
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA microfluidic platform reveals differential response of regulatory T cells to micropatterned costimulation arrays†
Joung-Hyun
Lee
a,
Michael L.
Dustin
b and
Lance C.
Kam
*a
aDepartment of Biomedical Engineering, Columbia University in the City of New York, New York, USA. E-mail: lance.kam@columbia.edu
bKennedy Institute of Rheumatology, Nuffield Department of Orthopaedics, Rheumatology, and Musculoskeletal Sciences, University of Oxford, Oxford, UK
First published on 16th September 2015
Abstract
T cells are key mediators of adaptive immunity. However, the overall immune response is often directed by minor subpopulations of this heterogeneous family of cells, owing to specificity of activation and amplification of functional response. Knowledge of differences in signaling and function between T cell subtypes is far from complete, but is clearly needed for understanding and ultimately leveraging this branch of the adaptive immune response. This report investigates differences in cell response to micropatterned surfaces by conventional and regulatory T cells. Specifically, the ability of cells to respond to the microscale geometry of TCR/CD3 and CD28 engagement is made possible using a magnetic-microfluidic device that overcomes limitations in imaging efficiency associated with conventional microscopy equipment. This device can be readily assembled onto micropatterned surfaces while maintaining the activity of proteins and other biomolecules necessary for such studies. In operation, a target population of cells is tagged using paramagnetic beads, and then trapped in a divergent magnetic field within the chamber. Following washing, the target cells are released to interact with a designated surface. Characterization of this system with mouse CD4+ T cells demonstrated a 50-fold increase in target-to-background cell purity, with an 80% collection efficiency. Applying this approach to CD4+CD25+ regulatory T cells, it is then demonstrated that these rare cells respond less selectively to micro-scale features of anti-CD3 antibodies than CD4+CD25− conventional T cells, revealing a difference in balance between TCR/CD3 and LFA-1-based adhesion. PKC-θ localized to the distal pole of regulatory T cells, away from the cell–substrate interface, suggests a mechanism for differential regulation of TCR/LFA-1-based adhesion. Moreover, specificity of cell adhesion to anti-CD3 features was dependent on the relative position of anti-CD28 signaling within the cell–substrate interface, revealing an important role for coincidence of TCR and costimulatory pathway in triggering regulatory T cell function.
Insight, innovation, integrationThe subcellular organization of signaling proteins has an important and increasingly recognized role in determining cell function. Multicomponent, micropatterned surfaces have emerged as a powerful platform for studying this aspect of cellular physiology, but the inherent inefficiencies of conventional microscopy platforms limit their use of cells of limited availability. This report combines a magnetic-microfluidic system with protein micropatterned surfaces to investigate artificial immune synapses formed by regulatory T cells, a rare subtype that plays important roles in suppressing adaptive immune function. This platform dramatically improves purity and collection efficiency of target cells, making possible studies on differences in function and protein localization between regulatory and conventional T cells. |
Introduction
T cells are key mediators of the adaptive immune response, carrying out a wide range of functions such as production of inflammatory cytokines and killing of target cells. There are correspondingly multiple subtypes of T cells, each specializing in a select set of functions. Accordingly, overall immune response is often driven by small subpopulations of cells; for example, regulatory T cells (Tregs), which comprise normally 1% of circulating T cells, temper the reactive T cell response.1–3 As these subtypes are largely derived from a common precursor (thymocytes), a contemporary challenge is to understand the similarities and differences in intracellular signaling that distinguish each one. An emerging arena for these comparisons is in the localization of signaling proteins at both the subcellular level and the smaller micrometer scale within the immune synapse (IS), a specialized area of contact between T cells and antigen-presenting cells (APCs) which focuses communication between these partners.4–6 For example, Zanin-Zhorov and coworkers reported7 that PKC-θ is sequestered away from the IS and concentrated at the distal pole of Tregs interacting with APCs. This localization of PKC-θ correlates with Tregs' suppressive function, as relocalization of PKC-θ towards proximal position of the IS by tumor necrosis factor-α (TNF-α) correlated with inhibition of suppression.7,8 At the smaller scale of the IS, Tseng and coworkers have shown that microscale coincidence of CD80 relative to T cell receptor (TCR) correlates with activation of conventional T cells.9,10 This growing body of knowledge, gained using microscopy- and surface engineering-based techniques, reveals that the microscale organization of signaling proteins within the IS influences T cell activation.11–13 However, application of these techniques to rare cell populations, such as Tregs, remains a challenge due to both the low frequency of these targets in a cell population and the low efficiency of observing cells under conventional microscopy conditions; less than 1% of a starting sample of cells are analyzed in such systems due to the small observation areas associated with high-magnification imaging.We report here a microfluidic system that provides high efficiency capture and microscopy-based analysis of target cells as they interact with engineered surfaces. While conventional magnetic bead-based columns or fluorescence-activated cell sorting (FACS) methods are available for isolating cells with high precision and efficiency, these methods need large starting number/volume of cells, i.e., at least a few hundred thousand of cells in at least a submilliliter volume. Besides, the practical portion of the isolated cells that are viewed under the microscope is extremely small due to dead volumes associated with commercially available flow chambers (such as ibidi μ-slides, Ibidi). On the other hand, our microfluidic system deals with not only smaller number of cells in less than a few tens of microliters but also concentrate the isolated cells on the field of view on a microscope. This platform builds on, but is distinguished from, technologies developed by other groups to study rare cell populations, such as microfluidic cell sorters,14 epithelial cell adhesion molecule (EpCAM)-based adhesion using pillar arrays,15–17 size-based microfluidic separation,18 and combined microfluidic/micromagnetic systems to capture and release target cells.19 In particular, the “CTC chip” developed by Nagrath and colleagues,15 which uses EpCAM based adhesion and optimum shear stress, has emerged as a gold standard in the field. This method, however, is limited for our proposed application by low purity (about 50–65%) and moderate capturing yield (about 65%). The topology of the CTC chip capture surface also hinders its use in high-resolution microscopy. The platform reported here combines microfluidics with magnetic field-based capture of target cells which have been tagged with paramagnetic beads. Magnetic capture has been used extensively to isolate target cell populations from bulk preparations. By integrating this approach into a microfluidic chamber (Fig. 1), and using an external magnet to apply a field during an initial capture step, we optimize the delivery of target cells within a microscopy observation area. The simplicity of this device allows its use with complex surfaces containing micropatterned proteins, lipid bilayers, or other biological and non-biological materials. The functional attributes of the platform are thus (1) a one-step isolation, seeding and stimulation of a small portion of positively labeled T cells among other abundant cell types, (2) an integration of delicate but experimentally important surfaces, and (3) a precise collection/release of small number of cells, i.e., ∼1000 cells which is about 1/100 fold of conventional method.
