germany. E-mail: michael.buchmeiser@ipoc.uni-stuttgart.de; Fax: +49 (0)-711-685-64050
bInstitut für Organische Chemie, Universität Stuttgart, Pfaffenwaldring 55, D-70569 Stuttgart, Germany
cInstitut für Textilchemie und Chemiefasern (ITCF) Denkendorf, Körschtalstr. 26, D-73770 Denkendorf, Germany
First published on 19th March 2015
Abstract
Three tailored ansa-type Zr(IV)- and Hf(IV)-complexes, Zr-1, Hf-1 and Zr-2, all bridged by a dimethylsilylene group and bearing both a 6-[2-(BR2)phenyl]pyrid-2-yl motif (R = ethyl, mesityl) and an η1/η5-bound ligand with different Lewis base character and steric demand, have been synthesized. Their structures have been determined by single-crystal X-ray diffraction analysis. Upon activation with methylalumoxane (MAO), Zr-1, Hf-1 and Zr-2 are capable of polymerizing norborn-2-ene (NBE) via ring-opening metathesis polymerization (ROMP). In the MAO-activated copolymerization of ethylene (E) with NBE; however, pure vinyl insertion polymerization (VIP)-derived poly(NBE)-co-poly(E) is obtained by Zr-1, Hf-1 and the model pre-catalyst (Zr-3), which does not contain the borylamino motif. In contrast, Zr-2 yields copolymers containing both ROMP- and VIP-derived poly(NBE)ROMP-co-poly(NBE)VIP-co-poly(E) units via an α-H elimination process. Variable-temperature 11B NMR measurements allow for identifying tri- and tetracoordinated borane species. The propensity of catalysts Zr-1, Hf-1, Zr-2 and Zr-3 to switch from VIP to ROMP is discussed on the basis of their structural differences. A detailed NMR study of the reaction of Zr-1, Zr-2, Hf-1 and in particular of dimethylsilylene-bis(6-[2-(diethylboryl)phenyl]pyrid-2-ylamido)ZrCl2 (Zr-4) with MAO and with MAO/NBE clarifies the reaction cascade from VIP to ROMP and allows for establishing a general concept for a tailored switch from VIP to ROMP within the same polymer chain, allowing for tandem VIP–ROMP copolymerization of E with NBE.
Introduction
Tailor-made pre-catalysts containing the 6-[2-(diethylboryl)phenyl]pyrid-2-yl motif, e.g., (η5-tetramethylcyclopentadienyl)dimethylsilylene-(6-[2-(diethylboryl)phenyl]pyrid-2-ylamido)TiCl2 or dimethylsilylene-bis(6-[2-(diethylboryl)phenyl]pyrid-2-ylamido)ZrCl2, activated by methylalumoxane (MAO) are capable of forming multi-block copolymers from ethylene (E) and norborn-2-ene (NBE) with both ring-opening metathesis (ROMP)- and vinyl insertion polymerization (VIP)-derived poly(NBE) sequences within one single polymer chain.1–3 The structure of these unique polyolefins was confirmed by NMR; narrow polydispersity indexes (PDIs ≤ 1.3), single peaks in the GPC and the absence of glass transitions (Tg) that could be assigned to pure poly(NBE)ROMP further support the proposed polymer structures. These tailored cyclic olefin copolymers bear the potential for functionalization through polymer-analogous reactions at the double bonds present in the copolymers.4–9 If successful, this approach would offer an alternative to currently investigated copolymerizations of ethylene with functional monomers such as acrylates.10–14 In terms of polymer synthesis, the ligand system at the group 4 metals as such was designed in a way that it can switch from VIP to ROMP via abstraction of the α-proton from the growing polymer chain by the nitrogen at the pyrid-2-yl moiety. This proton can then in principle re-add to the metal alkylidene. The α-H elimination/addition process is controlled by the temperature-dependent dissociation propensity of the N–B bond where the boryl group and α-hydrogen at the growing polymer chain compete for the lone pair of the pyridyl group. However, apart from the 6-[2-(R2B)phenyl]pyrid-2-ylamido]-motif, the additional structural requirements that allow for the desired tandem polymerization are not clear.15,16 Within that context, it also appears desirable to tailor the dissociation temperature of the N–B bond, which is ∼60 °C for dimethylsilylene-bis(6-[2-(diethylboryl)phenyl]pyrid-2-ylamido)ZrCl2 (Zr-4).3 This can be achieved, e.g. either by increasing the sterics or the electron density at the boron. Since fluorenyl ligands are less Lewis basic compared to their cyclopentadienyl or tetramethylcyclopentadienyl counterparts, fluorenyl-based metal complexes bear the potential of increasing the electrophilicity of the metal and thus enhance polymerization activity.17–20 In addition, introduction of the bulky fluorenyl moiety is known to have an effect on the stereoselectivity of the polymers produced.20–22Based on these considerations, group 4 metal complexes containing the 6-[2-(diethylboryl)phenyl]pyrid-2-yl or the 6-[2-(dimesitylboryl)phenyl]pyrid-2-yl motif and a fluorenyl or tetramethylcyclopentadienyl ligand, all bridged by a dimethylsilylene group, were prepared in order to examine the polymerization behavior of the modified catalysts for potentially new polymer structures and to clarify the mechanism of the switch from VIP to ROMP.
Results
Synthesis of pre-catalysts Zr-1 and Hf-1
6-[2-(Diethylboryl)phenyl]pyridine-2-amine (1)2,3 and 9-(chlorodimethylsilyl)-9H-fluorene (2)23–25 were prepared as described in the literature. The synthetic routes to Zr-1 and Hf-1 are shown in Scheme 1. Deprotonation of 1 by n-butyllithium (n-BuLi) in diethyl ether followed by reaction with 2 yielded N-((9H-fluoren-9-yl)dimethylsilyl)-6-(2-(diethylboryl)phenyl)-pyridin-2-amine (3). For single-crystal X-ray analysis, see Fig. S1, ESI.† Treatment of 3 with two equivalents of n-BuLi in toluene, followed by recrystallization from tetrahydrofuran–pentane26 allowed isolating the ionic lithium complex (4) in 80% yield. For single-crystal X-ray analysis, see Fig. S2, ESI.† Direct transmetalation of 4 with ZrCl4·2THF or HfCl4·2THF provided the corresponding dichloro complexes Zr-1 and Hf-1. Crystals suitable for X-ray crystallography were obtained from CH2Cl2–pentane for Zr-1 and toluene–pentane for Hf-1. Zr-1 and Hf-1 crystalize both in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . Zr-1: a = 884.24(6), b = 1246.31(9), c = 1610.75(12) pm, α = 105.517°, β = 91.645°, γ = 106.554°, Z = 2; Hf-1: a = 881.19(5), b = 1246.37(7), c = 1612.23(9) pm, α = 105.491°, β = 91.532°, γ = 106.268°, Z = 2. Both single crystal X-ray studies on Zr-1 and Hf-1 (Fig. 1 and 2) show that one THF is bound to the metal center leading to a trigonal bipyramidal (TB) structure with the amino-nitrogen and oxygen in the apical positions. Disordered traces of THF and CH2Cl2 in Zr-1 or THF in Hf-1 stem from solvents involved in the corresponding synthesis or recrystallization process.
. Zr-1: a = 884.24(6), b = 1246.31(9), c = 1610.75(12) pm, α = 105.517°, β = 91.645°, γ = 106.554°, Z = 2; Hf-1: a = 881.19(5), b = 1246.37(7), c = 1612.23(9) pm, α = 105.491°, β = 91.532°, γ = 106.268°, Z = 2. Both single crystal X-ray studies on Zr-1 and Hf-1 (Fig. 1 and 2) show that one THF is bound to the metal center leading to a trigonal bipyramidal (TB) structure with the amino-nitrogen and oxygen in the apical positions. Disordered traces of THF and CH2Cl2 in Zr-1 or THF in Hf-1 stem from solvents involved in the corresponding synthesis or recrystallization process.
 | ||
| Scheme 1 Synthesis of pre-catalysts Zr-1 and Hf-1. | ||
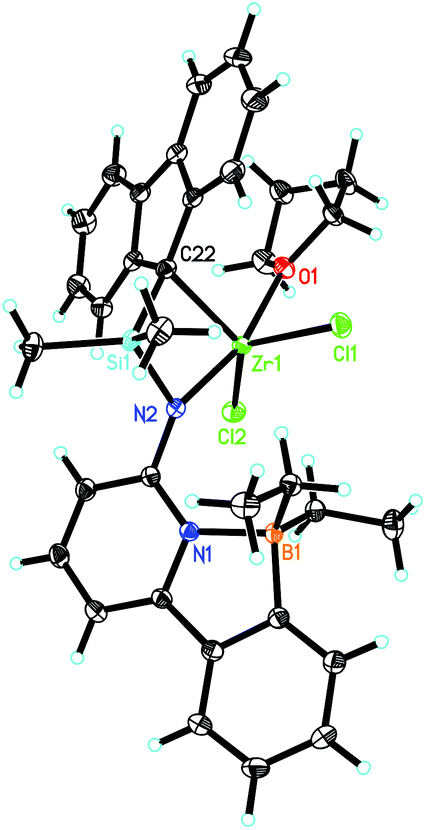 | ||
| Fig. 1 Single-crystal X-ray structure of Zr-1. Selected bond lengths [pm] and angles [°]: Zr(1)–N(2) 207.95(10), Zr(1)–O(1) 225.02(9), Zr(1)–C(22) 227.04(12), Zr(1)–Cl(1) 237.79(4), Zr(1)–Cl(2) 241.51(3), N(1)–B(1) 165.20(17); N(2)–Zr(1)–O(1) 164.84(4), N(2)–Zr(1)–C(22) 76.12(4), O(1)–Zr(1)–C(22) 92.57(4), N(2)–Zr(1)–Cl(1) 104.63(3), O(1)–Zr(1)–Cl(1) 86.91(3), C(22)–Zr(1)–Cl(1) 99.18(3), N(2)–Zr(1)–Cl(2) 95.21, O(1)–Zr(1)–Cl(2) 84.64(3), C(22)–Zr(1)–Cl(2) 131.36(3), Cl(1)–Zr(1)–Cl(2) 128.976(13). | ||
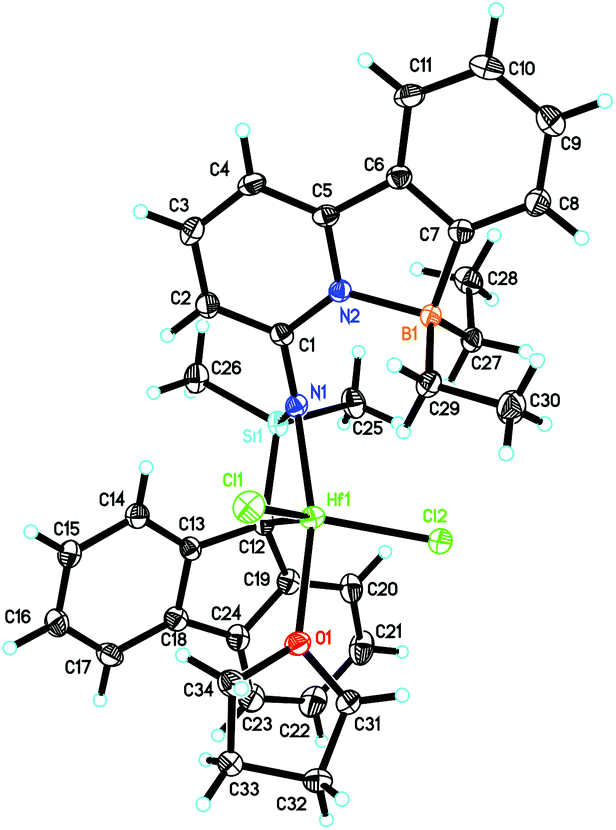 | ||
| Fig. 2 Single-crystal X-ray structure of Hf-1. Selected bond lengths [pm] and angles [°]: Hf(1)–N(1) 207.1(2), Hf(1)–O(1) 222.29(18), Hf(1)–C(12) 224.6(2), Hf(1)–Cl(2) 234.41(7), Hf(1)–Cl(1) 239.39(7), Hf(1)–C(13) 276.7(2), Hf(1)–Si(1) 289.54(7), B(1)–N(2) 164.6(3); N(1)–Hf(1)–O(1) 164.14(8), N(1)–Hf(1)–C(12) 76.43(9), O(1)–Hf(1)–C(12) 91.65(8), N(1)–Hf(1)–Cl(2) 104.79(6), O(1)–Hf(1)–Cl(2) 87.66(5), C(12)–Hf(1)–Cl(2) 101.61(7), N(1)–Hf(1)–Cl(1) 95.24(6), O(1)–Hf(1)–Cl(1) 84.49(5), C(12)–Hf(1)–Cl(1) 130.49(7), Cl(2)–Hf(1)–Cl(1) 127.39(3). | ||
An η1-bonding of the fluorenyl moiety is observed for both Zr-1 and Hf-1. This η1-bonding fashion of the fluorenyl ligand is very unusual in group 4 complexes where usually an η5-binding mode of a fluorenyl ligand is observed.17,27–29 The M–N(amino) and M–C(fluorenyl) distances are 207.9/227.0 pm for Zr-1 and 207.1/224.6 pm for Hf-1 and are thus similar to the distances found in comparable complexes, e.g. in Me2Si(η1-C29H36)(η1-N-tBu)MCl2(Et2O)2 (M = Zr, Hf).30 The B–N bond length is 165.2 pm for Zr-1, which is slightly longer than the one in dimethylsilylene-bis(6-[2-(diethylboryl)phenyl]pyrid-2-ylamido)ZrCl2 (Zr-4, 163.5 pm).3 An explanation for this slight lengthening of the N–B bond is an increased electrophilic character of the metal in Zr-1 caused by the weaker electron-donation ability of the η1-bound fluorenyl ligand. This results in a reduced electron density at the pyridine, which directly translates into a decreased coordination ability of the pyridine nitrogen to the boron and consequently a longer intramolecular N–B bond (Fig. 3).
