Cyclodextrin- and calixarene-based polycationic amphiphiles as gene delivery systems: a structure–activity relationship study†
Received
16th October 2014
, Accepted 17th November 2014
First published on 19th November 2014
Abstract
Multi-head/multi-tail facial amphiphiles built on cyclodextrin (CD) and calixarene (CA) scaffolds are paradigmatic examples of monodisperse gene delivery systems. The possibility to precisely control the architectural features at the molecular level offers unprecedented opportunities for conducting structure–activity relationship studies. A major requirement for those channels is the design of a sufficiently diverse ensemble of compounds for parallel evaluation of their capabilities to condense DNA into transfection nanoparticles where the gene material is protected from the environment. Here we have undertaken the preparation of an oriented library of β-cyclodextrin (βCD) and calix[4]arene (CA4) vectors with facial amphiphilic character designed to ascertain the effect of the cationic head nature (aminothiourea-, arginine- or guanidine-type groups) and the macrocyclic platform on the abilities to complex plasmid DNA (pDNA) and in the efficiency of the resulting nanocomplexes to transfect cells in vitro. The hydrophobic domain, formed by hexanoyl or hexyl chains, remains constant in each series, matching the overall structure found to be optimal in previous studies. DLS, TEM and AFM data support that all the compounds self-assemble in the presence of pDNA through a process that involves initially electrostatic interactions followed by formation of βCD or CA4 bilayers between the oligonucleotide filaments. Spherical transfectious nanoparticles that are monomolecular in DNA are thus obtained. Evaluation in epithelial COS-7 and human rhabdomyosarcoma RD-4 cells evidenced the importance of having primary amino groups in the vector to warrant high levels of transfection, probably because of their buffering capacity. The results indicate that the optimal cationic head depends on the macrocyclic core, aminothiourea groups being preferred in the βCD series and arginine groups in the CA4 series. Whereas the transfection efficiency relationships remain essentially unchanged within each series, irrespective of the cell type, the optimal platform (βD or CA4) strongly depends on the cell type. The results illustrate the potential of monodisperse vector prototypes and diversity-oriented strategies on identifying the optimal candidates for gene therapy applications.
Introduction
Gene therapy represents a potent tool for the therapeutic treatment of a broad range of genetic and acquired diseases. After some controversial failures, the last few years have witnessed the first really successful applications of this technique, opening the possibility to fight pathologies like severe immunodeficiencies, the Wiskott–Aldrich syndrome, beta-thalassaemia, haemophilia, adrenoleukodystrophy and several types of cancer,1 with an ever increasing number of clinical trials under way.2 In most cases, the delivery of genetic material aimed at the correction of defects in the patients’ cell genome is performed by using viruses as vectors in the context of an “ex vivo” strategy. Adenoviruses and retroviruses are properly modified to eliminate their infectiousness and to incorporate the therapeutic sequences. Their efficiency in transfecting cells, due to their own nature, does the rest. Yet, the use of viral vectors is still accompanied by some not negligible risks, such as violent adverse immune responses and genotoxicity,3 and limits like complicate and expensive preparation processes, production in rather scarce quantities, restricted dimensions of the nucleic acid filaments that can be transported or possible compromised bioavailability because of their large molecular size. These not yet resolved problems fuelled the research for alternatives that materialized, in the years, in the development of non-viral gene vectors based initially on cationic lipids and then on cationic polymers, dendrimers and nanoparticles.4 Among them, some have shown relevant efficiency and relatively low toxicity and are widely used in transfection protocols. Nevertheless, although some outstanding examples are on record,5 the systems proposed so far cannot really replace viruses. For these reasons, the efforts in the development of new molecules and formulations able to deliver nucleic acids into cells with increasing efficiency and safety as of today are still ongoing.
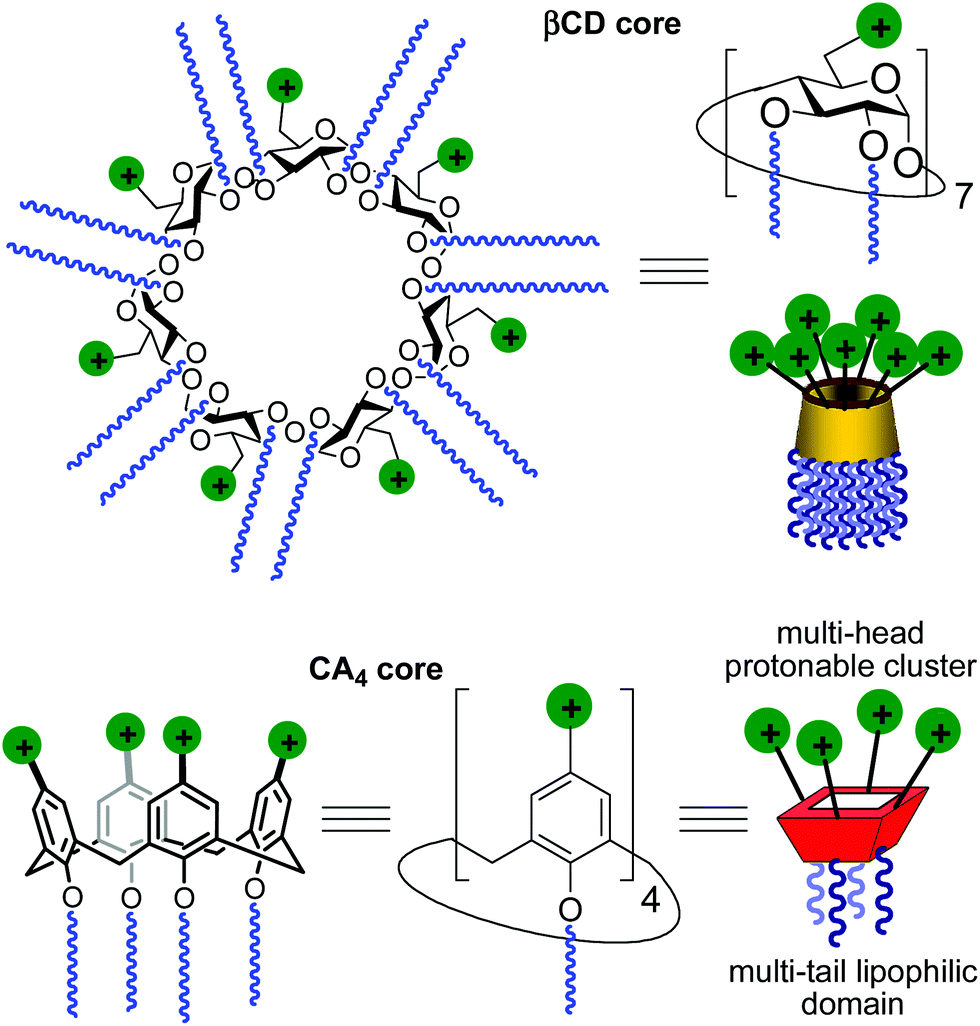
A main difficulty in non-viral gene carrier optimization strategies is the multicomponent or polydisperse nature and random conformational properties of most of the systems that are currently available, which handicaps establishing reliable relationships between chemical structure and transfection efficiency. The design of monodisperse, molecularly well-defined gene vector prototypes, while more challenging, offers unprecedented opportunities in this respect. In recent years, cyclodextrins (CDs) and calixarenes (CAs) have been proposed as central frameworks allowing the controlled incorporation of nucleic acid complexation elements.6 While some positive results using polycationic hydrophilic derivatives have been reported,7 we and others have shown that endowing the vector architecture with facial amphiphilicity,8 by installing segregated clusters of cationizable and hydrophobic groups at opposite rims in the macrocyclic cores, significantly improves the nucleic acid condensation abilities and the transfection efficiency of the resulting supramolecular nanocomplexes (Fig. 1).9–12
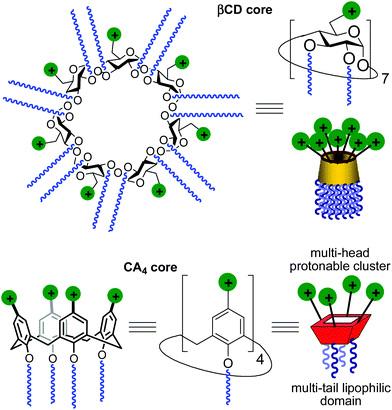 |
| | Fig. 1 Schematic representation of the amphiphilic polycationic β-cyclodextrin (βCD) and calix[4]arene (CA4) gene vector prototypes in the optimal skirt-type and upper-rim protonable cone arrangements, respectively. | |
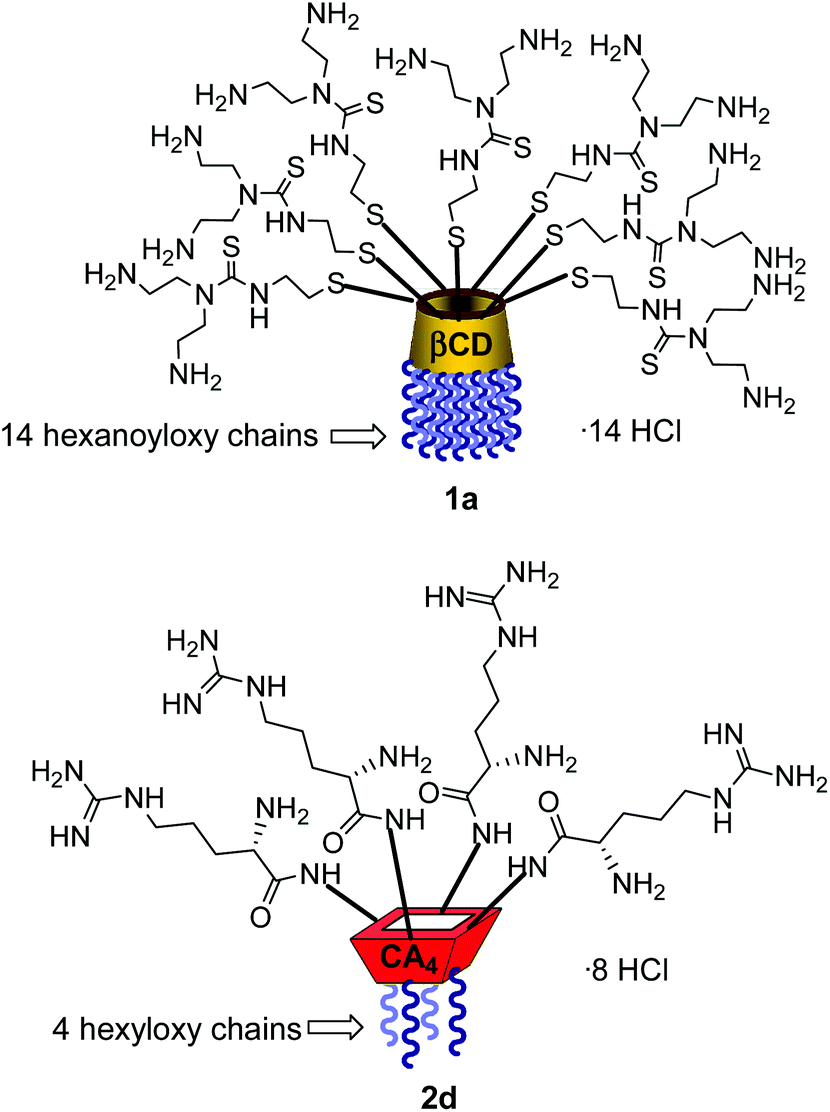
Structure–activity relationship studies independently conducted in both series of compounds allowed identification of some favourable structural features. Thus, β-cyclodextrin (βCD)-based architectures were generally superior to αCD or γCD derivatives13 and dispositions having the cationic heads at the primary face and the lipophilic tails at the secondary hydroxyls (skirt-type arrangement)14 performed better than analogues exhibiting the reversal orientation.15 In the polycationic amphiphilic CA family, the reports point to the supremacy of the calix[4]arene (CA4) core in the cone conformation with the protonable groups at the upper rim.16 The combination of thiourea and amine groups at the cationic domain was found optimal for βCD derivatives,17 whereas arginine clustering imparted the highest DNA delivery efficiency in the CA4 series.16 Copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) and amine–isothiocyanate coupling were implemented for “click” multiconjugation, both ligation chemistries warranting full homogeneity. Interestingly, in both types of macrocycles incorporation of linear six-carbon chains into the hydrophobic domain provided the best results. The tetradecacationic βCD derivative 1a, displaying a dendritic presentation of primary amino groups and fourteen hexanoyl chains at the secondary hydroxyls, and the tetraarginine-CA4 conjugate 2d, bearing four hexyl ether substituents, were identified as lead compounds within each category (Fig. 2).
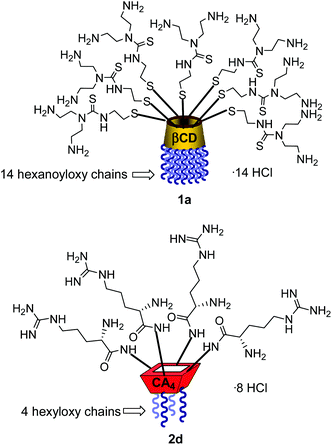 |
| | Fig. 2 Schematic representation of the lead βCD and CA4 vectors 1a and 2d. | |
In our ongoing efforts to develop CD and CA-based artificial viruses for drug and gene delivery,16 evaluating the properties of structurally related series of CD and CA vectors in the same cell systems was highly sought. By combining our expertise on the chemistry and supramolecular properties of both archetypes we are in the position to underpin whether or not the macrocyclic platform determines the optimal architectural requirements for gene therapy applications.
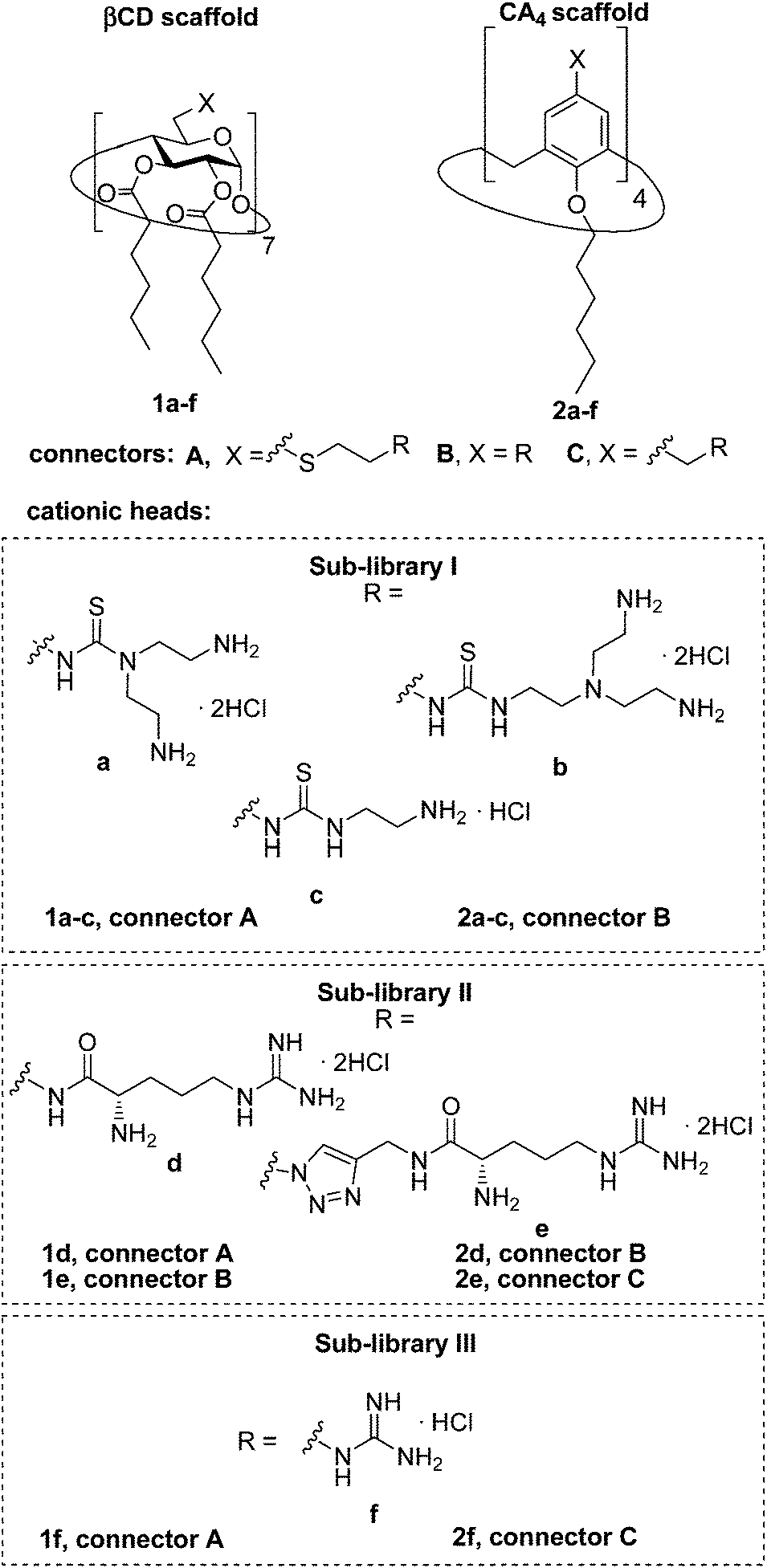
Herein we report the synthesis of a collection of polycationic amphiphilic βCD (1a–f) and CA4 (2a–f) homogeneously functionalized with the same polar heads at the narrower and upper rim, respectively, and distinguished by the cationic species introduced: ammonium, arginine and guanidinium groups (sub-libraries I–III, Fig. 3). The lipophilic domains, in contrast, were maintained unvaried within a series, namely hexanoyl ester groups for the βCD derivatives and hexyl ether chains for the CA4 counterparts. The relative orientation of the multi-head and multi-tail domains thus remains constant within each family. A parallel evaluation of the ability of these regioselectively functionalized macrocycles to condense pDNA into discrete cationic nanoparticles and of their in vitro transfection capabilities toward the COS-7 African green monkey kidney epithelial and RD-4 human rhabdomyosarcoma cell lines have been conducted.