 | ||
| Fig. 1 A magnetic microfluidic chamber for improved cell microscopy. (a) Device design. (b) Schematics of top and side views of the red boxed area in (a). (c) Capture and isolation of paramagnetic beads-labeled cells are carried out using a diverging magnetic field, applied during cell loading. In this implementation, the magnetic field is applied using a stack of permanent magnets. When collection is finished, the magnet is removed and cells are released onto antibody patterns of interest (shown in red box in panel a and b). | ||
As an initial demonstration of this platform, we investigated the interaction of regulatory T cells with multicomponent, micropatterned surfaces containing independent patterns of antibodies that engage and activate CD3 (a key signaling component of the T cell receptor (TCR) complex), and CD28 (which provides a costimulatory signal essential for functional activation). These activation/costimulation sites are surrounded and separated by intercellular adhesion molecule-1 (ICAM-1), which provides an adhesive signal to T cells through the integrin lymphocyte function-associated antigen-1 (LFA-1). Previous studies demonstrated that micrometer-scale features of anti-CD3 (i.e., antibodies that bind and activate CD3) stop migration and direct adhesion of conventional, reactive T cells on these surfaces.11,12,20 Moreover, the microscale organization of CD28 engagement, both independently and relative to CD3 activation, can be recognized by CD4+ T cells.11,12 Understanding how regulatory T cells differ from conventional T cells with respect to these behaviors will provide new perspectives on the mechanisms that balance adaptive immunity. These insights may provide new strategies for engineering the adaptive immune system, including therapies directed towards cancer.21
Method
Device fabrication
Established microfluidics fabrication techniques22,23 were used to create the devices illustrated in Fig. 1, consisting of a poly(dimethyl siloxane) (PDMS, Sylgard 184, Dow Corning) chamber to be affixed to a glass coverslip. The basic device layout consists of a main channel measuring 800 μm × 150 μm × 5 cm (W × D × L), which converges with a washing/side channel at right angle (Fig. 1a). An area reserved for microscopy-based imaging was located downstream of the channel junction shown in the red box in Fig. 1a and b. Thin PDMS channels22 were molded from silicon wafers containing patterns of SU 8 (Microchem) by spin coating mixed, but not cured, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (base:crosslinker) PDMS at 350 rpm for 30 s in order to achieve 250 μm in thickness. Upon curing at 60 °C for 2 h, 5 mm-thick, 5:1 PDMS pieces were bonded to the previous thin PDMS layer at both the inlet and outlet areas. Holes for fluidic tubing connections were subsequently punched through the PDMS structure. The prepared PDMS negative channel housing can be stored for several months. For use in experiments, the channel was bonded to a glass slide or coverslip (2.5 × 7.5 cm2) by plasma or corona treatment of the PDMS housing.
:1 (base:crosslinker) PDMS at 350 rpm for 30 s in order to achieve 250 μm in thickness. Upon curing at 60 °C for 2 h, 5 mm-thick, 5:1 PDMS pieces were bonded to the previous thin PDMS layer at both the inlet and outlet areas. Holes for fluidic tubing connections were subsequently punched through the PDMS structure. The prepared PDMS negative channel housing can be stored for several months. For use in experiments, the channel was bonded to a glass slide or coverslip (2.5 × 7.5 cm2) by plasma or corona treatment of the PDMS housing.
Substrate preparation
Glass coverslips were patterned with mouse anti-CD3 and anti-CD28 via microcontact printing methods; detailed stamping procedures are described elsewhere.11 In brief, a negative array of poly(methyl methacrylate) (PMMA) dot patterns of 2 or 1 μm in diameter, 10 or 5 μm in pitch and 1 μm in height on silicon wafer was used to obtain positive PDMS dot stamps. Total 25 μg mL−1 of mouse anti-CD3 and anti-CD28 antibodies (anti-mouse CD3e, clone 145-2C11, eBioscience 14-0031-86 and anti-mouse CD28, clone 37.51, eBioscience 14-0281-86) at ratio of 1:3 by weight were prepared in PBS and left wet on the PDMS stamps of 1 cm2 for at least 30 min until stamping. Prior to stamping, stamps were washed in PBS and DI water followed by gentle N2 blow. To stamp, the stamps were let sit on coverslips for approximately 5 min with a weight on. The stamped glass coverslips were bonded immediately with the prepared PDMS housing as described above and the channels were filled with 2 μg mL−1 mouse ICAM-1 (recombinant mouse ICAM-1/CD54 Fc chimera, R&D Systems 796IC) for 1 h followed by replacement with 5% bovine serum albumin (BSA) for another hour before adding cells.
Cells
CD4+ T cells were first isolated from spleens of C57BL/6 mice by positive selection (CD4 microbeads, Miltenyi Biotec 130-049-021). For visualization, bead-bound CD4+ T cells were fluorescently labeled with green CFSE stain (CellTrace™ CFSE Cell Proliferation Kit, Invitrogen C34554) and the rest of the splenocytes labeled with red CMTPX (CellTracker™ Red CMTPX, Invitrogen C34552). For modeling rare cell isolation, CFSE-labeled CD4+ T cells were spiked into CMTPX-labeled CD4− splenocytes with 1, 10 or 25% of CFSE-cells in total cell concentration. When seeding, cells were resuspended at a concentration of 1 million total cells per milliliter in RPMI-1640 medium (Invitrogen 12633-020) supplemented with 10% FBS, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, Gibco), 2 mM L-glutamine (Gibco), 50 μM of β-mercaptoethanol, and 50 U mL−1 penicillin and 50 μg mL−1 streptomycin (complete medium).For studies of regulatory T cells, cells were labeled with paramagnetic beads for microfluidic isolation by slight modification from the protocol of CD4+CD25+ regulatory T cell isolation kit (Miltenyi Biotec 130-091-301). In brief, CD4+ T cells were isolated by negative selection from spleens of C57BL/6 mice. CD4+CD25+ cells were further labeled with CD25-PE antibody, followed by indirect magnetic labeling of CD4+CD25+ cells with anti-PE beads. This step results in mixture of untouched CD4+CD25− T cells (denotes as “Tconvs” from here) and PE- and beads-labeled CD4+CD25+ T cells (Tregs). This cell mixture was used for in situ isolation and investigation of Tconvs and Tregs on antibody patterned coverslips bonded to microfluidic channels. The purity of collected Tregs in channels was determined by counting of PE-labeled cells out of all the collected cells. Separately, in order to confirm the percentage of Foxp3hi T cells in the CD4+CD25+ T cells compared to CD4+CD25− T cells, the CD4+CD25+ T cells were isolated by MACS columns (Miltenyi Biotec), fixed and permeabilized by True-Nuclear transcription factor buffer set (BioLegend), and stained with anti-Foxp3 (BioLegend 320012) and subsequently analyzed by FACS.