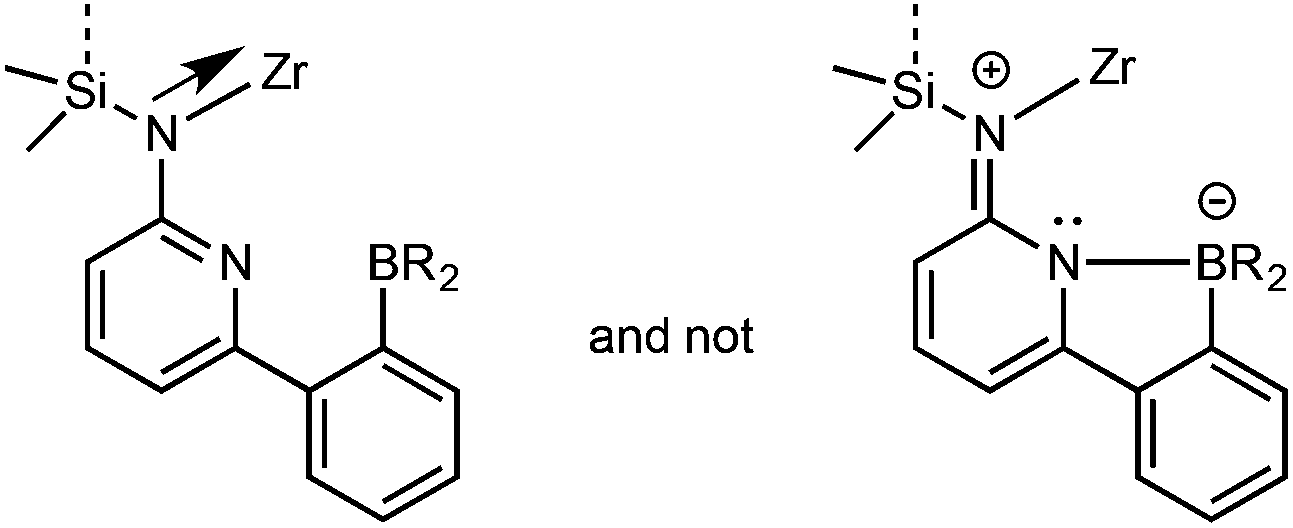 | ||
| Fig. 3 Preferred resonance structure in Zr-1. | ||
Synthesis of pre-catalyst Zr-2
As described in Scheme 2, 6-[2-(dimesitylboryl)phenyl]pyridin-2-amine (5)31,32 was prepared in analogy to compound 1 in 42% isolated yield.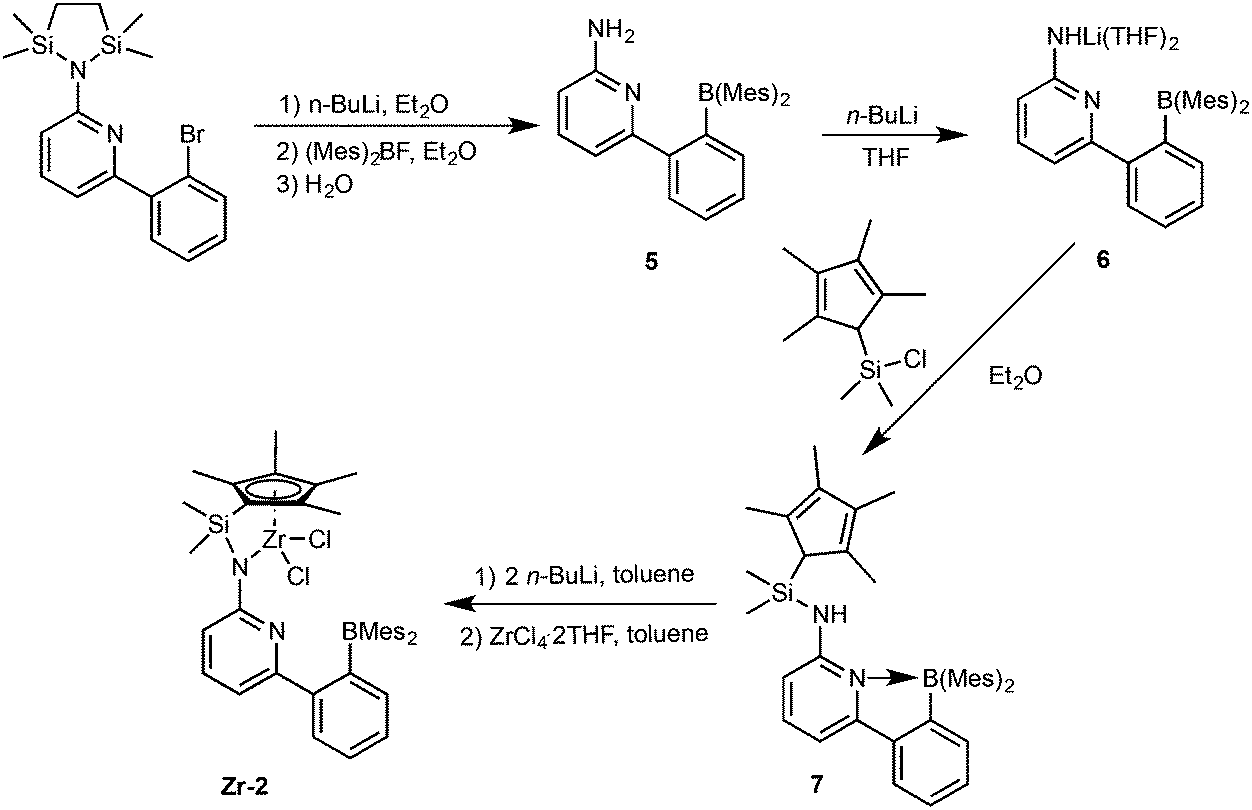 | ||
| Scheme 2 Synthesis of pre-catalyst Zr-2. | ||
Deprotonation of 5 in THF produced the lithium complex 6 with two THF molecules coordinated to lithium. Subsequent reaction with chlorodimethyl(2,3,4,5-tetramethylcyclopenta-2,4-dienyl)silane and recrystallization from diethyl ether–pentane allowed isolating compound 7 in 94% yield. Double deprotonation of 7 and combination with ZrCl4·2THF provided Zr-2 in 32% isolated yield. Crystals suitable for single-crystal X-ray diffraction were obtained from toluene–pentane. Zr-2 (Fig. 4) crystalizes in the triclinic space group P with a = 993.61(7), b = 1247.23(8), c = 1590.00(11) pm, α = 84.293°, β = 73.108°, γ = 83.935°, Z = 2.
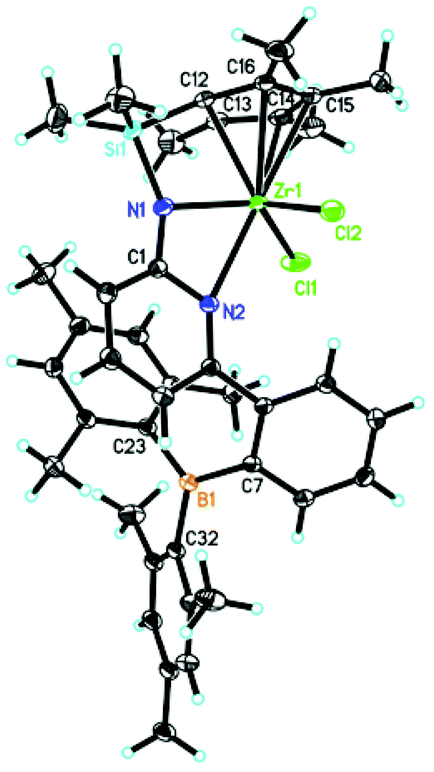 | ||
| Fig. 4 Single-crystal X-ray structure of Zr-2. Selected bond lengths [pm] and angles [°]: Zr(1)–N(1) 210.85(10), Zr(1)–Cl(1) 239.60(3), Zr(1)–Cl(2) 242.19(4), Zr(1)–C(13) 245.26(11), Zr(1)–C(12) 245.60(11), Zr(1)–N(2) 249.61(9), Zr(1)–C(16) 254.07(11), Zr(1)–C(14) 256.92(12), Zr(1)–C(15) 258.66(12); N(1)–Zr(1)–Cl(1) 124.37(3), N(1)–Zr(1)–Cl(2) 108.44(3), Cl(1)–Zr(1)–Cl(2) 109.597(13), N(1)–Zr(1)–N(2) 57.42(3), Cl(1)–Zr(1)–N(2) 86.51(2), Cl(2)–Zr(1)–N(2) 86.10(2). | ||
The cyclopentadienyl ligand is η5-bound to Zr(1); the Zr(1)–Namide distance is 210.85(10) pm and thus longer than in Zr-1 (207.9 pm). In stark contrast to Zr-1, the nitrogen in the pyridyl ring is coordinated to zirconium (Zr(1)–N(2) 249.61(9) pm) and no coordination to the boron atom exists, at least in the solid state. For the structure in solution, vide infra. The absence of any N–B bonding in Zr-2 is attributed to the pronounced steric demand of the two mesityl groups at boron compared to the two small ethyl groups in Zr-1. The absence of any coordinated solvent to the metal is attributable to a weakly but still significantly intramolecular stabilization of the electrophilic metal center by the pyridine ring through the N(2)–Zr(1) bond, which becomes also evident from its single crystal X-ray structure.
Synthesis of pre-catalyst Zr-3
The borylamine-free model catalyst Zr-3 was prepared as is shown in Scheme 3. 6-(2-(2-Propyl)phenyl)pyrid-2-ylamine was prepared via Pd-mediated coupling of 2-amino-6-bromopyridine with 2-isopropylphenylboronic acid, Deprotonation with n-BuLi followed by reaction with compound 2 yielded ligand 8. Double deprotonation of 8 yielded the lithium complex 9, which could not be isolated as a stable compound. However, in situ formation and reaction with ZrCl4·2THF allowed isolating analytically pure Zr-3.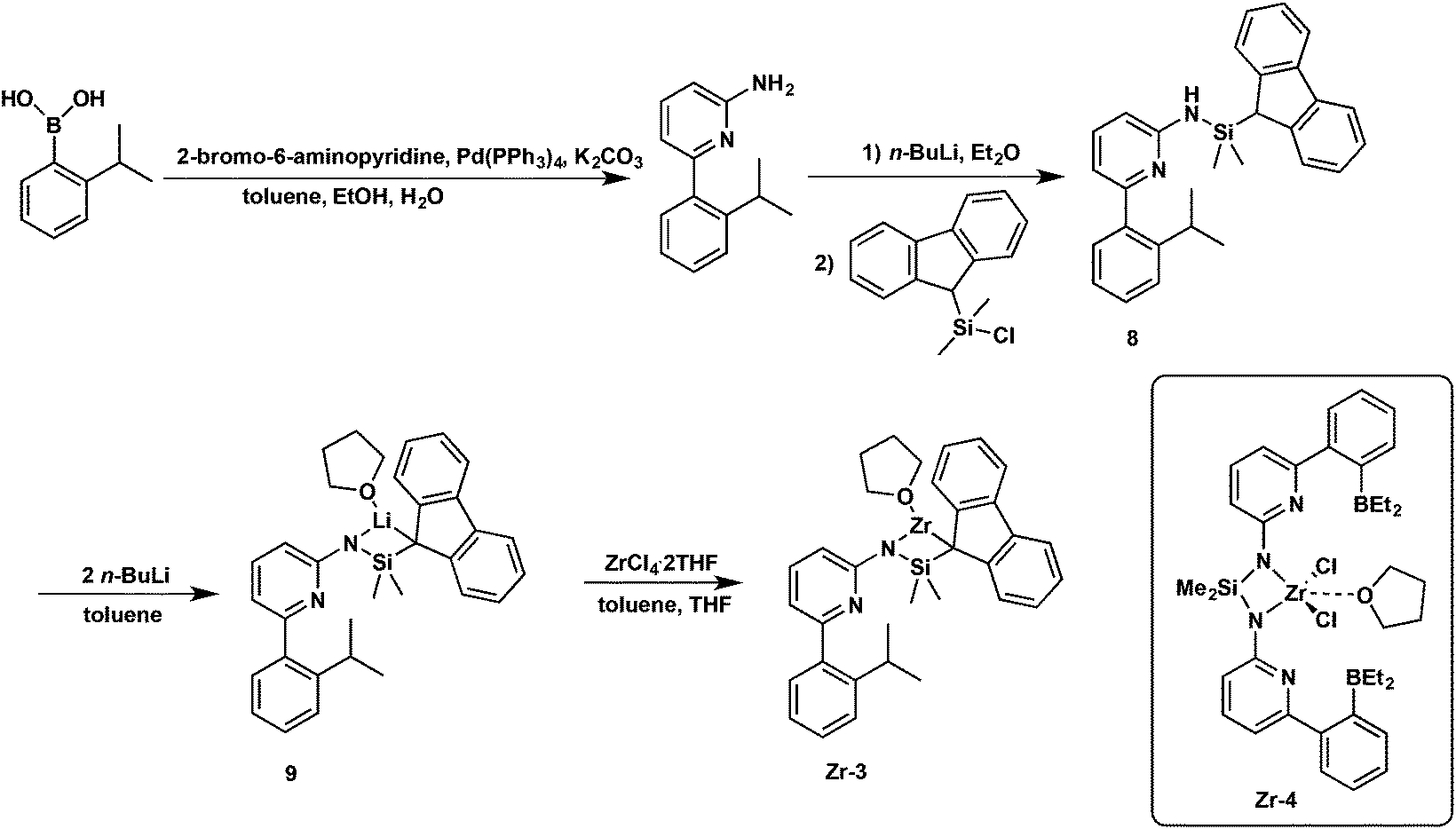 | ||
| Scheme 3 Synthesis of pre-catalyst Zr-3. | ||
Variable-temperature 11B NMR measurements
11B NMR measurements33–35 at different temperatures on Zr-1 and Zr-2 were carried out in a temperature range of 0–80 °C. Zr-1 shows a sharp peak at δ ∼ 6 ppm (Fig. S3†), which is typical for tetracoordinated borane in accordance with its single-crystal X-ray structure.Once the temperature is increased from 0 to 80 °C, the signal shifts from δ = 6.3 to 4.7 pm. The pyridine nitrogen remains bound to the boron forming a Lewis acid/base pair that does not dissociate up to 80 °C, which is indicative of a strong binding and a high-energy barrier to break the N–B bond. After the addition of MAO, the 11B NMR shows the signal of tetracoordinated borane (δ ∼ −5 ppm) and, above 30 °C, a new (weak) boron peak at δ ∼ 87 ppm (Fig. S4†), which can be assigned to tricoordinated borane. Clearly, in the presence of MAO the N–B bond starts to dissociate above 30 °C, though to a low degree. Upon addition of NBE to a toluene-d8 solution of Zr-1/MAO, a very weak peak at δ ∼ 85 ppm, which can be assigned to tricoordinated borane, becomes visible (Fig. S5†). Evidently, the system contains predominantly tetracoordinated borane moieties. For implications on reactivity, vide infra.