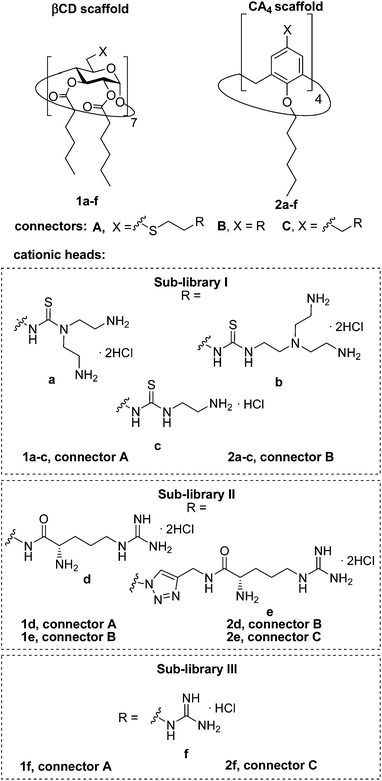 |
| | Fig. 3 Structures of the polycationic amphiphilic βCD and CA4 derivatives synthesized and evaluated in this work. | |
Results and discussion
Synthesis
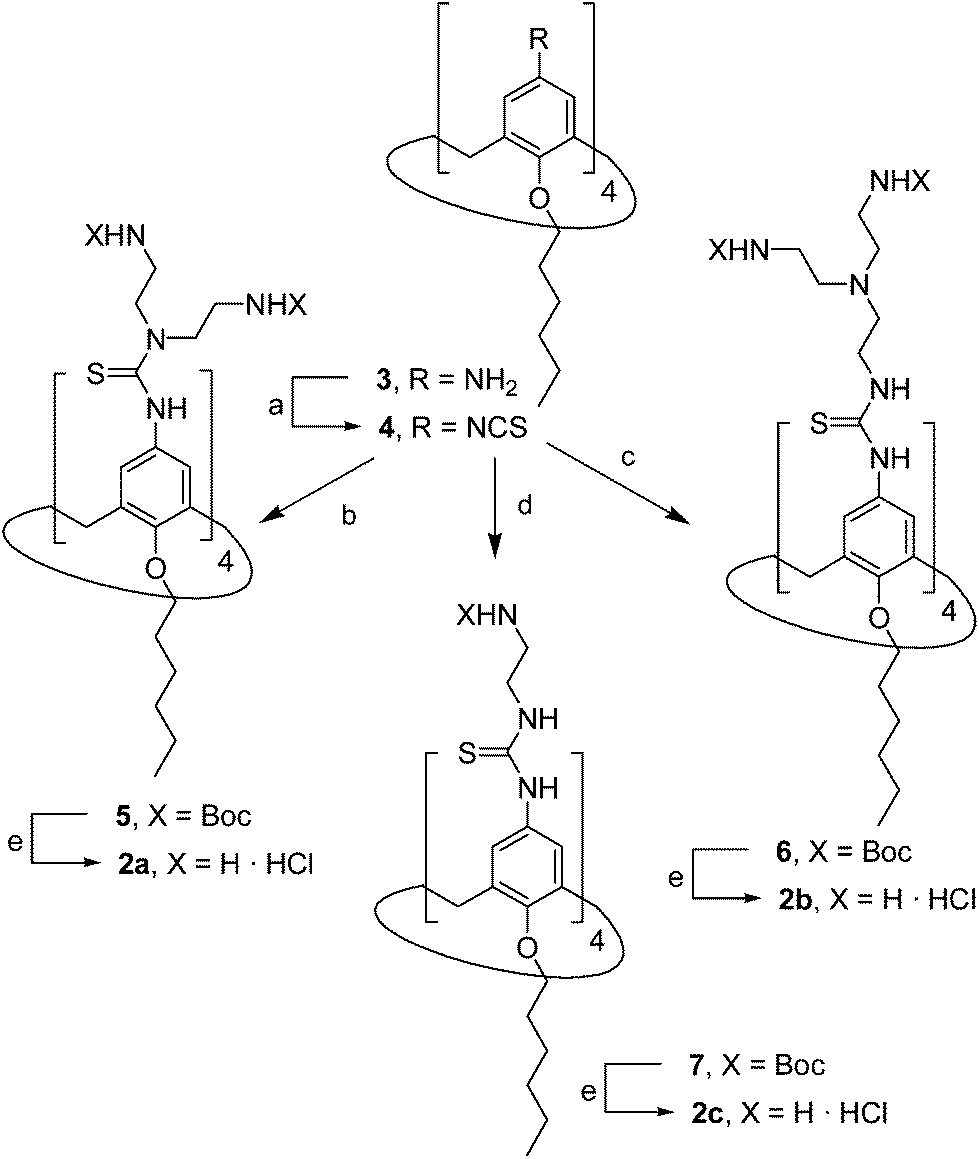
The known polycationic amphiphilic βCD derivatives 1a–c, bearing aminothiourea head groups, have been previously shown to exhibit promising transfection capabilities both in vitro9b and, in the case of 1a, also in vivo.14d,f For this reason, they were selected as the βCD representatives in sub-library I for the purpose of this study. The synthesis of the new analogues 2a–c (Scheme 1), incorporating the same cationic heads in the CA4 series, started from the known 5,11,17,23-tetraamino-25,26,27,28-tetrakis(n-hexyloxy)calix[4]arene 3,10b which was transformed into the pivotal tetraisothiocyanate 4 using an excess of the isothiocyanation reagent carbon disulfide/bis(tert-butyl) carbonate.18 Reaction of 4 with bis(2-tert-butyloxycarbonylaminoethyl) amine (→5), bis(2-tert-butyloxycarbonylaminoethyl) 2-aminoethyl amine (→6) and 2-tert-butyloxycarbonylaminoethylamine (→7), in the presence of triethylamine, and the removal of the carbamate protecting group in the thiourea adducts by trifluoroacetic acid (TFA)-promoted hydrolysis in the presence of triethylsilane (TES) afforded the target CA4 facial amphiphiles 2a–c in high yield. The final compounds were isolated as the corresponding octa- (2a and 2b) or tetra-hydrochloride salts (2c) after repeated dissolution/evaporation cycles from methanolic HCl and final lyophilisation (Scheme 1).
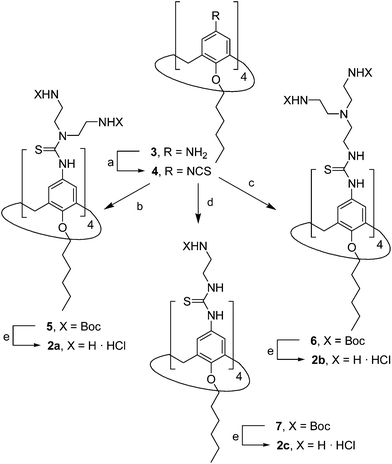 |
| | Scheme 1 Synthesis of polyaminothioureido CA4 derivatives 2a–c. Reagents and conditions: (a) (i) CS2, Et3N, EtOH, rt, 2 h; (ii) Boc2O, DMAP, EtOH, 0 °C → rt, 2 h, 88%; (b) NH(CH2CH2NHBoc)2, Et3N, DCM, rt, 12 h, quantitative; (c) H2NCH2CH2N(CH2CH2NHBoc)2, Et3N, DCM, rt, 12 h, 99%; (d) NH2CH2CH2NHBoc, Et3N, DCM, rt, 1.2 h, 99%; (e) (i) TFA–TES–DCM, 5 min, rt; (ii) HCl, 99%. | |
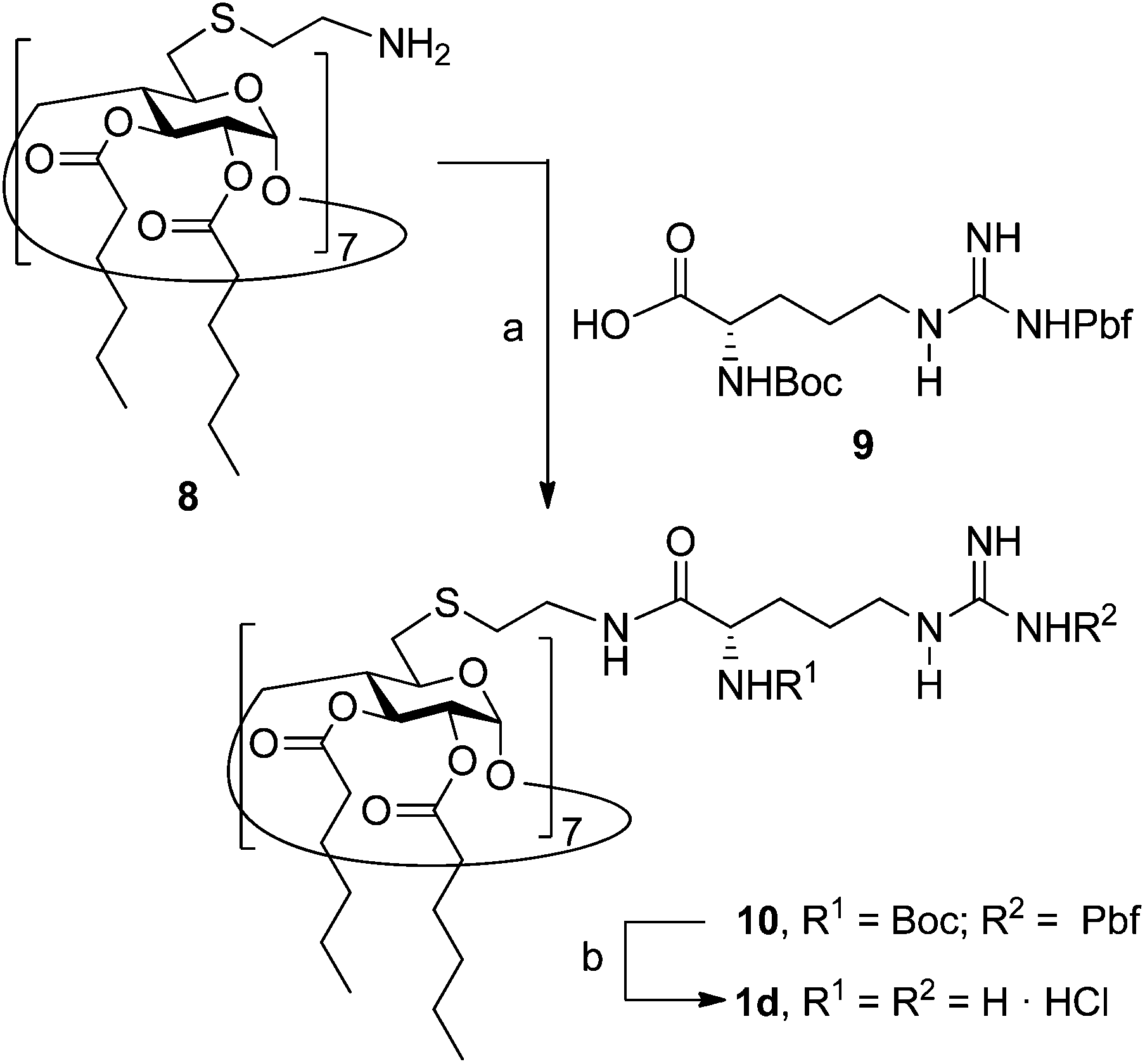
The lead calixarene derivative 2d was the reference compound that inspired sub-library II. The homologous-βCD representative 1d was obtained by hepta-amidation of the per-(C-6)-cysteaminyl-per-(O-2,O-3)-hexanoyl βCD precursor 8, accessible in only three steps from βCD,9b,19 with the commercial protected arginine derivative 9 (→10) and final deprotection (Scheme 2).
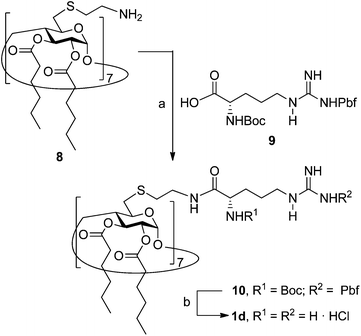 |
| | Scheme 2 Synthesis of amide-linked polyarginine βCD derivative 1d. Reagents and conditions: (a) HBTU, DIPEA, DCM, rt, 12 h, 68%; (b) (i) 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5:2.5 TFA–TIS–DCM, rt, 1 h; (ii) 0.1 M HCl, quantitative. Pbf: 2,2,4,6,7-pentamethyl-dihydrobenzofuran-5-sulfonyl. HBTU: o-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate. DIPEA: Diisopropyl ethyl amine. TIS: triisopropylsilane. :2.5:2.5 TFA–TIS–DCM, rt, 1 h; (ii) 0.1 M HCl, quantitative. Pbf: 2,2,4,6,7-pentamethyl-dihydrobenzofuran-5-sulfonyl. HBTU: o-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate. DIPEA: Diisopropyl ethyl amine. TIS: triisopropylsilane. | |
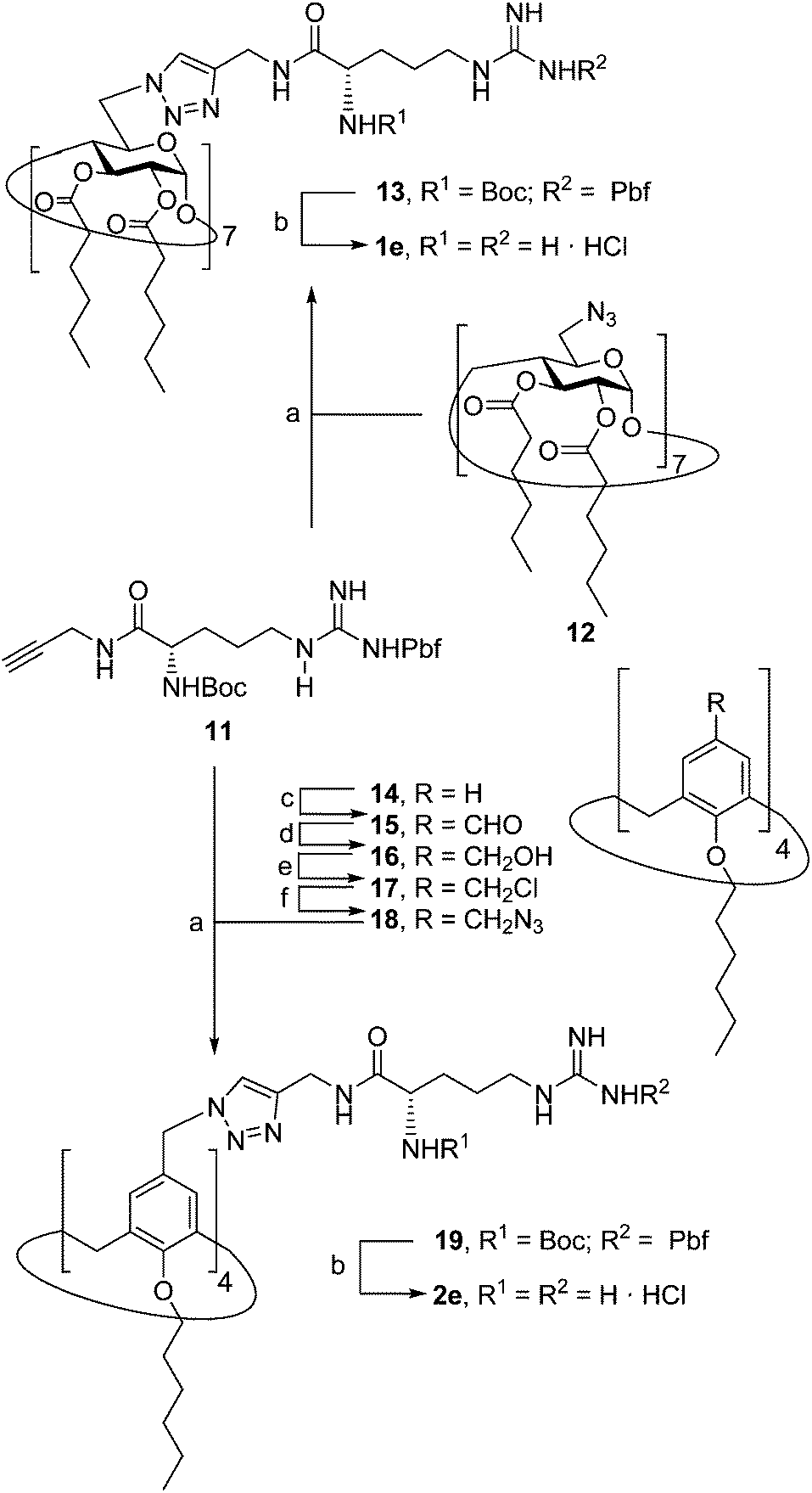
The presence of the cysteaminyl connector releases the steric constrain at the primary βCD rim and warrants homogeneous multiconjugation even for hyperbranched architectures.20 Sub-library II has been further enriched by synthesizing the corresponding 1,2,3-triazol-linked isosters 1e and 2e (Scheme 3). Whereas the resulting triazole segment is considered to be isosteric of the amide functionality, it imparts a higher rigidity thereby influencing the conformational properties. Moreover, the triazole moiety might actively participate in DNA complexation and release by behaving as the hydrogen bonding acceptor, and/or intercalating DNA, thereby affecting transfection efficiency.21 The key “click” multiconjugation step involved the copper(I)-catalyzed azide–alkyne cycloaddition reaction (CuAAC)22 of the known hexanoylated heptaazide βCD 129c or the hexyloxy tetraazide CA4 derivative 18 with the propargyl-armed arginine derivative 11 (→13 and 19, respectively). Alkyne 11 was prepared by standard amide coupling of the commercial protected amino acid 9 with propargylamine, whereas installation of the azidomethyl substituents onto the upper rim of the CA4 scaffold, to access tetraazide 18, was accomplished in four-steps from the tetrahexyl ether precursor 1423 through a reaction sequence involving formylation (→15), reduction (→16), replacement of the resulting primary hydroxyl groups into chloro groups (→17) and nucleophilic displacement of the latter by an azide anion. Acid-promoted removal of the carbamate and sulfonyl protecting groups in the triazole adducts 13 and 19 afforded the target polycationic amphiphilic clicked clusters 1e and 2e (Scheme 3).