Cell seeding and target cell enrichment in microchannels
Prior to seeding, the dual inlet channels on the device were filled with warm media using syringe pumps while an inlet of a main channel was left with no fluidic connection. Cells (labeled as described above) to be loaded into the chamber were resuspended in a 5 μL volume, and then manually seeded using micropipetters into the inlet of the main channel. Once cells were loaded, the inlet of the main channel was plugged with a solid rod and complete medium was flowed in through the side-washing channel. Schematic operation procedures are shown in Fig. 1c. A set of permanent magnets (rod type NdFeB magnets, d = 1/8′′, K&J Magnetic) suspended over the device was brought down into contact with the top of the channel. Using syringe pumps, cell suspension was flowed past the magnet until sufficient numbers of target cells were collected and the rest of non-target cells washed away, which takes about 15 min typically; then medium flow was stopped and the magnet was removed so that the collected target cells dropped to the bottom surface on which antibodies were patterned. Cells were then incubated for 30 min on the antibody arrays and then analyzed for collection efficiency or subjected to further immunostaining. A custom-built stage-top incubation chamber was employed to maintain 37 °C and 5% CO2 during the live cell microscopy phase.Collection efficiency in Fig. 2b was obtained by the number of captured cells by the magnet at a FOV (at 10× objective) compared to total number of cells passed during the collection procedure, which is extrapolated from number of total cells at a FOV at the beginning of the collection procedure. Purity after collection was determined by comparing the resulting number of target cells to non-target cells in a FOV after collection was finished.
 | ||
| Fig. 2 Device performance. (a) Number density of target, CFSE-labeled CD4+ T cells and non-target, CMTPX-labeled splenocytes per area over time. Target cells comprised 25, 10 and 1% of initial population. Progression of cell collection was described as time passed since the washing step began, that is immediately after placement of magnets onto the device. (b) Collection efficiency and resulting purity. (c) Fluorescent microscopic images of target and non-target cells before and after collection, using a 1% initial population of target cell. Panel c was obtained using a 10× objective, with the field of view covering the complete channel width. Data in panels a and b represent mean ± s.d. from at least three separate experiments for each condition. | ||
While Tregs were isolated using the microfluidic system, Tconvs were isolated using MACS columns from bulk of splenocytes to remove small portion of Tregs. Subsequently, Tconvs were seeded in the channels, and ultimately tested as same manner as Tregs.
Real-time observation of cell motility
Bright-field and interference reflection microscopy (IRM) images of cells and fluorescent images of patterns (in blue) were obtained using 60× objective in time-lapsed manner for 30 min with 20 s intervals immediately after seeding. Movie S1 (ESI†) was obtained in the same manner. Mean velocity (μm min−1) of cells walking or stopping on patterns was analyzed from IRM images using Track Object App in Metamorph software (Olympus).Immunochemistry and analysis
Quantitative data of differential adhesion behaviors of Tregs compared to Tconvs on patterns (Fig. 3c and d) were obtained and analyzed by counting cells on and off the patterns from multiple bright field and IRM images of cells overlaid with fluorescent images of patterns at 100× objective lens after fixing and washing. Unadhered cells were washed off during this washing step by using syringe pumps at a flow rate of 10 μL min−1. As IRM images indicate true interaction of cells with surfaces, cells without proper foot print in IRM was considered as non-reactive and excluded from the analysis.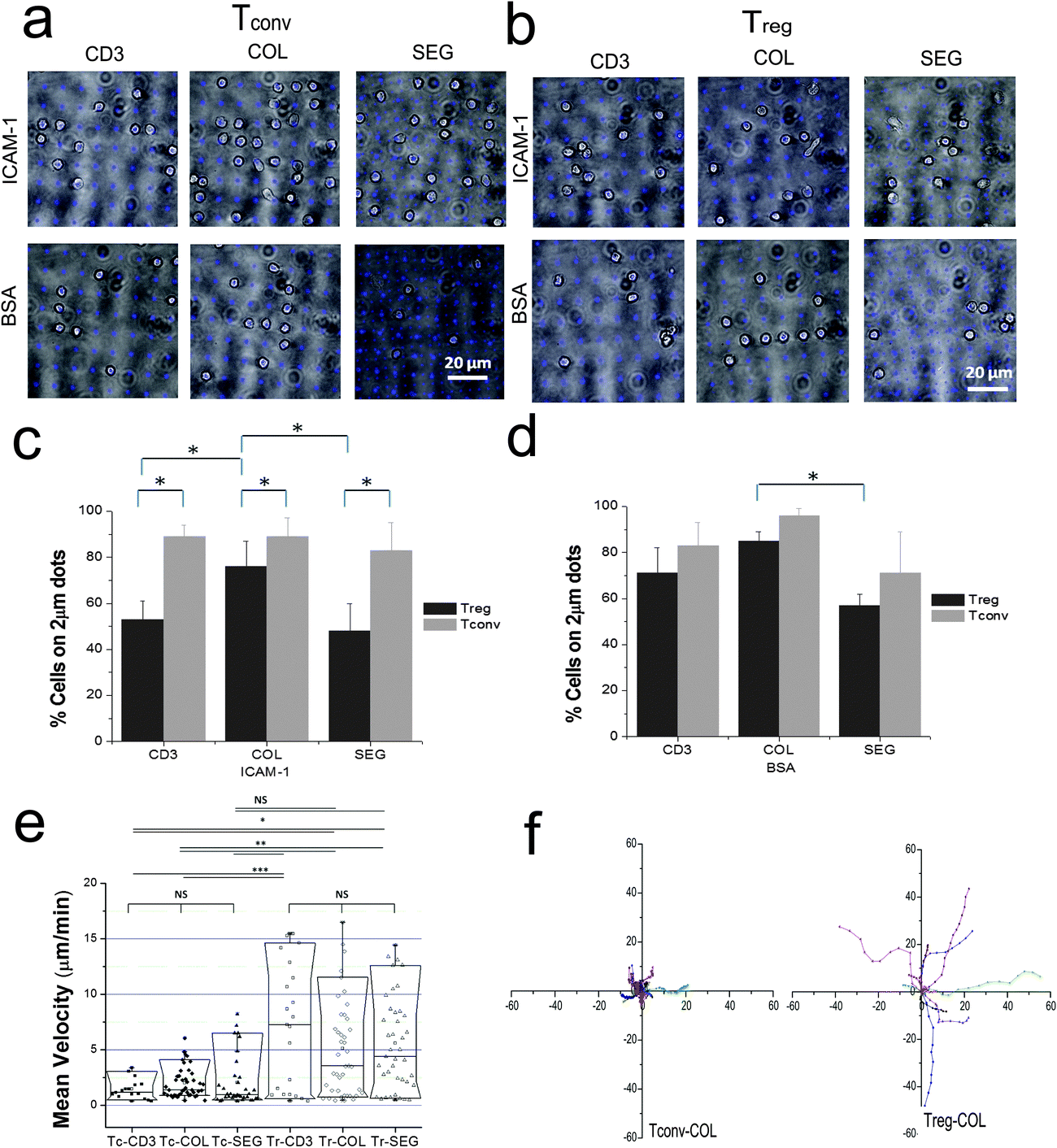 | ||
| Fig. 3 Regulatory T cells exhibit differential adhesion than conventional T cells. (a and b) Overlay of bright field images of Tconv (panel a) and Treg cells (panel b) with patterns of anti-CD3/anti-CD28 patterns (blue). Background areas surrounding and separating these features were backfilled with ICAM-1 or BSA. (c and d) Percentage of cells adhering on 2 μm anti-CD3 dots as a function of pattern and background, specifically ICAM-1 (c) or BSA (d). (e) Mean velocity of cells for 30 min from seeding on patterns. (f) Representative cell-tracking diagrams of Tconvs and Tregs on COL patterns. X and Y axis indicate cell position from origin (μm) during the 5 min. Data in panels c–e are mean ± s.d. from greater than 15 images (each covering a 80 μm × 80 μm area FOV by 100× objective) collected within the observation region from duplicate samples of three experiments. *P < 0.05, **P < 0.01, or ***P < 0.001 by ANOVA followed by Tukey's method. | ||
For observation of protein localization, cells were fixed and permeabilized by using 4% paraformaldehyde and 0.1% Triton X-100 after 30 min incubation on patterns in channels and then stained by using standard immunofluorescence techniques24 by flowing reagents in and washing with PBS through the channels by using syringe pumps at a flow rate of 10 μL min−1. A polyclonal antibody to PKC-θ (sc-212; C-terminus, Santa Cruz Biotechnology) was used in 5% BSA, followed by Alexa Fluor 568 donkey anti-rabbit IgG (Invitrogen A10042). Actin clusters were stained with Texas Red®-X phalloidin (Life Technologies T7471) stock solution in 5% BSA.