In ligand 7, the N–B bond exists in solution as evidenced by the chemical shift for boron at δ = −0.52 ppm in the 11B NMR (Fig. S6†), which is assignable to a tetracoordinated borane (vide infra).33–35 In contrast to the solid-state structure, in solution above 10 °C both free and nitrogen-coordinated borane are observed for Zr-2 in toluene-d8 as indicated by two peaks around δ ∼ 76 (tricoordinated borane) and ∼5 ppm (tetracoordinated borane) in the 11B NMR spectrum (Fig. S7†). These findings clearly show that in solution there exists an equilibrium between a closed and open structure for Zr-2. The 11B NMR of Zr-2/MAO and Zr-2/MAO/NBE (Fig. S8 and S9†) display two boron peaks similar to those of Zr-2 except that the tetracoordinated species become more abundant.
The 11B NMR of Hf-1 shows the signals for tetracoordinated borane at δ = 6 and 4 ppm above 25 °C (Fig. S10†); the absence of any tricoordinated borane again indicates a strong N–B bond. The 11B NMR of Hf-1/MAO is characterized by one signal for tricoordinated borane at δ = 87 ppm and two signals for tetracoordinated borane at δ = 4 and −2 ppm (Fig. S11†). For Hf-1/MAO/NBE two sets of signals at δ ∼ 4 and 1 ppm, both typical for tetracoordinated borane and at δ ∼ 87 ppm for tricoordinated borane is observed (Fig. S12†). The peak at δ ∼ 1 ppm is invisible while a new peak at δ ∼ 85 ppm is observed at T ≥ 60 °C.
Norborn-2-ene (NBE) homopolymerization and copolymerization of ethylene (E) with NBE
Catalysts Zr-1, Zr-2, Zr-3 and Hf-1 were activated with MAO and used in the homopolymerization of NBE. While the model catalyst Zr-3 did not produce any appreciable amount of poly-NBE, Zr-1, Zr-2 and Hf-1 allowed for the synthesis of the target polymer (Table 1).| # | Cat. | T (°C) | Productivityb |
M
n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c (g mol−1) c (g mol−1) |
PDIc |
T
gd (°C) |
cis (%) | Type |
|---|---|---|---|---|---|---|---|---|
|
a 100 mL Schlenk flask, total volume of reaction mixture: 50 mL of toluene, [catalyst] = 1 × 10−4 mol L−1, catalyst:MAO:NBE = 1:2000:10000, t = 1 h.
b Activity in kg of polymer mol−1catalyst h−1.
c HT-GPC in 1,2,4-trichlorobenzene vs. PS.
d Measured by DSC.
e Determined by 13C NMR analysis in 1,1,2,2-tetrachloroethane-d2.
|
||||||||
| 1 | Zr-1 | 50 | 5 | 40000 |
1.2 | 35 | 75 | ROMP |
| 2 | Zr-1 | 65 | 6 | 160000 |
1.4 | 48 | 90 | ROMP |
| 3 | Hf-1 | 50 | 42 | 120000 |
2.1 | 55 | 96 | VIP:ROMP = 0.3:100 |
| 4 | Hf-1 | 65 | 18 | 52000 |
2.6 | 55 | 90 | VIP:ROMP = 0.9:100 |
| 5 | Zr-2 | 50 | 5 | 120000 |
2.5 | 45 | 86 | ROMP |
| 6 | Zr-2 | 65 | 7 | 200000 |
1.5 | 37 | 95 | ROMP |
Both Zr-1 and Zr-2 produced high molecular weight poly(NBE), albeit with low productivity. Interestingly, polymers with an extraordinary high cis-content (up to 95%) were obtained (Fig. S13–S16, ESI†). The measured Tg values are in line with those for high cis poly(NBE). Importantly, poly(NBE) prepared by the action of Hf-1 contains both ROMP and, to a very minor extent, VIP-derived sequences (Fig. S17 and S18 ESI†). Also, productivities are substantially higher than those for Zr-1 and Zr-2. Also in line with a VIP-derived polymer block, the Tg values are higher than those of Zr-1 and Zr-2-derived poly(NBE). The data presented here clearly show that catalysts Zr-1, Zr-2, Hf-1 containing the 6-[2-(R2B)phenyl]pyrid-2-ylamido motif are ROMP active, while systems that lack this motif are not (Zr-3).
Next, to check for their VIP/ROMP propensity, catalysts Zr-1, Zr-2, Zr-3 and Hf-1, all activated by MAO, were used in the copolymerization of E with NBE. Polymerization results are summarized in Table 2.
| # | Cat. | M:MAO:NBE |
T/p (°C)/bar | A |
C
ROMPc (mol-%) |
C
VIPc (mol%) |
M
nd (g mol−1) |
PDId |
T
me (°C) |
|---|---|---|---|---|---|---|---|---|---|
| a 250 mL of toluene (including the volume of monomer), t = 1 h, [catalyst] = 2 × 10−5 mol L−1. b Activity in kg of polymer mol−1catalyst h−1 bar−1. c NBE content (ROMP and VIP) in the copolymer [mol%] as estimated by 13C NMR spectroscopy. d HT-GPC in 1,2,4-trichlorobenzene vs. PS. e Measured by DSC. | |||||||||
| 1 | Zr-1 | 1:2000:10000 |
50/2 | 55 | 0 | 14.2 | 290000 |
1.9 | 123 |
| 2 | Zr-1 | 1:2000:10000 |
50/4 | 45 | 0 | 29.3 | >6000000 |
— | 128 |
| 3 | Zr-1 | 1:2000:20000 |
50/2 | 10 | 0 | 6.6 | 330000 |
4.1 | 123 |
| 4 | Zr-1 | 1:2000:20000 |
50/4 | 9 | 0 | 6.8 | >6000000 |
— | 124 |
| 5 | Zr-1 | 1:2000:20000 |
50/6 | 27 | 0 | 10.7 | 360000 |
3.2 | 124 |
| 6 | Zr-1 | 1:2000:20000 |
65/4 | 24 | 0 | 4.3 | 210000 |
3.6 | 123 |
| 7 | Zr-1 | 1:2000:20000 |
80/4 | 19 | 0 | 7.3 | 150000 |
4.8 | 127 |
| 8 | Hf-1 | 1:2000:10000 |
50/4 | 7 | 0 | 6.5 | >6000000 |
— | 126 |
| 9 | Hf-1 | 1:2000:20000 |
50/4 | 6 | 0 | 18.1 | 430000 |
3.7 | 126 |
| 10 | Zr-3 | 1:2000:10000 |
50/4 | 73 | 0 | 4.1 | >6000000 |
— | 129 |
| 11 | Zr-3 | 1:2000:20000 |
50/4 | 68 | 0 | 3.0 | >6000000 |
— | 129 |
| 12 | Zr-2 | 1:2000:10000 |
50/4 | 1 | 6 | 3 | >6000000 |
— | 130 |
| 13 | Zr-2 | 1:2000:20000 |
30/4 | 2 | 11 | 8 | >6000000 |
— | 131 |
| 14 | Zr-2 | 1:2000:20000 |
50/4 | 1 | 21 | 7 | >6000000 |
— | 129 |
| 15 | Zr-2 | 1:2000:20000 |
65/4 | 6 | 0 | 2.7 | >6000000 |
— | 132 |
| 16 | Zr-2 | 1:2000:20000 |
80/4 | 4 | 0 | 2.5 | >6000000 |
— | 129 |
With Zr-1/MAO (Table 2, entries 1–7), activity decreased with increasing NBE concentration. This effect can be counterbalanced by an increase in E-pressure. Up to 29.3 mol% of NBE could be incorporated into the copolymer. The 13C NMR spectrum (Fig. 5) shows the characteristic signals for both alternating (E-NBE-E-NBE) and isolated sequences (E-NBE-E-E) at δ = 47.8, 47.2 (C2/C3), 42.0, 41.5 (C1/C4), 33.0 (C7) and 30.7–29.7 ppm (C5/C6, PE).36,37 While one cannot distinguish between alternating syndiotactic (alt-st) and isolated NBE sequences and therefore not judge on the extent of syndiotacticity, the alternating isotactic (alt-it) units are those that must be expected from a centrosymmetric catalyst such as Zr-1. Signals for NBE diads (E-NBE-NBE-E) or even NBE triads (E-NBE-NBE-NBE-E) were absent. None of these copolymers showed signals for ROMP-derived poly(NBE). For the copolymers obtained by the catalytic system Hf-1/MAO, up to 18.1 mol% VIP-derived poly(NBE) was realized at high NBE concentration (Table 2, entry 9, Fig. S19†). Generally, catalytic activities were lower than those for Zr-1/MAO. Again, no signals for ROMP-derived poly(NBE) were found in these copolymers. As anticipated, the aminoborane motif-free catalyst Zr-3 also produced only VIP-derived poly(NBE)-co-poly(E), albeit with a lower NBE incorporation (Table 2, entries 10 and 11, Fig. S20†) compared to Zr-1 under identical conditions (Table 2, entries 2 and 4). However, activities were higher than those by Zr-1/MAO, probably due to the sterically less encumbered nature of Zr-3.
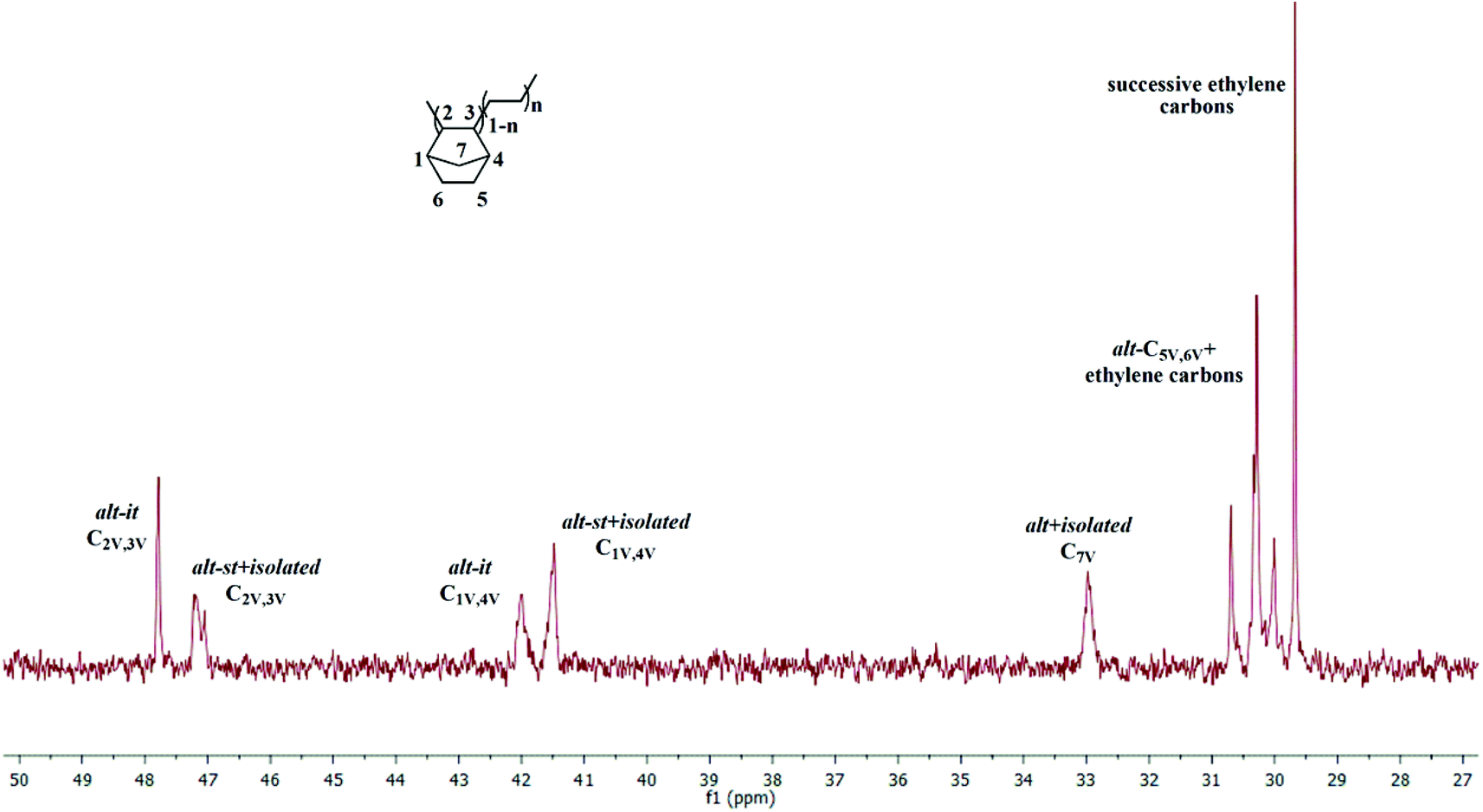 | ||
| Fig. 5 13C NMR spectrum of poly(E)-co-poly(NBE)VIP produced by Zr-1/MAO (Table 2, entry 2) (in 1,1,2,2-tetrachloroethane-d2). | ||
In contrast to Zr-1 and Hf-1, Zr-2 allows the synthesis of copolymers containing blocks of both ROMP- and VIP-derived poly(NBE) sequences in the same polymer chain. At low NBE concentration ([NBE] = 0.2 mol L−1), poly(NBE)ROMP-co-poly(NBE)VIP-co-poly(E) with a block ratio of 6:3:91 (Table 2, entry 12, Fig. S21†) was obtained. An increase in NBE concentration ([NBE] = 0.4 mol L−1) resulted in a further increase in the proportion of ROMP- and VIP-derived poly(NBE) units with ratios of 11:8:81 and 21:7:72 (Table 2, entries 13 and 14, Fig. S22 and S23†) at 30 and 50 °C, respectively. An increase in polymerization temperature to 65 °C and 80 °C produced VIP-derived poly(NBE)-co-poly(E) with 2.7 mol% and 2.5 mol% NBE incorporation (Fig. S24†). Notably, the molecular weights of these copolymers were all >6000000 g mol−1. Such high molecular weights are indicative for polymerizations with almost no β-hydride elimination or transfer to monomer.