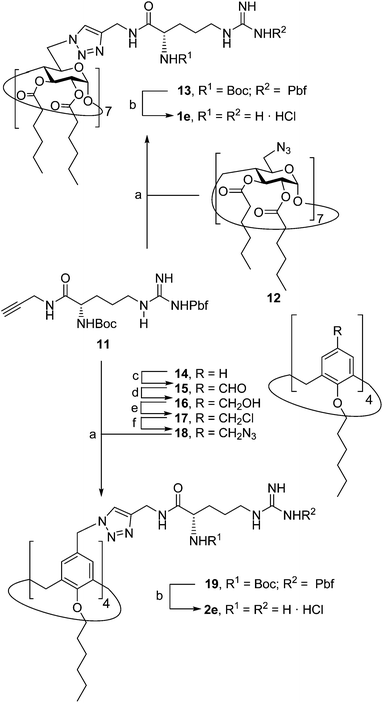 |
| | Scheme 3 Synthesis of triazol-linked polyarginine βCD and CA4 “clicked” clusters 1e and 2e. Reagents and conditions: (a) CuI·P(OEt)3, DIPEA, acetone, reflux, 48 h, 99% (for 13) or 24 h, 71% (for 19); (b) (i) 95:2.5:2.5 TFA–TIS–H2O, rt, 1 h; (ii) 0.1. M HCl, quantitative; (c) (i) hexamethylenetetramine, TFA, reflux, 2 d; (ii) HCl, 3 h, 96%; (d) NaBH4, EtOH, 0 °C → rt, 18 h, 86%; (e) SOCl2, DCM, 4 h, rt, 95%; (f) NaN3, DMF, 16 h, 40 °C, 95%. | |
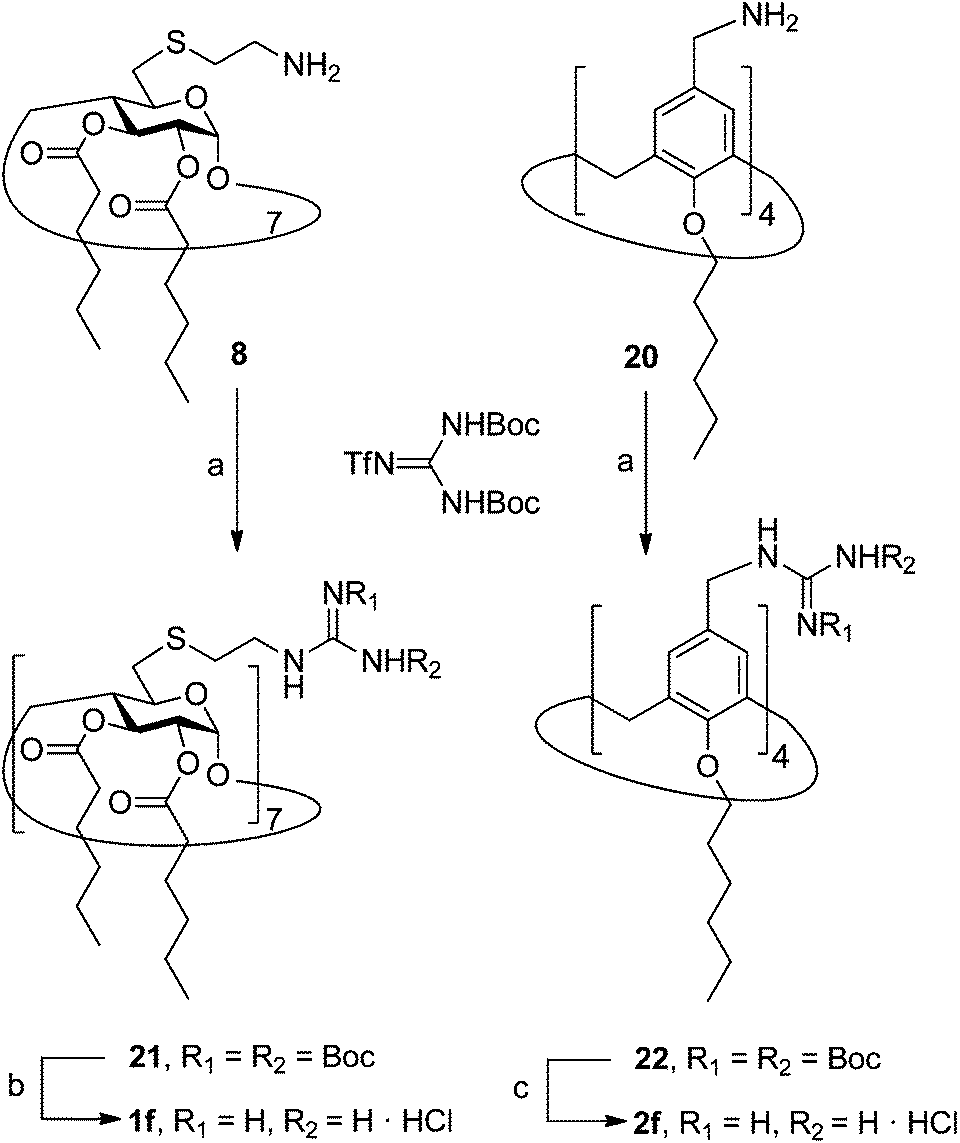
In order to evaluate possible synergies between the amino and the guanidino groups of polyarginine facial amphiphiles in DNA complexation and delivery, the inclusion in our study of the polyguanidine analogues 1f and 2f, grouped in sub-library III, as controls lacking any amine functionality was considered appropriate. Their synthesis was achieved by guanidinylation of the βCD heptaamine 8 and the CA4 tetraamine derivative 20, obtained by the reduction of tetraazide 18 (see Experimental), respectively, with bis-Boc-triflyl guanidine and the final hydrolysis of the carbamate protecting groups (Scheme 4).
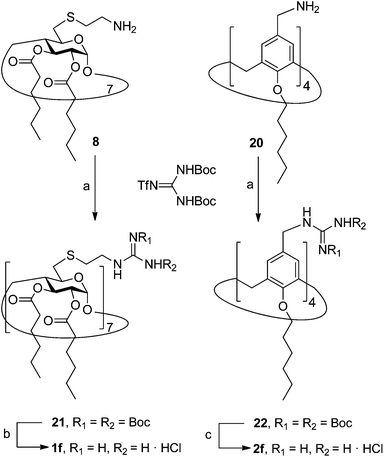 |
| | Scheme 4 Synthesis of polyguanidino βCD and CA4 derivatives 1f and 2f. Reagents and conditions: (a) Et3N, DCM, rt, 12 h, 99% (for 21) or 48 h, 91% (for 22); (b) (i) 1:1 TFA–DCM, rt, 3 h; (ii) 0.1 M HCl, 99%; (c) TES, dioxane, 0.1 M HCl, rt, 24 h, quantitative. | |
pDNA complexation abilities and nanocomplex characterization
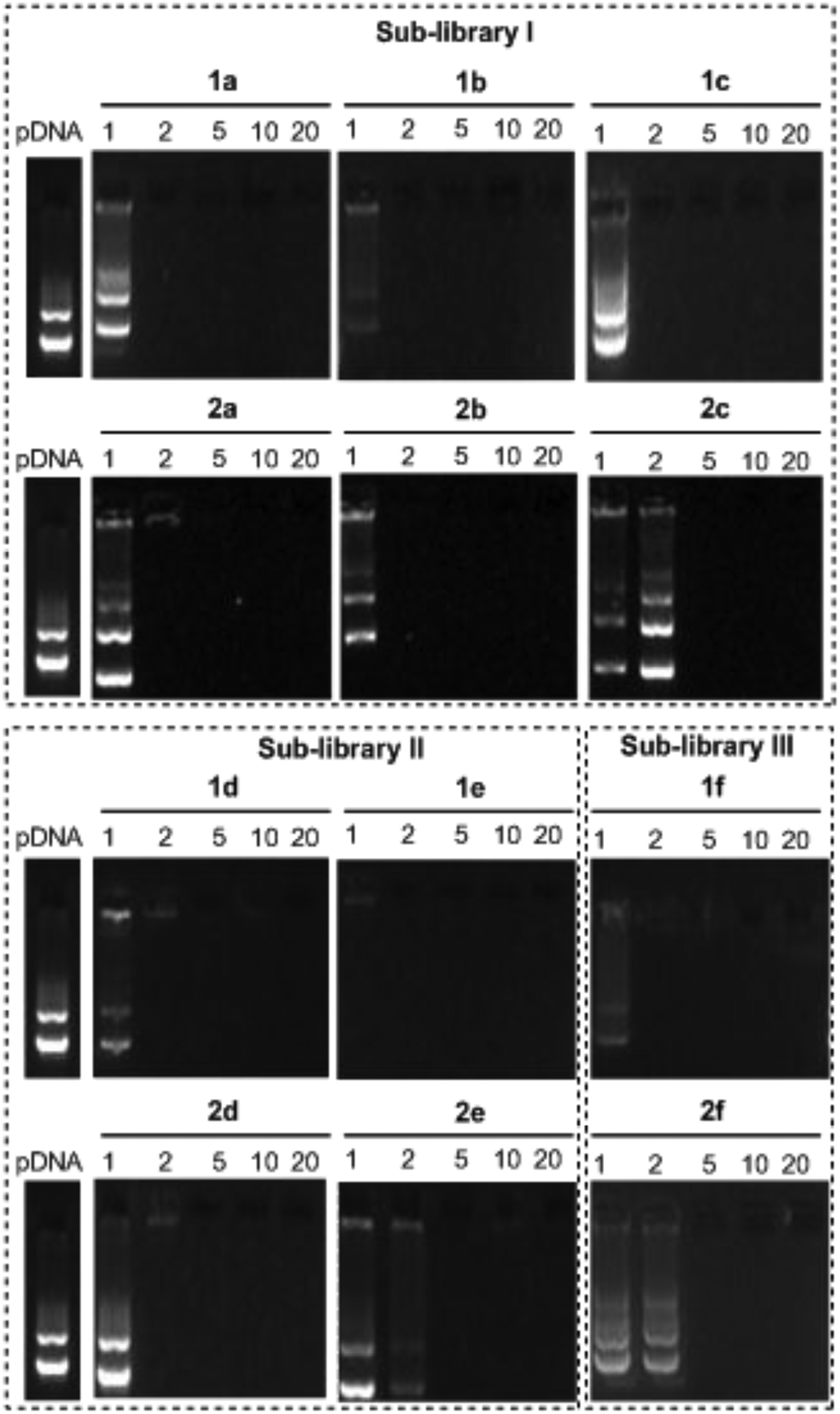
The ability of the βCD- and CA4-based polycationic amphiphiles 1a–f and 2a–f to complex and protect DNA was first examined by electrophoresis mobility shift assays (EMSA) and by determining their capability to prevent intercalation of GelRed™ (Biotium) used to stain the DNA filaments. The luciferase-encoding plasmid DNA (pDNA) pTG11236 (pCMV-SV40-luciferase-SV40pA, 5739 base pairs) was employed in this experimental setting at protonable nitrogen/phosphorous (N/P) ratios 1, 2, 5, 10 and 20. The data (Fig. 4) evidenced that all the tested derivatives were able to fully complex and protect pDNA at N/P > 2 as indicated by the absence of free mobile plasmid in the corresponding lanes. As a general trend, sub-library I compounds 1a,b or 2a,b, displaying a dendritic presentation of primary amino groups, were more efficient at condensing pDNA than the linear aminothioureido derivatives 1c or 2c. In sub-library II, amide connectors (1d or 2d) performed better than the triazol linkers (1e or 2e). Overall, βCD-scaffolded polycations achieved full neutralization of pDNA and fully blocked GelRed™ intercalation at lower N/P ratios as compared with the respective CA4 counterparts.
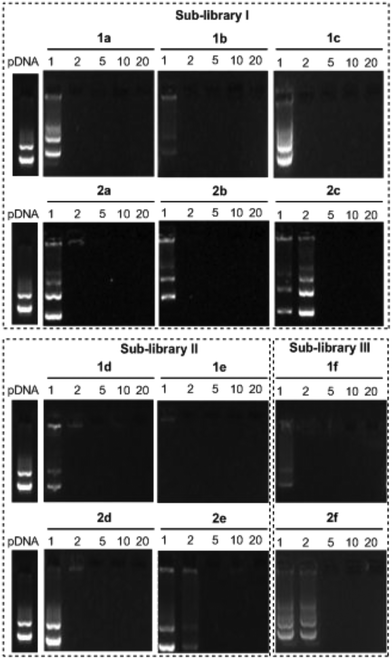 |
| | Fig. 4 Electrophoresis mobility shift assays (EMSA) for pDNA complexes formulated with 1a–f and 2a–f at different N/P ratios (1, 2, 5, 10 and 20). GelRed™ was used as the staining reagent. Naked pDNA was used as the negative control. | |
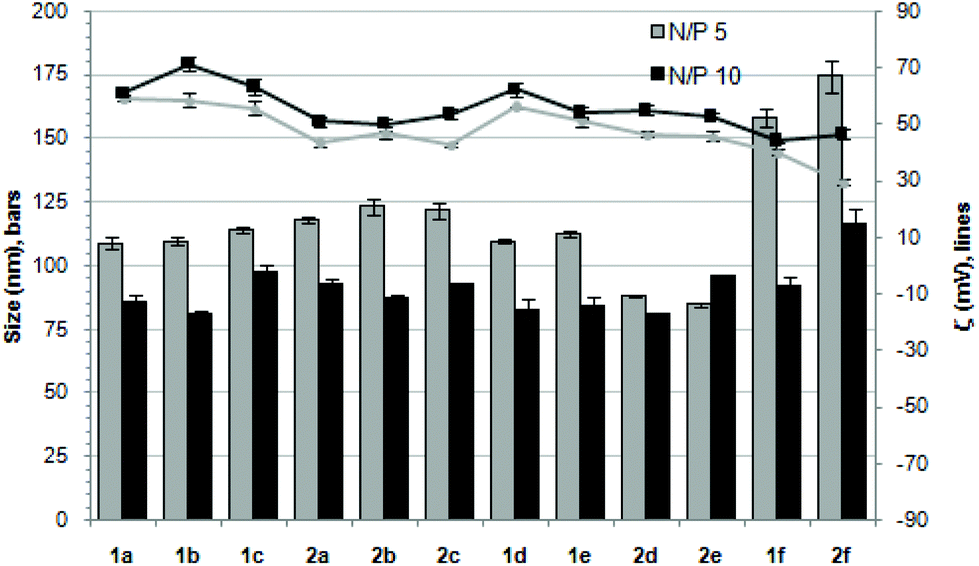
Nanocondensates formulated with 1a–f or 2a–f and the pTG11236 plasmid at N/P 5 and 10, for which EMSA indicated full pDNA complexation and protection, were characterized by dynamic light scattering (DLS) to determine the average hydrodynamic size and by mixed-mode measurement-phase analysis light scattering (M3-PALS) to measure the ζ-potential (Fig. 5). Apart from the two guanidino macrocycles 1f and 2f at N/P = 5 that formed aggregates of 160 and 175 nm, respectively, all the other compounds gave rise to supramolecular species of similar size in the range of 80–120 nm hydrodynamic diameter with a rather low polydispersity. The observed size decrease on going from N/P = 5 to N/P = 10 for most of the nanoparticles, in spite of the equivalent results obtained in EMSA, is indicative of different compaction states. Accordingly, the ζ-potential, which is positive for both N/P values, was higher at N/P = 10 (Fig. 5 and Tables S1 and S2 in the ESI†). No significant changes in size or ζ-potential were observed at higher N/P ratios (data not shown).
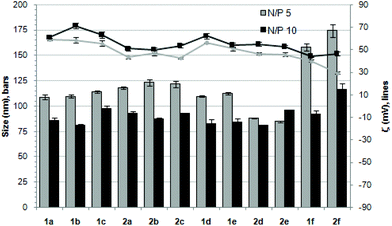 |
| | Fig. 5 Hydrodynamic diameter (bars, nm) and ζ potential (lines, mV) of complexes pDNA–polycationic amphiphilic derivatives determined by DLS and M3-PALs analysis. | |
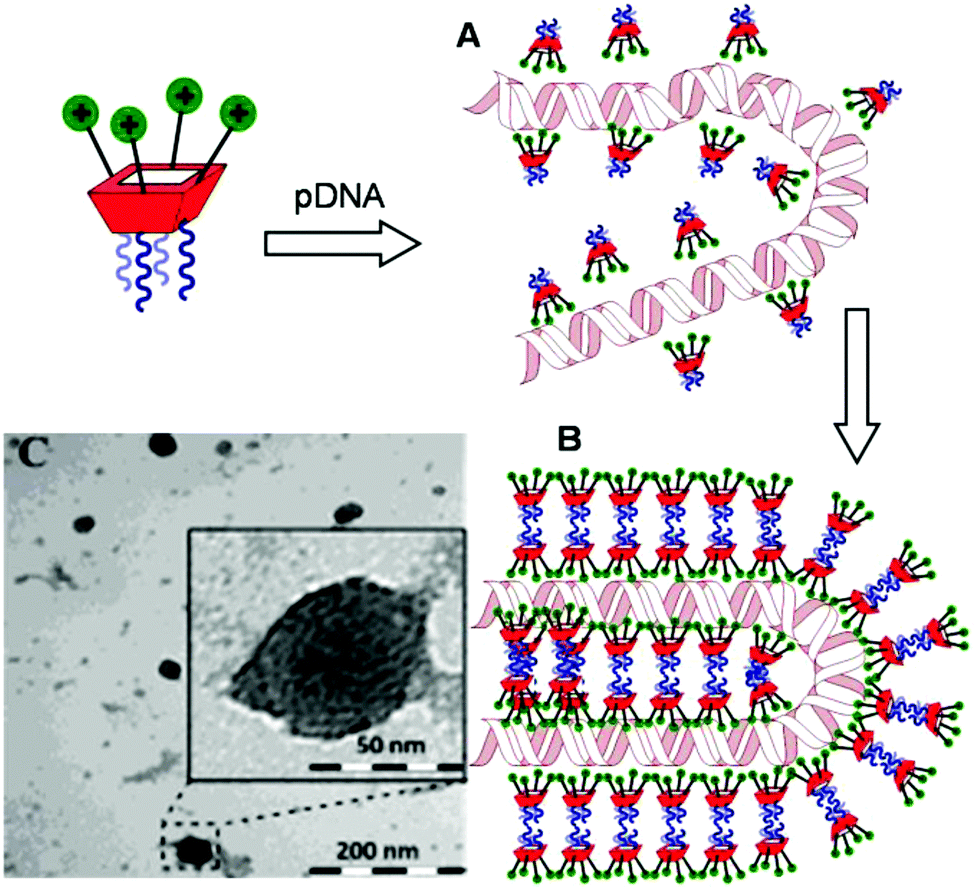
Transmission electron microscopy (TEM) of the nanocomplexes formulated at N/P 10 confirmed their relatively small size and low polydispersity (Fig. 6). As previously observed for amphiphilic βCD aminothiourea polycations,13,14b a snake-like ultra-thin structure revealing an alternating arrangement of high (dark) and low (light) electron density regions was apparent, independently of the sub-library or the macrocyclic scaffold. The dark regions account for the DNA chain, whereas the lighter regions probably correspond to bilayers of the facial amphiphile. This scenario strongly suggests a compaction mechanism involving the polynucleotide chain acting as a template for the alignment of the cationic clusters, a process driven initially by electrostatic interactions. Zipping of the bilayers with simultaneous expulsion of hydration water must then take place through hydrophobic contacts implying the hydrophobic domains, leading to the final nanocondensates (Fig. 6).