Statistics
Data were analyzed by using ANOVA followed by Tukey test. When presented as box plots, the whiskers and box elements correspond to 5, 25, 50, 75, and 95 percentiles. All samples from a single experiment were processed in one session. Experiments were carried out at least three times.Results
Device performance
Using conventional approaches for microscopy preparation, only a small fraction of cells in a starting sample is typically subjected to final analysis. We address this limitation by using a microchannel system to direct the volume of a cell suspension over the microscopy field of view and a diverging magnetic field to further enrich and concentrate paramagnetically-labeled target cells within this region. Following the aforementioned operating mode, the PDMS channel was affixed to a working surface (a coverslip containing a designated area of interest), then flooded with media or buffer. A set of permanent magnets was applied over the collection/observation area (red boxed area downstream of the junction between the main and side streams in Fig. 1a and b). Cells were loaded into the chamber and flowed past the collection area by adding media into the side channel; target cells were therefore trapped in this region while non-target cells were washed out of the chamber. The trapping force produced by the magnetic field must balance the drag force25,26 experienced by cells to achieve successful collection of target cells of high purity within a reasonable amount of time. The magnetic field was then released, which allowed cells to settle onto the observation area of the substrate. The performance of this system was first characterized using samples containing a mix of CFSE-labeled, target CD4+ T cells and CMTPX-labeled splenocytes (at 1, 10 or 25% of initial CD4+ T cells at a total cell concentration of approximately 1 million per mL). Target cells were co-labeled with magnetic beads coated with anti-CD4. A 5 μL volume of this mix was loaded into the chamber, and washed with media at a flow rate of 0.35 μL min−1. The number of target cells and non-target cells in a 800 μm × 800 μm collecting/observation area was counted over the progression of collection/washing (Fig. 2a). Starting with 25% of the target cell population, the number density of target cells per area increased five-fold over the course of collection (Fig. 2a, top panel); meanwhile the density of CMTPX-cells ultimately decreased with washing. The target cells retained in the observation area corresponds to a three-fold increase in cell purity (to 80% target vs. total cells from an initial 25%) and 94% cell collection efficiency (Fig. 2b). The benefit of this collection approach was more evident with lower initial fraction of target cells; at 10% of initial CFSE-cells, the purity of CFSE-labeled target cells increased six-fold to 65%, with an 81% of collection efficiency (Fig. 2a and b). At 1% initial concentration, the magnetic capture system provided a fifty-fold increase in target cell purity, and high collection efficiency close to 100% (Fig. 2a and b). Furthermore, residual non-target cells were found at the corner area of the channels due to the low shear rate in this region (bottom right panel of Fig. 2c); the purity of target cells in the main observation area was close to 100% (top right panel of Fig. 2c). Data in the following sections was collected using higher magnification objectives and were limited to this high-purity area.Regulatory T cells exhibit different responses to micropatterned surfaces than conventional T cells
The magnetic-microfluidic device was then used to investigate spatially resolved signaling in Treg cells. These experiments focused on protein-micropatterned surfaces, which present localized activation signals to cells and allow the study of how cell function changes in response to complex environments.11,12,20 The simple design and assembly of the microchannel allows mounting onto these surfaces while retaining the biological activities of the patterned proteins. These substrates contained arrays of costimulation sites, each consisting of a central, 2 μm diameter circle presenting antibody that engages and activates the epsilon chain of CD3 (denoted here as anti-CD3), a core signaling component of the TCR complex. These are surrounded by the LFA-1 integrin ligand ICAM-1. Superimposed on this basic layout are patterns of antibodies to CD28 (anti-CD28). In one configuration, anti-CD28 is patterned with anti-CD3, presenting a colocalized (COL) feature. In a second configuration, anti-CD28 is patterned as a set of four, 1 μm diameter dots surrounding a central anti-CD3 feature, presenting a segregated (SEG) configuration; the full methods for preparing these surfaces, including steps taken to maintain a consistent per-area concentration of each antibody across the various patterns, are detailed in previous reports.11,12 These studies showed that CD4+ T cells cease migration upon encountering anti-CD3 dots, selectively adhering to and aligning with the costimulation sites. Moreover, activation of these cells is sensitive to the differences in spatial organization of the COL and SEG patterns.11,12The response of regulatory T cells to micropatterned surfaces was analyzed by magnetically labeling CD25+ cells in mixed CD4+ T cell populations, which were then loaded into the microfluidic system. The purity of Tregs isolated in this manner was confirmed by fluorescence imaging of CD25+ cells and was consistently above 90%. It was separately confirmed by FACS that these CD4+CD25+ T cells isolated using magnetically labeled anti-CD25 beads were predominantly CD25hiFoxp3hi (74.1%, while 90% of CD25hi), indicative of regulatory T cells, and are denoted as Tregs here; conversely, in populations of CD4+CD25− T cells (denoted as Tconvs), approximately 0.1% of the cells were CD25hiFoxp3hi.