In order to shed light on the polymer structure, the 13C NMR spectrum of poly(NBE)ROMP-co-poly(NBE)VIP-co-poly(E) was compared to the one of poly(NBE)ROMP and poly(NBE)VIP-co-poly(E) (Fig. 6). Signals at δ = 47.0 (C2,3), 41.5 (C1,4), 32.9 (C7) ppm are assignable to alt-st/isolated VIP-derived E-NBE sequences while the one at δ = 29.7 ppm corresponded to PE sequences. Signals at δ = 47.8 and 41.9 ppm, which could be assigned to alt-it E-NBE diads, were absent. Most importantly, signals at δ = 134.2, 42.8, 38.8 and 33.5 ppm that can unambiguously be assigned to poly(NBE)ROMP were observed even after the extraction by THF, which is known to dissolve pure ROMP-derived poly(NBE).
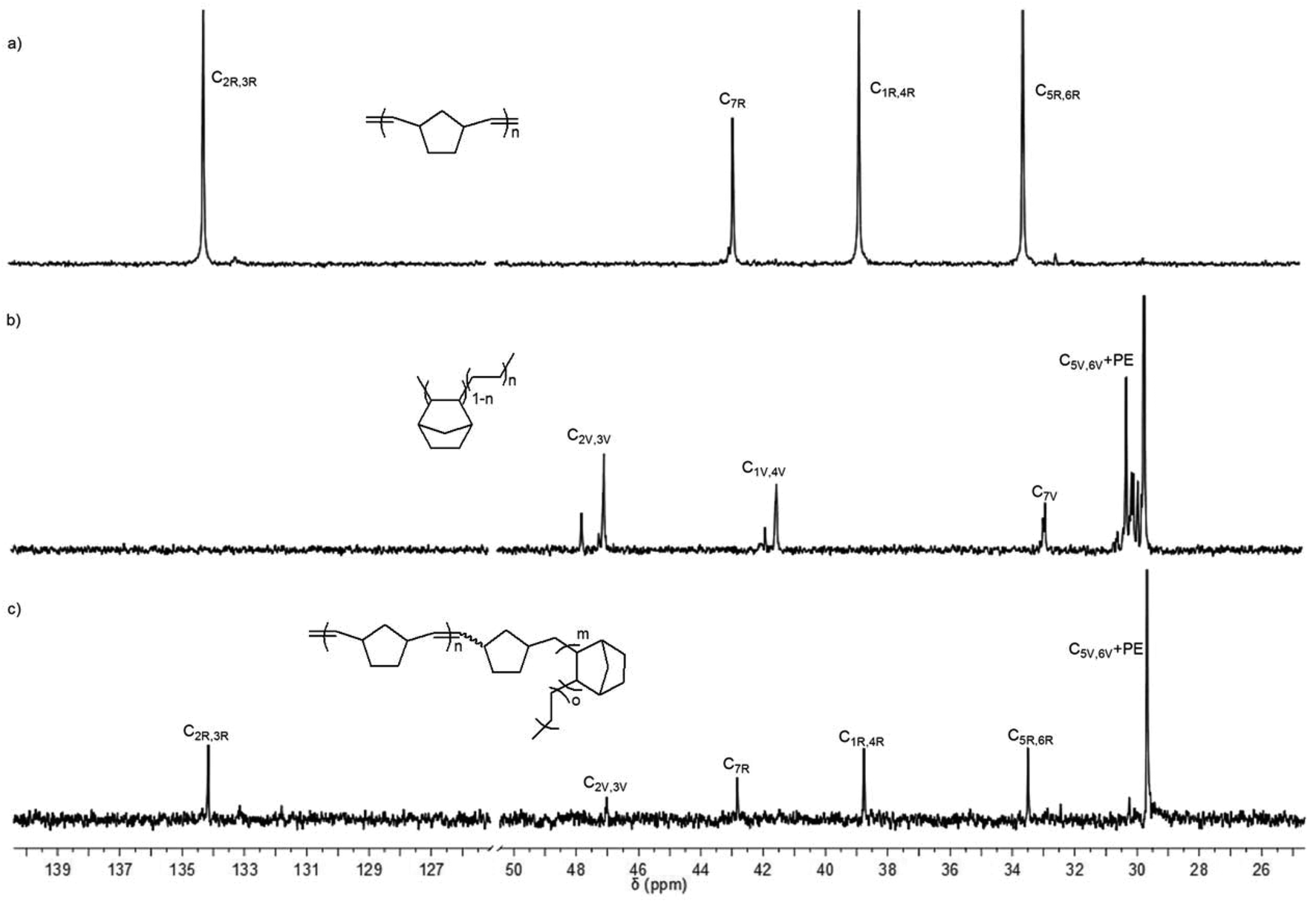 | ||
| Fig. 6 13C NMR spectra of (a) poly(NBE)ROMP, (b) poly(NBE)VIP-co-poly(E) (Table 1, entry 1) and (c) poly(NBE)ROMP-co-poly(NBE)VIP-co-poly(E) (Table 1, entry 14) in 1,1,2,2-tetrachloroethane-d2. | ||
All together, poly(NBE)ROMP-co-poly(NBE)VIP sequences must be present in the same polymer chain, which suggests the incorporation of ROMP-derived poly(NBE) units in the chain through an α-H elimination process. Further evidence comes from the absence of any glass transition attributable to a poly(NBE)ROMP homopolymer. Most vinylic carbons in the segment of the ROMP-type poly(NBE), surprisingly, were observed in the cis-configuration, signals at δ = 133.1 or 43.2 ppm belonging to trans-C2,3 or C1,4 repeat units were almost invisible. For mechanistic implications, vide infra.
In contrast to poly(NBE)ROMP-co-poly(NBE)VIP-co-poly(E) prepared by the action of (η5-tetramethylcyclopentadienyl)dimethylsilyl(6-[2-(diethylboryl)phenyl]pyrid-2-yl)amido-TiCl2,2Zr-2 containing the more bulky 6-[2-(dimesitylboryl)phenyl]pyrid-2-ylamido motif allows only for lower NBE incorporation, which further supports the argument about sterics. Also in line with increased sterics, higher molecular weights (Mn > 6000000 g mol−1) were obtained with Zr-2/MAO.
Discussion
As outlined above, both tri- and tetracoordinated boranes are present in Hf-1/MAO and Hf-1/MAO/NBE (Fig. S11 and S12†). Once MAO is added to Hf-1, the formation of methane occurs in the temperature range of 25–80 °C (Fig. S25†). At 60 °C, ethylene becomes visible, too. Notably, at 25 °C, a weak but detectable signal for a Hf-alkylidene at δ = 8.60 ppm (d, J = 8 Hz) can be observed. A terminal vinyl group can be identified at δ = 4.92 and 5.73 ppm in the range of 25–80 °C (Fig. 7 and S26b†). The signal at δ = 5.73 ppm can clearly be assigned to a CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CH2–R species (for a comparison with 1-octene see Fig. S26a†). The 1H NMR spectrum of Hf-1/MAO/NBE shows weak but detectable signals assignable to a Hf-methylidene at 25 °C at δ = 8.58 (d, J = 8 Hz) and 8.49 ppm (d, J = 8 Hz, Fig. S27†). Ethylene (δ = 5.24 ppm at 40 °C) can only be observed at T > 25 °C. Methane (δ = 0.16 ppm at 25 °C) is produced in the range of 25–80 °C. The above-mentioned terminal vinyl group at δ = 4.92 and 5.73 ppm (CH2CH–CH2–R species) remains visible in the range of 25–80 °C (Fig. S28†).
CH–CH2–R species (for a comparison with 1-octene see Fig. S26a†). The 1H NMR spectrum of Hf-1/MAO/NBE shows weak but detectable signals assignable to a Hf-methylidene at 25 °C at δ = 8.58 (d, J = 8 Hz) and 8.49 ppm (d, J = 8 Hz, Fig. S27†). Ethylene (δ = 5.24 ppm at 40 °C) can only be observed at T > 25 °C. Methane (δ = 0.16 ppm at 25 °C) is produced in the range of 25–80 °C. The above-mentioned terminal vinyl group at δ = 4.92 and 5.73 ppm (CH2CH–CH2–R species) remains visible in the range of 25–80 °C (Fig. S28†).
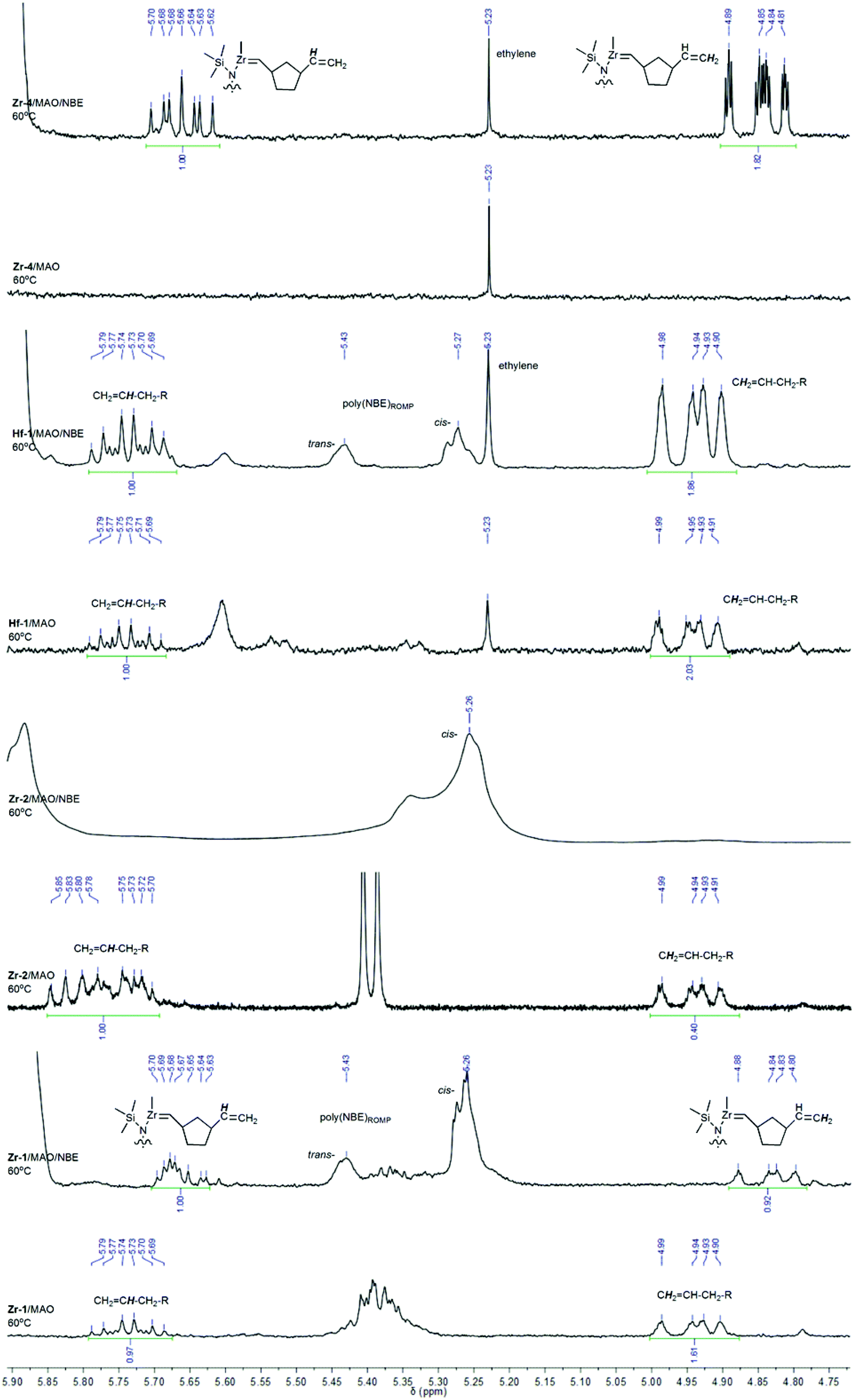 | ||
| Fig. 7 1H NMR spectra of catalyst/MAO and catalyst/MAO/NBE in toluene-d8 at 60 °C (4.7–5.9 ppm). | ||
It changes only slightly in intensity while ROMP-derived poly(NBE) (δ = 5.26 (cis) and 5.43 ppm (trans)) formed slowly but steadily. Thus, upon activation with MAO, Hf-1 must form a VIP-active, cationic species that quickly consumes the E that is present in the system. α-Hydrogen elimination results in the formation of a Hf-alkylidene, which upon reaction with E forms a Hf-methylidene and vinyl-terminated oligoethylenes. These two reaction products are visible in the spectrum. The Hf-methylidene then starts the ROMP of NBE. The low ROMP propensity of Hf-1 explains why only VIP-derived poly(NBE)-co-poly(E) is produced in the copolymerization of E with NBE.