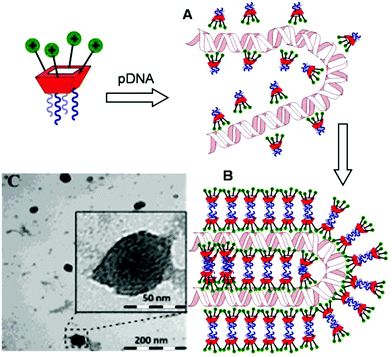 |
| | Fig. 6 Schematic representation of the proposed mechanism for pDNA complexation by polycationic amphiphilic macrocycles (the general cartoon for CA4 derivatives in Fig. 1 has been used) involving electrostatically-driven templating (A) and bilayer zipping/nanocondensation (B). The TEM micrograph corresponding to the nanocomplexes formulated with compound 2d at N/P 10, is also presented (C). The typical snake-like ultra-thin structure can be appreciated in the insert. | |
Atomic force microscopy (AFM) further confirmed the size, spherical shape and monomolecular DNA character of the nanocomplexes formulated with 1a–f or 2a–f, irrespective of the nature of the protonable groups or the scaffold. Most interestingly, this technique allowed the monitoring of DNA compaction by increasing concentrations of the vector. The green fluorescence protein (GFP)-encoding plasmid pEGFP-C1 (4731 bps) was used for this purpose. As an example, Fig. 7 shows the images obtained for sub-library III polyguanidine derivatives 1f and 2f. At N/P 0.5 complexation is incomplete and free pDNA filaments of about 0.5 μm in length can be observed together with partially shrunk plasmids. At N/P 5 all DNA molecules appear as individual nanocondensates.
 |
| | Fig. 7 AFM images recorded in tapping mode on air showing the effects induced on EGFP-C1 plasmid DNA folding by incubation with polyguanidino βCD and CA4 derivatives 1f and 2f at N/P 0.5 (A and C) or N/P 5 (B and D). | |
In vitro transfection of COS-7 cells
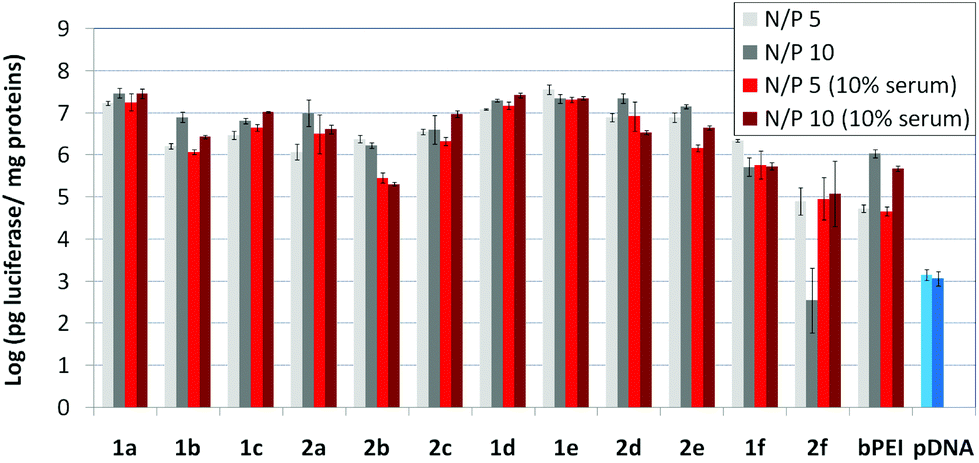
The βCD and CA4:pDNA nanocomplexes (CDplexes and calixplexes) obtained by formulation of 1a–f or 2a–f and the luciferase-encoding plasmid pTG11236 at N/P 5 and 10, were first tested in gene delivery assays towards the COS-7 cell line. For comparative purposes, naked pDNA and polyplexes formulated with commercial branched polyethyleneimine (bPEI) were used as negative and positive controls, respectively. The suitability of the vectors for potential systemic applications was assessed by additionally performing experiments in the presence of 10% serum. The total amount of luciferase expressed was normalized to the total amount of proteins produced by untreated cells. Transfection efficiencies are thus presented as logarithm of picograms of luciferase with respect to the number of milligrams of total proteins (Fig. 8).
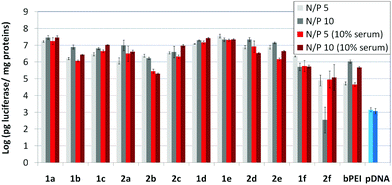 |
| | Fig. 8
In vitro transfection efficiency in COS-7 cells for CDplexes and calixplexes formulated with 1a–f and 2a–f and pTG11236 plasmid at N/P 5 and 10, in the absence or in the presence of serum (10%). Polyplexes formulated with branched PEI (bPEI) and naked pDNA in the absence (light blue) and presence (dark blue) of 10% serum were included as positive and negative controls, respectively. | |
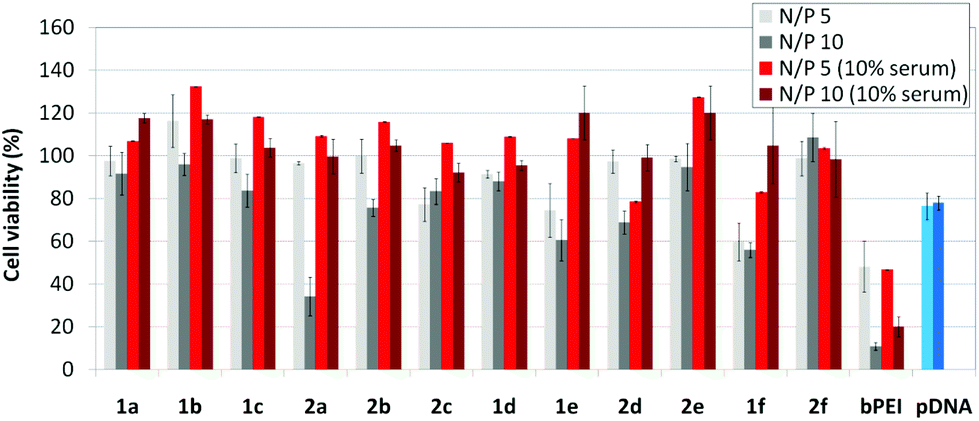
Barring the guanidine CA4 cluster 2f, all the βCD:pDNA and CA4:pDNA formulations showed transfection efficiencies that compare favourably with the results obtained for bPEI polyplexes and all, without exception, exhibited more favorable cell viabilities (Fig. 9). Within sub-library I and III derivatives, βCD-scaffolded vectors 1a–c and 1f proved superior to the homologous CA4 partners 2a–c and 2f, but excepting for the latter pair, differences remained within one order of magnitude. In the group of vectors displaying arginine groups (sub-library II) the influence of the scaffold is less evident. On the other hand, in the CA4 series the presence of amide connectors (compound 2d) was somehow more favourable as compared to triazol linkers (compound 2e). Indeed, compounds 1d, 1e and 2d were as efficient as the lead aminothiourea representative 1a and similarly preserved the transfection capabilities in serum-containing medium. The most striking observation is the dramatic drop in transfection efficiency on comparing sub-libraries II and III. The compounds bearing the simple guanidinium units showed by far the lowest transfection efficacy among all the synthesized compounds. This result suggests that the presence in the vectors of nitrogen atoms with the ability to reversibly shift from protonated to neutral state in a physiological pH window is advantageous, probably by imparting buffering capabilities to the corresponding nanoparticles, thereby facilitating endosome escape through the so-called proton sponge mechanism.24
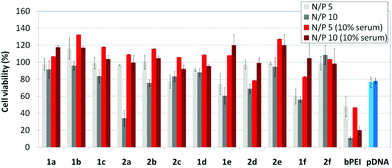 |
| | Fig. 9 Cell viability of COS-7 cells in the presence of CDplexes and calixplexes formulated with 1a–f and 2a–f and pTG11236 plasmid at N/P 5 and 10, in the absence or in the presence of serum (10%). Data for polyplexes formulated with branched PEI (bPEI) and naked pDNA in the absence (light blue) and presence (dark blue) of 10% serum are also included. | |
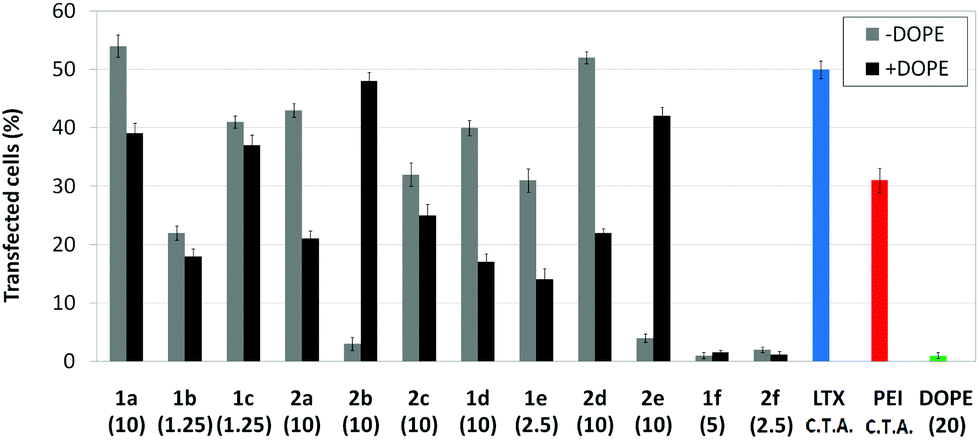
Transfection efficiency towards COS-7 cells was also determined using the GFP-encoding plasmid pEGFP-C1 and directly monitoring by fluorescence microscopy the expression of the GFP protein into the cells as a consequence of successful transfection. Rather than the amount of protein produced, this experiment aims at evaluating the proportion of cells that is effectively transfected by each polycationic cluster:pDNA formulation. Transfection efficiency is then reported as percentage of transfected cells (Fig. 11). Given that cytotoxicity has a strong impact in this assay, for each vector the concentration producing the best compromise between cell viability and transfection efficiency in the range of 1.25–10 μM, was used. At concentrations lower than those selected the cell viability was comparable or higher but the percentage of transfected cells was lower while at higher concentrations cytotoxicity was too high. Cell viabilities, determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method (see the ESI† for Experimental details) at the concentrations studied in this experimental setting, are collected in Fig. 10. In addition to PEI-formulated polyplexes, lipoplexes formulated with commercial Lipofectamine® (LTX) were included as positive controls. Parallel experiments were also conducted in the presence of dioleoylphosphatidylethanolamine (DOPE), a commonly used transfection adjuvant25 that has been previously found to improve the transfection efficiency of CA4-scaffolded polycationic clusters in some cases.10a–c,16 Cells treated only with DOPE were then used as negative controls.
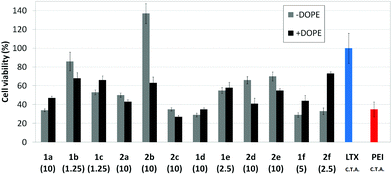 |
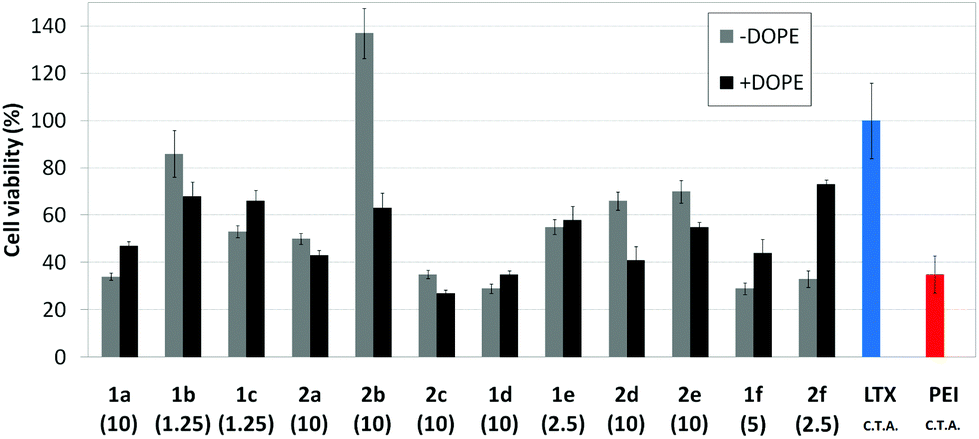
| | Fig. 10 Cell viability of COS-7 cells (MTT determination) in the presence of nanocomplexes formulated with the pEGFP-C1 and βCD or CA4 polycations 1a–f or 2a–f at their optimal concentration (μM) with and without DOPE (1:2 vector–DOPE molar ratio). Data for lipoplexes formulated with Lipofectamine® (LTX), and polyplexes formulated with polyethyleneimine (PEI) are also shown. C.T.A.: commercial transfection agent. | |
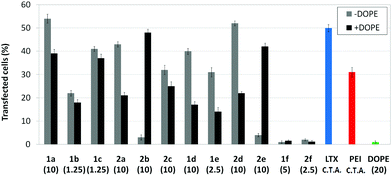 |
| | Fig. 11
In vitro transfection efficiency in COS-7 cells in the presence of nanocomplexes formulated with the pEGFP-C1 and βCD or CA4 polycations 1a–f or 2a–f at their optimal concentration (μM) with and without DOPE (1:2 vector–DOPE molar ratio). Data for lipoplexes formulated with Lipofectamine® (LTX), and polyplexes formulated with polyethyleneimine (PEI) are also shown. C.T.A.: commercial transfection agent. | |
Overall, the results obtained using this evaluation protocol (Fig. 11) qualitatively paralleled those previously obtained with the luciferase-encoding plasmid. Thus, the two guanidino derivatives 1f and 2f included in sub-library III exhibited the poorer transfection abilities among all vectors assayed, with a percentage of transfected cells close to zero. In the polyaminothiourea series (sub-library I) βCD-scaffolded derivatives 1a–c proved superior to the corresponding CA4 analogues 2a–c bearing identical cationic heads, whereas in sub-library II amide-linked derivatives 1d and 2d provided higher transfection efficiencies as compared with triazol-linked analogues 1e and 2e. In any case, only the nanocomplexes formulated with the two lead compounds in sublibraries I and II, namely the dendritic βCD aminothiourea 1a and the amide-linked CA4 tetraarginine derivative 2d, rivalled lipoplexes formulated with LTX. Co-formulation with DOPE was detrimental in most cases with the notable exceptions of the calixplexes obtained from compounds 2b and 2e, for which a quite significant enhancement in the percentage of transfected COS-7 cells was observed.
In vitro transfection of the human rhabdomyosarcoma RD-4 cell line
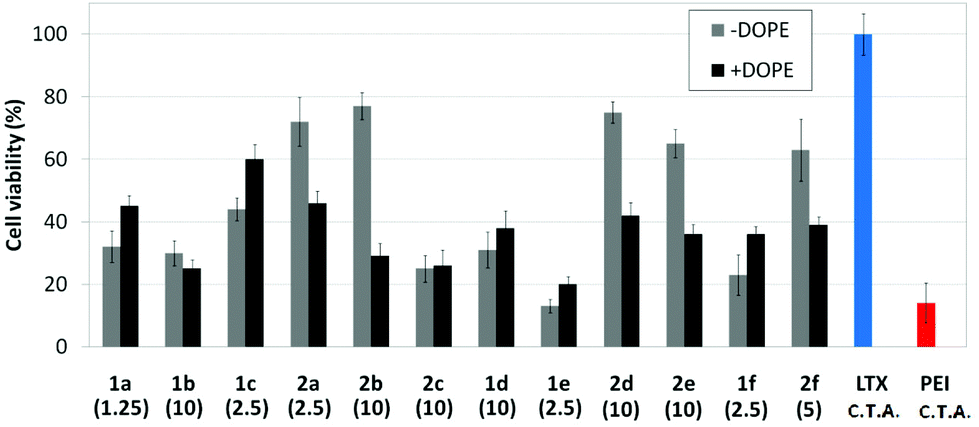
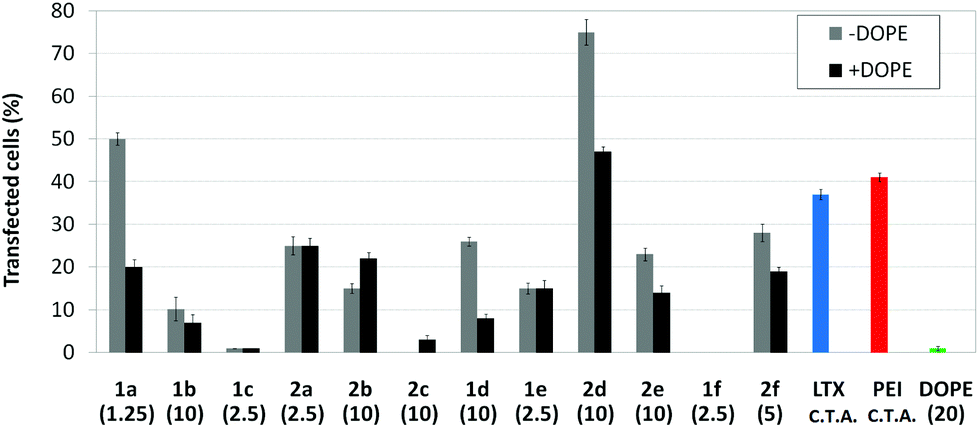
Human rhabdomyosarcoma RD-4 cells are a kind of cancer cells of connective tissues. The difficulties of the treatment and the impossibility, in many cases, of removing the tumor make this cell line of high medical relevance for gene therapy. Moreover, it is a cell line which is particularly very difficult to transfect. The protocol based on the use of the pEGFP-C1 and fluorescence microscopy monitoring of the percentage of cells expressing GFP after treatment with the nanocomplexes formulated with the optimal concentration of each molecular vector, eventually co-formulated with DOPE, was applied. This choice is consistent with previous studies on the ability of calixplexes to mediate transfection in this particular cell line.16 The corresponding cell viability and transfection data are collected in Fig. 12 and 13, respectively. Formulations prepared with LTX and PEI were used as positive controls whereas parallel experiments with DOPE alone were conducted as a negative control.