Captured cells were released onto the surfaces and allowed to interact for 30 minutes. Movie S1 (ESI†) shows how Tconvs recognize patterns and interact with them. Majority of Tconvs found patterns readily and formed nice adhesion on the dots (Fig. 3a and c). Unlike Tconvs, Tregs were found not only on the anti-CD3/anti-CD28 costimulation sites, but also the intervening open regions of ICAM-1 (Fig. 3b, top row). This lower selectivity of attachment to the 2 μm, anti-CD3 sites is expressed quantitatively in Fig. 3c. Intriguingly, Treg adhesion to anti-CD3 was higher for COL patterns than SEG or CD3, indicating that these cells respond to the spatial colocalization of anti-CD28 engagement. The role of ICAM-1 was further tested by replacing this adhesion ligand with BSA (Fig. 3cvs. d) to eliminate nonspecific adhesion of cells to glass surface when ICAM-1 was removed. Tregs showed a higher selectivity to anti-CD3 features on surfaces with a BSA background than on ICAM-1.
The difference in recognition of anti-CD3 by Tconvs and Tregs was also seen in their respective motilities. As shown in Fig. 3e and f, Tconvs were slow and stationary, and stayed on the dots upon recognition while Tregs were mobile and continuously exploring the surfaces. Notably, the mean velocities of the both cell types were not significantly influenced by the presence or relative position of CD28 costimulation. Tconvs showed a significant difference in mean velocity on CD3 or COL patterns when compared to Tregs, but not on SEG patterns. Fig. 3f shows that Tregs moved much farther from they initially land than Tconvs on COL patterns and this trend was not different on other patterns types (data not shown). These results indicate that the lower selectivity of Tregs shown in Fig. 3b and c is not simply due to insufficient time for Tregs to encounter a feature of anti-CD3.
The off-pattern adhesion of Tregs suggests a significant response of these cells to ICAM-1. In support of this, Tregs often formed larger cell–substrate interfaces than Tconv, as measured by interference reflection microscopy (IRM). While the interface of Tconvs with these surfaces demonstrated consistent cell adhesion area (∼3–6 μm in diameter based on IRM images in Fig. 4a), 20–40% of Tregs spread to 7 μm or larger in diameter in IRM images (CD3 only in Fig. 4b and e). When the ICAM-1 background was replaced with BSA, this sub-population with large contact areas was lost (Fig. 4evs. f). In fact, Tregs barely spread on surfaces with a BSA background (Fig. 4d, IRM), supporting the concept that these cells interact significantly with ICAM-1. In contrast, adhesion levels and spreading of Tconvs were only similar or slightly smaller on BSA compared to the ICAM-1 background (e.g., 5 μm and 3 μm on ICAM-1 and BSA surface, respectively, as shown in Fig. 4a and c), implying a lesser role of ICAM-1 in adhesion of Tconv compared to Treg.
 | ||
| Fig. 4 Regulatory T cells form fewer actin clusters than conventional T cells. (a–d) Actin cluster formation in Tconv (panels a and c) and Treg (b and d) cells on anti-CD3, COL, or SEG patterns with ICAM-1 background (a and b) or BSA (c and d) background. Actin is shown in green and patterns are in blue. IRM image (internal reflection microscopy) indicates cell adhesion area. (e and f) Adhesion area of Tconvs and Tregs on different patterns of anti-CD3 and anti-CD28 against an ICAM-1 (e) or BSA (f) background, as measured from IRM images. Cells were classified by diameter, with “Normal” = 3–6 μm, “xs” < 3 μm, and “xL” > 6 μm. Data in panels e and f are from a representative experiment, including >50 cells per condition from three independent experiments. (g and h) Percentage of cells showing actin clusters near the 2 μm anti-CD3 dots of each pattern type with ICAM-1 background (panel g) or BSA background (panel f). Data represent mean ± s.d. Experiment has been repeated at least 3 times. *P < 0.05, ANOVA with multiple comparisons carried out using Tukey's method. | ||
The higher motility and lower selectivity of attachment on micropatterned surfaces suggested that Treg interaction is not strongly dependent on CD3 signaling as previously reported Tregs' lower avidity to anti-CD3.27 In this direction, we examined the distribution of actin cytoskeletal structures at the cell–substrate interface, specifically the presence of actin clusters associated with sites of CD3 engagement which is an indicative of functional activation.28 These dense clusters were evident in Tconv cells against a more diffused cytoskeletal background, as shown in Fig. 4a. Actin clusters were present in roughly 75% of cells on these surfaces, organized along the rim of the 2 μm circular features containing anti-CD3 in all pattern types (Fig. 4a and g). These clusters were also seen in cells on surfaces on which ICAM-1 was replaced with BSA (Fig. 4c and h), indicating that their formation was associated with CD3 signaling and independent of LFA-1 engagement. Tregs, in contrast, showed only occasional actin clusters on the micropatterned surfaces on either ICAM-1 or BSA background (Fig. 4g and h) reflecting a lack of sustained TCR signaling or activation of this pathway.
Microscale organization of PKC-θ responds to costimulation geometry
As a potential mechanism for the differences in cell adhesion and actin cluster formation, we examined the subcellular localization of PKC-θ in response to pattern geometry. PKC-θ plays key roles in T cell adhesion and activation, mediating signaling from both the TCR and LFA-1.29,30 PKC-θ has also been shown to associate with CD28 at cell–cell interfaces.9 We thus sought to examine if the subcellular distribution of PKC-θ in Tregs is sensitive to the presence and microscale organization of CD28 engagement and activation.However, PKC-θ in mouse Tregs interacting with micropatterned anti-CD3, anti-CD28, and ICAM-1 was largely sequestered to the distal pole, away from the cell–substrate interface7,31,32 regardless of pattern type (Fig. 5a, z-stack), in sharp contrast to Tconvs in which PKC-θ was concentrated in the proximal position to this artificial IS (Fig. 5b). The differences by cell and pattern types are quantified in Fig. 5c. The presence or relative position of CD28 engagement had little effect on this proximal or distal localization of this protein in Tconvs or Tregs. Instead, lateral correlation of proximal PKC-θ to anti-CD3 signal as a function of CD28 colocalization to CD3 was analyzed using Tconvs (Fig. 5d), represented by Pearson's correlation coefficient. Unfortunately, localization of PKC-θ away from the proximal cell–substrate interface made quantitative correlation analysis impractical in Tregs.
 | ||
| Fig. 5 PKC-θ is differentially localized in regulatory and conventional T cells. (a and b) Lateral and axial distribution of PKC-θ in Treg (panel a) and Tconv (b) cells as a function of surface micropattern. (c) Ratio of proximal to distal location of PKC-θ in Tconv and Treg cells. (d) Correlation between proximal PKC-θ and the 2 μm anti-CD3 dots in Tconv on each pattern type. PKC-θ (green) was visualized by immunostaining after 30 min of cell–substrate interaction. Micropatterned anti-CD3/anti-CD28 is shown in blue. In panels a and b, xy-plane showed cell–surface interface and yz- and xz-plane showed 3D-reconstruction of z-stack images of PKC-θ axial distribution. For panels c and d, data represent greater than 15 cells of each cell type from duplicate samples, and experiments were repeated three times. *P < 0.01, by ANOVA followed by Tukey's method. | ||
Pearson's correlation coefficient (r) is one of the most standard procedures to correlate fluorescent intensity distribution between channels.33,34 The correlation coefficient was 0.71 for COL, 0.52 for SEG and 0.42 for CD3 patterns, respectively, indicating PKC-θ was strongly correlated with COL patterns but less strongly correlated with CD3 or SEG patterns in Tconvs. This means that PKC-θ in Tconvs was preferentially localized with regions presenting anti-CD3, while the degree of correlation depended on the presence and organization of anti-CD28. This may be explained by that PKC-θ is recruited to IS upon activation and located in the cSMAC/pSMAC junction in CD28 costimulatory-dependent manner.31,35 In addition, the results further implies that PKC-θ localization in IS might be modulated by control of CD28 using patterning of anti-CD28.