As outlined above, the boron in Zr-1 is fully coordinated to the nitrogen both in the solid state and in solution. Upon addition of MAO or MAO/NBE, the borane remains mostly tetracoordinated, however, some tricoordinated species become visible, too (Fig. S4 and S5†). This rather small amount of tricoordinated borane translates into a small amount of free pyridine, which in the presence of MAO is capable of starting the ROMP of NBE at elevated temperatures (Fig. S13 and S14†). However, in case ethylene is present at the same time, no ROMP-derived structures are observed. Obviously, similar to Hf-1, most of the catalyst forms a cationic alkyl complex after activation with MAO, resulting in poly(NBE)VIP-co-poly(E). Upon treatment of Zr-1 with MAO at −60 °C, methane (δ = 0.26 ppm at −60 °C, 0.17 ppm at 20 °C) starts to evolve, a process that becomes more visible with increasing temperature. Clearly, the corresponding cationic RR′Zr+–CH2–Al(CH3)–O– species forms from RR′Zr+–CH3 and MAO (Fig. S29 and S30†). Zr-1/MAO at 60 °C also shows vinyl-terminated oligoethylenes (Fig. 7). In the presence of NBE, small amounts of poly(NBE)ROMP starts to form at −10 °C (cis![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :trans = 80:20) again accompanied by the formation of methane (Fig. S31 and S32†). No Zr-alkylidene is observed, probably because its concentration is too low. Zr-1/MAO/NBE at 60 °C again shows terminal vinyl groups, however, with somewhat different chemical shift and multiplicity for the signal at δ = 5.67 ppm (Fig. 7). In addition, numerous signals for poly(NBE)ROMP become visible (Fig. 7). This terminal vinyl group is believed to result from the reaction of a Zr-methylidene with NBE. For a detailed discussion of this reaction sequence with Zr-4, vide infra.
:trans = 80:20) again accompanied by the formation of methane (Fig. S31 and S32†). No Zr-alkylidene is observed, probably because its concentration is too low. Zr-1/MAO/NBE at 60 °C again shows terminal vinyl groups, however, with somewhat different chemical shift and multiplicity for the signal at δ = 5.67 ppm (Fig. 7). In addition, numerous signals for poly(NBE)ROMP become visible (Fig. 7). This terminal vinyl group is believed to result from the reaction of a Zr-methylidene with NBE. For a detailed discussion of this reaction sequence with Zr-4, vide infra.
In contrast to Zr-1, the pyridyl group in Zr-2 is not coordinated to the boron in the solid state. In solution, equally substantial and comparable amounts of tri- and tetracoordinated boranes are observed, both in the absence and presence of MAO and NBE (Fig. S7–S9†). Upon activation with MAO an active cationic species forms via release of methane (Fig. S33 and S34†). Larger fractions of free pyridine favor α-H-elimination and in the presence of NBE and predominantly cis-poly(NBE)ROMP forms even at −50 °C (Fig. S35†). This is why Zr-2/MAO in the presence of both E and NBE produces poly(NBE)VIP-co-poly(NBE)ROMP-co-poly(E). No methane is visible even at elevated temperature. Again, no Zr-alkylidene is observed. With Zr-4, which contains two 6-[2-(diethylboryl)phenyl]pyrid-2-yl groups (Scheme 3), the reaction sequence from a VIP- to a ROMP-active species can be followed best. There, homopolymerization of NBE at 60 °C by MAO-activated Zr-4 applying a ratio of Zr-4/MAO/NBE = 1:2000:20000 results in poly(NBE)ROMP (Mn = 93000 g mol−1, PDI = 1.6, cis:trans = 10:90, Fig. S36†). Copolymerization of E with NBE at 60 °C using Zr-4/MAO/NBE in a ratio of 1:2000:20000, pethylene = 4 bar reportedly yielded poly(NBE)ROMP-co-poly(NBE)VIP-co-poly(E) = 3:13:84 (Mn = 230000 g mol−1, PDI = 1.6).3Zr-4/MAO at 60 °C shows no terminal vinyls but ethylene (Fig. 7). In the presence of NBE, a terminal vinyl group adjacent to a 1,3-cyclopentylen ring, –(c-1,3-C5H8)–CHCH2, is observed (Fig. 7). The signal for the terminal CH-group at δ = 5.67 ppm (ddd, J = 7.1 Hz, J = 10.1 Hz, J = 17.4 Hz) unambiguously proofs this structural motif. Since the signals for poly(NBE)ROMP are completely absent, this structural motif cannot result from a cross metathesis of a ROMP-active, propagating Zr-alkylidene but must stem from the reaction of a Zr-methylidene with NBE.
Following the reaction of Zr-4 with MAO and NBE by NMR suggests that upon addition of MAO to Zr-4 in toluene-d8 in a molar ratio of Zr-4:MAO = 1:30 at −60 °C, the cationic species II (Scheme 4) forms. Potentially, it can be stabilized by the coordination of the pyridine nitrogen. II can further react with MAO to produce III (Scheme 4) and methane (δ = 0.17 ppm at 20 °C, Fig. S37 and S38†).38–41 At −20 °C, the dormant species III, which is inactive in polymerization, becomes visible in the 1H NMR (δCH2 = 0.82 ppm at 20 °C). A further increase in temperature to 20 °C results in α-H elimination and a ROMP-active Zr-alkylidene (V, Scheme 4) forms from II as evidenced by the alkylidene signal visible at δ = 8.6 ppm in the 1H NMR (Fig. S37†).3,42–44 In line with that, at 30 °C, 11B NMR shows apart from the parent tetracoordinated B atom (δ = 2.6 ppm) the formation of a tricoordinated species at δ = 86.7 ppm (Fig. S39 and S40†) indicating the dissociation of the N–B bond. This strongly suggests that the opening of the N–B bond generates a sufficient fraction of free pyridine moiety that induces α-H elimination. Substantial fractions of this ROMP-active Zr-alkylidene (V) experience bimolecular decomposition, resulting in the formation of ethylene at T ≥ 30 °C (Scheme 4). Thus, the signal for ethylene is clearly observed at δ = 5.25 ppm in the 1H NMR at T ≥ 30 °C (Fig. S37†). For Zr-4/MAO/NBE (Zr-4:MAO:NBE = 1:30:10), a zirconium alkylidene (Zr = CHR)42,44–46 is observed in the 1H NMR at δ = 8.5 ppm at 20 °C (Fig. S41 and S42†). In case Zr-4 (or Zr-2) starts first vinyl insertion copolymerization of E with NBE, insertion of NBE followed by α–H elimination promoted by the pyridine nitrogen through a six-membered transition state produces the Zr-alkylidene VI-P from V-P (Scheme 4), which is the ROMP-active species. Competition between the insertion of NBE and the re-formation of the N–B bond is proposed. As observed for Zr-1/MAO/NBE, a terminal vinyl end group (VI, Scheme 4) formed via reaction of ZrCH2 with NBE can be clearly seen in the 1H NMR at δ = 4.86 and 5.67 ppm (Fig. S43†). Concomitantly, apart from tetracoordinated borane at δ = 2.8 ppm, tricoordinated borane can be observed by 11B NMR at δ = 86.8 ppm at T ≥ 30 °C (Fig. S44†). Notably, at T ≥ 60 °C, a second tricoordinated B species appears in the 11B NMR at δ = 84.3 ppm and grows in intensity with increasing temperature. So far, this tricoordinated B-species cannot be assigned to a specific intermediate. High NBE concentrations were found to stabilize the metal alkylidene in both Zr-2 and Zr-4 and to encourage ROMP of NBE resulting in high proportion of ROMP-derived poly(NBE) units in the polymer (Table 2, entries 12 and 14). Vice versa, ethylene pressures > 4 bar shift the reaction from ROMP towards VIP.1–3,15,16
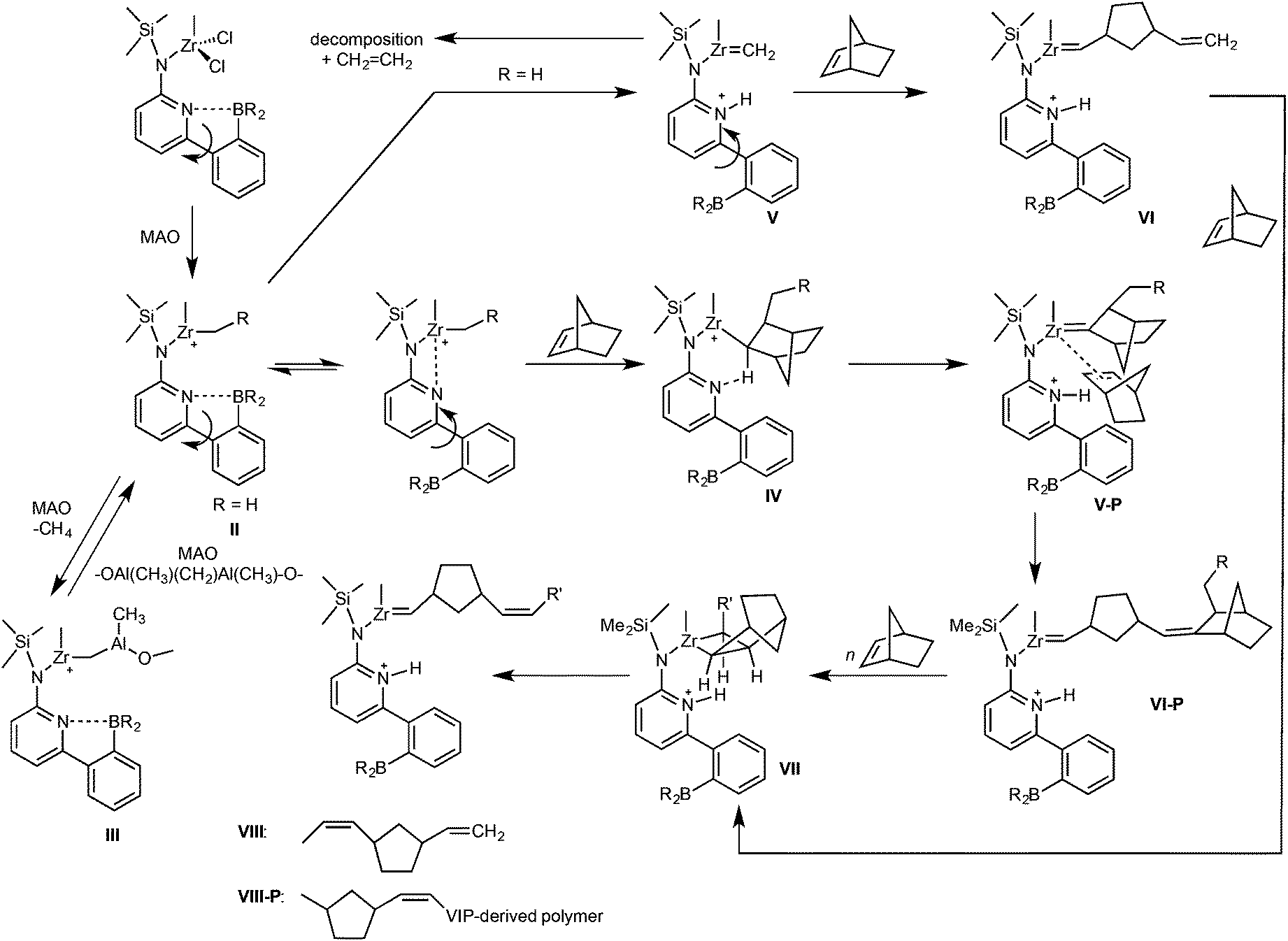 | ||
| Scheme 4 Proposed mechanism for the switch from VIP to ROMP. P = polymer. | ||
All together, the data presented here are in line with our previous proposal,2,3,15,16 which show that only high NBE concentrations promote the ROMP process. Notably, Zr-alkylidenes cannot be isolated unless hexacoordinated species containing chelating ligands are formed.45 As surmised earlier,15 a crowded ligand sphere around the metal as found in Zr-2 favors this α-H elimination process, i.e. the switch from VIP to ROMP. The low propensity of pyridine to coordinate to boron in Zr-2 clearly stems from the sterics provided by the mesityl groups, which together with the η5-tetramethylcyclopentadienyl (Cp*) ligand simply prevents any extensive coordination. This and the fact that Zr-3 without a 6-(2-BR2-phenyl)pyrid-2-yl group does not show any ROMP-activity, neither for NBE nor for E-NBE, strongly support an involvement of the pyridyl group and an α-H abstraction process triggered by the pyridine. In view of these sterics, it is also not surprising at all that Hf-1 with minor steric constraints around the metal shows no tendency to switch from VIP to ROMP in the presence of E regardless of VIP- and ROMP-derived poly(NBE) obtained in NBE homopolymerization (Fig. S17 and S18†). Finally, the high cis-selectivity of Zr-2 is a result of the large dimesitylboryl group, which forces NBE to add to VI-P in the outlined way to from VII (Scheme 4, Fig. S15 and S16†).