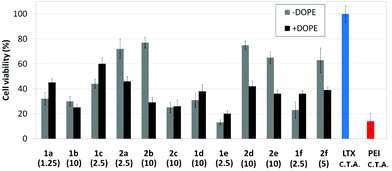 |
| | Fig. 12 Cell viability of RD-4 cells (MTT determination) in the presence of nanocomplexes formulated with the pEGFP-C1 and βCD or CA4 polycations 1a–f or 2a–f at their optimal concentration (μM) with and without DOPE (1:2 vector–DOPE molar ratio). Data for lipoplexes formulated with Lipofectamine® (LTX), and polyplexes formulated with polyethyleneimine (PEI) are also shown. C.T.A.: commercial transfection agent. | |
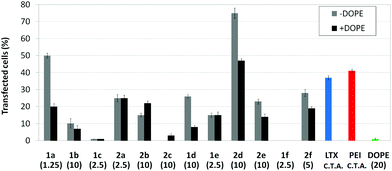 |
| | Fig. 13
In vitro transfection efficiency in RD-4 cells in the presence of nanocomplexes formulated with the pEGFP-C1 and βCD or CA4 polycations 1a–f or 2a–f at their optimal concentration (μM) with and without DOPE (1:2 vector–DOPE molar ratio). Data for lipoplexes formulated with Lipofectamine® (LTX), and polyplexes formulated with polyethyleneimine (PEI) are also shown. C.T.A.: commercial transfection agent. | |
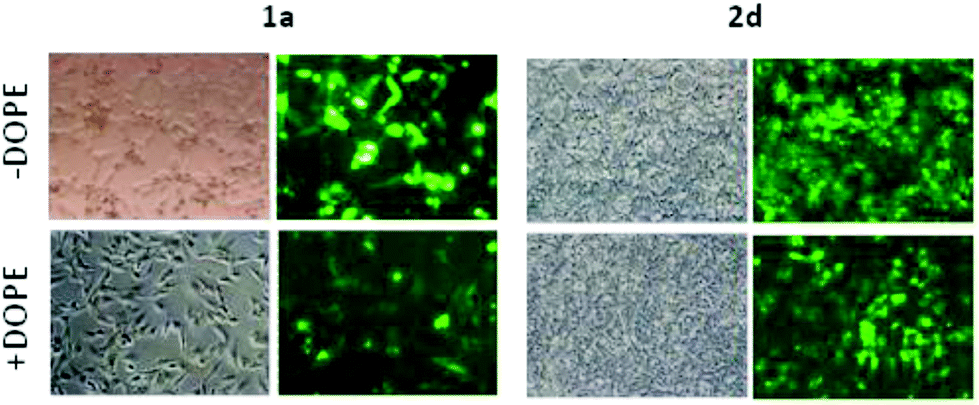
Most of the nanocomplexes formulated with the βCD or CA4 polycationic amphiphiles 1a–f or 2a–f led to GFP expression in the RD-4 cell line, but only the lead compound in each series, namely the dendritic aminothiourea βCD derivative 1a and the arginine CA4 derivative 2d, performed better than the references LTX and PEI. Both 1a and 2d showed their best efficiency when formulated without the adjuvant (Fig. 14). Actually, as already observed in COS-7 cells, the incorporation of DOPE was generally detrimental for the activity of the tested ligands. The data confirms the high transfection efficiency of the argininocalixarene 2d in RD-4 cells. While in the experiments with COS-7 cells differences in transfection efficiencies between vectors in the tested library remained modest, in the case of RD-4 cells the transfection efficiency of the argininocalixarene 2d stands out of the rest, with an exceptional 75% of cells successfully transfected. The dendritic aminothiourea βCD derivative 1a, which was the best performing system in the COS-7 cell line, achieved 50% of RD-4 cells transfected, meaning that the optimal vector depends on the target cell. In any case, with few exceptions, such as the significant transfection level observed for the tetraguanidine CA4 derivative 2f in this assay, the data within a series follow the same trends already observed in COS-7 cells, but differences are quantitatively more pronounced. Thus, the proportion of transfected cells drops from 50% or 25% to almost zero on going from the dendritic tetradecacationic derivatives 1a or octacationic 2a to the linear heptacationic 1c or tetracationic 2c analogues. A high cationic density thus seems to be critical for the successful transfection of RD-4 cells.
 |
| | Fig. 14 Optical (left columns) and fluorescent (right columns) microscopy images of human RD-4 cells transfected (in green) upon treatment with EGFP-C1 pDNA (1 nM) formulated with polyaminothiourea βCD derivative 1a and polyarginine CA4 derivative 2d. | |
When comparing homologous βCD and CA4 derivatives in each sub-library, barring the 1a/2a pair the calixarene derivatives were generally superior to the cyclodextrins vectors, which is the opposite situation to that encountered in COS-7 cells. It has been previously advanced that βCD facial amphiphiles can interact with cholesterol in the cell membrane through inclusion complex formation and that this interaction facilitates caveolin-mediated endocytosis (CME) of the corresponding CDplexes.13,14a Indeed, CME has been shown to be by far the most productive internalization route for CDplexes.14a Since caveolin is down-regulated in rhabdomyosarcoma,26 it can be expected that the efficiency of βCD-based vector will be decreased in RD-4 as compared with COS-7 cells. It is interesting to speculate that calixplexes can enter the cell through alternative caveolin-independent routes that remain productive regarding transfection, which may be at the origin of the outstanding result obtained with compound 2d. Exploring this hypothesis is currently underway in our laboratories.
Conclusions
In summary, we have demonstrated that the approach based in the installation of counter-directional multi-head/multi-tail cationizable/O-hexanoyl or O-hexyl domains onto a β-cyclodextrin or a calix[4]arene platform provides facial amphiphiles with gene delivery capability. Total control of the homogeneity at the molecular level is warranted in homologous series of compounds, allowing reliable structure–activity relationship studies. The components of the three sub-libraries considered in this study, namely compounds featuring aminothiourea, arginine and guanidine clusters were all able to condense pDNA into self-assembled nanocomplexes through a process that involves electrostatic vector–DNA interactions and hydrophobic vector–vector interactions, resulting in a well-ordered arrangement of alternated DNA chains and vector bilayers. As a general trend, increasing the density of protonable groups had a beneficial impact in transfection capabilities provided that amino groups, with buffering capabilities, were present in the structure. The effect of the macrocyclic core on this was more evident when comparing different cell lines. In epithelial COS-7 cells from apes, βCD formulated CDplexes exhibited a higher transfection efficiency than the homologous CA4-formulated calixplexes, while the reverse situation was encountered in human RD-4 cells. This switch can be tentatively ascribed to the operation of different cell uptake mechanisms that affect in a dissimilar manner the fate of CDplexes and calixplexes. In any case, this work provides clues for the rational design of new molecular gene delivery systems and validates the strategy based on systematic structural modifications in CD and CA-based facial amphiphiles and structure–activity relationship studies for the identification of optimal candidates for gene therapy applications.
Experimental
General methods
Bis(2-tert-butyloxycarbonylaminoethyl) amine and bis(2-tert-butyloxycarbonylaminoethyl) 2-aminoethyl amine were obtained according to literature procedures.9b Optical rotations were measured at 20 ± 2 °C in 1 dm tubes on a Jasco P-2000 polarimeter. 1H (and 13C NMR) spectra were recorded at 500 (125.7) MHz with a Bruker 500 DRX magnet. 1D TOCSY, 2D COSY, HMQC and HSQC experiments were used to assist on NMR assignments. Thin-layer chromatography (TLC) was carried out on aluminum sheets coated with silica gel 60 F254 Merck with visualization by UV light and by charring with ethanolic 10% H2SO4 or 0.1% ninhydrin. Column chromatography was carried out on Silica Gel 60. ESI mass spectra were recorded on a Bruker Daltonics Esquire6000™ ion-trap mass spectrometer; in some cases, CuI was added as the cationizing agent. MALDI-TOF mass spectra were registered in a Bruker Daltonics Autoflex instrument in the linear positive mode with pulse ion extraction; 2,5-dihydroxybenzoic acid (DHB) was used as a desorption matrix. Elemental analyses were carried out at the Instituto de Investigaciones Químicas (Sevilla, Spain).
5,11,17,23-Tetraisothiocyanate-25,26,27,28-tetrakis(n-hexyloxy)calix[4]arene (4).
To a solution of 5,11,17,23-tetraamino-25,26,27,28-tetrakis(n-hexyloxy)calix[4]arene (3, 150 mg, 0.183 mmol) in absolute EtOH (4 mL), CS2 (440 μL, 7.3 mmol) and Et3N (102 μL, 0.73 mmol) were added. The reaction mixture was stirred for 2 h at rt and then cooled in an ice bath. A solution of Boc2O (158 mg, 0.72 mmol) in absolute EtOH (1 mL) was added followed by the addition of a catalytic amount of DMAP (1.8 mg, 15 μmol). The reaction mixture was kept in the ice bath for 20 min. Then it was allowed to reach rt and further stirred for 2 h. The solvent was removed under reduced pressure and the residue was purified by column chromatography (1:3 DCM–cyclohexane). Yield: 181 mg (88%); Rf = 0.67 (1:3 DCM–cyclohexane); 1H NMR (400 MHz, CDCl3): δ 6.56 (s, 8 H, Ar), 4.35 (d, J = 10.2 Hz, 4 H, ArCHaxAr), 3.83 (t, J = 5.7 Hz, 8 H, CH2-1Hex), 3.09 (d, J = 10.2 Hz, 4 H, ArCHeqAr), 1.89–1.77 (m, 8 H, CH2-2Hex), 1.41–1.28 (m, 24 H, CH2-3Hex, CH2-4Hex, CH2-5Hex), 0.97–0.84 (m, 12 H, CH3-6Hex); 13C NMR (100.6 MHz, CDCl3): δ 155.7, 135.9 (Ar), 134.3 (NCS), 125.7, 125.6 (Ar), 75.9 (C-1Hex), 32.1 (C-3Hex), 30.9 (ArCH2Ar), 30.2 (C-2Hex), 25.9 (C-4Hex), 22.9 (C-5Hex), 14.2 (C-6Hex); ESI-MS: m/z 1011.1 [M + Na]+; Anal. calcd for C56H68N4O4S4: C 67.98, H 6.93, N 5.66, S 12.96. Found: C 68.07, H 6.88, N 5.61, S 12.79.
5,11,17,23-Tetra-[N′,N′-bis-(2-(tert-butoxycarbonylamino)ethyl)-thioureido]-25,26,27,28-tetrakis(n-hexyloxy)calix[4]arene (5).
A solution of 4 (84.1 mg, 0.085 mmol) in dry DCM (3 mL) was added dropwise to a solution of bis(2-tert-butyloxycarbonylaminoethyl) amine (124 mg, 0.41 mmol) and Et3N (85 μL, 0.62 mmol) in dry DCM (3 mL). The mixture was stirred overnight at rt. The solvent was removed under vacuum and the residue was purified by column chromatography (100:1 → 99:1 DCM–MeOH). Yield: 187 mg (99%); Rf = 0.58 (9:1 DCM–MeOH); 1H NMR (500 MHz, CD3OD): δ 6.96 (bs, 8 H, Ar), 4.48 (d, J = 12.5 Hz, 4 H, ArCHaxAr), 4.05–3.70 (m, 24 H, CH2-1Hex, CH2NCS), 3.31 (bs, 16 H, CH2NHBoc), 3.14 (d, J = 12.5 Hz, 4 H, ArCHeqAr), 2.07 (m, 8 H, CH2-2Hex), 1.67–1.28 (m, 96 H, CMe3, CH2-3Hex, CH2-4Hex, CH2-5Hex), 1.05–0.95 (m, 12 H, CH3-6Hex); 13C NMR (125.7 MHz, CD3OD, 333 K): δ 183.2 (CS), 158.5 (CO), 155.3–127.3 (Ar), 80.7 (CMe3), 76.6 (C-1Hex), 52.4 (CH2NHCS), 39.7 (CH2NHBoc), 33.3 (C-3Hex), 32.0 (ArCH2Ar), 31.5 (C-2Hex), 29.0 (CMe3), 27.2 (C-4Hex), 23.9 (C-5Hex), 14.4 (C-6Hex); ESI-MS: m/z 2265.9 [M + Cu]+, 1144.9 [M + Cu + Na]2+; Anal. calcd for C112H184N16O20S4: C 61.06, H 8.42, N 10.17, S 5.82. Found: C 61.30, H 8.30, N 9.88, S 5.88.
5,11,17,23-Tetra-[bis-2-(aminoethyl)thioureido]-25,26,27,28-tetrakis-(n-hexyloxy)calix[4]arene octahydrochloride (2a).
A solution of 5 (25 mg, 11 μmol) in DCM–TFA–TES (87.5:10:2.5, 3 mL) was stirred at 0 °C for 2 h. The solvent was removed under reduced pressure. The residue was precipitated and washed with Et2O (5 mL). Then the solid was dissolved in an aqueous 0.1 M HCl solution and concentrated to give the product as hydrochloride. Yield: 19 mg (99%); 1H NMR (300 MHz, CD3OD): δ 6.86 (s, 8 H, Ar), 4.50 (d, J = 12.6 Hz, 4 H, ArCHaxAr), 4.09 (t, J = 6.7 Hz, 16 H, CH2NHCS), 3.96 (t, J = 7.8 Hz, 8 H, CH2-1Hex), 3.26 (t, J = 6.7 Hz, 16 H, CH2NH2), 3.16 (d, J = 12.6 Hz, 4 H, ArCHeqAr), 2.11–1.95 (m, 8 H, CH2-2Hex), 1.56–1.32 (m, 24 H, CH2-3Hex, CH2-4Hex, CH2-5Hex), 1.02–0.90 (m, 12 H, CH3-6Hex); 13C NMR (125.7 MHz, CD3OD, 313 K): δ 184.3 (CS), 155.5–127.9 (Ar), 76.9 (C-1Hex), 48.65 (CH2NHCS), 38.4 (CH2NH2), 33.4 (C-3Hex), 31.8 (ArCH2Ar), 31.6 (C-2Hex), 27.1 (C-4Hex), 23.9 (C-5Hex), 14.5 (C-6Hex); ESI-MS: m/z 1463.9 [M + Cu]+; Anal. calcd for C72H128Cl8N16O4S4: C 51.06, H 7.62, N 13.23, S 7.57. Found: C 50.77, H 7.59, N 12.91, S 7.19.
5,11,17,23-Tetra-[N′-(2-(N,N-bis-(2-tert-butoxycarbonylaminoethyl)amino)ethyl)thioureido]-25,26,27,28-tetrakis-(n-hexyloxy)calix[4]arene (6).
A solution of 4 (40 mg, 0.040 mmol) in dry DCM (3 mL) was added dropwise to a solution of bis(2-tert-butyloxycarbonylaminoethyl) 2-aminoethyl amine (67.4 mg, 0.19 mmol) and Et3N (40 μL, 0.28 mmol) in dry DCM (3 mL). The mixture was stirred overnight at rt. The solvent was removed under vacuum and the residue was purified by column chromatography (19:1 DCM–MeOH). Yield: 95 mg (99%); Rf = 0.56 (9:1 DCM–MeOH); 1H NMR (300 MHz, CD3OD): δ 6.721 (bs, 8 H, Ar), 4.47 (d, J = 12.9 Hz, 4 H, ArCHaxAr), 3.93 (m, 8 H, CH2-1Hex), 3.69–3.53 (m, 8 H, CH2NHCS), 3.20 (d, J = 13.3 Hz, 4 H, ArCHeqAr), 3.07 (t, J = 6.4 Hz, 16 H, CH2CH2NHBoc), 2.77–2.65 (m, 8 H, CH2CH2NHCS), 2.64–2.53 (m, 16 H, CH2CH2NHBoc), 2.06–1.89 (m, 8 H, CH2-2Hex), 1.62–1.25 (m, 96 H, CMe3, CH2-3Hex, CH2-4Hex, CH2-5Hex), 1.02–0.87 (m, 12 H, CH3-6Hex); 13C NMR (75 MHz, CD3OD, 313 K): δ 181.7 (CS), 158.4 (CO), 155.9–126.1 (Ar), 80.3 (CMe3), 76.6 (C-1Hex), 55.3 (CH2NHBoc), 54.4 (CH2CH2NHCS), 43.8 (CH2NHCS), 40.1 (CH2CH2NHBoc), 33.3 (C-3Hex), 32.0 (ArCH2Ar), 31.5 (C-2Hex), 29.0 (CMe3), 27.3 (C-4Hex), 23.9 (C-5Hex), 14.4 (C-6Hex); ESI-MS: m/z 2397.3 [M + Na]+, 1209.6 [M + 2Na]2+; Anal. calcd for C120H204N20O20S4: C 60.68, H 8.66, N 11.79, S 5.40. Found: C 60.52, H 8.58, N 11.62, S 5.24.
5,11,17,23-Tetra-[N′-(2-(N,N-bis-(2-aminoethyl)amino)ethyl)thioureido]-25,26,27,28-tetrakis-(n-hexyloxy)calix[4]arene (2b).
A solution of 6 (52 mg, 22 μmol) in TFA (0.4 mL) was stirred at rt for 5 min. H2O (10 mL) was added to it and the solution was freeze-dried. The residue was dissolved in 0.1 M HCl methanolic solution and freeze-dried to obtain the product as hydrochloride. Yield: 41 mg (99%); 1H NMR (300 MHz, CD3OD): δ 7.02 (bs, 8 H, Ar), 4.51 (d, J = 12.7 Hz, 4 H, ArCHaxAr), 4.18 (t, J = 7.2 Hz, 8 H, CH2-1Hex), 3.96 (t, J = 7.7 Hz, 8 H, CH2NHCS), 3.37–3.26 (m, 40 H, CH2CH2NHBoc, CH2CH2NHCS, CH2CH2NHBoc), 3.21 (d, 4 H, ArCHeqAr), 2.18–2.06 (m, 8 H, CH2-2Hex), 1.56–1.34 (m, 24 H, CH2-3Hex, CH2-4Hex, CH2-5Hex), 1.05–0.89 (m, 12 H, CH3-6Hex); 13C NMR (125.7 MHz, CD3OD): δ 183.9 (CS), 155.4–127.8 (CAr), 76.5 (C-1Hex), 51.5, 51.8 (CH2N(CH2CH2)2), 42.2 (CH2NHCS), 37.9 (CH2NH2), 32.5 (C-3Hex), 31.3 (ArCH2Ar), 30.8 (C-2Hex), 26.7 (C-4Hex), 23.4 (C-5Hex), 14.4 (C-6Hex); ESI-MS: m/z 1574.9 [M + H]+, 788.0 [M + 2H]2+; Anal. calcd for C80H152Cl12N20O4S4: C 47.76, H 7.62, N 13.92, S 6.38. Found: C 47.67, H 7.72, N 13.90, S 6.29.
5,11,17,23-Tetra-[N′-(2-(tert-butoxycarbonylamino)ethyl)thioureido]-25,26,27,28-tetrakis-(n-hexyloxy)calix[4]arene (7).
A solution of 4 (84.1 mg, 0.085 mmol) in dry DCM (3 mL) was added dropwise to a solution of commercial 2-tert-butyloxycarbonylaminoethylamine (67 mg, 0.41 mmol) and Et3N (85 μL, 0.62 mmol) in dry DCM (3 mL). The mixture was stirred overnight at rt, the solvent was removed under vacuum and the residue was purified by column chromatography (95:5 → 9:1 DCM–MeOH). Yield: 138 mg (99%); Rf = 0.61 (9:1 DCM–MeOH); 1H NMR (400 MHz, CD3OD): δ 6.68 (bs, 8 H, Ar), 4.46 (d, J = 12.7 Hz, 4 H, ArCHaxAr), 3.93 (t, J = 7.9 Hz, 8 H, CH2-1Hex), 3.65 (t, J = 6.0 Hz, 8 H, CH2NHCS), 3.34–3.16 (m, 12 H, ArCHeqAr, CH2NHBoc), 2.06–1.91 (m, 8 H, CH2-2Hex), 1.55–1.32 (m, 60 H, CH2-3Hex, CH2-4Hex, CH2-5Hex, CMe3), 1.01–0.92 (m, 12 H, CH3-6Hex); 13C NMR (100.6 MHz, CD3OD): δ 181.6 (CS), 158.8 (CO), 155.8–125.9 (Ar), 80.2 (CMe3), 76.7 (C-1Hex), 45.9 (CH2NHCS), 40.8 (CH2NHBoc), 33.4 (C-3Hex), 31.9 (ArCH2Ar), 31.6 (C-2Hex), 28.9 (CMe3), 27.3 (C-4Hex), 24.1 (C-5Hex), 14.5 (C-6Hex); ESI-MS: m/z 1651.8 [M + Na]+, 1667.8 [M + K]+; Anal. calcd for C84H132N12O12S4: C 61.88, H 8.16, N 10.31, S 7.87. Found: C 61.85, H 8.29, N 10.24, S 7.79.
5,11,17,23-Tetraaminoethylthioureido-25,26,27,28-tetrakis(n-hexyloxy)calix[4]arene (2c).