Discussion
It is increasingly recognized that the microscale organization of signaling and structural biomolecules is a critical determinant of cell function. A compelling example is found in the immune synapse; the archetypal structure consists of a central supramolecular activation complex (cSMAC) surrounded by a peripheral SMAC (pSMAC) containing TCR-pMHC and LFA-1-ICAM-1 complexes, respectively. Variations on this bullseye pattern have been found in different T cell–APC interactions.36–39 For example, while Th1 cells form the archetypic bullseye pattern, Th2 cells form an interface with multiple TCR-containing foci.38 Multifocal structures are also found in T cell interfaces with dendritic cells.37 An additional level of complexity is found in the organization of CD28.10,36 A rapidly expanding set of microscopy, molecular, and microfabrication tools has emerged to test the role of spatial organization on intracellular signaling and T cell activation. In particular, micropatterned surfaces presenting ligands to these receptors patterned in specific configurations provide direct control over IS geometry, including the production of configurations that reveal key aspects of spatially resolved signaling while not seen in physiological cell–cell interaction.11,12,20,40 Studies from our group have shown that conventional CD4+ T cells respond to the layout of CD3 and CD28 engagement, and identified a role of Lck in spatially coordinating the convergence of signaling between these pathways.11,12This study sought to understand the response of regulatory T cells using micropatterned surfaces controlling the layout of CD3, CD28, and LFA-1 signaling. The role of such interfacial structures is not well understood in Tregs, as these cells suppress the overall immune response through complex actions on Tconvs and APCs, using both cell–cell contact and soluble factors. By improving the efficiency of microscopy-based analysis, we were able to quantitatively examine Treg responses with micropatterned surfaces. Our result of significant off-pattern adhesion and higher motility of Tregs suggest a lower dependency of the stop signal provided by TCR signaling in regulatory compared to conventional T cells, and a stronger role of integrin-mediated, non-antigen-dependent interaction. Similarly, lower avidity of Tregs to anti-CD3 was reported previously due to low level of expression of CD3 and higher activation thresholds in Tregs.27 Furthermore, the observation of higher pattern selectivity on COL patterns compared to CD3 and SEG revealed that functional responses of mouse regulatory T cells are dependent on the relative position of CD3 and CD28 engagement. This is notable as activation of conventional mouse CD4+ T cells, measured with respect to IL-2 secretion, was insensitive to CD3–CD28 colocalization/separation.11,12 However, it is not clear whether this ability to read the spatial organization of these signals, an ability shared with human Tconvs,12 is related to the differences in Treg and Tconv physiology. Another possibility perhaps is that the effect of COL and SEG patterns on adhesion of Tconvs is overshadowed due to the effectiveness of anti-CD3 signal in halting Tconv migration. It is also recognized that a potential artifact of using beads to collect cells (i.e., positive selection) is cell activation prior to interaction with the surface. However, positively selected Tconvs showed selective adhesion to the anti-CD3 dots comparable to those prepared by negative selection (data not shown).
Subcellular localization of PKC-θ away from the cell–substrate interface31,32 provides an intriguing explanation for the lower adhesion selectivity and actin cluster formation observed in Tregs compared to Tconvs. Specifically, PKC-θ plays a central role in signaling downstream of TCR, including induction of F-actin polymerization through the WASP (Wiskott-Aldrich Syndrome protein) pathway;41,42 knockdown of PKC-θ produces hyperstable IS structures in Tconv cells.30,43 Our observation of lower actin cluster formation near sites of CD3 engagement in Tregs is consistent with a role of PKC-θ inducing actin dynamics, synapse breaking, and kinapse formation.21 It has also been reported that Tregs express higher LFA-1 than Tconvs,1,2,32 as an alternative or complementary mechanism for the lower specificity of adhesion to anti-CD3 features. While LFA-1 is functionally important for Tregs, the pathways connecting TCR and LFA-1 may be different.44,45 Until the immediate mechanisms for distal pole sequestration are identified and manipulated, establishing this role of PKC-θ will remain incomplete.
While the potential role that the relative position of signaling complexes may have on overall cell function is an area of increasing interest, general principles for describing and understanding this effect have been only at the early stages of development. Our results surprisingly showed that the differences in cell adhesion to anti-CD3 features, which is a high-level function, were not as apparent in earlier measures, namely PKC-θ localization, actin cluster formation, and cell motility. These are important in that they suggest that differences in cell signaling networks begin to manifest between these early readouts and higher functions of cells.
In this regard, the question we want to answer in the immediate future is if relocalization of PKC-θ from distal to proximal position in Tregs (e.g., by TNF-α as shown in Zanin-Zhorov7) would upregulate actin cluster formation and induce cytokine secretion depending on costimulation by CD28. Unfortunately, the platform we introduce is not well-suited for measuring longer-term functions, which are governed in part by cell–cell communication through soluble factors; concentrating cells within the field of view for microscopy consequently produces adjacent areas with few cells, which complicates analysis of longer-term cell function. We are developing a next-generation platform to examine such questions. Secondly, we speculate that Tregs might need stronger signals than Tconvs to be activated.27 We therefore intend to pursue developing surface patterns with higher concentration of antibodies.