Conclusions
In the copolymerization of E with NBE, Zr-2, which has mesityl substituents at the boron and a crowded ligand sphere around the metal, allows the cis-selective synthesis of copolymers containing both ROMP- and VIP-derived poly(NBE) units within a single polymer chain resulting in a poly(NBE)ROMP-co-poly(NBE)VIP-co-poly(E) structure through a switch from VIP to ROMP. In the presence of E, both Zr-1 and Hf-1 show no tendency to undergo α-H elimination producing solely VIP-derived poly(NBE)-co-poly(E) with up to 29.3 mol% NBE incorporation. We attribute the low propensity of both Zr-1 and Hf-1 to promote ROMP to the instability of any metal alkylidene in the presence of E. As expected and in line with previous findings, the aminoborane-free model catalyst Zr-3 results in the formation of poly(NBE)VIP-co-poly(E) copolymers, which again supports the crucial role of the 6-[2-(R2-boryl)phenyl]pyrid-2-ylamido ligand in the α-H abstraction process. Variable-temperature 1H and 11B NMR allow correlating the propensity of a pre-catalyst activated by MAO to form tricoordinated borane with its propensity to produce ROMP-derived structures inside the copolymer. A detailed study on Zr-4 offers insight into important intermediates and confirms the proposed mechanism. Current efforts concentrate on detailed NMR studies and 13C-labelled NBE to shed more light on the role of borylamino group. The studies outlined here clearly support the formation of a Zr-methylidene in the ROMP of NBE via pyridine-induced α-H abstraction. Besides of the 6-(2-BR2-phenyl)pyrid-2-yl motif,1,16 the proposed crucial role of the bulky tetramethylcyclopentadienyl (Cp*) moiety in the α-H abstraction in ansa-type half sandwich complexes has been confirmed. Apart from sterically enforcing a switch from VIP to ROMP, the Cp* ligand as a six-electron donor can sterically and maybe also electronically stabilize44 the metal alkylidene and thus promote the ROMP of cyclic olefins. Whether the strong Lewis base character of the Cp* moiety also favors α-proton abstraction remains speculative. Other complexes bearing the 6-[2-BR2-phenyl]pyrid-2-yl motif like Zr-1 can in principle also be capable of promoting the ROMP of NBE but might be too unstable in the presence of E to allow for any ROMP-derived poly(NBE) structures under such conditions. While the fundamentals for a switch from VIP to ROMP have now been clarified, the low polymerization activity remains a challenge. The highly constrained geometry of the Zr-2, the resulting low propensity to undergo β-hydride elimination together with the possibility to undergo cross-metathesis with E accounts for a very low productivity. One key to higher activities seems therefore the suppression of cross-metathesis of the propagation alkylidenes with E. Progress in this issue will be reported in due course.Experimental
All manipulations were conducted by using standard Schlenk or dry box techniques under an atmosphere of Argon or nitrogen unless specified otherwise. Deuterated solvents for NMR were freeze–pump–thaw degassed and stored inside a glove box. Benzene-d6, toluene-d8 and tetrahydrofuran-d8 were dried and distilled from sodium/benzophenone; CD2Cl2 and CDCl3 were dried and distilled from P2O5; C2D2Cl4 was distilled from calcium hydride. Regular solvents, i.e. diethyl ether, toluene, THF, n-pentane, CH2Cl2 were dried and deoxygenated by sparging with N2, followed by passing through a triple-column solvent purification system (MBraun, Garching, Germany). Commercially available reagents for synthesis were used without further purification. Celite was dried in vacuo at 180 °C for two days prior to use. Methylalumoxane (MAO, 10 wt% solution in toluene) was purchased from Sigma-Aldrich. The toluene was removed and the residue was dried in vacuo at 80 °C overnight to remove any free AlMe3 and stored inside a glove box. Ethylene gas was purified by passing through columns filled with Cu-catalyst (BASF R3-11G) and 3 Å molecular sieves before use. All homopolymerization reactions of NBE were carried out in Schlenk tubes under an inert atmosphere. All copolymerizations of ethylene with NBE were performed in a Büchi–Uster pressure reactor equipped with a Huber thermostat. The feed of ethylene was kept constant and ethylene consumption was monitored by a Büchi pressflow bpc 6010 flow controller.NMR data were recorded at 400 MHz for 1H and 100 MHz for 13C on a Bruker Avance III 400 spectrometer at 25 °C unless noted otherwise and reported in ppm relative to tetramethylsilane (TMS). All NMR data of the homo-/copolymers were measured at 100 °C except where noted. FTIR spectra were measured on a Perkin-Elmer 881 spectrometer with ATR technology.
Molecular weights and molecular weight distributions were measured by high-temperature gel permeation chromatography (HT-GPC) on an Agilent PL-GPC 220 system equipped with three consecutive PLgel 5 μm MIXED-C 300 × 7.5 mm columns in 1,2,4-trichlorobenzene at 160 °C. The flow rate was set to 1 mL min−1. The GPC system was calibrated with narrow polystyrene standards in the range of 162–6035000 g mol−1 (Easi Vial-red, yellow and green, Fig. S45†), which were all purchased from Polymer Labs. DSC data were recorded by heating under a nitrogen atmosphere on a Perkin–Elmer DSC7 differential scanning calorimeter.
2-Isopropylphenylboronic acid
To a solution of 1-bromo-2-isopropylbenzene (19.9 g, 0.10 mol) in 200 mL of THF cooled to −78 °C was added n-BuLi (66 mL, 1.6 M in hexane, 0.11 mol) and the resulting solution was stirred for another 15 minutes at −78 °C. A solution of tris(2-propyl)borate (62 mL, 0.27 mol) in 60 mL of THF was added drop wise. Afterwards, the resulting mixture was stirred for a further hour at −78 °C and then allowed to the room temperature for another 1 h. The system was quenched with aqueous HCl and extracted with ethyl acetate. After removal of the solvents, the crude was recrystallized from CH2Cl2. Yield: 9.88 g (60.2%). 1H NMR (DMSO-d6): δ = 0.45 (d, 6H, 3JHH = 6.9 Hz, isopropyl), 1.35 (s, 2H, B(OH)2), 2.50 (m, 1H, isopropyl), 6.37 (t, 1H, 3JHH = 7.0 Hz, H–C5 of phenyl), 6.51 (1H, d, 3JHH = 7.6 Hz, H–C3 of phenyl), 6.54 (t, 1H, 3JHH = 7.4 Hz, H–C4 of phenyl) 6.57 (d, 1H, 3JHH = 7.2 Hz, H–C6 of phenyl). 13C NMR (DMSO-d6): δ = 24.4 (CH3 of isopropyl), 32.6 (CH of isopropyl), 124.1 (C3,5 of phenyl), 124.6 (C4 of phenyl), 128.6 (C6 of phenyl), 132.2(C1 of phenyl), 151.5 (C2 of phenyl).6-(2-Isopropylphenyl)pyridin-2-amine
A solution of 2-amino-6-bromopyridine (8.65 g, 0.05 mol) and Pd(PPh3)4 (0.30 g, 0.26 mmol) in toluene (240 mL) was stirred for 10 minutes. 2-Isopropylphenylboronic acid (8.20 g, 0.05 mol), ethanol (90 mL), sodium carbonate (10.6 g, 0.10 mol) and water (120 mL) were added successively. The degassed mixture was heated at reflux overnight, then cooled to room temperature and the aqueous phase was extracted with ethyl acetate. The combined extracts were dried over magnesium sulphate and concentrated in vacuo to give a solid. This solid was subjected to chromatography on silica gel using the mixture (ethyl acetate–CH2Cl2–pentane = 1:1:5, 2 or 3 drops of triethylamine per 1000 mL of solvents) as eluent. Yield: 10.3 g (97.2%). 1H NMR (CDCl3): δ = 1.19 (d, 6H, 3JHH = 6.9 Hz, isopropyl), 3.20 (m, 1H, isopropyl), 4.52 (s, 2H, NH2), 6.45 (d, 1H, 3JHH = 8.2 Hz, PyH), 6.70 (d, 1H, 3JHH = 7.3 Hz, PyH), 7.22 (m, 1H, PyH), 7.27 (dd, 1H, 3JHH = 3.3, 9.9 Hz, ArH), 7.35 (m, 1H, ArH), 7.39 (d, 1H, 3JHH = 7.3 Hz, ArH), 7.48 (t, 1H, 3JHH = 7.8 Hz, ArH). 13C NMR (benzene-d6): δ = 24.3, 29.4, 106.6, 114.5, 125.5, 125.7, 128.4, 129.4, 137.8, 140.2, 146.5, 157.9, 158.9. FT-IR (ATR mode, cm−1): ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) = 3461 (m), 3292 (m), 3127 (s), 2962 (m), 1629 (s), 1591 (w), 1567 (w), 1461 (s), 1354 (m), 1262 (m), 1052 (w), 985 (m), 800 (m), 757 (s). MS (ESI) [m/z] calcd for C14H16N2: 212.1, found 213.1 ([M + H] +, 75%), 235.1 ([M + Na ]+, 25%).
= 3461 (m), 3292 (m), 3127 (s), 2962 (m), 1629 (s), 1591 (w), 1567 (w), 1461 (s), 1354 (m), 1262 (m), 1052 (w), 985 (m), 800 (m), 757 (s). MS (ESI) [m/z] calcd for C14H16N2: 212.1, found 213.1 ([M + H] +, 75%), 235.1 ([M + Na ]+, 25%).
Compound 3
To a solution of 1 (1.9 g, 8.0 mmol) in pentane–diethyl ether (10:1) was added n-butyllithium (1.6 M in hexane, 5.0 mL, 8.0 mmol) at −35 °C and the mixture was warmed to room temperature and stirred for 2 h. The precipitate was filtered and washed with cold pentane–diethyl ether (10:1) and dried in vacuo to give a light-yellow solid. Yield: 2.2 g (86.4%).
The suspension of 1·Li(Et2O) (1.59 g, 5.0 mmol) in diethyl ether (50 mL) at −35 °C was added to a solution of 2 (1.29 g, 5.0 mmol) in diethyl ether (13 mL). After the mixture was stirred overnight at room temperature, LiCl was filtered off and diethyl ether was removed in vacuo to give a white crude solid, which was recrystallized from CH2Cl2 and pentane to yield white crystals. Yield: 1.9 g (82.5%). 1H NMR (C6D6): δ = 7.81–7.76 (m, 1H), 7.73 (dd, J = 6.9, 1.1 Hz, 2H), 7.61 (dt, J = 7.7, 0.8 Hz, 1H), 7.47–7.38 (m, 3H), 7.30–7.19 (m, 5H), 6.98 (t, J = 7.9 Hz, 1H), 6.80 (dd, J = 7.6, 0.9 Hz, 1H), 6.02 (dd, J = 8.3, 0.9 Hz, 1H), 5.69 (s, 1H), 3.82 (s, 1H), 1.26 (dq, J = 15.4, 7.7 Hz, 2H), 1.03–0.88 (m, 2H), 0.74 (t, J = 7.6 Hz, 6H), 0.00–0.15 (m, 6H). 1H NMR (CD2Cl2): δ = 7.89 (d, J = 7.6 Hz, 2H), 7.74–7.70 (m, 1H), 7.65 (t, J = 7.9 Hz, 1H), 7.57 (dd, J = 7.5, 0.9 Hz, 2H), 7.48–7.44 (m, 1H), 7.43–7.37 (m, 2H), 7.37–7.29 (m, 3H), 7.24 (ddd, J = 15.2, 7.5, 1.0 Hz, 2H), 6.44 (dd, J = 8.3, 0.8 Hz, 1H), 5.43 (s, 1H), 4.25 (s, 1H), 0.71 (dq, J = 15.4, 7.7 Hz, 2H), 0.50 (tt, J = 14.5, 7.4 Hz, 2H), 0.31–0.22 (m, 12H). 13C NMR (C6D6): δ = 163.6, 158.1, 155.9, 143.7, 141.4, 140.2, 137.7, 130.4, 129.5, 126.9, 126.6, 125.6, 124.7, 121.3, 120.7, 107.8, 106.3, 41.9, 15.1, 10.4, −3.8. ESI-MS: m/z calcd for C30H33BN2Si: 460.25; found: 461.26 [M + H]+. Elemental analysis calcd (%) for C30H33BN2Si: C 78.25, H 7.22, N 6.08; found: C 78.03, H 7.26, N 6.04. FTIR (ATR mode): 3378 (s), 2940 (m), 2857 (m), 1622 (s), 1571 (vs), 1486 (vs), 1447 (s), 1377 (s), 1255 (s), 1171(s), 833 (vs), 801 (vs), 734 (vs).