A solution of 7 (40 mg, 25 μmol) in DCM–TFA–TES (87.5:10:2.5, 0.5 mL) was stirred at 0 °C for 2 h. The solvent was removed and the residue was precipitated and washed with Et2O. Then the solid was dissolved in an aqueous 0.1 M HCl solution and concentrated to yield the product as hydrochloride. Yield: 33.8 mg (99%); 1H NMR (500 MHz, CD3OD): δ 6.72 (bs, 8 H, Ar), 4.47 (d, J = 13.2 Hz, 4 H, ArCHaxAr), 3.98–3.85 (m, 16 H, CH2-1Hex, CH2NHCS), 3.26–3.13 (m, 12 H, ArCHeqAr, CH2NH2), 2.02–1.88 (m, 8 H, CH2-2Hex), 1.56–1.35 (m, 24 H, CH2-3Hex, CH2-4Hex, CH2-5Hex), 0.99–0.91 (m, 12 H, CH3-6Hex); 13C NMR (75 MHz, CD3OD): δ 182.8 (CS), 156.0–125.7 (Ar), 76.7 (C-1Hex), 42.8 (CH2NHCS), 40.8 (CH2NH2), 33.4 (C-3Hex), 31.9 (ArCH2Ar), 31.5 (C-2Hex), 27.3 (C-4Hex), 24.0 (C-5Hex), 14.5 (C-6Hex); ESI-MS: m/z 1291.4 [M + Cu]+; Anal. calcd for C64H100N12O4S4·4HCl: C 55.88, H 7.62, N 12.22, S 9.32. Found: C 55.59, H 7.56, N 11.92, S 9.03.
Heptakis[6-(2-(Nα-tert-butoxycarbonyl-Nω-(2,2,4,6,7-pentamethyldihydrobenzofurane-5-sulfonyl)-L-arginine-N-amide)ethylthio)-2,3-di-O-hexanoyl]cyclomaltoheptaose (10).
To a solution of heptakis[2,3-di-O-hexanoyl-6-(2-aminoethylthio)]cyclomaltoheptaose heptahydrochloride (8, 154, 60 mg, 19 μmol) in dry DCM (4 mL), under an Ar atmosphere, DIPEA (90 μL, 53 μmol), Nα-tert-butoxycarbonylamino-Nω-(2,2,4,6,7-pentamethyldihydro-benzofurane-5-sulfonyl)-L-arginine (9, Nα-Boc-Nω-Pbf-L-arginine) (104 mg, 198 μmol) and O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU) (75 mg, 198 μmol) were added and the mixture was stirred overnight. The reaction mixture was washed with an aqueous solution of NaHCO3 (8 mL), the organic layer was dried (MgSO4), filtered and concentrated. The residue was purified by column chromatography (25:1 → 18:1 DCM–MeOH). Yield: 83 mg (68%); Rf = 0.43 (17:1 DCM–MeOH). [α]D = +31.9 (c 1 in MeOH); 1H NMR (500 MHz, CD3OD): δ 5.54 (t, 7 H, J2,3 = J3,4 = 9.5 Hz, H-3), 5.36 (d, J1,2 = 3.7 Hz, 7 H, H-1), 5.02 (dd, 7 H, H-2), 4.41 (m, 7 H, H-5), 4.33–4.23 (m, 7 H, CH-2Arg), 4.12 (m, 7 H, H-4), 3.76–3.67 (m, 7 H, CH2NHCO), 3.64–3.55 (m, 7 H, CH2NCO), 3.48–3.28 (s, 28 H, CH2-5Arg, H6a, H6b), 3.17 (s, 14 H, MePbf), 3.07–2–96 (m, 14 H, CH2S), 2.77 (s, 21 H, MePbf), 2.71 (s, 21 H, MePbf), 2.57–2.38 (m, 28 H, CH2-2Hex), 2.26 (s, 21 H, MePbf), 2.03–1.93 (m, 7 H, CH2-3Arg), 1.91–1.71 (m, 49 H, CH2-3Arg, CH2-4Arg, CH2-3Hex), 1.63 (bs, 42 H, CMe2Pbf), 1.61 (bs, 63 H, CMe3), 1.59–1.44 (m, 56 H, CH2-4Hex, CH2-5Hex), 1.20–1.05 (m, 42 H, CH3-6Hex); 13C NMR (75 MHz, CD3OD): δ 174.9, 174.7 (CO ester), 173.5 (CO amide), 159.9 (CN), 158.1 (CO carbamate), 157.7–118.4, (CqPbf), 98.2 (C-1), 87.7 (CMe3), 80.7 (CMe2Pbf), 71.7 (C-4), 73.5 (C-5), 71.7, 71.6 (C-2, C-3), 55.8 (CH2-2Arg), 44.1 (CH2Pbf), 41.5 (CH-5Arg), 40.5 (CH2NHCO), 35.1, 35.0 (C-2Hex, C-6), 34.1 (CH2S), 32.6, 32.5 (C-4Hex), 30.9 (CH2-3Arg), 28.9, 28.8 (CMe2Pbf, CMe3), 27.1 (CH2-4Arg), 25.6 (C-3Hex), 23.6 (C-5Hex), 19.8, 18.6 (MePbf), 14.6 (C-6Hex), 12.7 (MePbf); ESI-MS: m/z 3263.3 [M + 2Na]2+, 2182.6 [M + 3Na]3+; Anal. calcd for C308H497N35O84S14: C 57.06, H 7.73, N 7.56, S 6.92. Found: C 56.88, H 7.64, N 7.34, S 6.63.
Heptakis[6-(2-(L-arginine-N-amide)ethylthio)-2,3-di-O-hexanoyl]cyclomaltoheptaose tetradecahydrochloride (1d).
A solution of 10 (37.5 mg, 6 μmol) in TFA–TIS–H2O (95:2.5:2.5, 4 mL) was stirred at rt for 1 h. The solvent was removed under reduced pressure and coevaporated several times with H2O. The residue was dissolved in an aqueous 0.1 M HCl solution and freeze-dried to obtain the product as hydrochloride. Yield: 41 mg (99%); [α]D = +9.7 (c 1.0 in DMSO); 1H NMR (500 MHz, DMSO-d6, 323 K): δ 8.96 (bs, 7 H, NHCO), 8.51–8.29 (m, 14 H, NH2Arg), 7.93–7.77 (bs, 7 H, NH guanidine), 7.44–7.09 (m, 21 H, NH, NH2 guanidine), 5.25 (t, J2,3 = J3,4 = 9.0 Hz, 7 H, H-3), 5.09 (bs, 7 H, H-1), 4.69 (bd, 7 H, H-2), 4.17–4.07 (m, 7 H, H-5), 3.99–3.83 (m, 14 H, CH2-5Arg, H-4), 3.43–3.32 (bs, 14 H, CH2N), 3.29–3.19 (bs, 7 H, CH2-5Arg), 3.15–2.99 (m, 14 H, H-6a, H-6b), 2.87–2.66 (m, 14 H, CH2S), 2.44–2.29 (m, 7 H, CH2-2Hex), 2.26–2.11 (m, 7 H, CH2-2Hex), 1.92–1.80 (m, 14 H, CH2-3Arg), 1.70–1.58 (m, 14 H, CH2-4Arg), 1.58–1.45 (m, 28 H, CH2-3Hex), 1.35–1.19 (bs, 56 H, CH2-4Hex, CH2-5Hex), 0.93–0.78 (m, 42 H, CH3-6Hex); 13C NMR (125.7 MHz, DMSO-d6, 323 K): δ 175.7, 174.5 (CO ester), 177.8 (CO amide), 161.2 (CN), 99.4 (C-1), 81.3 (C-4), 74.3 (C-5), 73.1 (C-2), 72.9 (C-3), 55.1 (CH2-2Arg), 43.7 (CH2-5Arg), 42.4 (CH2NHCO), 36.5, 36.3 (C-2Hex, C-6), 35.3 (CH2S), 33.9, 33.8 (C-4Hex), 31.4 (CH2-3Arg), 27.2 (CH2-4Arg), 26.9, 26.8 (C-3Hex), 24.9, 24.8 (C-5Hex), 16.65, 16.63 (C-6Hex); MALDI-MS: m/z 4017.86 [M + H]+; Anal. calcd for C182H343Cl14N35O49S7·7H2O: C 46.98, H 7.73, N 10.54, S 4.82. Found: C 47.01, H 7.50, N 10.48, S 4.75.
N
α-tert-Butoxycarbonyl-Nω-(2,2,4,6,7-pentamethyldihydrobenzofurane-5-sulfonyl)-L-arginine-N-propargylamide (11).
A mixture of Nα-Boc-Nω-Pbf-L-arginine (1 g, 1.9 mmol), DIPEA (0.78 mL, 4.5 mmol) and HBTU (865 mg, 2.28 mmol) were dissolved in dry DMF (15 mL), under an Ar atmosphere. After stirring for 30 min at rt, propargylamine (0.146 mL, 2.28 mmol) was added and the mixture was stirred for 15 h. The solvent was removed under reduced pressure and the residue was dissolved in EtOAc (15 mL), the organic layer was washed with aqueous saturated NaHCO3 (15 mL), H2O (15 mL), dried (MgSO4), filtered and concentrated. The residue was purified by column chromatography (1:2 → 3:1 EtOAc–cyclohexane). Yield: 935 mg (87%); Rf = 0.41 (19:1 DCM–MeOH); [α]D = +0.9 (c 1.0 in MeOH). 1H NMR (500 MHz, CDCl3): δ 7.34 (bs, 1 H, NHCO), 6.27 (m, 3 H, NH), 5.59 (bs, 1 H, NHBoc), 4.20 (bs, 1 H, CH-2Arg), 3.99 (m, 2 H, CH2-alkyne), 3.27 (bs, 2 H, CH2-5Arg), 2.96 (s, 2 H, CH2Pbf), 2.51 (s, 3 H, MePbf), 2.50 (s, 3 H, MePbf), 2.18 (t, J = 2.5 Hz, 1 H, CH-alkyne), 2.09 (s, 3 H, MePbf), 1.81 (m, 2 H, CH2-3Arg), 1.60 (m, 2 H, CH2-4Arg), 1.46 (s, 6 H, CMe2Pbf), 1.41 (s, 9 H, CMe3); 13C NMR (100.6 MHz, CDCl3): δ 165.9 (CO amide), 159.0 (CAr), 156.8 (CN), 156.1 (CO carbamate), 138.3–117.6 (CAr), 86.4 (CMe2), 80.2 (CMe3), 79.6 (Cq alkyne), 71.4 (CH alkyne), 60.4 (CHNHBoc), 43.2 (CH2Pbf, CH2-5Arg), 29.9 (CH2-4Arg), 29.1 (CH2-6Arg), 28.6 (CMe2), 28.3 (CMe2), 19.3, 17.9, 12.5 (MePbf); ESI-MS: m/z 586.3 [M + Na]+, 602.1 [M + K]+; Anal. calcd for C27H41N5O6S: C 57.53, H 7.33, N 12.42, S 5.69. Found: C 57.58 H 7.24, N 12.31, S 5.36.
Heptakis[6-deoxy-2,3-di-O-hexanoyl-6-(4-(Nα-tert-butoxycarbonyl-Nω-(2,2,4,6,7-pentamethyldihydrobenzofurane-5-sulfonyl)-L-arginine-N-amidomethyl)-1H-1,2,3-triazol-1-yl)]cyclomaltoheptaose (13).
To a solution of heptakis[6-azido-6-deoxy-2,3-di-O-hexanoyl]cyclomaltoheptaose (12, 88 mg, 33 μmol) in acetone (5 mL), Nα-tert-12-Nω-(2,2,4,6,7-pentamethyldihydrobenzofurane-5-sulfonyl)-L-arginine-N-propargylamide (11, 142 mg, 0.25 mmol), N,N-diisopropylethylamine (38 μL, 0.24 mmol) and CuI·P(OEt)3 (8.0 mg, 25 μmol) were added. The reaction mixture was refluxed for 48 h. The solvent was removed under reduced pressure and the residue was purified by column chromatography (20:1 → 9:1 DCM–MeOH). Yield: quantitative; Rf = 0.60 (9:1 DCM–MeOH); [α]D = +29.7 (c 1.0 in MeOH); 1H NMR (500 MHz, DMSO-d6) δ 8.12 (bs, 7 H, CHtriazole), 7.84 (bs, 7 H, Ar), 6.69 (bs, 7 H, NH2 guanidine), 6.55 (bs, 14 H, NH2 guanidine), 5.48–5.35 (m, 14 H, H-3, H-1), 4.74–4.59 (m, 21 H, CH2NHCO amide, H-2), 4.57–4.54 (m, 7 H, H-5), 4.41–4.36 (m, 7 H, H-6a), 4.23–4.11 (m, 7 H, H-6b), 3.95 (m, 7 H, CHArg), 3.61 (bt, 7 H, H-4), 3.09 (m, 14 H, CH2-5Arg), 2.96 (s, 14 H, CH2Pbf), 2.51 (s, 21 H, CH3 Pbf), 2.46 (s, 21 H, CH3 Pbf), 2.28–2.26 (m, 7 H, CH2-2Hex), 2.27–2.10 (m, 7 H, CH2-2Hex), 2.03 (s, 21 H, CH3 Pbf), 1.66–1.18 (217 H, CH2-2Arg, CH2-4Arg, CH2-3Hex, CH2-4Hex, CH2-5Hex, CH3 Pbf, C(CH3)3), 0.93–0.80 (bs, 21 H, CH3-6Hex); 13C NMR (125.7 MHz, DMSO-d6) δ 175.3, 175.1 (CO ester), 174.3 (CO amide), 160.6 (CN guanidine), 159.3 (CO carbamate), 147.6 (C-4triazole), 140.3–134.6 (Cq Pbf), 127.8 (C-5 triazole), 127.3, 119.3 (CqPbf), 99.0 (C-1), 89.2 (CMe3), 81.3 (CMe2 Pbf), 79.6 (C-4), 72.8 (C-3), 72.5 (C-5), 72.3 (C-2), 57.2 (CH2-2Arg), 52.6 (CH2NHCO), 45.7 (CH2Pbf), 42.4 (CH-5-Arg), 37.4 (C-6), 36.4, 36.3 (C-2Hex), 33.9, 33.7 (C-4Hex), 32.0 (CH2-3Arg), 31.3, 31.2 (CMe2Pbf, CMe3), 28.7 (CH2-4Arg), 26.9, 26.8 (C-3Hex), 24.8, 24.9 (C-5Hex), 21.8, 20.5 (Me Pbf), 16.6 (C-6Hex), 15.2 (MePbf); ESI-MS: m/z 3336.4 [M + 2Na]2+; Anal. calcd for C315H490N56O84S7: C 57.06, H 7.45, N 11.83, S 3.39. Found: C 56.78, H 7.20, N 11.47, S 2.92.
Heptakis[6-deoxy-2,3-di-O-hexanoyl-6-(4-L-arginine-N-amidomethyl)-1H-1,2,3-triazol-1-yl)]cyclomaltoheptaose tetradecahydrochloride (1e).
A solution of 13 (45.2 mg, 7 μmol), in TFA–TIS–H2O (95:2.5:2.5, 4.5 mL) was stirred at rt for 2 h. The solvent was removed under reduced pressure and coevaporated several times with water. The residue was dissolved in a 0.1 M HCl solution and freeze-dried to obtain the product as hydrochloride. Yield: 35.7 mg (quantitative); [α]D = +46.5 (c 1.0 in DMF); 1H NMR (500 MHz, DMSO-d6) δ 8.99 (bs, 7 H, NHamide), 7.99 (m, 21 H, CHtriazole, NH2 arginine), 7.18 (bs, 21 H, NH2 guanidine, NH guanidine), 5.41–5.37 (m, 14 H, H-3, H-1), 4.68–4.63 (m, 21 H, H-2, CH2NHCO amide), 4.51 (m, 7 H, H-5), 4.36–4.34 (m, 7 H, H-6a), 4.23–4.21 (m, 7 H, H-6b), 3.92 (m, 7 H, CHArg), 3.72 (bt, 7 H, H-4), 3.19 (m, 14 H, CH2-5Arg), 2.37–1.93 (m, 28 H, CH2-2Hex), 2.27–2.10 (m, 14 H, CH2-2Hex), 1.86–1.79 (m, 14 H, CH2-2Arg), 1.48–1.38 (m, 42 H, CH2-4Arg, CH2-3Hex), 1.37–2.20 (m, 56 H, CH2-4Hex, CH2-5Hex), 0.94–0.80 (bs, 21 H, CH3-6Hex); 13C NMR (125.7 MHz, DMSO-d6): δ 172.8, 171.8 (CO ester), 169.1 (CO amide), 157.7 (CN guanidine), 144.2 (C-4triazole), 125.7 (C-5triazole), 102.5 (C-1), 77.3 (C-4), 70.3 (C-3), 70.1 (C-5), 69.7 (C-2), 52.5 (CH-2Arg), 49.9 (CH2NHCO), 40.7 (CH-5-Arg), 34.9 (C-6), 33.8, 33.7 (C-4Hex), 31.2, 31.1 (C-4Hex), 28.5 (CH2-3Arg), 24.3, 24.2 (C-3Hex, CH2-4Arg), 22.2, 22.1 (C-5Hex), 13.9 (C-6Hex); MALDI-MS: m/z 4163.60 [M + H]+; Anal. calcd for C189H329Cl14N56O49: C 48.57, H 7.25, Cl 10.62, N 16.78, O 16.78. Found: C 48.33, H 6.82, N 16.49.
5,11,17,23-Tetraformyl-25,26,27,28-tetrakis(n-hexyloxy)calix[4]arene (15).
A solution of hexamethylenetetramine (16.57 g, 118.3 mmol) in TFA (150 mL) was stirred at 100 °C for 10 min. Then, 25,26,27,28-tetrakis(n-hexyloxy)calix[4]arene (14, 2.5 g, 3.29 mmol) was added and the mixture was stirred at reflux for 2 days. The reaction was quenched by the addition of HCl 1 M (400 mL) and stirred for 3 h. The aqueous layer was extracted twice with DCM (250 mL); the combined organic phases were washed with a saturated aqueous solution of NaHCO3 (200 mL) and brine (200 mL), dried (Na2SO4) and filtered. The solvent was removed under reduced pressure. The product was purified by crystallization from hexane (50 mL). Yield: 2.75 g (96%); Rf = 0.20 (2:3 EtOAc–cyclohexane); 1H NMR (300 MHz, CDCl3) δ 9.58 (s, 4 H, CHO), 7.15 (s, 8 H, Ar), 4.49 (d, J = 13.8 Hz, 4 H, ArCHaxAr), 3.96 (t, J = 7.5 Hz, 8 H, CH2-1Hex), 3.34 (d, J = 13.8 Hz, 4 H, ArCHeqAr), 2.00–1.80 (m, 8 H, CH2-2Hex), 1.50–1.20 (m, 24 H, CH2-3Hex, CH2-4Hex, CH2-5Hex), 1.00–0.80 (m, 12 H, CH3-6Hex); 13C NMR (100.6 MHz, CDCl3) δ 191.3 (CHO), 162.0–130.2 (Ar), 75.8 (C-1Hex), 31.9 (C-3Hex), 30.9 (ArCH2Ar), 30.3 (C-2Hex), 25.8 (C-4Hex), 22.8 (C-5Hex), 14.0 (C-6Hex); ESI-MS: m/z 895.9 [M + Na]+; Anal. calcd for C56H72O8: C 77.03, H 8.31. Found: C 76.78, H 8.04.
5,11,17,23-Tetrahydroxymethyl-25,26,27,28-tetrakis(n-hexyloxy)calix[4]arene (16).