Finally, we propose that this magnetic-microfluidic system will facilitate studies of T cell subclasses that are of less abundance than regulatory T cells. We note that purity of bead-labeled, target cells was higher in the middle of the microfluidic channel than near the edges, approaching 99% for regulatory T cells. Successful capture of rare cells was dependent on establishing a balance between trapping force supplied by a divergent magnetic field and the drag force (Fd) produced by microfluidic flow,25,26 given
| Fd = πηrν, | (1) |
Conclusions
Owing to the role of specific cell subtypes in adaptive immunity, it has been of particular importance to study rare populations such as CD4+CD25+ regulatory T cells (Tregs). We have developed a magnetic microfluidic sorting platform aimed at enhancing the efficacy of microscopy-based analysis of these rare cells on biologically active substrates. We have achieved 50-fold increase in target-to-background cell purity with 80% collection efficiency from 1% initial target cell composition. Through this platform, we were able to successfully isolate Tregs from mouse splenic CD4+ T cells and compare and contrast their behaviors on micropatterned anti-CD3 and costimulating anti-CD28 antibodies that are colocalized or segregated to each other. Intriguingly, Tregs were less responsive to anti-CD3 patterns than CD4+CD25− Tconvs and showed only occasional actin clusters compared Tconvs; both of which reflect a lack of sustained TCR signaling or activation of TCR-LFA-1 pathway. We confirmed that protein kinase C-θ (PKC-θ) localized preferentially to the distal position of the IS in Tregs while being highly localized to the proximal position of the IS in Tconvs. A plausible explanation might be that distal localization of PKC-θ in Tregs may inhibit actin polymerization through the WASP pathway; however, this cannot be fully resolved until an immediate mechanism for distal pole localization of PKC-θ is identified and controlled. By improving the efficiency of microscopy-based cell analysis, the platform presented here will facilitate studies of rare T cell subtypes and other difficult-to-obtain cells.Acknowledgements
This work was supported in part by the National Institutes of Health – R01AI088377, U24AI118669 (L.C.K.), PN2EY016586 (L.C.K. and M.L.D), and F32AI100496 (J.H.L.). M.L.D. is a Principal Research Fellow of the Wellcome Trust and Senior Research Fellow of the Kennedy Trust.References
- S. Sakaguchi, T. Yamaguchi, T. Nomura and M. Ono, Regulatory T Cells and Immune Tolerance, Cell, 2008, 133, 775–787 CrossRef CAS PubMed.
- S. Z. Josefowicz, L.-F. Lu and A. Y. Rudensky, Regulatory T Cells: Mechanisms of Differentiation and Function, Annu. Rev. Immunol., 2014, 30, 531–564 CrossRef PubMed.
- E. M. Shevach, Mechanisms of Foxp3+ T Regulatory Cell-Mediated Suppression, Immunity, 2009, 30, 636–645 CrossRef CAS PubMed.
- C. R. F. Monks, B. A. Freiberg, H. Kupfer, N. Sciaky and A. Kupfer, Three-dimensional segregation of supramolecular activation clusters in T cells, Nature, 1998, 395, 82–86 CrossRef CAS PubMed.
- A. Grakoui, S. K. Bromley, C. Sumen, M. M. Davis, A. S. Shaw, P. M. Allen and M. L. Dustin, The immunological synapse: A molecular machine controlling T cell activation, Science, 1999, 285, 221–227 CrossRef CAS.
- M. L. Dustin, S. Y. Tseng, R. Varma and G. Campi, T cell-dendritic cell immunological synapses, Curr. Opin. Immunol., 2006, 18, 512–516 CrossRef CAS PubMed.
- A. Zanin-Zhorov, Y. Ding, S. Kumari, M. Attur, K. L. Hippen, M. Brown, B. R. Blazar, S. B. Abramson, J. J. Lafaille and M. L. Dustin, Protein kinase C-θ mediates negative feedback on regulatory T cell function, Science, 2010, 328, 372–376 CrossRef CAS PubMed.
- K. T. Roybal and C. Wulfing, Inhibiting the inhibitor of the inhibitor: blocking PKC-θ to enhance regulatory T cell function, Sci. Signaling, 2010, 3, pe24 CrossRef PubMed.
- S.-Y. Tseng, M. Liu and M. L. Dustin, CD80 cytoplasmic domain controls localization of CD28, CTLA-4 and PKC-θ in the immunological synapse, J. Immunol., 2005, 175, 7829–7836 CrossRef CAS.
- S. Y. Tseng, J. C. Waite, M. Liu, S. Vardhana and M. L. Dustin, T cell-dendritic cell immunological synapses contain TCR-dependent CD28-CD80 clusters that recruit protein kinase C-θ, J. Immunol., 2008, 181, 4852–4863 CrossRef CAS.
- K. Shen, V. K. Thomas, M. L. Dustin and L. C. Kam, Micropatterning of costimulatory ligands enhances CD4+ T cell function, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7791–7796 CrossRef CAS PubMed.
- K. T. Bashour, J. Tsai, K. Shen, J.-H. Lee, E. Sun, M. C. Milone, M. L. Dustin and L. C. Kam, Cross Talk between CD3 and CD28 Is Spatially Modulated by Protein Lateral Mobility, Mol. Cell. Biol., 2014, 34, 955–964 CrossRef PubMed.
- K. D. Mossman, G. Campi, J. T. Groves and M. L. Dustin, Altered TCR Signaling from Geometrically Repatterned Immunological Synapses, Science, 2005, 310, 1191–1193 CrossRef CAS PubMed.
- L. S. L. Cheung, X. Zheng, A. Stopa, J. C. Baygents, R. Guzman, J. A. Schroeder, R. L. Heimark and Y. Zohar, Detachment of captured cancer cells under flow acceleration in a bio-functionalized microchannel, Lab Chip, 2009, 9, 1721–1731 RSC.
- S. Nagrath, L. V. Sequist, S. Maheswaran, D. W. Bell, D. Irimia, L. Ulkus, M. R. Smith, E. L. Kwak, S. Digumarthy, A. Muzikansky, P. Ryan, U. J. Balis, R. G. Tompkins, D. A. Haber and M. Toner, Isolation of rare circulating tumour cells in cancer patients by microchip technology, Nature, 2007, 450, 1235–1239 CrossRef CAS PubMed.
- Y.-T. Lu, L. Zhao, Q. Shen, M. A. Garcia, D. Wu, S. Hou, M. Song, X. Xu, W.-H. OuYang, W. W. L. OuYang, J. Lichterman, Z. Luo, X. Xuan, J. Huang, L. W. K. Chung, M. Rettig, H.-R. Tseng, C. Shao and E. M. Posadas, NanoVelcro Chip for CTC enumeration in prostate cancer patients, Methods, 2013, 64, 144–152 CrossRef CAS PubMed.
- G. Shi, W. Cui, M. Benchimol, Y.-T. Liu, R. F. Mattrey, R. Mukthavaram, S. Kesari, S. C. Esener and D. Simberg, Isolation of Rare Tumor Cells from Blood Cells with Buoyant Immuno-Microbubbles, PLoS One, 2013, 8, e58017 CAS.
- W. Sun, C. Jia, T. Huang, W. Sheng, G. Li, H. Zhang, F. Jing, Q. Jin, J. Zhao, G. Li and Z. Zhang, High-Performance Size-Based Microdevice for the Detection Of Circulating Tumor Cells from Peripheral Blood in Rectal Cancer Patients, PLoS One, 2013, 8, e75865 CAS.
- J. H. Kang, S. Krause, H. Tobin, A. Mammoto, M. Kanapathipillai and D. E. Ingber, A combined micromagnetic-microfluidic device for rapid capture and culture of rare circulating tumor cells, Lab Chip, 2012, 12, 2175–2181 RSC.