Compound 4
To a solution of 3 (1.125 g, 2.44 mmol) in 15 mL toluene at −35 °C were added 2 equivalents of n-BuLi (1.6 M in hexane, 3.05 mL, 4.88 mmol). After the solution was stirred 2 h at room temperature, the solvent was removed in vacuo and the resulting orange residue was recrystallized from THF and pentane at −35 °C to give yellow crystals. Yield: 1.63 g (80.7%). 1H NMR (C6D6): δ = 8.27–8.16 (m, 4H), 7.82 (t, J = 6.5 Hz, 2H), 7.48–7.40 (m, 1H), 7.37–7.27 (m, 4H), 7.09–7.01 (m, 2H), 6.93 (dd, J = 8.6, 0.8 Hz, 1H), 6.83 (dd, J = 7.0, 0.9 Hz, 1H), 2.95 (q, J = 6.5 Hz, 20H, THF), 1.23–1.08 (m, 20H, THF), 1.07–0.92 (m, 16H). 13C NMR (C6D6): δ = 163.8, 157.8, 140.6, 140.3, 137.0, 128.9, 125.0, 124.8, 122.9, 120.6, 120.1, 120.0, 115.7, 114.6, 98.6, 81.5, 67.6 (s, C–O, THF), 25.3 (s, C–O, THF), 13.2, 11.1, 3.4. Elemental analysis calcd (%) for C50H71BN2O5SiLi2(THF): C 71.67, H 8.80, N 3.10; found: C 71.64, H 8.48, N 3.46.Complex Zr-1
A solution of 4 (1.43 g, 1.58 mmol) in toluene (15 mL) at −35 °C was added to a solution of ZrCl4·2THF (596 mg, 1.58 mmol) in THF and the resulting orange–red mixture was stirred at room temperature for 36 h. LiCl was filtered off and all solvents was removed in vacuo. The resulting red residue was dissolved in toluene (10 mL) and stirred for 15 min, followed by the removal of any insoluble solid via filtration through celite. After the removal of the solvent, the red solid was recrystallized from CH2Cl2 and pentane at −35 °C. Yield: 220 mg (18.2%). 1H NMR (CD2Cl2): δ = 8.10 (d, J = 7.6 Hz, 2H), 8.07–7.95 (m, 3H), 7.84 (d, J = 7.7 Hz, 1H), 7.67 (dd, J = 7.7, 0.9 Hz, 1H), 7.55 (dt, J = 15.0, 4.1 Hz, 3H), 7.44–7.33 (m, 3H), 7.28 (td, J = 7.5, 1.2 Hz, 1H), 7.09 (dd, J = 8.1, 0.8 Hz, 1H), 3.00 (s, 4H, THF), 1.49 (t, J = 6.6 Hz, 4H, THF), 0.77 (d, J = 59.3 Hz, 10H), 0.47 (dd, J = 8.3, 6.4 Hz, 6H). 1H NMR (C6D6): δ = 8.24 (d, J = 7.7 Hz, 2H), 7.89 (d, J = 7.2 Hz, 1H), 7.78 (d, J = 7.6 Hz, 2H), 7.66 (d, J = 7.7 Hz, 1H), 7.50–7.37 (m, 3H), 7.27 (td, J = 7.6, 1.1 Hz, 1H), 7.19 (ddd, J = 7.8, 4.2, 2.3 Hz, 3H), 7.06 (dd, J = 7.7, 1.0 Hz, 1H), 6.96 (dd, J = 8.0, 0.8 Hz, 1H), 2.55 (s, 4H), 1.53 (s, 4H), 1.05 (s, 6H), 0.60 (s, 10H). 13C NMR (C6D6): δ = 165.2, 162.5, 159.3, 140.3, 137.1, 130.2, 129.7, 126.8, 125.4, 123.7, 122.0, 121.4, 120.3, 116.3, 111.2, 99.4, 76.4 (THF), 25.1 (THF), 11.9, 1.4, −0.1. 13C NMR (CD2Cl2): δ = 162.5, 158.8, 140.9, 137.5, 137.2, 129.7, 129.3, 127.2, 125.3, 124.1, 121.8, 121.3, 120.7, 117.0, 111.4, 99.7, 75.9 (THF), 25.9 (THF), 11.2, 4.1, 4.0, −0.1. Elemental analysis calcd (%) for C34H39BCl2N2OSiZr (THF)1/4(CH2Cl2)1/4: C 57.84, H 5.71, N 3.83; found: C 57.87, H 5.65, N 3.94. Crystals suitable for single-crystal X-ray analysis were obtained by recrystallization from CH2Cl2–pentane.Complex Hf-1
A solution of 4 (400 mg, 0.442 mmol) in toluene (15 mL) at −35 °C was added to a suspension of HfCl4·2THF (205 mg, 0.442 mmol) in toluene; then the yellow mixture was stirred at room temperature for 24 h. Lithium chloride was filtered off and the solvent was removed in vacuo. The resulting red residue was dissolved in toluene and any insoluble solid was filtered off through a pad of celite. After the removal of the solvent, the red solid was recrystallized from toluene and pentane at −35 °C. Yield: 35 mg (9.2%). 1H NMR (C6D6): δ = 8.19 (d, J = 48.0 Hz, 2H), 7.92 (d, J = 7.2 Hz, 1H), 7.83 (d, J = 7.4 Hz, 2H), 7.68 (d, J = 7.7 Hz, 1H), 7.48–7.39 (m, 3H), 7.28 (td, J = 7.5, 1.2 Hz, 2H), 7.22 (t, J = 7.8 Hz, 1H), 7.06 (dd, J = 7.7, 1.0 Hz, 1H), 7.04–7.00 (m, 1H), 2.50 (d, 3H), 1.64 (d, 2H), 1.03 (s, 6H), 0.54 (s, 4H), 0.20 (s, 4H). 13C NMR (C6D6): δ = 165.4, 162.5, 159.3, 145.6, 140.1, 139.3, 138.0, 137.2, 136.3, 130.2, 129.6, 129.3, 128.1, 126.8, 125.3, 124.2, 123.6, 123.0, 121.4, 120.7, 119.1, 117.9, 111.3, 96.1, 77.3, 25.2, 10.4, 2.6, 1.4. Elemental analysis calcd (%) for C34H39BCl2N2OSiHf (THF)1/2: C 52.99, H 5.31, N 3.43; found: C 53.07, H 5.35, N 3.53. Crystals suitable for single-crystal X-ray analysis were grown from toluene–pentane.Compound 5
To a solution of 2-(2-bromophenyl)-6-(2,2,5,5-tetramethyl-1,2,5-azadisilolidin-1-yl)pyridine (2.0, 5.1 mmol) in diethyl ether was slowly added a solution of n-butyl lithium (2.5 M, 2.45 mL, 6.1 mmol) in hexane at −78 °C and the mixture was stirred for 1.5 h at this temperature. A solution of dimesitylboron fluoride (2.06 g, 7.66 mmol) in THF (10 mL) was added and the mixture was stirred for a further hour at −78 °C, then gradually warmed to room temperature and stirred overnight. Ice water was added to hydrolyze the protecting group and the solution was stirred for 30 min. The mixture was diluted with ethyl acetate, washed with water and brine and dried over MgSO4. After removal of the solvent, the residue was subjected to chromatography on silica gel eluting with hexane–ethyl acetate (9:1) to yield the target product.31,32 Yield: 0.97 g (45.5%). 1H NMR (THF-d8): δ = 7.81–7.72 (m, 1H), 7.71–7.62 (m, 2H), 7.33 (dd, J = 7.5, 1.1 Hz, 1H), 7.13–6.99 (m, 2H), 6.56 (s, 4H), 6.42 (dd, J = 8.3, 1.1 Hz, 1H), 6.28 (s, 2H, NH2), 2.10 (s, 6H), 2.00–1.76 (m, 12H). 13C NMR (THF-d8): δ = 159.2, 158.2, 144.1, 141.8, 136.8, 134.0, 131.0, 129.5, 125.2, 122.2, 110.1, 105.4, 67.2, 25.1, 20.6. ESI-MS: m/z calcd for C29H31BN2: 418.26; found: 419.26 [M + H]+. FTIR (ATR mode): 3446 (m), 3348 (s), 2907 (m), 1633 (s), 1603 (vs), 1566 (s), 1493 (s), 1444 (s), 846 (m), 753 (vs), 729 (m), 702 (s).
Compound 6
To a solution of 5 (900 mg, 2.15 mmol) in 25 mL of THF at −35 °C was added n-BuLi (1.6 M in hexane, 1.34 mL, 2.15 mmol). After the solution was stirred 3 h at room temperature, the solvent was removed in vacuo and the resulting orange residue was washed twice with pentane to give a yellow solid, which was used without any further purification. Due to its sensitivity and instability, only the 1H NMR spectrum was recorded. 1H NMR (C6D6): δ = 8.04 (d, J = 7.3 Hz, 1H), 7.67 (d, J = 7.4 Hz, 1H), 7.20 (dd, J = 12.1, 5.9 Hz, 1H), 7.15–7.10 (m, 1H), 7.04 (t, J = 7.7 Hz, 1H), 6.71 (t, J = 23.4 Hz, 5H), 6.07 (d, J = 8.4 Hz, 1H), 4.79 (s, 1H), 3.20 (d, J = 6.1 Hz, 8H), 2.16 (s, 18H), 1.35–1.15 (m, 8H).Compound 7
To a solution of 6 (1.977 g, 3.47 mmol) in diethyl ether (50 mL) at −35 °C was added a solution of chlorodimethyl(2,3,4,5-tetramethylcyclopenta-2,4-dienyl)silane (768 μL, 3.47 mmol) in diethyl ether (5 mL). After the mixture was stirred overnight at room temperature, lithium chloride was filtered off and diethyl ether was removed in vacuo to yield a pink crude solid, which was subsequently recrystallized from diethyl ether and pentane to give a white solid. Yield: 1.70 g (82.1%). 1H NMR (C6D6): δ = 8.08 (d, J = 7.7 Hz, 1H), 7.54 (d, J = 7.5 Hz, 1H), 7.13 (dd, J = 7.5, 1.1 Hz, 1H), 7.07–6.97 (m, 2H), 6.85 (dd, J = 7.6, 1.0 Hz, 1H), 6.75 (s, 4H), 6.41 (s, 1H, NH), 6.26 (dd, J = 8.3, 1.0 Hz, 1H), 2.89 (s, 1H), 2.16 (d, J = 7.3 Hz, 18H), 1.68 (s, 6H), 1.52 (s, 6H), −0.06 (s, 6H). 13C NMR (C6D6): δ = 159.6, 159.2, 142.2, 140.6, 137.9, 137.2, 135.9, 134.5, 131.8, 131.1, 130.7, 129.5, 128.1, 125.2, 122.2, 110.0, 106.0, 54.2, 25.3, 20.8, 13.8, 11.2, −3.6. Elemental analysis calcd (%) for C40H49BN2Si: C 80.51, H 8.28, N 4.69; found C 80.41, H 8.43, N 4.43. ESI-MS: m/z calcd for C40H49BN2Si: 596.38; found. 619.38 [M + Na]+. FTIR (ATR mode): 3291 (m), 2913 (m), 1624 (m), 1560 (s), 1490 (vs), 1438 (s), 845 (s), 820 (vs), 756 (vs), 699 (s).Complex Zr-2
To a solution of 7 (1.0 g, 1.676 mmol) in toluene (10 mL) at −35 °C were added 2 equivalents of n-BuLi (1.6 M in hexane, 2.1 mL, 3.35 mmol). The solution was allowed to warm to room temperature and stirred for 2 h, then re-chilled to −35 °C and finally added to a solution of ZrCl4·2THF (632 mg, 1.676 mmol) in toluene (10 mL). The resulting brown mixture was stirred at room temperature for 36 h. LiCl was filtered off and the solvent was removed in vacuo. The resulting earth-yellow residue was dissolved in toluene and filtered off insoluble solid. After the removal of the solvent, the solid was recrystallized from toluene and pentane at −35 °C. Yield: 412 mg (32.5%). 1H NMR (C6D6): δ = 9.19 (dd, J = 7.8, 0.7 Hz, 1H), 7.58 (dd, J = 7.5, 0.9 Hz, 1H), 7.50 (td, J = 7.6, 1.4 Hz, 1H), 7.10 (td, J = 7.5, 1.2 Hz, 1H), 6.82–6.72 (m, 2H), 6.64 (s, 4H), 5.34 (dd, J = 7.5, 1.4 Hz, 1H), 2.16 (s, 12H), 2.09 (t, J = 4.7 Hz, 18H), 0.41 (s, 6H). 13C NMR (C6D6): δ = 162.7, 157.7, 148.7, 143.9, 143.5, 141.4, 139.3, 138.6, 135.9, 131.8, 131.6, 131.3, 130.5, 129.0, 128.8, 120.2, 106.7, 102.6, 24.2, 21.3, 14.5, 11.9, 2.8. Elemental analysis calcd (%) for C40H47BCl2N2OSiZr: C 63.48, H 6.26, N 3.70; found C 63.13, H 6.42, N 3.62. Crystals suitable for single-crystal X-ray analysis were grown from toluene–pentane.Compound 8
To a solution of 6-(2-isopropylphenyl)pyridin-2-amine (1.06 g, 5.0 mmol) in diethyl ether (25 mL) was added a butyllithium solution (1.6 M in hexane, 3.12 mL, 5.0 mmol) at −35 °C. After 1 min, a precipitate formed and the mixture was allowed to warm to room temperature and stirred for another 10 minutes. The suspension was cooled to −35 °C again and a solution of 2 (1.29 g, 5.00 mmol) in diethyl ether (20 mL) was added. The mixture was warmed to room temperature and stirred for another 2 h. The precipitate was filtered off and the resulting filtrate was dried in vacuo. The residue was recrystallized from diethyl ether and pentane to obtain a green solid. Yield: 1.98 g (91.1%). 1H NMR (CDCl3): δ = 7.85 (d, J = 7.5 Hz, 6H), 7.52 (t, J = 7.8 Hz, 3H), 7.43 (d, J = 7.5 Hz, 6H), 7.41–7.36 (m, 4H), 7.36–7.30 (m, 11H), 7.26–7.18 (m, 8H), 6.74 (d, J = 7.4 Hz, 3H), 6.50 (d, J = 8.2 Hz, 3H), 4.56 (s, 3H), 4.19 (s, 3H), 3.42 (dq, J = 13.7, 6.8 Hz, 3H), 1.17 (d, J = 6.9 Hz, 18H), 0.02–0.05 (m, 18H). 1H NMR (C6D6): δ = 7.77 (d, J = 7.6 Hz, 6H), 7.48–7.39 (m, 9H), 7.29–7.22 (m, 10H), 7.08 (td, J = 7.4, 1.4 Hz, 3H), 6.65 (dd, J = 7.4, 0.8 Hz, 3H), 5.96 (dd, J = 8.2, 0.8 Hz, 3H), 4.78 (s, 3H), 3.71–3.53 (m, 6H), 1.14 (d, J = 6.9 Hz, 18H), −0.01–0.09 (m, 17H). 13C NMR (CDCl3): δ = 159.2, 159.1, 146.4, 145.1, 140.8, 140.7, 137.8, 129.7, 128.2, 126.2, 125.5, 125.4, 124.5, 120.0, 114.6, 108.1, 77.2, 42.0, 29.2, 24.3, −3.5. ESI-MS: m/z calcd for C29H30N2Si: 434.22; found 435.22 [M + H]+. FTIR (ATR mode): 3404 (m), 2957 (m), 1588 (s), 1571 (s), 1460 (s), 1493 (s), 1371 (s), 1294 (s), 1036 (s), 855 (s), 801 (vs), 740 (vs).Compound 9