To a suspension of 15 (910 g, 1.04 mmol) in absolute EtOH (50 mL) at 0 °C, NaBH4 (0.24 g, 6.25 mmol) was added. The mixture was stirred at rt for 18 h. Then, 1 M HCl (20 mL) was added and the solvents were evaporated under reduced pressure. EtOAc (50 mL) was added to this and the organic layer was washed with a saturated aqueous solution of NaHCO3 (50 mL) and H2O (50 mL), dried over Na2SO4, and concentrated. Yield: 788 mg (86%); Rf = 0.16 (19:1 DCM–MeOH); 1H NMR (300 MHz, CD3OD) δ 6.66 (s, 8 H, Ar), 4.46 (d, J = 13.2 Hz, 4 H, ArCHaxAr), 4.24 (s, 8 H, CH2OH), 3.90 (t, J = 7.2 Hz, 8 H, CH2-1Hex), 3.15 (d, J = 13.2 Hz, 4 H, ArCHeqAr), 2.00–1.87 (m, 8 H, CH2-2Hex), 1.52–1.34 (m, 24 H, CH2-3Hex, CH2-4Hex, CH2-5Hex), 0.95 (t, J = 6.6 Hz, 12 H, CH3-6Hex); 13C NMR (100 MHz, CD3OD) δ 157.1–128.3 (Ar), 76.0 (C-1Hex), 65.1 (CH2OH), 33.1 (C-3Hex), 32.1 (ArCH2Ar), 31.5 (C-2Hex), 27.3 (C-4Hex), 24.0 (C-5Hex), 14.5 (C-6Hex); ESI-MS: m/z 903.8 [M + Na]+; Anal. calcd for C56H80O8: C 76.33, H 9.15. Found: C 76.41, H 9.20.
5,11,17,23-Tetrachloromethyl-25,26,27,28-tetrakis(n-hexyloxy)calix[4]arene (17).
To a solution of 16 (600 mg, 0.68 mmol) in dry DCM (10 mL), SOCl2 (0.99 mL, 13.60 mmol) was added dropwise and the mixture was stirred at rt for 4 h. Then, the solvent was evaporated under reduced pressure. Yield: 622 mg (95%); 1H NMR (400 MHz, CDCl3) δ 6.64 (s, 8 H, Ar), 4.40 (d, J = 13.2 Hz, 4 H, ArCHaxAr), 4.29 (s, 8 H, CH2Cl), 3.88 (t, J = 7.6 Hz, 8 H, CH2-1Hex), 3.12 (d, J = 13.2 Hz, 4 H, ArCHeqAr), 1.95–1.80 (m, 8 H, CH2-2Hex), 1.49–1.19 (m, 24 H, CH2-3Hex, CH2-4Hex, CH2-5Hex), 1.01–0.77 (m, 12 H, CH3-6Hex); 13C NMR (100 MHz, CDCl3) δ 157.0–128.6 (Ar), 75.4 (C-1Hex), 46.6 (CH2Cl), 32.0 (C-3Hex), 30.9 (ArCH2Ar), 30.2 (C-2Hex), 25.9 (C-4Hex), 22.8 (C-5Hex), 14.1 (C-6Hex); ESI-MS: m/z 977.7 [M + Na]+; Anal. calcd for C56H76Cl4O4: C 70.43, H 8.02. Found: C 70.54, H 8.16.
5,11,17,23-Tetraazidomethyl-25,26,27,28-tetrakis(n-hexyloxy)calix[4]arene (18).
To a solution of 17 (192 mg, 0.20 mmol) in dry DMF (7 mL), NaN3 (78.5 mg) was added. The mixture was stirred, under an Ar atmosphere, at rt, overnight. Then, the solvent was evaporated and the crude was dissolved in EtOAc (15 mL). The organic layer was washed with H2O (15 mL), dried over MgSO4, filtered and concentrated. The residue was purified by column chromatography (cyclohexane → 19:1 cyclohexane–EtOAc). Yield: 186 mg (95%); Rf = 0.29 (19:1 cyclohexane–EtOAc); 1H NMR (300 MHz, CDCl3) δ 6.61 (bs, 8 H, Ar), 4.45 (d, J = 13.2 Hz, 4 H, ArCH2axAr), 3.95 (s, 8 H, CH2N3), 3.89 (t, J = 7.5 Hz, 8 H, CH2-1Hex), 3.16 (d, J = 13.2 Hz, 4 H, ArCH2eqAr), 1.98–1.85 (m, 8 H, CH2-2Hex), 1.48–1.30 (m, 24 H, CH2-3Hex, CH2-4Hex), 0.93 (t, J = 6.9 Hz, 12 H, CH3-6Hex); 13C NMR (75 MHz, CDCl3) δ 162.1–128.2 (Ar), 75.0 (C-1Hex), 53.8 (CH2N3), 31.7 (C-3Hex), 30.4 (ArCH2Ar), 29.9 (C-2Hex), 25.5 (C-4Hex), 22.4 (C-5Hex), 13.7 (C-6Hex); ESI-MS: m/z 1004.1 [M + Na]+, 1020.0 [M + K]+; Anal. calcd for C56H76N12O4: C 68.54, H 7.81, N 17.13, O 6.52. Found: C 68.60, H 7.67, N 17.09.
5,11,17,23-Tetra-[4-(Nα-tert-butoxycarbonyl-Nω-(2,2,4,6,7-pentamethyldihydro-benzofurane-5-sulfonyl)-L-arginine-N-amidomethyl)-1H-1,2,3-triazol-1-methyl]-25,26,27,28-tetrakis(n-hexyloxy)calix[4]arene (19).
To a solution of 11 (35 mg, 35.7 μmol) in acetone (5 mL), 18 (104.5 mg, 0.185 mmol), DIPEA (25 μL, 0.143 mmol) and CuI·P(OEt)3 (5.1 mg, 14.3 μmol) were added. The reaction mixture was refluxed for 24 h. The solvent was removed under reduced pressure and the residue was purified by column chromatography (99:1 → 9:1 DCM–MeOH). Yield: 82 mg (71%); Rf = 0.65 (9:1 DCM–MeOH); [α]D = +2.69 (c 1.0 in MeOH), 1H NMR (500 MHz, CD3OD, 323 K): δ 7.79 (bs, 4 H, CHtriazole), 6.60 (bs, 8 H, Ar), 5.25 (bs, 8 H, CH2NHCO amide), 4.50–4.36 (m, 12 H, CH2Ar, ArCHaxAr), 4.02 (bs, 4 H, CH-2-Arg), 3.86 (t, J = 7.4 Hz, 8 H, CH2-1Hex), 3.19–3.02 (m, 12 H, ArCHeqAr, CH2-5-Arg), 2.96 (s, 8 H, CH2Pbf), 2.55 (s, 12 H, MePbf), 2.50 (s, 12 H, MePbf), 2.05 (s, 12 H, MePbf), 1.94–1.85 (m, 8 H, CH2-2Hex), 1.79–1.29 (m, 100 H, CH2-3Hex, CH2-4Hex, CH2-5Hex, CMe3, CMe2Pbf, CH2-3-Arg, CH2-4-Arg), 0.98–0.88 (m, 12 H, CH3-6Hex), 13C NMR (100.6 MHz, CD3OD): δ 173.5 (CO amide), 158.4 (CN guanidine), 156.4 (Ar), 156.2 (CO carbamate), 145.0 (C-4triazole), 138.0–128.8 (CqPbf, Ar), 127.9, 124.5 (Ar), 122.6 (C-5triazole), 116.9 (Ar), 86.2 (CMe2 Pbf), 79.1 (CMe3), 75.1 (C-1Hex), 54.2 (CH-2-Arg), 53.3 (CH2NHCO), 42.4 (MePbf), 39.8 (CH2-5-Arg), 34.3 (CH2Ar), 31.8 (C-3Hex), 30.1 (ArCH2Ar), 30.0 (C-2Hex), 29.0 (CH2-3-Arg), 27.2 (CMe2Pbf, CMe3), 25.8 (C-4Hex), 25.5 (CH2-4-Arg), 22.5 (C-5Hex), 18.2, 17.0 (MePbf), 13.0 (C-6Hex), 11.1 (MePbf); ESI-MS: m/z 1640.4 [M + 2Na]2+; Anal. calcd for C164H240N32O28S4: C 60.87, H 7.48, N 13.85, S 3.96. Found: C 60.80, H 7.43, N 13.89, S 3.82.
5,11,17,23-Tetra-(4-(L-arginine-N-amidomethyl)-1H-1,2,3-triazol-1-methyl)-25,26,27,28-tetrakis(n-hexyloxy)calix[4]arene octahydrochloride (2e).
A solution of 14 (20 mg, 6.18 μmol) in TFA–TIS–H2O (95:2.5:2.5, 2 mL) was stirred at rt for 1 h. The solvent was removed under reduced pressure and the residue was washed with EtOAc (15 mL). Then the solid was dissolved in 0.1 M HCl solution followed by evaporation under reduced pressure to obtain the product as hydrochloride. Yield: 13 mg (99%). [α]D = +21.4 (c 1.0 in MeOH); 1H NMR (300 MHz, CD3OD): δ 8.21 (bs, 4 H, CHtriazole), 6.71 (bs, 8 H, Ar), 5.39 (bs, 8 H, CH2NHCO amide), 4.75–4.50 (m, 8 H, ArCH2), 4.44 (d, J = 13.2 Hz, ArCHaxAr), 4.00 (t, J = 7.39 Hz, 4 H, CH-2-Arg), 3.85 (t, J = 7.4 Hz, 8 H, CH2-1Hex), 3.27–3.08 (m, 12 H, CH2-5-Arg, ArCHeqAr,), 2.08–1.82 (m, 16 H, CH2-3-Arg, CH2-4-Arg), 1.78–1.61 (m, 8 H, CH2-2Hex), 1.53–1.33 (m, 24 H, CH2-3Hex, CH2-4Hex, CH2-5Hex), 1.00–0.90 (m, 12 H, CH3-6Hex); 13C NMR (75 MHz, CD3OD): δ 170.2 (CO amide), 158.6 (CN guanidine), 158.2, 137.0 (Ar), 136.8 (C-4triazole), 130.0, 129.8 (Ar), 129.7 (C-5triazole), 76.6 (C-1Hex), 55.6 (CH-2-Arg), 54.1 (CH2NHCO), 41.8 (CH-5-Arg), 35.5 (CH2Ar), 33.3 (C-3Hex), 31.6 (ArCH2Ar), 31.5 (C-2Hex), 29.6 (CH2-3-Arg), 27.3 (C-4Hex), 25.4 (CH-4-Arg), 24.0 (C-5Hex), 14.5 (C-6Hex); ESI-MS: m/z 610.0 [M + 3H]3+, 914.4 [M + 2H]2+, 936.3 [M + 2Na]2+, 952.3 [M + 2K]2+; Anal. calcd for C92H152Cl8N32O8·4H2O: C 50.45, H 7.36, N 20.47. Found: C 50.48, H 7.34, N 20.51.
5,11,17,23-Tetraaminomethyl-25,26,27,28-tetrakis(n-hexyloxy)calix[4]arene tetrahydrochloride (20).
To a solution of 18 (300 mg, 0.306 mmol) in EtOAc–EtOH (1:1, 50 mL), a catalytic amount of Pd/C (30 mg) and 1 M HCl (4 mL) were added. Hydrogenation was carried out at 2 atm for 48 h. Then, the catalyst was filtered and the solvent was removed under reduced pressure. Yield: 313 mg (99%); 1H NMR (400 MHz, CD3OD): δ 6.87 (s, 8 H, Ar), 4.49 (d, J = 13.2 Hz, 4 H, ArCHaxAr), 3.92 (t, J = 7.2 Hz, 8 H, CH2-1Hex), 3.84 (s, 8 H, CH2NH2), 3.31 (m, 4 H, ArCHeqAr), 2.00–1.85 (m, 8 H, CH2-2Hex), 1.55–1.30 (m, 24 H, CH2-3Hex, CH2-4Hex, CH2-5Hex), 1.00–0.85 (m, 12 H, CH3-6Hex); 13C NMR (100.6 MHz, CD3OD): δ 157.0–126.7 (Ar), 75.2 (C-1Hex), 42.6 (CH2NH2), 31.9 (C-3Hex), 30.3 (ArCH2Ar), 30.1 (C-2Hex), 25.8 (C-4Hex), 22.5 (C-5Hex), 13.1 (C-6Hex); ESI-MS: m/z 877.6 [M + H]+, 899.8 [M + Na]+; Anal. calcd for C56H88Cl4N4O4: C 65.74, H 8.67, N 5.48. Found: C 66.91, H 8.37, N 5.62.
Heptakis[6-(2-(di-tert-butoxycarbonylguanidino)ethylthio)-2,3-di-O-hexanoyl]cyclomaltoheptaose (21).
To a solution of heptakis[2,3-di-O-hexanoyl-6-(2-aminoethylthio)]-cyclomaltoheptaose heptahydrochloride (8, 50 mg, 15 μmol) in dry DCM (5 mL), under an Ar atmosphere, Et3N (61 μL, 440 μmol) and N-N′-di-tert-butoxycarbonyl-N″-triflylguanidine (86 mg, 220 μmol) were added. The reaction mixture was stirred overnight. The reaction mixture was washed with an aqueous solution of 2 M KHSO4. The organic layer was washed with a saturated aqueous solution of NaHCO3, dried (MgSO4), filtered and concentrated. The residue was purified by column chromatography (DCM → 1:4 EtOAc–cyclohexane). Yield: 67 mg (99%); Rf = 0.75 (1:3 EtOAc–cyclohexane); [α]D = +30.7 (c 1.0 in MeOH); 1H NMR (300 MHz, CD3OD, 313 K): δ 5.33 (t, 7 H, J2,3 = J3,4 = 8.6 Hz, H-3), 5.14 (d, J1,2 = 3.7 Hz, 7 H, H-1), 4.88 (dd, 7 H, H-2), 4.26–4.15 (m, 7 H, H-5), 3.93 (t, 7 H, H-4), 3.78–3.53 (m, 14 H, H-6a, H-6b), 3.16 (bs, 14 H, CH2NH), 2.98–2.85 (m, 14 H, CH2S), 2.51–2.14 (m, 28 H, CH2-2Hex), 1.74–1.56 (m, 28 H, CH2-3Hex), 1.57–1.43 (bs, 126 H, CMe3), 1.45–1.24 (m, 56 H, CH2-4Hex, CH2-5Hex), 1.02–0.85 (m, 42 H, CH3-6Hex); 13C NMR (75 MHz, CD3OD, 313 K): δ 174.5, 173.4 (CO ester), 164.6 (CN), 157.3, 154.2 (CO carbamate), 98.6 (C-1), 84.5 (C-4), 80.4 (CMe3), 73.0 (C-5), 71.9 (C-3), 71.5 (C-2), 41.4 (C-6), 35.2, 35.0 (C-2Hex, CH2NH), 34.1 (CH2S), 32.6, 32.5 (C-4Hex), 28.9, 28.6 (CMe3), 25.6 (C-3Hex), 23.6, 23.5 (C-5Hex), 14.5 (C-6Hex); ESI-MS: m/z 2332.2 [M + 2Na]2+, 1562.7 [M + 3Na]3+; Anal. calcd for C217H371N21O70S7: C 56.43, H 8.10, N 6.37, S 4.86. Found: C 56.26, H 8.00, N 6.12, S 4.51.
Heptakis[6-(2-guanidinoethylthio)-2,3-di-O-hexanoyl]cyclomaltoheptaose heptahydrochloride (1f).
A solution of 16 (60 mg, 13 μmol) in DCM–TFA (1:1, 2 mL) was stirred at rt for 3 h. The solvent was eliminated under reduced pressure and coevaporated several times with water. The residue was dissolved in a 0.1 M HCl solution and freeze-dried to yield the product as hydrochloride. Yield: 45 mg (99%); [α]D = +68.3 (c 1.0 in DMF); 1H NMR (300 MHz, CD3OD): δ 5.39 (t, 7 H, J2,3 = J3,4 = 9.4 Hz, H-3), 5.18 (d, J1,2 = 3.51 Hz, 7 H, H-1), 4.86 (m, 7 H, H-2), 4.16–4.07 (m, 7 H, H-5), 3.96 (t, 7 H, H-4), 3.48 (t, 14 H, CH2NH), 3.20–3.11 (m, 14 H. H-6a, H-6b), 2.97–2.85 (m, 14 H, CH2S), 2.55–2.21 (m, 28 H, CH2-2Hex), 1.72–1.55 (m, 28 H, CH2-3Hex), 1.41–1.26 (m, 56 H, CH2-4Hex, CH2-5Hex), 0.99–0.87 (m, 42 H, CH3-6Hex); 13C NMR (125.7 MHz, DMSO-d6, 323 K): δ 172.2, 171.2 (CO), 156.8 (CN), 96.1 (C-1), 78.0 (C-4), 71.2 (C-5), 69.7 (C-3), 69.8 (C-2), 45.5 (C-6), 40.6 (CH2NH), 33.1, 32.9 (C-2Hex,), 32.1 (CH2S), 30.6, 30.4 (C-4Hex), 23.6, 23.5 (C-3Hex), 21.5 (C-5Hex), 13.3, 13.2 (C-6Hex); ESI-MS: m/z 1609.6 [M + 2H]2+, 1073.0 [M + 3H]3+, 805.1 [M + 4H]4+; Anal. calcd for C147H266Cl7N21O42S7·7H2O: C 49.06, H 7.84, N 8.17, S 6.24. Found: C 48.91, H 7.78, N 8.03, S 6.15.
5,11,17,23-Tetra-[N,N′-di-(tert-butoxycarbonyl)]-guanidinomethyl-25,26,27,28-tetrakis(n-hexyloxy)calix[4]arene (22).
To a solution of 20 (100 mg, 0.098 mmol) in dry DCM (5 mL), Et3N (137 μL, 0.98 mmol) was added. Then, a solution of N-N′-di-tert-butoxycarbonyl-N″-triflylguanidine (192 mg, 0.49 mmol) in dry DCM (1 mL) was added. The mixture was stirred at rt, under N2 atm, 48 h. The reaction mixture was washed with an aqueous solution of 2 M KHSO4. The organic layer was washed with a saturated aqueous solution of NaHCO3, dried (MgSO4), filtered and concentrated. The residue was purified by column chromatography (2:1 DCM–cyclohexane → 1:5 EtOAc–cyclohexane). Yield: 165 mg (91%); Rf = 0.24 (1:6 EtOAc–cyclohexane); 1H NMR (300 MHz, CDCl3): δ 7.74 (bs, 4 H, NH), 7.55 (bs, 4 H, NH), 6.60 (s, 8 H, Ar), 4.38 (d, J = 12.8 Hz, 4 H, ArCHaxAr), 4.27 (d, J = 4.4 Hz, 8 H, CH2NH), 3.84 (t, J = 7.6 Hz, 8 H, CH2-1Hex), 3.10 (d, J = 13.2 Hz, 4 H, ArCHeqAr), 1.98–1.85 (m, 8 H, CH2-2Hex), 1.52–1.28 (m, 96 H, CH2-3Hex, CH2-4Hex, CH2-5Hex, CMe3), 0.90 (t, J = 6.8 Hz, 12 H, CH3-6Hex); 13C NMR (75 MHz, CDCl3): δ 163.6 (CN), 156.1 (Ar), 155.7, 153.1 (CO carbamate), 135.1–127.9 (Ar), 86.0, 82.9 (CMe3), 75.4 (C-1Hex), 44.9 (CH2NH), 32.2, 31.6 (C-3Hex), 31.1 (ArCH2Ar), 30.3 (C-2Hex), 28.3, 27.1, 30.9 (CMe3), 26.0 (C-4Hex), 22.9, 22.6 (C-5Hex), 14.1 (C-6Hex); ESI-MS: m/z 1846.7 [M + H]+, 1868.7 [M + Na]+, 1885.7 [M + K]+; Anal. calcd for C100H156N12O20: C 65.05, H 8.52, N 9.10. Found: C 65.23, H 8.64, N 9.03.