- J. Doh and D. J. Irvine, Immunological synapse arrays: Patterned protein surfaces that modulate immunological synapse structure formation in T cells, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5700–5705 CrossRef CAS PubMed.
- M. L. Dustin, Cell adhesion molecules and actin cytoskeleton at immune synapses and kinapses, Curr. Opin. Cell Biol., 2007, 19, 529–533 CrossRef CAS PubMed.
- J. H. Lee, H. Wang, J. B. Kaplan and W. Y. Lee, Effects of Staphylococcus epidermidis on Osteoblast Adhesion and Viability on Ti Alloy Surface in Microfluidic Co-Culture Environment, Acta Biomater., 2010, 6, 4422–4429 CrossRef CAS PubMed.
- J. H. Lee, Y. Gu, H. Wang and W. Y. Lee, Microfluidic 3D bone tissue model for high-throughput evaluation of wound-healing and infection-preventing biomaterials, Biomaterials, 2012, 33, 999–1006 CrossRef CAS PubMed.
- E. Judokusumo, E. Tabdanov, S. Kumari, M. L. Dustin and L. C. Kam, Mechanosensing in T Lymphocyte Activation, Biophys. J., 2012, 102, L5–L7 CrossRef CAS PubMed.
- M. Tanase, N. Biais and M. Sheetz, Magnetic Tweezers in Cell Biology, Methods Cell Biol., 2007, 83, 473–493 CAS.
- H. Huang, C. Y. Dong, H. S. Kwon, J. D. Sutin, R. D. Kamm and P. T. C. So, Three-dimensional cellular deformation analysis with a two-photon magnetic manipulator workstation, Biophys. J., 2002, 82, 2211–2223 CrossRef CAS.
- J. M. Rojo, G. Ojeda, Y. Y. Acosta, M. Montes-Casado, G. Criado and P. Portoles, Characteristics of TCR/CD3 complex CD3{varepsilon} chains of regulatory CD4+ T (Treg) lymphocytes: role in Treg differentiation in vitro and impact on Tregin vivo, J. Leukocyte Biol., 2014, 95, 441–450 CrossRef PubMed.
- S. Kumari, D. Depoil, R. Martinelli, E. Judokusumo, G. Carmona, F. B. Gertler, L. C. Kam, C. V. Carman, J. K. Burkhardt, D. J. Irvine and M. L. Dustin, Actin foci facilitate activation of the phospholipase C-γ in primary T lymphocytes via the WASP pathway, eLife, 2015, e04953 Search PubMed.
- T. Letschka, V. Kollmann, C. Pfeifhofer-Obermair, C. Lutz-Nicoladoni, G. J. Obermair, F. Fresser, M. Leitges, N. Hermann-Kleiter, S. Kaminski and G. Baiser, PKC-θ selectively controls the adhesion-stimulating molecule Rap1, Immunobiology, 2008, 112, 4617–4627 CAS.
- T. N. Sims, T. J. Soos, H. S. Xenias, B. Dubin-Thaler, J. M. Hofman, J. C. Waite, T. O. Cameron, V. K. Thomas, R. Varma, C. H. Wiggins, M. P. Sheetz, D. R. Littman and M. L. Dustin, Opposing effects of PKC-θ and WASp on symmetry breaking and relocation of the immunological synapse, Cell, 2007, 129, 773–785 CrossRef CAS PubMed.
- A. Zanin-Zhorov, M. L. Dustin and B. R. Blazar, PKC-θ function at the immunological synapse: prospects for therapeutic targeting, Trends Immunol., 2011, 32, 358–363 CrossRef CAS PubMed.
- K.-F. Kong and A. Altman, In and out of the bull's eye: protein kinase Cs in the immunological synapse, Trends Immunol., 2013, 34, 234–242 CrossRef CAS PubMed.
- E. M. M. Manders, F. J. Verbeek and J. A. Aten, Measurement of co-localization of objects in dual-colour confocal images, J. Microsc., 1993, 169, 375–382 CrossRef PubMed.
- R. Taylor, Interpretation of the Correlation Coefficient: A Basic Review, J. Diagn. Med. Sonogr., 1990, 6, 35–39 CrossRef PubMed.
- T. Yokosuka and T. Saito, Dynamic regulation of T-cell costimulation through TCR-CD28 microclusters, Immunol. Rev., 2009, 229, 27–40 CrossRef CAS PubMed.
- S.-Y. Tseng and M. L. Dustin, T-cell activation: a multidimensional signaling network, Curr. Opin. Cell Biol., 2002, 14, 575–580 CrossRef CAS.
- C. Brossard, V. Feuillet, A. Schmitt, C. Randriamampita, M. Romao, G. Raposo and A. Trautmann, Multifocal structure of the T cell – Dendritic cell synapse, Eur. J. Immunol., 2005, 35, 1741–1753 CrossRef CAS PubMed.
- T. J. Thauland, Y. Koguchi, S. A. Wetzel, M. L. Dustin and D. C. Parker, Th1 and Th2 Cells Form Morphologically Distinct Immunological Synapses, J. Immunol., 2008, 181, 393–399 CrossRef CAS.
- T. J. Thauland and D. C. Parker, Diversity in immunological synapse structure, Immunology, 2010, 131, 466–472 CrossRef CAS PubMed.
- D. J. Irvine and J. Doh, Synthetic surfaces as artificial antigen presenting cells in the study of T cell receptor triggering and immunological synapse formation, Semin. Immunol., 2007, 19, 245–254 CrossRef CAS PubMed.
- Y. Sasahara, R. Rachid, M. J. Byrne, M. A. de la Fuente, R. T. Abraham, N. Ramesh and R. S. Geha, Mechanism of recruitment of WASP to the immunological synapse and of its activation following TCR ligation, Mol. Cell, 2002, 10, 1269–1281 CrossRef CAS.
- M. Thome, The Immunological Synapse and Actin Assembly: A Regulatory Role for PKCθ, Dev. Cell, 2003, 4, 3–5 CrossRef CAS.
- M. F. Krummel, Immunological Synapses: Breaking Up May Be Good to Do, Cell, 2007, 129, 653–655 CrossRef CAS PubMed.
- D. Q. Tran, D. D. Glass, G. Uzel, D. A. Darnell, C. Spalding, S. M. Holland and E. M. Shevach, Analysis of adhesion molecules, target cells, and role of IL-2 in human FOXP3+ regulatory T cell suppressor function, J. Immunol., 2009, 182, au2929–au2938 CrossRef PubMed.
- B. B. Au-Yeung, S. E. Levin, C. Zhang, L.-Y. Hsu, D. A. Cheng, N. Killeen, K. M. Shokat and A. Weiss, A genetically selective inhibitor demonstrates a function for the kinase Zap70 in regulatory T cells independent of its catalytic activity, Nat. Immunol., 2010, 11, 1085–1093 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ib00215j |
| This journal is © The Royal Society of Chemistry 2015 |