To a solution of 8 (1.5 g, 3.45 mmol) in toluene (7 mL) at −35 °C were added 2 equivalents of n-BuLi (1.6 M in hexane, 4.31 mL, 6.90 mmol). The solution was stirred for 2 h at room temperature, followed by the removal of the solvent. The resulting orange residue was recrystallized from THF and pentane at −35 °C to give yellow crystals, which turned out to be unstable even at low temperature. Yield: 1.83 g (72.5%). 1H NMR (THF-d8): δ = 7.82 (d, J = 7.5 Hz, 2H), 7.70 (d, J = 8.1 Hz, 2H), 7.11 (dd, J = 16.0, 7.6 Hz, 3H), 6.98 (t, J = 7.4 Hz, 1H), 6.76 (d, J = 24.1 Hz, 3H), 6.39 (s, 2H), 6.26 (d, J = 8.3 Hz, 1H), 5.67 (d, J = 6.2 Hz, 1H), 3.55 (d, J = 4.9 Hz, 3H), 3.44 (s, 1H), 1.69 (s, 4H), 1.09 (d, J = 6.9 Hz, 6H), 0.48 (s, 6H).Complex Zr-3
A solution of 9 (1.24 g, 1.694 mmol) in toluene (5 mL) at −35 °C was added to a solution of ZrCl4·2THF (639 mg, 1.694 mmol) in THF (10 mL); then the mixture was stirred at room temperature for 36 h. Lithium chloride was filtered off and all solvents were removed in vacuo. The resulting red residue was dissolved in CH2Cl2 and filtered through a pad of celite. After the removal of the solvent and recrystallization from CH2Cl2 and pentane at −35 °C, an orange solid was obtained. Yield: 283 mg (17.5%). 1H NMR (C6D6): δ = 8.05 (d, J = 8.1 Hz, 4H), 7.85–7.78 (m, 6H), 7.30 (ddd, J = 8.2, 7.0, 1.2 Hz, 4H), 7.21 (ddd, J = 8.0, 7.0, 1.0 Hz, 5H), 7.06–7.03 (m, 3H), 6.98 (dt, J = 9.9, 5.0 Hz, 2H), 6.44 (dd, J = 7.6, 0.9 Hz, 2H), 5.82 (dd, J = 8.2, 0.9 Hz, 2H), 3.07 (dt, J = 13.6, 6.8 Hz, 2H), 2.94 (s, 8H), 1.19 (d, J = 6.8 Hz, 12H), 0.88 (td, J = 6.7, 3.0 Hz, 9H), 0.64 (d, J = 3.4 Hz, 12H). 13C NMR (C6D6): δ = 165.7, 156.4, 146.8, 141.9, 140.8, 137.4, 134.6, 130.4, 129.4, 128.1, 126.5, 125.8, 125.4, 124.9, 123.3, 121.0, 117.1, 108.2, 83.1, 74.3, 30.4, 25.2, 24.7, 1.1. Elemental analysis calcd (%) for C36H40BCl2N2OSiZr (CH2Cl2) (toluene)1/4: C 61.00, H 5.73, N 3.98; found C 61.25, H 5.85, N 4.31.Variable-temperature 1H and 11B NMR
Manipulations were carried out inside a glove box under a nitrogen atmosphere. Catalyst:MAO:NBE = 1:30:10, [catalyst] = 0.08 mol L−1. MAO or MAO/NBE were dissolved in toluene-d8 and kept in an NMR tube, after cooling at −37 °C for 2 h, a chilled solution of the catalyst in toluene-d8 was added to the NMR tube. The total volume of the solution was ∼ 0.6 mL. The NMR tube was chilled in liquid nitrogen prior to 1H and 11B NMR measurements.
General procedure for NBE homopolymerization
All preparations were carried out inside a glove box. Defined amounts of NBE and MAO were dissolved in toluene (45 mL) in a Schlenk tube. The mixture was stirred for 5 min prior to the addition of a defined amount of catalyst in toluene (5 mL) and heated to the desired temperature. The reaction was quenched after 1 h by the addition of methanol (10 mL). The resulting mixture was poured into a solution of acidic methanol (500 mL of methanol containing 10 mL of concentrated HCl). The polymer was collected by filtration and washed with methanol (3 × 100 mL), then dried in vacuo at 50 °C for 2 days.General procedure for E-NBE copolymerization
Samples were prepared inside a glove box. Polymerization reactions were carried out in a Büchi glass reactor (500 mL), which was dried at 120 °C in vacuo for 2 h, cooled to 30 °C and purged with Ar gas prior to use. A mixture of monomer and MAO in toluene (ca. 245 mL) and a solution of the catalyst in toluene (ca. 5 mL) were quickly introduced into the reactor and stirred (300 rpm) at 30 °C. Ethylene gas was introduced once the mixture had reached the desired temperature. The reaction was quenched after 1 h by the addition of methanol (10 mL). The resulting mixture was poured into a solution of acidic methanol (500 mL of methanol containing 10 mL of concentrated HCl). The polymer was collected by filtration and washed with methanol (3 × 100 mL), then dried in vacuo at 50 °C for 2 days.Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (DFG, grant-nr: 2174/14-1) is gratefully acknowledged.Notes and references
- M. R. Buchmeiser, Curr. Org. Chem., 2013, 17, 2764–2775 CrossRef CAS.
- M. R. Buchmeiser, S. Camadanli, D. Wang, Y. Zou, U. Decker, C. Kühnel and I. Reinhardt, Angew. Chem., Int. Ed., 2011, 123, 3628–3633 CrossRef.
- Y. Zou, D. Wang, K. Wurst, C. Kühnel, I. Reinhardt, U. Decker, V. Gurram, S. Camadanli and M. R. Buchmeiser, Chem. – Eur. J., 2011, 17, 13832–13846 CrossRef CAS PubMed.
- B. P. Carrow and K. Nozaki, Macromolecules, 2014, 47, 2541–2555 CrossRef CAS.
- T. C. M. Chung, Macromolecules, 2013, 46, 6671–6698 CrossRef CAS.
- M. Ciftci, P. Batat, A. L. Demirel, G. Xu, M. Buchmeiser and Y. Yagci, Macromolecules, 2013, 46, 6395–6401 CrossRef CAS.
- J.-Y. Dong and Y. Hu, Coord. Chem. Rev., 2006, 250, 47–65 CrossRef CAS PubMed.
- N. M. Franssen, J. N. Reek and B. de Bruin, Chem. Soc. Rev., 2013, 42, 5809–5832 RSC.
- G. Xu, D. Wang and M. R. Buchmeiser, Macromol. Rapid Commun., 2012, 33, 75–79 CrossRef CAS PubMed.
- Z. Jian, P. Wucher and S. Mecking, Organometallics, 2014, 33, 2879–2888 CrossRef CAS.
- H. Leicht, I. Göttker-Schnetmann and S. Mecking, Angew. Chem., Int. Ed., 2013, 125, 4055–4058 CrossRef.
- K. E. Allen, J. Campos, O. Daugulis and M. Brookhart, ACS Catal., 2015, 5, 456–464 CrossRef CAS.
- A. C. Gottfried and M. Brookhart, Macromolecules, 2003, 36, 3085–3100 CrossRef.
- S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169–1203 CrossRef CAS PubMed.
- G. V. Narayana, G. Xu, D. Wang, W. Frey and M. R. Buchmeiser, ChemPlusChem, 2014, 79, 151–162 CrossRef CAS.
- G. Xu, G. V. Narayana, M. Speiser, D. Wang, Y. Zou and M. R. Buchmeiser, Macromol. Chem. Phys., 2014, 215, 893–899 CrossRef CAS.
- H. G. Alt and E. Samuel, Chem. Soc. Rev., 1998, 27, 323–329 RSC.
- G. W. Coates, Chem. Rev., 2000, 100, 1223–1252 CrossRef CAS PubMed.
- T. Senda, H. Hanaoka, S. Nakahara, Y. Oda, H. Tsurugi and K. Mashima, Macromolecules, 2010, 43, 2299–2306 CrossRef CAS.
- S. Gentil, M. Dietz, N. Pirio, P. Meunier, J. C. Gallucci, F. Gallou and L. A. Paquette, Organometallics, 2002, 21, 5162–5166 CrossRef CAS.
- A. A. H. Van Der Zeijden, C. Mattheis and R. Fröhlich, Chem. Ber., 1997, 130, 1231–1234 CrossRef CAS.
- J. A. Ewen, Macromol. Symp., 1995, 89, 181–196 CrossRef CAS.
- J. Okuda, F. J. Schattenmann, S. Wocadlo and W. Massa, Organometallics, 1995, 14, 789–795 CrossRef CAS.
- L. J. Irwin, J. H. Reibenspies and S. A. Miller, J. Am. Chem. Soc., 2004, 126, 16716–16717 CrossRef CAS PubMed.
- S. A. Miller and J. E. Bercaw, Organometallics, 2006, 25, 3576–3592 CrossRef CAS.
- E. Kirillov, L. Toupet, C. W. Lehmann, A. Razavi and J.-F. Carpentier, Organometallics, 2003, 22, 4467–4479 CrossRef CAS.
- M. A. Schmid, H. G. Alt and W. Milius, J. Organomet. Chem., 1995, 501, 101–106 CrossRef CAS.
- Stereoselective Polymerization With Single-Site Catalysts, ed. C. J. Price, L. J. Irwin, D. A. Aubry and S. A. Miller, CRC Press, Boca Raton, FL, 2008 Search PubMed.
- R. Tanaka, I. Kamei, Z. Cai, Y. Nakayama and T. Shiono, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 685–691 CrossRef CAS.
- L. J. Irwin, J. H. Reibenspies and S. A. Miller, Polyhedron, 2005, 24, 1314–1324 CrossRef CAS PubMed.
- H. Amarne, C. Baik, S. K. Murphy and S. Wang, Chem. – Eur. J., 2010, 16, 4750–4761 CrossRef CAS PubMed.
- Y.-L. Rao, H. Amarne, J.-S. Lu and S. Wang, Dalton Trans., 2013, 42, 638–644 RSC.
- S. Hermanek, Chem. Rev., 1992, 92, 325–362 CrossRef CAS.
- F. Teixidor, M. A. Flores, C. Viñas, R. Sillanpää and R. Kivekäs, J. Am. Chem. Soc., 2000, 122, 1963–1973 CrossRef CAS.
- Y.-L. Rao, H. Amarne, S.-B. Zhao, T. M. McCormick, S. Martić, Y. Sun, R.-Y. Wang and S. Wang, J. Am. Chem. Soc., 2008, 130, 12898–12900 CrossRef CAS PubMed.
- I. Tritto, L. Boggioni, C. Zampa and D. R. Ferro, Macromolecules, 2005, 38, 9910–9919 CrossRef CAS.
- P. Sudhakar, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 444–452 CrossRef CAS.
- D. E. Babushkin, N. V. Semiloenova, V. N. Panchenko, A. P. Sobolov, V. A. Zakharov and E. P. Talsi, Macromol. Chem. Phys., 1997, 198, 3845–3854 CrossRef CAS.
- D. Cam and U. Giannini, Makromol. Chem., 1992, 193, 1049–1055 CrossRef CAS.
- W. Kaminsky and R. Steiger, Polyhedron, 1988, 7, 2375–2381 CrossRef CAS.
- W. Kaminsky, Macromol. Chem. Phys., 1996, 197, 3907–3945 CrossRef CAS.
- J. Schwartz and K. I. Gell, J. Organomet. Chem., 1980, 184, C1–C2 CrossRef CAS.
- M. D. Fryzuk and S. S. H. Mao, J. Am. Chem. Soc., 1993, 115, 5336–5337 CrossRef CAS.
- M. D. Fryzuk, P. B. Duval, S. S. S. H. Mao, M. J. Zaworotko and L. R. MacGillivray, J. Am. Chem. Soc., 1999, 121, 2478–2487 CrossRef CAS.
- M. Kamitani, B. Pinter, C.-H. Chen, M. Pink and D. J. Mindiola, Angew. Chem., Int. Ed., 2014, 126, 11093–11095 CrossRef.
- F. M. Hartner Jr., J. Schwartz and S. M. Clift, J. Am. Chem. Soc., 1983, 105, 640–641 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Single crystal X-ray structures of compounds 3 and 4, 1H-, 13C-, 11B-NMR spectra, temperature-dependent NMR measurements, NMR spectra of catalysts and polymers, details of single crystal X-ray analysis of Zr-1, Zr-2 and Hf-1. CCDC 1050437, 1050441, 1050440, 1050434 and 1050442. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5py00303b |
| This journal is © The Royal Society of Chemistry 2015 |