5,11,17,23-Tetraguanidinomethyl-25,26,27,28-tetrakis(n-hexyloxy)calix[4]arene tetrahydrochloride (2f).
To a solution of 17 (69 mg, 0.037 mmol) in dry dioxane (7 mL), TES (59 μL, 0.37 mmol) and 37% HCl (500 μL) were added. The mixture was stirred for 24 h at rt. Then, the solvents were evaporated under reduced pressure. The product was precipitated with Et2O (6 mL) and dried under reduced pressure. Yield: 44 mg (99%); 1H NMR (400 MHz, CD3OD): δ 6.75 (s, 8 H, Ar), 4.49 (d, J = 13.2 Hz, 4 H, ArCHaxAr), 4.15 (s, 8 H, CH2NH), 3.93 (t, J = 7.2 Hz, 8 H, CH2-1Hex), 3.23 (d, J = 13.2 Hz, 4 H, ArCHeqAr), 2.05–1.95 (m, 8 H, CH2-2Hex), 1.55–1.31 (m, 24 H, CH2-3Hex, CH2-4Hex, CH2-5Hex), 1.01–0.93 (m, 12 H, CH3-6Hex); 13C NMR (100.6 MHz, CD3OD): δ 158.3 (CN), 157.6–129.0 (Ar), 76.5 (C-1Hex), 45.9 (CH2NH), 33.6 (C-3Hex), 31.9 (ArCH2Ar), 31.5 (C-2Hex), 27.3 (C-4Hex), 24.0 (C-5Hex), 14.5 (C-6Hex), ESI-MS: m/z 1045.89 [M + H]+; Anal. calcd for C60H96Cl4N12O4: C 60.49, H 8.12, N 14.11. Found: C 60.13, H 7.95, N 13.89.
Acknowledgements
L.G.-Y. is an FPU fellow. This study was financially supported by the Spanish Ministerio de Economía y Competitividad (contract numbers SAF2013-44021-R and CTQ2010-15848), the Junta de Andalucia (contract number FQM2012-1467), the Italian Ministry of Instruction, University and Research (MIUR, PRIN2010JMAZML MultiNanoIta), the COST actions MultiGlycoNano (CM1102) and Supramolecular Chemistry in Water (CM1005), and the European Regional Development Funds (FEDER and FSE). Technical assistance from the research support services of the University of Seville (CITIUS) and Centro Interdipartimentale di Misure (CIM) of Parma University is also acknowledged.
Notes and references
-
(a) A.. Aiuti, F. Cattaneo, S. Galimberti, U. Benninghoff, B. Cassani, L. Callegaro, S. Scaramazza, G. Andolfi, M. Mirolo, I. Brigida, A. Tabucchi, F. Carlucci, M. Eibl, M. Aker, S. Slavin, H. Al-Mousa, A. Al Ghonaium, A. Ferster, A. Duppenthaler, L. Notarangelo, U. Wintergerst, R. H. Buckley, M. Bregni, S. Marktel, M. G. Valsecchi, P. Rossi, F. Ciceri, R. Miniero, C. Bordignon and M. G. N. Roncarolo, Engl. J. Med., 2009, 360, 447 CrossRef CAS PubMed;
(b) F. Mavilio, G. Pellegrini, S. Ferrari, F. Di Nunzio, E. Di Iorio, A. Recchia, G. Maruggi, G. Ferrari, E. Provasi, C. Bonini, S. Capurro, A. Conti, C. Magnoni, A. Giannetti and M. De Luca, Nat. Med., 2006, 12, 1397 CrossRef CAS PubMed;
(c) S. L. Murphy and K. A. High, Br. J. Hematol., 2008, 140, 479 CrossRef CAS PubMed;
(d) P. Arumugam and P. Malik, Hematol. Am. Soc. Hematol.Educ. Program, 2010, 445 CrossRef PubMed;
(e) M. De Luca, G. Pellegrini and F. Mavilio, Br. J. Dermatol., 2009, 161, 19 CrossRef CAS PubMed;
(f) A. Biffi, E. Montini, L. Lorioli, M. Cesani, F. Fumagalli, T. Plati, C. Baldoli, S. Martino, A. Calabria, S. Canale, F. Benedicenti, G. Vallanti, L. Biasco, S. Leo, N. Kabbara, G. Zanetti, W. Rizzo, N. A. Mehta, M. Cicalese, M. Casiraghi, J. Boelens, U. Del Carro, D. Dow, M. Schmidt, A. Assanelli, V. Neduva, C. Di Serio, E. Stupka, J. Gardner, C. von Kalle, C. Bordignon, F. Ciceri, A. Rovelli, M. Roncarolo, A. Aiuti, M. Sessa and L. Naldini, Science, 2013, 341, 1233158 CrossRef PubMed;
(g) A. Aiuti, L. Biasco, S. Scaramuzza, F. Ferrua, M. Cicalese, C. Baricordi, F. Dionisio, A. Calabria, S. Giannelli, M. Castiello, M. Bosticardo, C. Evangelio, A. Assanelli, M. Casiraghi, S. Di Nunzio, L. Callegaro, C. Benati, P. Rizzardi, D. Pellin, C. Di Serio, M. Schmidt, C. Von Kalle, J. Gardner, N. Mehta, V. Neduva, D. Dow, A. Galy, R. Miniero, A. Finocchi, A. Metin, P. Banerjee, J. Orange, S. Galimberti, M. Valsecchi, A. Biffi, E. Montini, A. Villa, F. Ciceri, M. Roncarolo and L. Naldini, Science, 2013, 341, 853 CrossRef PubMed;
(h) M. Morille, C. Passirani, A. Vonarbourg, A. Clavreul and J. P. Benoit, Biomaterials, 2008, 29, 3477 CrossRef CAS PubMed.
- Gene Therapy Clinical Trials Worldwide provided by the J. Gene Med. ( http:// www.wiley.com/legacy/wileychi/genmed/clinical/).
- R. Srinivas, S. Samanta and A. Chaudhuri, Chem. Soc. Rev., 2009, 38, 3326–3338 RSC.
-
(a) S. D. Li and L. Huang, J. Controlled Release, 2007, 123, 181 CrossRef CAS PubMed;
(b) M. A. Mintzer and E. E. Simanek, Chem. Rev., 2009, 109, 259 CrossRef CAS PubMed.
-
(a) A. Rajala, Y. Wang, Y. Zhu, M. Ranjo-Bishop, J.-X. Ma, C. Mao and R. V. S. Rajala, Nano Lett., 2014, 9, 5257 CrossRef PubMed;
(b) M. E. Davis, Mol. Pharmaceutics, 2009, 6, 659 CrossRef CAS PubMed.
-
(a) C. Ortiz Mellet, J. M. García Fernández and J. M. Benito, Chem. Soc. Rev., 2011, 40, 1586 RSC;
(b) J. M. García Fernández, J. M. Benito and C. Ortiz Mellet, Pure Appl. Chem., 2013, 85, 1825 CrossRef PubMed;
(c) L. Wang, L. Li, Y. Fan and H. Wang, Adv. Mater., 2013, 25, 3888 CrossRef CAS PubMed.
-
(a) S. Srinivasachari, K. M. Fichter and T. M. Reineke, J. Am. Chem. Soc., 2008, 130, 4618 CrossRef CAS PubMed;
(b) N. Mourtzis, M. Paravatou, I. M. Mavridis, M. L. Roberts and K. Yannakopoulou, Chem. – Eur. J., 2008, 14, 4188 CrossRef CAS PubMed;
(c) S. Menuel, S. Fontanay, I. Clarot, R. E. Duval, L. Diez and A. Marsura, Bioconjugate Chem., 2008, 19, 2357–2362 CrossRef CAS PubMed;
(d) R. Lalor, J. L. DiGesso, A. Mueller and S. E. Matthews, Chem. Commun., 2007, 4907 RSC;
(e) V. Bennevault-Celton, A. Urbach, O. Martin, C. Pichon, P. Guégan and P. Midoux, Bioconjugate Chem., 2011, 22, 2404 CrossRef CAS PubMed.
- For a review, see:
(a) C. Ortiz Mellet, J. M. Benito and J. M. García Fernández, Chem. – Eur. J., 2010, 16, 6728 CrossRef CAS PubMed. For additional recent examples of polycationic facial amphiphiles based on macrocyclic scaffolds, see:
(b) Y. Yao, M. Xue, J. Chen, M. Zhang and F. Huang, J. Am. Chem. Soc., 2012, 134, 15712 CrossRef CAS PubMed;
(c) Y. Yao, M. Xue, Z. Zhang, M. Zhang, Y. Wang and F. Huang, Chem. Sci., 2013, 4, 3667 RSC.
-
(a) A. Díaz-Moscoso, P. Balbuena, M. Gómez-García, C. Ortiz Mellet, J. M. Benito, L. Le Gourrièrec, C. Di Giorgio, P. Vierling, A. Mazzaglia, N. Micalli, J. Defaye and J. M. García Fernández, Chem. Commun., 2008, 2001 RSC;
(b) A. Díaz-Moscoso, L. Le Gourrirec, M. Gómez-García, J. M. Benito, P. Balbuena, F. Ortega-Caballero, N. Guilloteau, C. Di Giorgio, P. Vierling, J. Defaye, C. Ortiz Mellet and J. M. García Fernández, Chem. – Eur. J., 2009, 15, 12871 CrossRef PubMed;
(c) A. Méndez-Ardoy, M. Gómez-García, C. Ortiz Mellet, N. Sevillano, M. D. Girón, R. Salto, F. Santoyo-González and J. M. García Fernández, Org. Biomol. Chem., 2009, 7, 2681 RSC;
(d) A. Martínez, C. Bienvenu, J. L. Jiménez Blanco, P. Vierling, C. Ortiz Mellet, J. M. García Fernández and C. Di Giorgio, J. Org. Chem., 2013, 78, 8143 CrossRef PubMed.
-
(a) M. Dudič, A. Colombo, F. Sansone, A. Casnati, G. Donofrio and R. Ungaro, Tetrahedron, 2004, 60, 11613 CrossRef PubMed;
(b) F. Sansone, M. Dudic, G. Donofrio, C. Rivetti, L. Baldini, A. Casnati, S. Cellai and R. Ungaro, J. Am. Chem. Soc., 2006, 128, 14528 CrossRef CAS PubMed;
(c) V. Bagnacani, F. Sansone, G. Donofrio, L. Baldini, A. Casnati and R. Ungaro, Org. Lett., 2008, 10, 3953 CrossRef CAS PubMed;
(d) V. Bagnacani, V. F. L. Fantuzzi, F. Sansone, G. Donofrio, A. Casnati and R. Ungaro, Bioconjugate Chem., 2012, 23, 993 CrossRef CAS PubMed.
-
(a) A. M. O'Mahony, B. M. D. C. Godinho, J. Ogier, M. Devocelle, R. Darcy, J. F. Cryan and C. M. O'Driscoll, ACS Chem. Neurosci., 2012, 3, 744 CrossRef PubMed;
(b) V. Villari, A. Mazzaglia, R. Darcy, C. M. O'Driscoll and N. Micalli, Biomacromolecules, 2013, 14, 811 CrossRef CAS PubMed;
(c) A. M. O'Mahony, S. Desgranges, J. Ogier, A. Quinlan, M. Devocelle, R. Darcy, J. F. Cryan and C. M. O'Driscoll, Pharm. Res., 2013, 30, 1086 CrossRef PubMed;
(d) J. McCarthy, M. J. O'Neill, L. Bourre, D. Walsh, A. Quinlan, G. Hurley, J. Ogier, F. Shanahan, S. Melgar, R. Darcy and C. M. O'Driscoll, J. Controlled Release, 2013, 168, 28 CrossRef CAS PubMed;
(e) B. M. D. C. Godinho, D. J. McCarthy, C. Torres-Fuentes, C. J. Beltrán, J. McCarthy, A. Quinlan, J. R. Ogier, R. Darcy, C. M. O'Driscoll and J. F. Cryan, Biomaterials, 2014, 489 CrossRef CAS PubMed.
- R. V. Rodik, A. S. Klymchenko, N. Jain, S. I. Miroshnichenko, L. Richert, V. I. Kalchenko and Y. Mély, Chem. – Eur. J., 2011, 17, 5526 CrossRef CAS PubMed.
- C. Bienvenu, A. Martínez, J. L. Jiménez Blanco, C. Di Giorgio, P. Vierling, C. Ortiz Mellet, J. Defaye and J. M. García Fernández, Org. Biomol. Chem., 2012, 10, 5570 CAS.
-
(a) A. Díaz-Moscoso, D. Vercauteren, J. Rejman, J. M. Benito, C. Ortiz Mellet, S. C. De Smedt and J. M. García Fernández, J. Controlled Release, 2010, 143, 318 CrossRef PubMed;
(b) A. Díaz-Moscoso, N. Guilloteau, C. Bienvenu, A. Méndez-Ardoy, J. L. Blanco, J. M. Benito, L. Le Gourriérec, C. Di Giorgio, P. Vierling, J. Defaye, C. Ortiz Mellet and J. M. García Fernández, Biomaterials, 2011, 32, 7263 CrossRef PubMed;
(c) A. Méndez-Ardoy, N. Guilloteau, C. Di Giorgio, P. Vierling, F. Santoyo-González, C. Ortiz Mellet and J. M. García Fernández, J. Org. Chem., 2011, 76, 5882 CrossRef PubMed;
(d) A. Méndez-Ardoy, K. Urbiola, C. Aranda, C. Ortiz-Mellet, J. M. García-Fernández and C. Tros de Ilarduya, Nanomedicine, 2011, 6, 1697 CrossRef PubMed;
(e) N. Symens, A. Méndez-Ardoy, A. Díaz-Moscoso, E. Sánchez-Fernández, K. Remaut, J. Demeester, J. M. García Fernández, S. C. De Smedt and J. Rejman, Bioconjugate Chem., 2012, 23, 1276 CrossRef CAS PubMed;
(f) C. Aranda, K. Urbiola, A. Méndez Ardoy, J. M. García Fernández, C. Ortiz Mellet and C. Tros de Ilarduya, Eur. J. Pharm. Biopharm., 2013, 85, 390 CrossRef CAS PubMed.
- F. Ortega-Caballero, C. Ortiz Mellet, L. Le Gourriérec, N. Guilloteau, C. Di Giorgio, P. Vierling, J. Defaye and J. M. García Fernández, Org. Lett., 2008, 10, 5143 CrossRef CAS PubMed.
- V. Bagnacani, V. Franceschi, M. Bassi, M. Lomazzi, G. Donofrio, F. Sansone, A. Casnati and R. Ungaro, Nat. Commun., 2013, 4, 1721 CrossRef PubMed.
-
(a) L. Gallego-Yerga, M. Lomazzi, F. Sansone, C. Ortiz Mellet, A. Casnati and J. M. García Fernández, Chem. Commun., 2014, 50, 7440 RSC;
(b) L. Gallego-Yerga, M. J. González-Alvárez, N. Mayordomo, F. Santoyo-González, J. M. Benito, C. Ortiz Mellet, F. Mendicuti and J. M. García Fernández, Chem. – Eur. J., 2014, 20, 6622 CrossRef CAS PubMed.
- H. Munch, J. S. Hansen, M. Pittelkow, J. Christensen and U. Boas, Tetrahedron Lett., 2008, 49, 3117 CrossRef CAS PubMed.
- M. Gómez-García, J. M. Benito, D. Rodríguez-Lucena, J.-X. Yu, K. Chmurski, C. Ortiz Mellet, R. Gutiérrez-Gallego, A. Maestre, J. Defaye and J. M. García Fernández, J. Am. Chem. Soc., 2005, 127, 7970 CrossRef PubMed.
-
(a) E. M. Aguilar Moncayo, N. Guilloteau, C. Bienvenu, J. L. Jiménez Blanco, C. Di Giorgio, P. Vierling, J. M. Benito, C. Ortiz Mellet and J. M. García Fernández, New J. Chem., 2014, 38, 5215 RSC;
(b) M. Gómez-García, J. M. Benito, A. P. Butera, C. Ortiz Mellet, J. M. García Fernández and J. L. Jiménez Blanco, J. Org. Chem., 2012, 77, 1273 CrossRef PubMed;
(c) M. Gómez-Garcia, J. M. Benito, R. Gutiérrez-Gallego, A. Maestre, C. Ortiz Mellet, J. M. Garcia Fernández and J. L. Jiménez Blanco, Org. Biomol. Chem., 2010, 8, 1849 RSC.
-
(a) S. Srinivasachari, Y. Liu, G. Zhang, L. Prevette and T. M. Reineke, J. Am. Chem. Soc., 2006, 128, 8176 CrossRef CAS PubMed;
(b) P. Midoux, C. Pichon, J. J. Yaouanc and P. A. Jaffres, Br. J. Pharmacol., 2009, 157, 166 CrossRef CAS PubMed.
-
(a) M. Meldal and C. W. Tornoe, Chem. Rev., 2008, 108, 2952 CrossRef CAS PubMed;
(b) P. Cintas, A. Barge, S. Tagliapietra, L. Boffa and G. Cravotto, Nat. Protoc., 2010, 5, 607 CrossRef CAS PubMed;
(c) C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef CAS PubMed;
(d) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS.
- H. Ihm and K. Paek, Bull. Korean Chem. Soc., 1995, 16, 71 CAS.
-
(a) M.-H. Louis, S. Dutoit, Y. Denoux, P. Erbacher, E. Deslandes, J.-P. Behr, P. Gauduchon and L. Poulain, Cancer Gene Ther., 2006, 13, 367 CrossRef CAS PubMed;
(b) O. Boussif, F. Lezoualch, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J.-P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7297 CrossRef CAS.
-
(a) L. Wasungu and D. Hoekstra, J. Controlled Release, 2006, 116, 255 CrossRef CAS PubMed;
(b) H. Farhood, N. Serbina and L. Huang, Biochim. Biophys. Acta, 1995, 1235, 289 CrossRef.
- S. Rossi, P. L. Poliani, C. Missale, E. Monti and A. Fanzani, J. Cell. Mol. Med., 2011, 15, 2553 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental protocols for nanocomplex formulation and characterization, cell cultures, evaluation of cell viability and transfection efficiency, NMR and ESI-MS spectra of newly synthesized compounds. See DOI: 10.1039/c4ob02204a |
|
| This journal is © The Royal Society of Chemistry 2015 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b and
José M.
García Fernández
*d
b and
José M.
García Fernández
*d